

Abstract
In metastatic melanoma, with a dismal survival rate and propensity for treatment resistance and recurrence, it is critical to establish biomarkers that better predict treatment response and disease severity. The melanoma glycome, composed of complex carbohydrates termed glycans, is an under investigated area of research though is gaining momentum in the cancer biomarker and therapeutics field. Novel findings suggest that glycans play a major role in influencing melanoma progression and could be exploited for prognosticating metastatic activity and/or as therapeutic targets. In this review, we discuss the role of aberrant glycosylation, particularly the specialized function of β1,6 N-acetylglucosaminyltransferase 2 (GCNT2), in melanoma pathogenesis and summarize mechanisms of GCNT2 regulation to illuminate its potential as a predictive marker and therapeutic target.
A. Introduction
Metastatic melanoma has a poor 5-year survival rate of 22.5%1. While immunotherapy, particularly anti-CTLA4 and anti-PD1/PD-L1, has significantly improved survival, relapse and therapy resistance endure. There is thus a pressing need to elucidate novel therapeutic targets as well as biomarkers to help predict therapy resistance and improve treatment outcomes. Even in patients experiencing clinical remission, relapse often occurs due to the presence of extra-cutaneous micrometastases that characteristically exhibit immunoevasion and encourage disease progression2, 3. There have been a variety of melanocytic-specific markers that positively identify melanoma cells, however, there is still a lack of adequate biomarkers to track and predict aggressive metastatic development, including those for treatment-resistant, immunoevasive cell subpopulations.
It is increasingly recognized that aberrant cell surface glycosylation is a hallmark of cancer, including skin cancers4–8. Melanoma-specific glycosylation, in particular, has not been thoroughly exploited for the identification of glycan biomarkers or factors that impact metastasis. In melanomas, recent efforts have focused on key alterations in major glycosyltransferases, namely the I-antigen-forming β1,6 N-acetylglucosaminyltransferase 2, GCNT29, 10. Notably, GCNT2 appears to have a unique role in the suppression of melanoma growth, which is paradoxical to its tumor-promoting activity in other malignancies11. The loss of GCNT2 expression and corresponding loss of I-antigen in melanoma cells has profound effects on key oncogenic signaling pathways, including integrin-mediated signaling pathways critical for metastasis10. Interestingly, GCNT2 has also been found to be a direct target of microphthalmia transcription factor (MITF) transcription3, suggesting GCNT2 may be a melanocytic-lineage determinant related to MITF levels and melanoma metastasis. It is therefore of therapeutic importance to clarify the consequences of GCNT2 loss and its mode of regulation in metastatic melanoma. Here, we dissect the role of glycosylation in melanoma development and focus on GCNT2 as well as the intricacies of GCNT2 regulation to highlight its promise as a prognostic marker and therapeutic target.
B. Glycosylation signature in melanomas and loss of I-antigen
Glycosylation, one of the most common post-translational modifications, involves the sequential addition of monosaccharides to form complex carbohydrates known as glycans. These glycan components on cellular proteins are critical for protein function and localization12–15. Cell surface protein glycosylation occurs in the cell’s secretory pathway, comprised of the endoplasmic reticulum (ER) and Golgi apparatus12, 15. . Glycosylation is critical to physiologic processes, including embryogenesis, fertilization, and metabolism. Alternatively, dysregulation or aberrant glycosylation has been linked to multiple pathologies, such as autoimmunity and cancer. There are two major types of protein glycosylation: 1.) Asparagine (N)-linked glycosylation, which transfers glycans to asparagine residues with a peptide consensus sequence of N – X – Serine/Threonine; and 2.) Serine/threonine (O)-linked glycosylation, the attachment of glycans to serine and threonine residues12, 15–17. Nearly 200 known human glycosyltransferases, responsible for the addition of specific glycans to proteins, and glycosidases, responsible for the trimming of glycans, work in concert within the ER and Golgi apparatus to synthesize and process the glycans to their final structure12, 18. These enzymes are highly specific to their substrates and corresponding linkages of sugar moieties that give rise to a glycan19. For example, of the various sialyltransferases, β-galactoside α−2,6-sialyltransferase 1 and 2 (ST6Gal-1 and −2) add a sialic acid to the carbon-6 position of galactose5, 20; similarly, among fucosyltransferases, some target carbons on galactose while others catalyze the transfer of fucose to N-acetylglucosamine21. Regarding I-antigen formation and topic of this review, the GCNT2 enzyme catalyzes the transfer of N-acetylglucosamine to the carbon-6 position of galactose11. While the above enzymes are just a few examples of the vast array of known glycosyltransferases, further details on glycan biosynthesis and structure have been reviewed extensively elsewhere19, 22, 23.
In melanoma, altered glycosylation has been documented to have a profound effect on metastatic processes24–27. For example, metastatic melanomas feature elevated levels of mannosyl-ß1,6 N-acetylglucosaminyltransferase 5 (MGAT5) and corresponding β1–6 N-glycan branching product on the adhesion receptor αVβ3 integrin, which enhances tumor angiogenesis and metastasis24, 25. Moreover, in murine melanoma cell lines, highly metastatic subclones express increased cell surface α2–6 sialylation, while poorly metastasizing subclones display increased α1–3 fucosylation26, 27. Additionally, analysis of melanoma patient samples implicates α1,6 fucosyltransferase FUT8 as a driver of metastasis9. More recently, our laboratory has established that expression of GCNT2, which is known for enzymatic conversion of linear i-antigens on neonatal erythrocytes into the adult I blood group antigen28–31, is progressively lost in metastatic melanomas, resulting aggressive tumor growth10. Mutations in GCNT2 have previously been linked to the persistence of i-antigen in adult erythrocytes and a cause of congenital cataracts with an autosomal recessive pattern of inheritance. The effects of GCNT2 and its mutant forms in cancer, including melanomas, however, remain unclear. Through targeting N-glycans, O-glycans, and glycolipids, GCNT2 has been shown to both activate and suppress cancer progression, functioning as an oncogene in breast, colon, esophageal, and prostate cancer, while acting as a tumor suppressor in melanoma cells10, 11, 32, 33. This dichotomy may be explained by cell type-specific regulation of GCNT2 expression at the epigenetic, transcriptional, post-transcriptional, or post-translational levels.
C. GCNT2 as a tumor-associated glycosyltransferase
Genetic alterations in the GCNT2 locus, classically associated with dysfunctional expression of branched I-antigen and reversal to linear i-antigen in adult erythrocytes29, have pathological significance in which autosomal recessive defects result in congenital cataracts in select populations28, 30, 31. In addition, alterations in GCNT2 expression have now been associated with multiple malignancies, with high GCNT2 promoting tumor progression in a majority of investigated cancers (Figure 1A). Studies of esophageal squamous cell carcinoma (ESCC), prostate cancer, metastatic breast cancer, and colon cancer demonstrate that GCNT2 expression is associated with an invasive, metastatic phenotype, wherein overexpression of GCNT2 promotes tumor progression via enhanced cell detachment, migration, proliferation, and invasion13, 14, 32, 34. Mechanistically, GCNT2 appears to be involved in the epithelial-mesenchymal transition (EMT), a critical step in tumor invasion, metastasis, treatment resistance, and recurrence. GCNT2 overexpression in mammary epithelial cells and ESCC cell lines results in downregulation of epithelial markers, including E-cadherin, and upregulation of mesenchymal markers, including vimentin, fibronectin, and/or N-cadherin13, 14. Further, induction of EMT via treatment with transforming growth factor-beta 1 (TGF-β1) in mammary glands or epidermal growth factor (EGF)/basic fibroblast growth factor (bFGF) in colon cancer cells leads to upregulation of GCNT2, confirming the association between GCNT2 and EMT driven by cancer type-specific pathways13, 34. Interestingly, further studies of colorectal cancer (CRC) show suppressed GCNT2 levels due to promoter hypermethylation in primary CRC tissue compared to normal mucosa, suggesting a tumor suppressive role of GCNT233. However, as CRC progresses to an advanced stage, the GCNT2 promoter is hypomethylated, resulting in elevated GCNT2 expression that positively correlates with stage, metastasis, and depth of invasion33. In concordance with its putative pro-invasive and pro-metastatic potential, GCNT2 expression is proposed as an inverse correlate marker in prostate cancer and ESCC, where patients with high GCNT2 expression have significantly lower survival rates than those with low GCNT2 expression14, 32. Data from studies on endometrial carcinoma (EC), however, demonstrate that while GCNT2 is positively correlated with the expression of key signaling molecules vital for EC development, high GCNT2 expression in these tissues significantly increased survival rates compared to low expression35. Additionally, it is important to note that some tumor types are characterized instead by low GCNT2 expression. For example, GCNT2 downregulation has been demonstrated in diffuse and intestinal-type gastric carcinoma compared to normal tissue36, and low GCNT2 expression has also been shown to be relevant for disease progression in cutaneous T-cell lymphoma and adult T-cell lymphoma/leukemia37. In fact, the most extensive evidence of GCNT2 acting as a tumor suppressor rather than an oncogenic driver has been provided by metastatic melanoma research, wherein loss of GCNT2 enhances tumor progression10.
Figure 1. Role of GCNT2 in cancer progression.
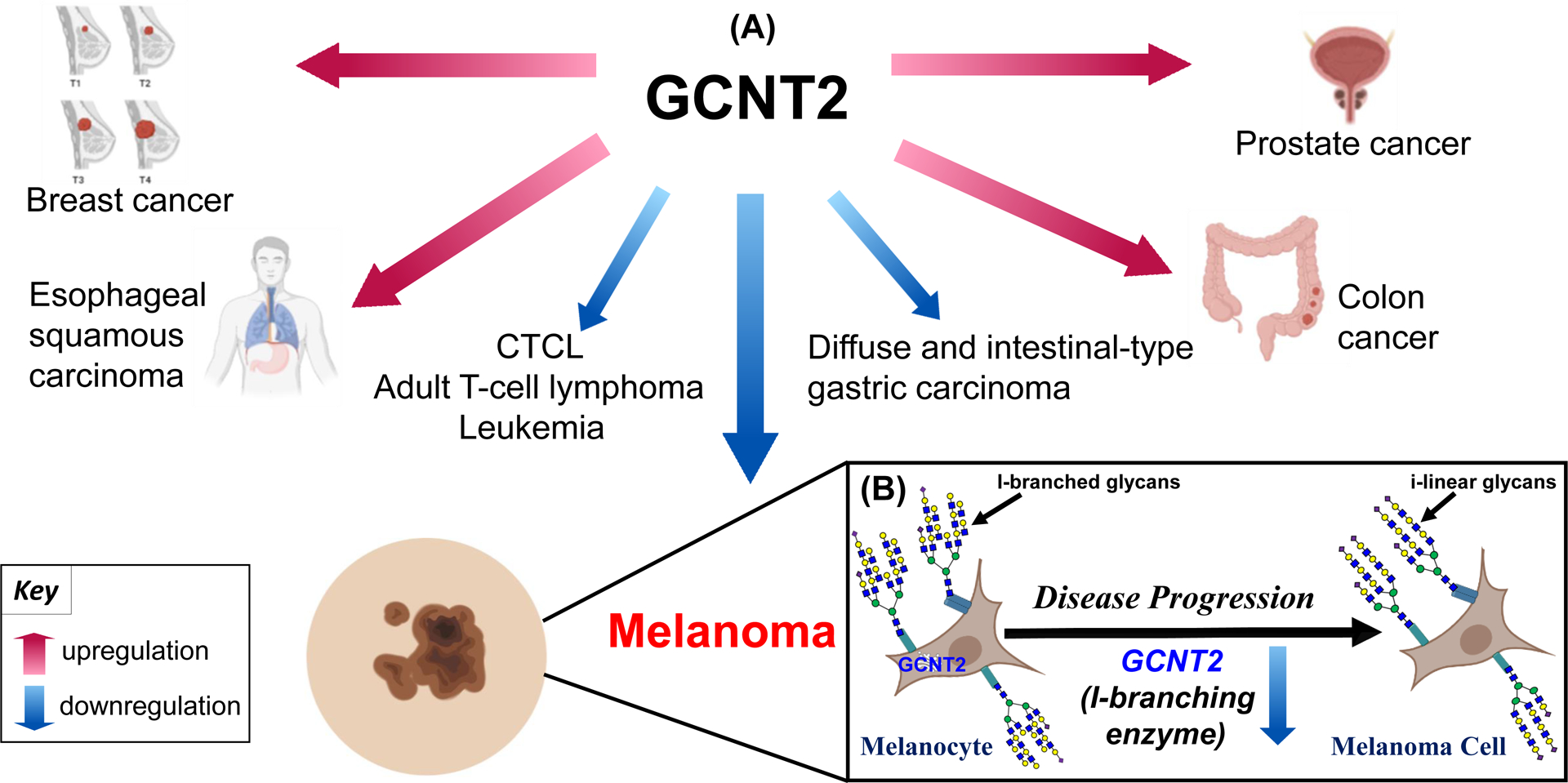
Dysregulated GCNT2 expression in various cancers (A). Metastatic melanoma displays a unique progressive loss of GCNT2 and GCNT2-synthesized I-branched glycans that correlates with disease progression (B). Created with BioRender.com and Reactome64.
Compared to normal melanocytes, primary and metastatic melanoma cells display a progressive loss of GCNT2 and GCNT2-synthesized I-antigen glycans that in turn leads to increased cell growth, proliferation, and survival through integrin and insulin-like growth factor 1 receptor (IGF1R)-mediated pathways10 (Figure 1B). Alternatively, enforced overexpression of GCNT2 in metastatic melanoma cell lines leads to a significant decrease in tumor growth, thereby establishing the potent role of GCNT2 in hindering tumor formation10. In addition, GCNT2-synthesized I-antigen glycans, otherwise referred to as I-branches, also decrease binding of melanoma cells to galectin-3 (Gal-3), a key pro-melanoma molecule that enhances cell proliferation and migration along with suppressing T-cell immune surveillance within the tumor microenvironment10, 38. The identification of GCNT2/I-branch-dependent alterations in major pro-neoplastic signaling cascades as well as in pro-melanoma Gal-3/glycan interactions implicates GCNT2 regulation as a critical determinant of melanoma progression. Further investigation will invariably illuminate the complexities of GCNT2/I-branching-dependent signaling alteration in melanoma progression and support the clinical development of promising new therapies. For example, clinical trials are already underway to test Gal-3 inhibitors as an adjunct to immune checkpoint inhibitors (ICI) such as ipilimumab39, where it is thought that hindering the pro-melanoma and immunosuppressive effects of Gal-3 may increase patient response to ICI therapy38, 40. However, further stratifying patients by high or low GCNT2/I-branching may provide a more personalized therapeutic approach, where patients with aggressive melanomas characterized by low GCNT2 and increased linear i-antigen/Gal-3 binding may see a greater benefit.
With melanoma among few exceptions where GCNT2 expression is associated with hindered tumor growth, it is important to elucidate the cause of this aberration. While GCNT2 acts to promote tumor growth in most neoplasms of epithelial origin, key differences in melanocytes compared to epithelial cells may explain the behavior of GCNT2 as a tumor suppressor in melanoma. Derived from neural crest cells, melanocytes are dendritic in shape and function mainly to produce and distribute melanin pigment. Melanocytes are characterized by the expression of specific proteins, including tyrosinase, tyrosinase-related proteins-1 and −2, melanosomal matrix proteins (Pmel17, MART-1), and MITF41, that are critical for proper differentiation and maturation. Tyrosinase, for example, is an essential regulator of melanogenesis, and MITF is a key melanocyte transcription factor involved in multiple processes, including cell proliferation, formation of dendrites, and anti-apoptotic gene expression41. Importantly, mentioned in detail later in the review, MITF also directly transcribes GCNT2. As differentiation of melanocytes generally leads to loss of their proliferative potential41, exploring mechanisms of melanocyte proliferation control is key to understanding neoplastic progression to melanoma. In fact, melanocyte proliferation and differentiation control is a complex and incompletely understood process that is heavily influenced by several components, including endogenous growth factors, hormones, and fatty acids secreted by surrounding keratinocytes/fibroblasts as well as distant organs and environmental factors such as ultraviolet and ionizing radiation41, 42. Considering their specialized development and adaptations to respond to precise but numerous stimuli, it is apparent that melanocytes have distinct interactions with their microenvironment that lead to alterations in gene regulation, hereby possibly explaining the tumor suppressive role of GCNT2 in melanoma. Given the varying and tumor-specific roles of GCNT2, it is evident that the regulation of this gene is both nuanced and cell type-specific, thus meriting further investigation.
D. The GCNT2 gene
GCNT2 has been referenced in the literature under many aliases, including IGNT, ULG3, II, NACGT1, NAGCT1, GCNT5, CCAT, CTRCT13, bA360O19.2, and bA421M1.1 (NCBI Gene ID: 2651)43. While GCNT2C is occasionally listed as a synonym, it is more appropriately used to reference isoform C, discussed below. The GCNT2 gene is located on the short arm of chromosome 6 between positions 24.3 and 24.2, base pairs 10,521,282 to 10,629,367 (OMIM: *600429)44. Different transcript variants have been demonstrated to encode three tissue-specific messenger RNA (mRNA) isoforms: GCNT2A, GCNT2B, and GCNT2C. Significant expression of GCNT2A has been reported in prostate, cerebellum, and fetal brain, though complete absence of GCNT2A in brain tissue has also been documented29, 45. On the other hand, GCNT2B appears to be expressed in all tissues, and it is the only isoform present in the epithelial cells of the lens30, 45, 46. Expression profiles of GCNT2C vary but generally show consistently high expression in the heart and bone marrow and varying expression in the brain, prostate, stomach, and small intestine29, 45. Notably, it is responsible for I-antigen expression on adult erythrocytes and is the only isoform present in reticulocytes29, 45, 46. Differential expression of GCNT2 isoforms is also observed in cancer. GCNT2A is the major isoform expressed in prostate cancer and metastatic breast cancer cell lines13, 32, while in primary CRC, all three isoforms are present, though at a lower level compared to normal mucosa33. Induction of EMT in CRC, however, is accompanied by a dramatic increase in GCNT2 isoforms, particularly isoform A34. Finally, in metastatic melanoma cells, GCNT2 expression is generally lost10, but whether a specific isoform is characteristically lost remains unknown.
E. Mechanisms of GCNT2 expression control
GCNT2 methylation.
In accordance with the canonical role of GCNT2C in erythroid cell development and the switch from fetal i-linear to adult I-branched glycans, fetal erythroblasts harbor hypermethylation of the GCNT2C core promoter that eventually transitions to significant hypomethylation in adult erythroblasts47. Pertaining to neoplasia, GCNT2 hypomethylation has also been linked to tumor development in aldosterone-producing adenomas (APAs)48 as well as UVB-induced mouse skin papillomas and carcinomas49. This hypomethylation and consequential increased expression of GCNT2 associated with primary tumor formation is consistent with the previously discussed oncogenic potential of GCNT2 in several malignancies; however, the association between GCNT2 methylation and cancer development appears to depend on cancer stage. Contrary to previous findings in skin carcinomas and APAs, primary CRC tissue shows decreased expression of all three GCNT2 variants compared to normal mucosa, a finding which is attributed to hypermethylation of the GCNT2B variant in particular33, 50. Alternatively, examination of tissue from more advanced CRC as well as the corresponding normal mucosa shows hypomethylation rather than hypermethylation of GCNT2B in both samples, with the methylation levels in normal tissues assumed to reflect that of the tumor tissues in these cases (Figure 2A and B)33. Low levels of methylation were most associated with lymph node metastasis and stage but also showed correlation with depth of invasion and distant metastasis, demonstrating that methylation status and thereby expression level of GCNT2 can serve as a prognostic marker in CRC, where GCNT2 promoter hypomethylation predicts increased metastasis33.
Figure 2. Known mechanisms of GCNT2 regulation.
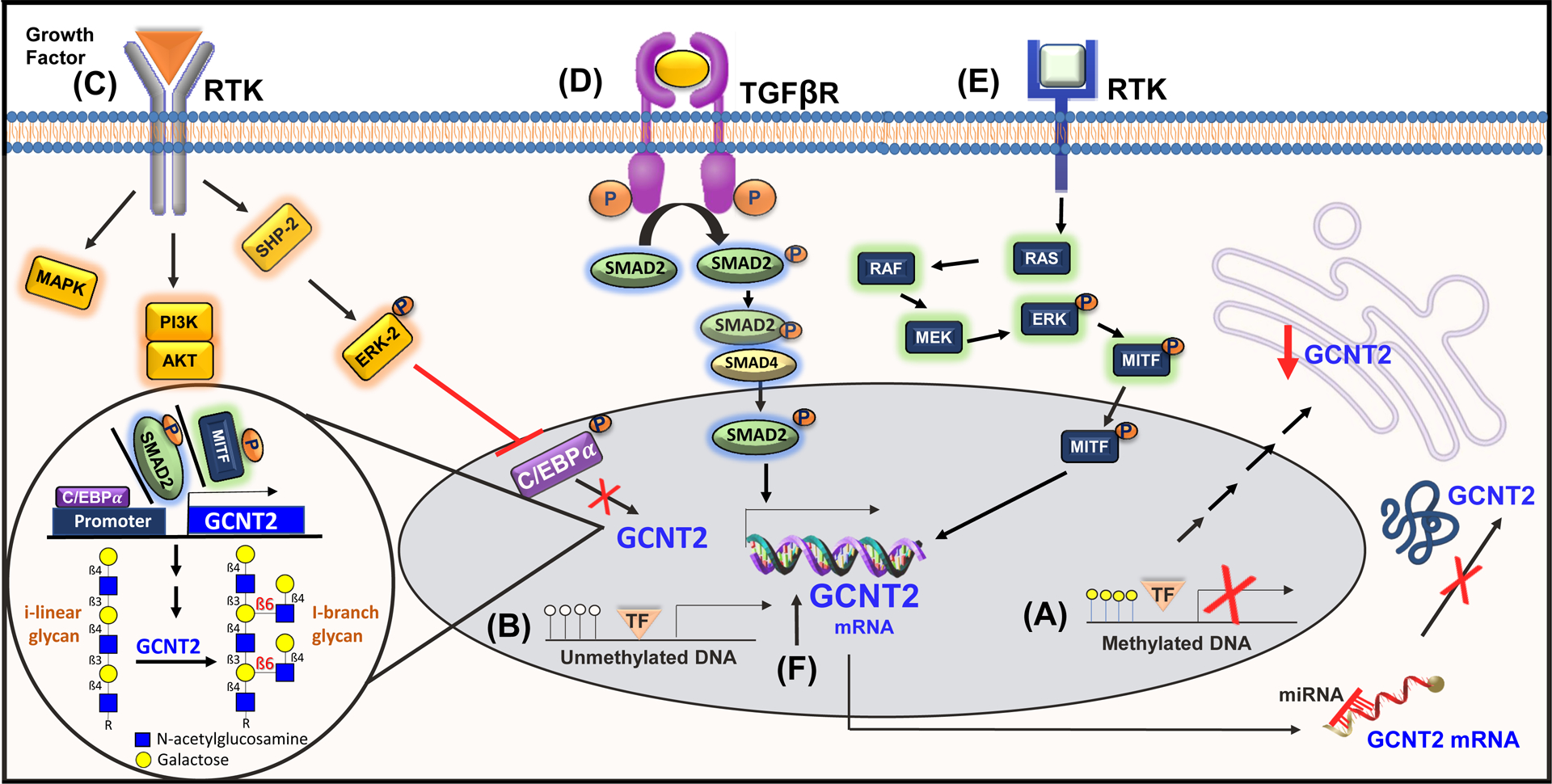
Epigenetic control via hypermethylation (A) or hypomethylation (B) of the GCNT2 promoter. Transcriptional regulation of GCNT2 mediated by prominent signaling cascades (C, D, E). miRNA-dependent repression of GCNT2 (F). Created with BioRender.com and Reactome64.
Transcriptional regulation.
The transcription factor (TF) CCAT/enhancer binding protein α (C/EBPα) has been studied extensively in relation to I-antigen/I-branch formation during erythroid cell differentiation. I-antigen formation has been specifically attributed to the GCNT2C variant, confirmed both by the exclusive presence of GCNT2C in reticulocytes and by experiments in human erythroleukemia K562 cells with intrinsically low I-antigen, where induction of erythroid differentiation leads to a marked increase in I-antigen along with GCNT2C expression51. Chromatin immunoprecipitation analysis, in fact, reveals that C/EBPα helps mediate this transition by binding the GCNT2C promoter to enhance transcription and I-antigen formation51. This erythroid-specific promoter contains binding sites for TFs OCT-2, SP1, and C/EBPα, though only C/EBPα has been shown to significantly induce I-antigen expression51. Importantly, the phosphorylation status of C/EBPα can alter its binding affinity to the GCNT2C promoter region46, 51–53. Specifically, dephosphorylation of Ser21 is associated with enhanced binding of C/EBPα to the GCNT2C promoter, while phosphorylation diminishes the ability of C/EBPα to induce GCNT2C expression and I-antigen formation52, 53. SHP2 and ERK2 have been identified to function in concert as negative regulators of this process, where ERK2 works upstream of C/EBPα to regulate its Ser21 phosphorylation, and SHP2 works upstream of ERK2 to regulate both ERK2 and C/EBPα phosphorylation (Figure 2C)53. The malignancy-associated transforming growth factor-β (TGF-β)/SMAD pathway is also of interest, enhancing GCNT2 transcription in breast cancer studies via activation of cofactor pSMAD2 (Figure 2D)13. TGF-β is among the proteins with a paradoxical role in malignancy, acting first as a potent tumor suppressor and later as a promoter of cell migration, invasion, and metastasis54, evidenced by its induction of EMT in breast cancer13, 55. Finally, while studies of transcription factors regulating GCNT2 in melanomas are ongoing, MITF has been found to directly transcribe GCNT2 (Figure 2E)3. This data is supported by the finding that normal human melanocytes with robust MITF expression present with high GCNT2 expression levels compared to metastatic melanoma cell lines10.
MicroRNAs and post-transcriptional regulation.
MicroRNA (miRNA) are a class of small non-coding RNAs that act as post-transcriptional regulators of gene expression. miRNA function by assembling into effector complexes known as RNA-induced silencing complexes (RISCs), which bind mainly to the 3’ untranslated region (UTR) but also to the 5’ UTR and other coding regions of their target mRNAs to hinder translation or induce deadenylation and subsequent degradation56–58. miRNAs are known to play an important role in embryonic development and cell differentiation, serving as negative regulators of at least 30% of protein-coding genes59, 60. The let-7 family of miRNAs, in particular, is important in erythrocyte development, known to be involved in the regulation of globin gene expression61. Of the let-7 family, let-7a and let-7b, specifically, are expressed significantly more than other members in peripheral blood cells, with the highest expression observed in reticulocytes62. In addition to the expected interference with globin genes, however, tough decoy targeted inhibition of let-7a produces decreased levels of GCNT2 mRNA, establishing a role for let-7a miRNA in the regulation of GCNT2 expression62. Furthermore, in pancreatic islet-like cell clusters, highly expressed miR-186, miR-199a, and miR-339 also downregulate the expression of GCNT2, among other target genes, compared to undifferentiated cells63. In colon cancer studies, miR-199a-5p and miR-199b-5p have been confirmed via RISC-trap assays to target the GCNT2 3’UTR directly34. Moreover, EMT induction in colon cancer cells leads to downregulation of miR-199a-5p and miR-199b-5p along with previously documented GCNT2 upregulation34. As constitutive expression of miR-199a/b in colon cancer cell lines both attenuates the production of GCNT2 mRNA and I-antigen as well as suppresses cell migration, invasion, and proliferation, this lends strong support to the idea that miR-199 downregulation is responsible, in part, for the EMT-associated GCNT2 induction observed in colon cancer (Figure 2F)34, 64.
Post-translational regulation.
In silico protein analysis shows that the GCNT2 enzyme interacts with the E3 ubiquitin-protein ligase TRIM6865, 66. Ubiquitination is a cellular process that marks proteins for degradation through activating (E1), conjugating (E2), and ligating (E3) enzymes that work together to add ubiquitin to substrates67. The tripartite motif (TRIM) proteins are a subfamily of RING type E3 ubiquitin ligases, which function both to bring E2-ubiquitin conjugates together with their substrates as well as aid in the transfer of ubiquitin from the E2 enzyme to the substrate68. TRIM proteins are thought to regulate cancer progression via degradation of tumor suppressor or oncogene products, among other processes67, 69. Evidence of interaction between TRIM68 and GCNT2 suggests that post-translational ubiquitination may be another method by which GCNT2 expression is regulated. In addition, GCNT2A appears to undergo N-linked glycosylation at the asparagine residue in the 41st position (Asn41), which is important for cellular localization (UniProtKB accession number: Q8NOV5)70–72. Altered glycosylation, in turn, may lead to improper localization and disrupt GCNT2 enzyme function by blocking access to substrates. In fact, N-glycosylation modifications have been demonstrated to be crucial regulators of function in other glycoproteins involved in cancer pathogenesis, including CD147 and the O-mannose kinase SGK19673, 74. Therefore, altering the glycosylation at Asn41 may be another opportunity for cells to execute GCNT2 regulation and influence cancer progression, though it is not yet known whether the absence of this glycosylation has significant functional consequences.
F. Conclusions
Notwithstanding current progress in novel targeted therapies and the advent of immunotherapy, curbing disease relapse and treatment resistance in metastatic melanoma remains an unmet objective. Establishing superior therapeutic targets and predictive biomarkers is, therefore, a critical goal in melanoma research. Multiple diagnostic markers exist for melanoma, such as MelanA/MART-1 and S100, but there are few that actually prognosticate disease severity and/or therapeutic response75. Importantly, as aberrant glycan expression plays a significant role in melanoma progression, our intents continue to focus on these relatively understudied molecular beacons. As described in this review, glycosylation is an integral part of cellular signaling that is hijacked by cancer cells, including melanoma cells, to promote growth and metastasis. We believe that understanding the cancer glycome can provide new molecular targets for monitoring disease progression and for therapeutic exploitation. In melanoma, we speculate that I-antigen-forming glycosyltransferase GCNT2 is a potential novel biomarker for disease progression. Studies using immunohistological methods to detect loss of GCNT2/I-antigen expression are underway to evaluate the predictive relationships between loss of GCNT2/I-antigen and metastatic activity, response to targeted therapy, and/or response to immunotherapy. Further investigation into the mechanisms of GCNT2/I-antigen-dependent tumor suppression in metastatic melanoma, namely the mode(s) of GCNT2 gene regulation, is also ongoing and could reveal ancillary therapeutic approaches, particularly within the Gal-3/i-linear-antigen axis to improve ICI efficacy and generate options for personalized cancer treatment.
What is already known about this topic?
Sporadic studies on the glycobiology of cancer share a common theme that glycan/lectin-mediated interactions between host and tumor cells impact cancer progression.
What does this study add?
Studying the glycome on melanoma cells can yield new insights in melanoma progression.
β1,6 N-acetylglucosaminyltransferase 2 (GCNT2) and its I-antigen product are emerging glycobiological factors in melanoma biomarker and therapeutics development.
Acknowledgements
We thank Ms. Nicole Izhakoff (Herbert Wertheim College of Medicine, Florida International University) for her assistance during the early formative stages of this review.
Funding: This research and support for this review was funded by the National Institutes of Health (NIH)/National Cancer Institute (NCI) Alliance of Glycobiologists for Cancer Research: Biological Tumor Glycomics Laboratory (U01 CA225644 to CJ Dimitroff) and the NIH/National Institute of Allergy and Infectious Diseases (NIAID) (R21 AI146368 to CJ Dimitroff). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
References
- 1.Melanoma Survival Rates Washington, D.C.: Melanoma Research Alliance; [Available from: https://www.curemelanoma.org/about-melanoma/melanoma-staging/melanoma-survival-rates/. [Google Scholar]
- 2.Klemen ND, Wang M, Feingold PL, Cooper K, Pavri SN, Han D, et al. Patterns of failure after immunotherapy with checkpoint inhibitors predict durable progression-free survival after local therapy for metastatic melanoma. Journal for ImmunoTherapy of Cancer. 2019;7(1):196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoek KS, Schlegel NC, Eichhoff OM, Widmer DS, Praetorius C, Einarsson SO, et al. Novel MITF targets identified using a two-step DNA microarray strategy. Pigment Cell Melanoma Res. 2008;21(6):665–76. [DOI] [PubMed] [Google Scholar]
- 4.Tang L, Chen X, Zhang X, Guo Y, Su J, Zhang J, et al. N-Glycosylation in progression of skin cancer. Medical Oncology. 2019;36(6):50. [DOI] [PubMed] [Google Scholar]
- 5.Chakraborty A, Dorsett KA, Trummell HQ, Yang ES, Oliver PG, Bonner JA, et al. ST6Gal-I sialyltransferase promotes chemoresistance in pancreatic ductal adenocarcinoma by abrogating gemcitabine-mediated DNA damage. J Biol Chem. 2018;293(3):984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz MJ, Holdbrooks AT, Chakraborty A, Grizzle WE, Landen CN, Buchsbaum DJ, et al. The Tumor-Associated Glycosyltransferase ST6Gal-I Regulates Stem Cell Transcription Factors and Confers a Cancer Stem Cell Phenotype. Cancer Res. 2016;76(13):3978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hakomori S Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 1996;56(23):5309–18. [PubMed] [Google Scholar]
- 8.Hakomori S Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A. 2002;99(16):10231–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal P, Fontanals-Cirera B, Sokolova E, Jacob S, Vaiana CA, Argibay D, et al. A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer cell. 2017;31(6):804–19.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sweeney JG, Liang J, Antonopoulos A, Giovannone N, Kang S, Mondala TS, et al. Loss of GCNT2/I-branched glycans enhances melanoma growth and survival. Nature Communications. 2018;9(1):3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dimitroff CJ. I-branched carbohydrates as emerging effectors of malignant progression. Proceedings of the National Academy of Sciences. 2019;116(28):13729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohtsubo K, Marth JD. Glycosylation in Cellular Mechanisms of Health and Disease. Cell. 2006;126(5):855–67. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Meng F, Wu S, Kreike B, Sethi S, Chen W, et al. Engagement of I-branching {beta}−1, 6-N-acetylglucosaminyltransferase 2 in breast cancer metastasis and TGF-{beta} signaling. Cancer Res. 2011;71(14):4846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng F, He Q, Cheng C, Pan J. GCNT2 induces epithelial-mesenchymal transition and promotes migration and invasion in esophageal squamous cell carcinoma cells. Cell Biochemistry and Function. 2019;37(1):42–51. [DOI] [PubMed] [Google Scholar]
- 15.Eichler J Protein glycosylation. Current Biology. 2019;29(7):R229–R31. [DOI] [PubMed] [Google Scholar]
- 16.Yan A, Lennarz WJ. Unraveling the Mechanism of Protein N-Glycosylation. Journal of Biological Chemistry. 2005;280(5):3121–4. [DOI] [PubMed] [Google Scholar]
- 17.Van den Steen P, Rudd PM, Dwek RA, Opdenakker G. Concepts and Principles of O-Linked Glycosylation. Critical Reviews in Biochemistry and Molecular Biology. 1998;33(3):151–208. [DOI] [PubMed] [Google Scholar]
- 18.Shimma Y-i, Saito F, Oosawa F, Jigami Y. Construction of a Library of Human Glycosyltransferases Immobilized in the Cell Wall of Saccharomyces cerevisiae. Applied and Environmental Microbiology. 2006;72(11):7003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rini JM, Esko JD. Glycosyltransferases and Glycan-Processing Enzymes. Essentials of Glycobiology. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2017. p. 65–75. [Google Scholar]
- 20.Cheng J, Wang R, Zhong G, Chen X, Cheng Y, Li W, et al. ST6GAL2 Downregulation Inhibits Cell Adhesion and Invasion and is Associated with Improved Patient Survival in Breast Cancer. Onco Targets Ther. 2020;13:903–14. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Schneider M, Al-Shareffi E, Haltiwanger RS. Biological functions of fucose in mammals. Glycobiology. 2017;27(7):601–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanley P, Taniguchi N, Aebi M. N-Glycans. Essentials of Glycobiology. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2017. p. 99–111. [Google Scholar]
- 23.Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pocheć E, Bubka M, Rydlewska M, Janik M, Pokrywka M, Lityńska A. Aberrant glycosylation of αvβ3 integrin is associated with melanoma progression. Anticancer Res. 2015;35(4):2093–103. [PubMed] [Google Scholar]
- 25.Pocheć E, Rydlewska M, Przybyło M, Lityńska A. Diverse expression of N-acetylglucosaminyltransferase V and complex-type β1,6-branched N-glycans in uveal and cutaneous melanoma cells. Acta Biochim Pol. 2015;62(2):323–8. [DOI] [PubMed] [Google Scholar]
- 26.Passaniti A, Hart GW. Cell surface sialylation and tumor metastasis. Metastatic potential of B16 melanoma variants correlates with their relative numbers of specific penultimate oligosaccharide structures. J Biol Chem. 1988;263(16):7591–603. [PubMed] [Google Scholar]
- 27.Finne J, Tao T-W, Burger MM. Carbohydrate Changes in Glycoproteins of a Poorly Metastasizing Wheat Germ Agglutinin-resistant Melanoma Clone. Cancer Research. 1980;40(7):2580. [PubMed] [Google Scholar]
- 28.Borck G, Kakar N, Hoch J, Friedrich K, Freudenberg J, Nürnberg G, et al. An Alu repeat-mediated genomic GCNT2 deletion underlies congenital cataracts and adult i blood group. Human Genetics. 2012;131(2):209–16. [DOI] [PubMed] [Google Scholar]
- 29.Inaba N, Hiruma T, Togayachi A, Iwasaki H, Wang X-H, Furukawa Y, et al. A novel I-branching β−1,6-N-acetylglucosaminyltransferase involved in human blood group I antigen expression. Blood. 2003;101(7):2870–6. [DOI] [PubMed] [Google Scholar]
- 30.Pras E, Raz J, Yahalom V, Frydman M, Garzozi HJ, Pras E, et al. A Nonsense Mutation in the Glucosaminyl (N-acetyl) Transferase 2 Gene (GCNT2): Association with Autosomal Recessive Congenital Cataracts. Investigative Ophthalmology & Visual Science. 2004;45(6):1940–5. [DOI] [PubMed] [Google Scholar]
- 31.Irum B, Khan SY, Ali M, Daud M, Kabir F, Rauf B, et al. Deletion at the GCNT2 Locus Causes Autosomal Recessive Congenital Cataracts. PLOS ONE. 2016;11(12):e0167562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikami J, Tobisawa Y, Yoneyama T, Hatakeyama S, Mori K, Hashimoto Y, et al. I-branching N-acetylglucosaminyltransferase regulates prostate cancer invasiveness by enhancing α5β1 integrin signaling. Cancer Sci. 2016;107(3):359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura K, Yamashita K, Sawaki H, Waraya M, Katoh H, Nakayama N, et al. Aberrant Methylation of GCNT2 Is Tightly Related to Lymph Node Metastasis of Primary CRC. Anticancer Research. 2015;35(3):1411–21. [PubMed] [Google Scholar]
- 34.Chao C-C, Wu P-H, Huang H-C, Chung H-Y, Chou Y-C, Cai B-H, et al. Downregulation of miR-199a/b-5p is associated with GCNT2 induction upon epithelial–mesenchymal transition in colon cancer. FEBS Letters. 2017;591(13):1902–17. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Hong Z, Ma H, Yu D, Qu P. Key factors mediated by PI3K signaling pathway and related genes in endometrial carcinoma. Journal of Bioenergetics and Biomembranes. 2020;52(6):465–73. [DOI] [PubMed] [Google Scholar]
- 36.Sexton RE, Hallak MNA, Uddin MH, Diab M, Azmi AS. Gastric Cancer Heterogeneity and Clinical Outcomes. Technol Cancer Res Treat. 2020;19:1533033820935477-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong Z, Zhu X, Li Y, Gan L, Chen H, Zhang W, et al. Oncogenomic analysis identifies novel biomarkers for tumor stage mycosis fungoides. Medicine (Baltimore). 2018;97(21):e10871–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farhad M, Rolig AS, Redmond WL. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology. 2018;7(6):e1434467–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ClinicalTrials.gov Identifier: NCT02117362- Galectin Inhibitor (GR-MD-02) and Ipilimumab in Patients With Metastatic Melanoma Bethesda (MD): National Library of Medicine (US); [Available from: https://clinicaltrials.gov/ct2/show/study/NCT02117362. [Google Scholar]
- 40.Chakraborty A, Dimitroff CJ. Cancer immunotherapy needs to learn how to stick to its guns. The Journal of Clinical Investigation. 2019;129(12):5089–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cichorek M, Wachulska M, Stasiewicz A, Tymińska A. Skin melanocytes: biology and development. Postepy Dermatol Alergol. 2013;30(1):30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirobe T How are proliferation and differentiation of melanocytes regulated? Pigment Cell & Melanoma Research. 2011;24(3):462–78. [DOI] [PubMed] [Google Scholar]
- 43.Gene. GCNT2 glucosaminyl (N-acetyl) transferase 2 (I blood group) [ Homo sapiens (human) ] Bethesda (MD): National Library of Medicine (US): National Center for Biotechnology Information; [updated August 18 2020. Available from: https://www.ncbi.nlm.nih.gov/gene/2651. [Google Scholar]
- 44.Online Mendelian Inheritance in Man. MIM Number: {*600429} GLUCOSAMINYL (N-ACETYL) TRANSFERASE 2, I-BRANCHING ENZYME; GCNT2 Baltimore, MD: Johns Hopkins University; [updated May 6 2015. Available from: https://omim.org/entry/600429. [Google Scholar]
- 45.Yu L-C, Twu Y-C, Chou M-L, Reid ME, Gray AR, Moulds JM, et al. The molecular genetics of the human I locus and molecular background explain the partial association of the adult i phenotype with congenital cataracts. Blood. 2003;101(6):2081–8. [DOI] [PubMed] [Google Scholar]
- 46.Yu L-C, Lin M. Molecular genetics of the blood group I system and the regulation of I antigen expression during erythropoiesis and granulopoiesis. Current Opinion in Hematology. 2011;18(6):421–6. [DOI] [PubMed] [Google Scholar]
- 47.Lessard S, Beaudoin M, Benkirane K, Lettre G. Comparison of DNA methylation profiles in human fetal and adult red blood cell progenitors. Genome Medicine. 2015;7(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murakami M, Yoshimoto T, Nakabayashi K, Tsuchiya K, Minami I, Bouchi R, et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. European Journal of Endocrinology. 2015;173(2):185. [DOI] [PubMed] [Google Scholar]
- 49.Yang AY, Lee JH, Shu L, Zhang C, Su Z-Y, Lu Y, et al. Genome-wide analysis of DNA methylation in UVB- and DMBA/TPA-induced mouse skin cancer models. Life Sci. 2014;113(1–2):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura K, Sawaki H, Yamashita K, Watanabe M, Narimatsu H. Identification of epigenetic silencing of GCNT2 expression by comprehensive real-time PCR screening in colorectal cancer. Journal of Clinical Oncology. 2014;32(3_suppl):506-. [Google Scholar]
- 51.Twu Y-C, Chen C-P, Hsieh C-Y, Tzeng C-H, Sun C-F, Wang S-H, et al. I branching formation in erythroid differentiation is regulated by transcription factor C/EBPα. Blood. 2007;110(13):4526–34. [DOI] [PubMed] [Google Scholar]
- 52.Twu Y-C, Hsieh C-Y, Lin M, Tzeng C-H, Sun C-F, Yu L-C. Phosphorylation status of transcription factor C/EBPα determines cell-surface poly-LacNAc branching (I antigen) formation in erythropoiesis and granulopoiesis. Blood. 2010;115(12):2491–9. [DOI] [PubMed] [Google Scholar]
- 53.Liao Y-J, Lee Y-H, Chang F-L, Ho H, Huang C-H, Twu Y-C. The SHP2-ERK2 signaling pathway regulates branched I antigen formation by controlling the binding of CCAAT/enhancer binding protein α to the IGnTC promoter region during erythroid differentiation. Transfusion. 2016;56(11):2691–702. [DOI] [PubMed] [Google Scholar]
- 54.Tian M, Schiemann WP. The TGF-beta paradox in human cancer: an update. Future Oncol. 2009;5(2):259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15(2):169–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nature Reviews Genetics. 2010;11(9):597–610. [DOI] [PubMed] [Google Scholar]
- 58.Catalanotto C, Cogoni C, Zardo G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int J Mol Sci. 2016;17(10):1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116(2):281–97. [DOI] [PubMed] [Google Scholar]
- 60.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee YT, de Vasconcellos JF, Yuan J, Byrnes C, Noh S-J, Meier ER, et al. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. 2013;122(6):1034–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Vasconcellos JF, Byrnes C, Lee YT, Allwardt JM, Kaushal M, Rabel A, et al. Tough decoy targeting of predominant let-7 miRNA species in adult human hematopoietic cells. Journal of Translational Medicine. 2017;15(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen B-Z, Yu S-L, Singh S, Kao L-P, Tsai Z-Y, Yang P-C, et al. Identification of microRNAs expressed highly in pancreatic islet-like cell clusters differentiated from human embryonic stem cells. Cell Biology International. 2011;35(1):29–37. [DOI] [PubMed] [Google Scholar]
- 64.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.neXtProt. GCNT2-Interactions: Swiss Institute of Bioinformatics; [updated July 17 2020. Available from: https://www.nextprot.org/entry/NX_Q8N0V5/interactions.
- 66.Zahn-Zabal M, Michel P-A, Gateau A, Nikitin F, Schaeffer M, Audot E, et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res. 2019;48(D1):D328–D34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nature Medicine. 2014;20(11):1242–53. [DOI] [PubMed] [Google Scholar]
- 68.Lips C, Ritterhoff T, Weber A, Janowska MK, Mustroph M, Sommer T, et al. Who with whom: functional coordination of E2 enzymes by RING E3 ligases during poly-ubiquitylation. The EMBO Journal.n/a(n/a):e104863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hatakeyama S TRIM proteins and cancer. Nature Reviews Cancer. 2011;11(11):792–804. [DOI] [PubMed] [Google Scholar]
- 70.neXtProt. GCNT2-Proteomics: Swiss Institute of Bioinformatics; [updated July 17 2020. Available from: https://www.nextprot.org/entry/NX_Q8N0V5/proteomics?isoform=NX_Q8N0V5-1.
- 71.UniProt. UniProtKB - Q8N0V5 (GNT2A_HUMAN): European Bioinformatics Institute, the Swiss Institute of Bioinformatics, and the Protein Information Resource; [updated October 7 2020. Available from: https://www.uniprot.org/uniprot/Q8N0V5.
- 72.Consortium TU. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2018;47(D1):D506–D15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li J-H, Huang W, Lin P, Wu B, Fu Z-G, Shen H-M, et al. N-linked glycosylation at Asn152 on CD147 affects protein folding and stability: promoting tumour metastasis in hepatocellular carcinoma. Scientific Reports. 2016;6(1):35210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu C, Zhang M, Bian L, Li Y, Yao Y, Li D. N-glycosylated SGK196 suppresses the metastasis of basal-like breast cancer cells. Oncogenesis. 2020;9(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davis LE, Shalin SC, Tackett AJ. Current state of melanoma diagnosis and treatment. Cancer Biol Ther. 2019;20(11):1366–79. [DOI] [PMC free article] [PubMed] [Google Scholar]