

Abstract
Random conjugations of chemotherapeutics to monoclonal antibodies result in heterogeneous antibody-drug conjugates (ADCs) with suboptimal pharmacological properties. We recently developed a new technology for facile generation of homogeneous ADCs by harnessing human CD38 catalytic domain and its dinucleotide-derived covalent inhibitor, termed ADP-ribosyl cyclase-enabled ADCs (ARC-ADCs). Herein we advance this technology by designing and synthesizing ARC-ADCs with customizable drug-to-antibody ratios (DARs). Through varying numbers and locations of CD38 fused to an antibody targeting human C-type lectin-like molecule-1 (hCLL-1), ARC-ADCs featuring DARs of 2 and 4 were rapidly generated via a single step with cytotoxic monomethyl auristatin F (MMAF) as payloads. In contrast to anti-hCLL-1 ARC-ADC carrying 2 drug molecules, anti-hCLL-1 ARC-ADC with a DAR of 4 shows highly potent activity in killing hCLL-1-positive acute myeloid leukemia (AML) cells both in vitro and in vivo. This work provides novel ADC candidates for combating AML and supports ARC-ADC as a general and versatile approach for producing site-specific ADCs with defined DARs.
Keywords: antibody-drug conjugate, acute myeloid leukemia, protein engineering, targeted therapy
Introduction
Antibody-drug conjugates (ADCs) can deliver small-molecule drugs covalently linked to the immunoglobulin scaffold to specific types of cells with surface-expressed target antigens.1 Due to limitations on conjugation strategies,2–4 all currently approved ADCs are heterogeneous with varying locations and numbers of payloads. Each ADC used in clinic contains multiple populations with different drug-to-antibody ratios (DARs) that perform distinctively in vivo,5, 6 raising efficacy and safety concerns.
To develop homogeneous ADCs, different technologies have been established such as utilizing engineered amino acids and carbohydrates, incorporating unnatural amino acids, grafting peptides, and fusing protein domains for creating handles for conjugation.7–22 By exploiting enzymatic activity of human CD38, a member of ADP-ribosyl cyclase family, we recently developed a new approach for single-step generation of site-specific ADCs, named ADP-ribosyl cyclase-enabled ADCs (ARC-ADCs).23 Through a 2′-Cl-arabinose nicotinamide adenine dinucleotide (2’-Cl-araNAD+)-based covalent inhibitor of CD38, cytotoxic monomethyl auristatin F (MMAF) could be covalently attached to antibodies genetically fused with CD38 via its catalytic glutamate 226 (Glu226) residue (Figure 1A). The resulting ARC-ADC with a DAR of 2 shows excellent stability and efficacy in treating breast cancer in preclinical models.23
Figure 1.
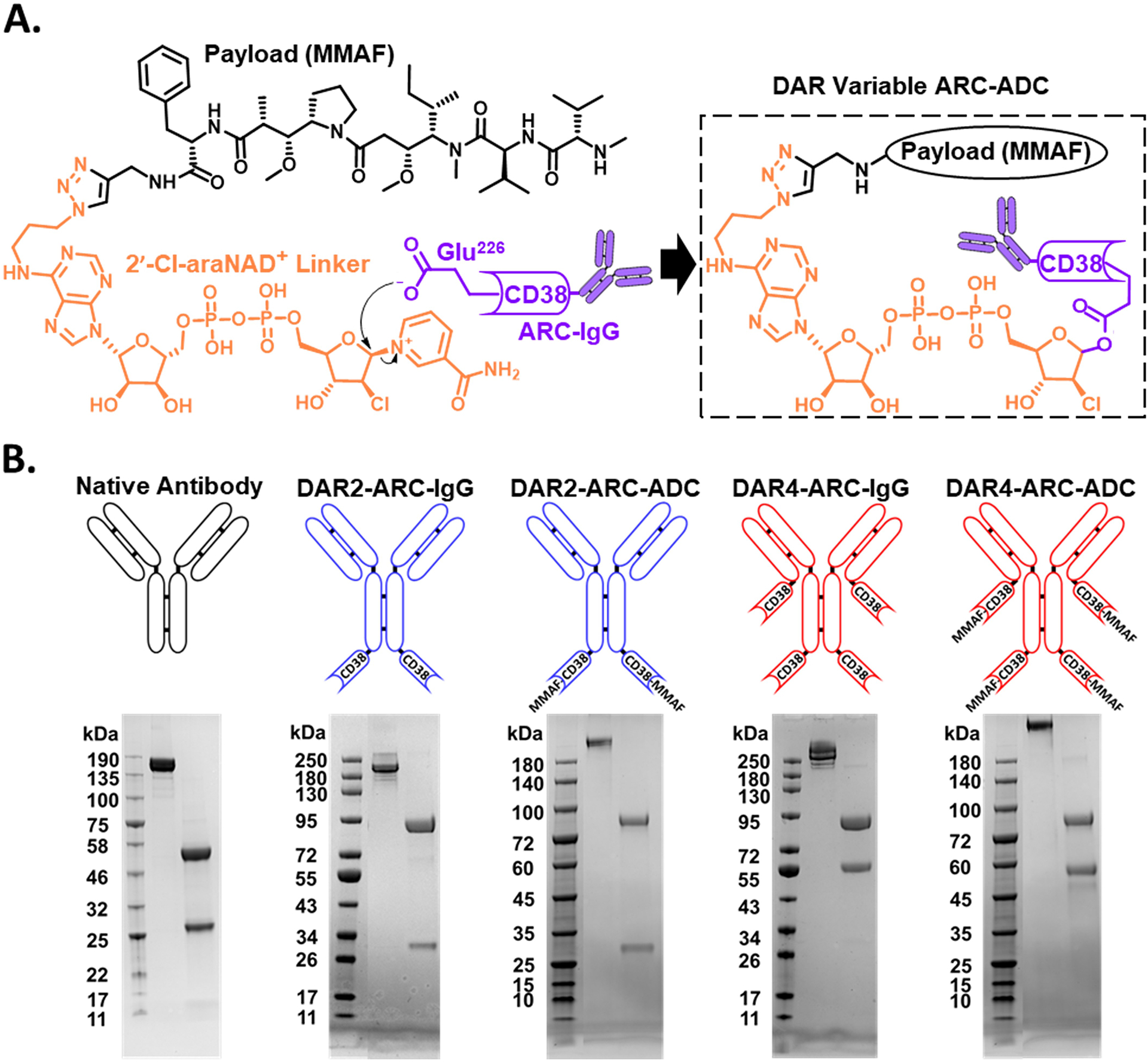
Homogeneous ARC-ADCs with variable DARs. (A) Schematic of 2′-Cl-araNAD+ linker-mediated site-specific antibody-drug conjugation. (B) Generation of anti-hCLL-1 IgG, anti-hCLL-1-CD38 C-fusions, and anti-hCLL-1 ARC-ADCs with DARs of 2 and 4. Lower panel: Coomassie-stained SDS-PAGE gels for purified anti-hCLL-1 IgG (native antibody), anti-hCLL-1-CD38 C-fusions (DAR2-ARC-IgG and DAR4-ARC-IgG), and anti-hCLL-1 ARC-ADCs.
Given the CD38-enabling nature for site-specific conjugation, we envisioned that ARC-ADCs may provide unique opportunities for facile production of homogeneous ADCs with customizable DARs. To test this notion, we designed and generated two forms of ARC-ADCs with DARs of 2 and 4, designated as DAR2-ARC-ADC and DAR4-ARC-ADC, respectively, which exhibit remarkable in vitro and in vivo efficacy against human acute myeloid leukemia (AML) cells through specifically targeting human C-type lectin-like molecule-1 (hCLL-1). AML is the most common type of acute leukemia in adults with 5-year survival rate below 30%.24 CLL-1 is frequently overexpressed in blasts and leukemia stem cells (LSCs) of AML patients,25 but absent on normal hematopoietic stem cells (HSCs) in bone marrow, representing a promising target for AML treatment.26–30 Our anti-hCLL-1 ARC-ADCs not only provide new therapeutic candidates for AML but also demonstrate ARC-ADC as a general approach for making homogeneous ADCs with tailored DARs.
Results
Since the DAR of an ARC-ADC is associated with the number of fused CD38 catalytic domain, fusing additional CD38 extracellular domains to the immunoglobulin scaffold may thus increase numbers of payloads, likely resulting in site-specific ADCs with enhanced potency. To this end, we genetically fused human CD38 enzymatic domain to C-termini of light chain (LC) and heavy chain (HC) of an anti-hCLL-1 monoclonal antibody 1075.7.31 The resulting HC-CD38 C-fusion construct was paired with LC or LC-CD38 C-fusion expression vector for transient transfection in mammalian cells for production of an anti-hCLL-1 IgG HC-CD38 C-fusion (denoted as DAR2-ARC-IgG) and an anti-hCLL-1 IgG HC-CD38 & LC-CD38 C-fusion (denoted as DAR4-ARC-IgG). Together with expressed native anti-hCLL-1 antibody, DAR2-ARC-IgG and DAR4-ARC-IgG were analyzed by Coomassie-stained SDS-PAGE gels (Figure 1B). The observed sizes of light and heavy chains for each construct are consistent with molecular designs. The yields are about 10 mg L−1 and 7 mg L−1 for DAR2-ARC-IgG and DAR4-ARC-IgG, respectively, lower than that of anti-hCLL-1 antibody (14 mg L−1).
Next, CD38 enzymatic activity and hCLL-1 binding affinity were examined for DAR2-ARC-IgG and DAR4-ARC-IgG. Fluorescence-based activity assays indicated that both fusion IgGs possess significantly higher catalytic activities than that of recombinant human CD38 extracellular domain, possibly due to improved stability (Figure 2A). Reactions catalyzed by DAR4-ARC-IgG show approximate 50% rate increase relative to those by DAR2-ARC-IgG, owing to two extra CD38 domains. As a control, native anti-hCLL-1 antibody gives no enzymatic activity. Enzyme linked immunosorbent assay (ELISA) analysis revealed tight binding to recombinant hCLL-1 for both DAR2-ARC-IgG and DAR4-ARC-IgG, comparable to that of native anti-hCLL-1 antibody (Figure 2B). These results support successful generation of anti-hCLL-1-CD38 fusions with robust CD38 enzymatic activity and high affinity to hCLL-1 antigen, allowing rapid generation of anti-hCLL-1 ARC-ADCs with distinct DARs. Additionally, ELISA indicated that in contrast to the anti-hCLL-1 antibody, DAR2-ARC-IgG and DAR4-ARC-IgG exhibit slightly increased or comparable binding affinities to human CD16a and C1q (Figure S1), which are Fc receptor and complement protein, respectively, involved in activation of antibody-dependent cellular cytotoxicity and the classical complement pathway. This suggests that genetic fusions of the CD38 catalytic domain to the C-termini of antibody heavy and light chains may have no adverse impact on functions of the antibody Fc region.
Figure 2.

Characterization of anti-hCLL-1-CD38 C-fusions and anti-hCLL-1 ARC-ADCs. (A) Analysis of ADP-ribosyl cyclase activity. Purified CD38 (20 nM), anti-hCLL-1 antibody (10 nM), DAR2-ARC-IgG (10 nM), or DAR4-ARC-IgG (10 nM) was incubated with 100 μM NGD+ in PBS to monitor ADPR cyclase activity based on the formation of fluorescent cyclic GDP-ribose at 410 nm. (B) ELISA analysis of binding to recombinant hCLL-1 extracellular domain. (C) Flow cytometric analysis of hCLL-1 expression on human U937 and KG1a cells. (D) In vitro cytotoxicity of DAR2-ARC-ADC and DAR4-ARC-ADC. U937 or KG1a cells were incubated for 72 hours at 37°C with 5% CO2 with various concentrations of ARC-ADCs, drug-linker conjugate, native anti-hCLL-1 antibody, and ARC-IgGs. Cell viability was determined by MTT assays with data for cells incubated with culture medium or 5 μM paclitaxel as 100% viable or 0% viable references, respectively. (E) Pharmacokinetics in mice for surrogate DAR2-ARC-ADC and DAR4-ARC-ADC with FITC as the payloads. Noncompartmental analysis was conducted based on the plasma concentrations of surrogate ARC-ADCs determined through a sandwich ELISA by utilizing an anti-FITC polyclonal antibody as capture antibody and an anti-human kappa light chain monoclonal antibody conjugated with HRP as detection antibody.
Anti-hCLL-1 ARC-ADCs were then generated by incubating DAR2-ARC-IgG or DAR4-ARC-IgG with synthesized drug-linker conjugate (2’-Cl-araNAD+-MMAF) on ice for 1 hour, resulting in DAR2-ARC-ADC and DAR4-ARC-ADC (Figure 1B). As a tubulin inhibitor, MMAF is a potent cytotoxic payload. Mass spectrometry confirmed the generation of anti-hCLL-1 ARC-ADCs with DARs of 2 and 4 (Figure S2). In vitro cytotoxicity of the anti-hCLL-1 ARC-ADCs was then evaluated using human AML cell lines U937 (CLL-1+) and KG1a (CLL-1−1) (Figure 2C–D). DAR4-ARC-ADC exhibits highly potent cytotoxicity (EC50 = 25 ± 8 pM) against U937 cells and DAR2-ARC-ADC displays relatively lower potency with an EC50 of 0.9 ± 0.4 nM (Figure 2D). The drug-linker and ARC-IgGs have much weaker or little cytotoxicity for U937 cells. In comparison, none of these agents reveals significant toxicity for KG1a cells. These results demonstrate outstanding potency and specificity of anti-hCLL-1 ARC-ADCs and markedly enhanced efficacy for DAR4-ARC-ADC.
To examine stability, surrogate ARC-ADCs were generated using Alexa Fluor 488 or fluorescein isothiocyanate (FITC) as the payloads. In-gel fluorescence-based imaging revealed considerable stability for both surrogate DAR2-ARC-ADC and DAR4-ARC-ADC incubated in fresh mouse plasma (Figure S3). Pharmacokinetic studies were then performed in mice. Following single-dose intravenous (i.v.) injections of FITC-conjugated surrogate ARC-ADCs, sandwich ELISA assays were conducted for collected plasma samples by using anti-FITC and anti-human kappa light chains antibodies as capture and detection reagents. The measured plasma concentrations indicated a half-life of 52.8 ± 9.8 hours for DAR2-ARC-ADC FITC and 27.4 ± 2.0 hours for DAR4-ARC-ADC FITC (Figure 2E), suggesting reduced stability for ARC-ADC with increased payloads.
Next, in vivo therapeutic efficacy of DAR2-ARC-ADC and DAR4-ARC-ADC were evaluated using AML mouse xenograft models. Immunodeficient mice were intravenously injected with firefly luciferase-labeled U937 cells via tail vein. Beginning on day 4 post U937 cell implantation, mice were treated with ARC-ADCs at a dose of 1 or 5 mg/kg every 72 hours for a total of 3 times through i.v. injections. PBS vehicle and DAR4-ARC-IgG (5 mg/kg) were included as controls. From day 2 post i.v. implantation of luciferase-expressing U937 cells, whole-body luminescence imaging was taken for each group of mice and continued on a weekly basis. IVIS imaging and quantified luminescence indicated rapid proliferation of U937 cells for mice treated by PBS vehicle and DAR4-ARC-IgG. By contrast, administered ARC-ADCs show significant anti-leukemia activity in dose- and DAR-dependent manners (Figures 3A–B and S4). Notably, DAR4-ARC-ADC at the dose of 5 mg/kg displays highly potent inhibition against the proliferation of engrafted U937 cells. In comparison to PBS- and DAR4-ARC-IgG-treated groups with median survivals of 13 days, DAR4-ARC-ADC-treated group at 5 mg/kg shows more than 100% increase of median survival (27 days) (Figure 3C). Consistent with IVIS imaging analysis, the median survivals for mice treated by DAR2-ARC-ADC and DAR4-ARC-ADC are dependent on dose and DAR. Same as the in vitro cytotoxicity studies, in vivo results support excellent anti-leukemia activities for anti-hCLL-1 ARC-ADCs and significantly improved efficacy for DAR4-ARC-ADC.
Figure 3.

In vivo efficacy and toxicity of DAR2-ARC-ADC and DAR4-ARC-ADC. (A) IVIS images of mice in different groups post inoculation of luciferase-expressing U937 cells. (B) Luminescence intensities for mice in different treatment groups after injecting luciferase-expressing U937 cells. Black arrows indicate treatments by i.v. injections. (C) Kaplan–Meier survival curves and medians of survival for different treatment groups. (D) Average body weight of mice in different groups throughout the in vivo study. (E)-(G) Plasma ALT activities (E) and creatinine concentrations (F) and percentages of mCD34+ mCD45+ cell population in total living cells of bone marrows (G) on day 13 after U937 implantation.
Toxicities of the administered anti-hCLL-1 ARC-ADCs on mice were also assessed. During the efficacy study, treatments with ARC-ADCs cause no negative impact on mice body weights in comparison to control groups (Figure 3D). On day 13 after U937 implantation, plasma alanine aminotransferase (ALT) activity and creatinine concentration and percentages of mouse CD34+ CD45+ cells in blood, bone marrow, and spleen samples were determined. No significant differences were observed among all groups (Figures 3E–G and S5). Taken together, these results show no apparent toxicity for anti-hCLL-1 ARC-ADCs.
In addition to IVIS imaging analysis, blood, bone marrow, and spleen were collected for all groups on day 13 post U937 implantation for flow cytometry. Consistent with the luminescence-based whole-body imaging, flow cytometric analysis indicate higher levels of engrafted U937 cells and lower percentages of mouse CD45+ (mCD45+) cells in bone marrow, spleen, and blood for mice treated by PBS and DAR4-ARC-IgG (Figure 4A–G). Less than 0.5% of hCLL-1+ cells were observed across 3 different types of tissues for mice treated by DAR2-ARC-ADC and DAR4-ARC-ADC at 5 mg/kg (Figure 4B–D), indicating their potent activities in suppressing U937 cells expansion. Relative to control groups, significantly higher percentages of mCD45+ were observed for mice treated by ARC-ADCs at both 1 and 5 mg/kg in bone marrow and at 5 mg/kg in spleen and blood (Figure 4F–G). Similar to the quantified luminescence intensities, the measured percentages of hCLL-1+ among different tissues for mice treated by ARC-ADCs inversely correlate with dose and DAR. In comparison to mice receiving DAR2-ARC-ADC at 1 mg/kg, DAR4-ARC-ADC-treated mice at 1 mg/kg revealed significantly lower percentages of hCLL-1+ cells in blood, bone marrow, and spleen (Figure 4B–D). Moreover, mice given with DAR4-ARC-ADC have higher levels of mCD45+ in bone marrow compared with DAR2-ARC-ADC-treated ones at both 1 and 5 mg/kg doses (Figure 4F). These results support remarkable efficacy for anti-hCLL-1 ARC-ADCs and considerably increased potency for DAR4-ARC-ADC.
Figure 4.

Flow cytometric analysis of tissue samples collected on day 13 post U937 implantation. (A) Representative flow cytometry data for hCLL-1 and mCD45 expression in blood, bone marrow, and spleen cells among different groups. Percentages were shown for hCLL-1+ mCD45− and mCD45+ hCLL-1− cells in total living cells. (B-D) Percentages of hCLL-1+ mCD45− cells in total living cells in blood (B), bone marrow (C), and spleen (D) samples among different groups. (E-G) Percentages of mCD45+ hCLL-1− cells in total living cell in blood (E), bone marrow (F), and spleen (G) among different groups.
Discussion
Anti-hCLL-1 ARC-ADCs were successfully generated with DARs of 2 and 4 by utilizing antibody-CD38 fusions coupled with an NAD+ analogue-derived drug linker. By specifically binding to hCLL-1 antigen, ARC-ADCs allow targeted delivery of tubulin inhibitor MMAF to human AML cells, resulting in potent in vitro and in vivo anti-leukemia activities. Despite reduced plasma half-life, DAR4-ARC-ADC exhibits notably enhanced potency in both cellular and animal AML models, revealing therapeutic benefits for developing homogeneous ADCs with increased DARs.
Genetic fusions of CD38 catalytic domains to an antibody enable facile production of site-specific ADCs through single-step enzymatic reactions. In addition to rapidly mediating drug conjugation at Glu226 of CD38, the 2′-Cl-araNAD+-based dinucleotide linker can stably carry payloads to target cells for rapid release upon internalization. The DARs of ARC-ADCs can possibly be adjusted by fusing additional CD38 domains to further increase efficacy. Meanwhile, attachments of CD38 to an immunoglobulin can potentially affect the overall stability of fusion proteins, binding affinity to target antigens, and tissue penetration.
DAR4-ARC-ADC was generated by fusing CD38 to C-termini of light and heavy chains of the anti-hCLL-1 antibody. Different formats of ARC-ADC with a DAR of 4 could be developed by placing CD38 at different positions. While having the same DAR, these ARC-ADC may possess distinguished physicochemical properties and biological activities, which requires further examination. By changing the fusion location or linkers used between the antibody and CD38 domain, stability and efficacy for ARC-ADCs could be further optimized. Importantly, ARC-ADCs with additionally increased DARs can potentially be synthesized and evaluated for pharmacological activities.
To understand how varied number of payloads affect cytotoxicity of ADCs, cellular uptake and drug release need to be quantitatively analyzed for ARC-ADCs with different DARs. The ARC-ADCs can also be extended to other types of payloads and antibodies specific for disease-associated antigens.
In summary, anti-hCLL-1 ARC-ADCs with DARs of 2 and 4 were generated through harnessing genetically fused CD38 catalytic activity. DAR4-ARC-ADC is highly potent in killing CLL-1-positive AML cells both in vitro and in vivo, providing a new candidate for AML targeted therapy. This study demonstrates ARC-ADC as a general and versatile approach for developing homogeneous ADCs with defined DARs.
Materials and Methods
Molecular Cloning.
Synthetic gBLOCK DNA fragment for recombinant human CLL-1 extracellular domain natural variant (Uniprot ID: Q5QGZ9, VAR_037669, amino acid I70-A265) with a 6*His tag fused at C terminus was purchased from Integrated DNA Technologies (IA, USA). The synthetic DNA fragment above was amplified by polymerase chain reaction (PCR) with primers incorporating DNA restriction enzyme cleavage sites for EcoRI and NheI at the 5’ and 3’ end, respectively. Primers used for amplification include CLL-1-Forward: 5’-CACGAATTCGATCGAGATGAAGAAGATGAACAAAC-3’; CLL-1-Reverse: 5’-CCAGCTAGCACTCACTAATGATGATGGTG-3’.
Synthetic gBLOCK DNA fragments for anti-human CLL-1 IgG light chain (LC) and heavy chain (HC) fragment of antigen binding (Fab) (clone 1075.7) were purchased from Integrated DNA Technologies (IA, USA). The anti-human CLL-1 IgG LC DNA fragment was amplified by PCR with primers incorporating DNA restriction enzyme cleavage sites for EcoRI and NheI at the 5’ and 3’ end, respectively. The full-length anti-human CLL-1 IgG HC was generated through overlap extension PCR using the anti-human CLL-1 Fab HC and human IgG1 Fc fragments using primers containing DNA restriction enzyme cleavage sites for EcoRI and NheI at the 5′ and 3′ end, respectively. Primers mentioned above include Anti-CLL-1 IgG LC F: 5’-CACGAATTCGGAGAACGTGCTCACCCAATCCCC-3’; Anti-CLL-1 IgG LC R: 5’-CCAGCTAGCACTTATCAACACTCTCCCCTGTTGAAGCTCTTTGTG-3’; Anti-CLL-1 IgG HC F1: 5’-CACGAATTCGGACATCCAGCTGCAGGAGAGCG-3’; Anti-CLL-1 IgG HC R1: 5’-CGCAAGATTTGGGTTCCACTTTCTTGTCCACCTTGGTGTTGCTG-3’; Anti-CLL-1 IgG HC F2: 5’-GGTGGACAAGAAAGTGGAACCCAAATCTTGCGACAAAACTCACACATG-3’; Anti-CLL-1 IgG HC R2: 5’-CCAGCTAGCACTTATCATTTACCCGGAGACAGGGAGAGGC-3’.
Acquired PCR products were then analyzed by DNA gel electrophoresis with the bands at target sizes excised and purified by DNA gel extraction kits (Zymo Research, CA, USA) to yield the target DNA fragment inserts. Purified DNA fragment inserts were first treated with DNA restriction enzyme EcoRI and NheI (New England Biolabs, MA, USA) per the manufacturer’s instructions and then cleaned by DNA cleaning & concentrating kits (Zymo Research, CA, USA). The treated and cleaned DNA fragments were then spliced in-frame to pFUSE expression vector backbone by T4 DNA ligase (New England Biolabs, MA, USA). Ligation products were utilized to transform DH10B Escherichia coli (E. coli) electro-competent cells, followed with positive selection for Zeocin-resistant colonies and DNA sequencing of the extracted plasmids.
The sequence-confirmed pFUSE expression vectors for anti-human CLL-1 IgG LC and anti-human CLL-1 IgG HC were utilized as DNA templates for constructing anti-CLL-1 IgG LC CD38 C-fusion and anti-CLL-1 IgG HC CD38 C-fusion pFUSE expression vectors, respectively. Synthetic gBLOCK DNA fragment of the extracellular domain of human CD38 (Uniprot ID: P28907, R45-I300) with four mutated asparagine residues (N100D, N164A, N129D, and N209D) and a flexible GGGGS linker at N-terminus was purchased from Integrated DNA Technologies (IA, USA). Overlap extension PCR reactions were performed to generate LC-CD38 C-fusion and HC-CD38 C-fusion fragments. Primers utilized for cloning include Anti-CLL-1 LC CD38 F1: 5’-GTCACGAATTCGGAGAACGTGCTC-3’; Anti-CLL-1 LC CD38 R1: 5’-CGCCACCCCCACACTCTCCCCTGTTGAAGCTCTTTG-3’; Anti-CLL-1 LC CD38 F2: 5’-GGAGAGTGTGGGGGTGGCGGAAGC-3’; Anti-CLL-1 LC CD38 R2: 5’-CTGGCCAGCTAGCACTTATCAGATCTC-3’; Anti-CLL-1 HC CD38 F1: 5’-CACGAATTCGGACATCCAGCTGCAGGAGAGCG-3’; Anti-CLL-1 HC CD38 R1: 5’-CGCAAGATTTGGGTTCCACTTTCTTGTCCACCTTGGTGTTGCTG-3’; Anti-CLL-1 HC CD38 F2: 5’-GGTGGACAAGAAAGTGGAACCCAAATCTTGCGACAAAACTCACACATG-3’; Anti-CLL-1 HC CD38 R2: 5’-CTGGCCAGCTAGCACTTATCAGATCTC-3’.
Acquired PCR products were then analyzed by DNA gel electrophoresis with the bands at target sizes excised and purified by DNA gel extraction kits (Zymo Research, CA, USA) to yield the target DNA fragment inserts. Purified DNA fragment inserts were first treated with DNA restriction enzyme EcoRI and NheI (New England Biolabs, MA, USA) per the manufacturer’s instructions and then cleaned by DNA cleaning & concentrating kits (Zymo Research, CA, USA). The treated and cleaned DNA fragments were then spliced in-frame to pFUSE expression vector backbone by T4 DNA ligase (New England Biolabs, MA, USA). Ligation products were utilized to transform DH10B E. coli electro-competent cells, followed with positive selection for Zeocin-resistant colonies and DNA sequencing of the extracted plasmids.
Protein Expression and Purification.
Recombinant human CD38 extracellular domain, human CLL-1 extracellular domain, anti-hCLL-1 IgG, anti-hCLL-1 IgG HC-CD38 C-fusion (DAR2-ARC-IgG), and anti-hCLL-1 IgG HC-CD38 & LC-CD38 C-fusion (DAR4-ARC-IgG) were all expressed in Expi293F cells (Thermo Fisher Scientific, MA, USA). Sequence-confirmed pFUSE expression vector for human CD38 (240 μg), CLL-1 (240 μg), or antibody light chain (120 μg) combined with heavy chain (120 μg) in 12 mL of Opti-MEM medium (Thermo Fisher Scientific, MA, USA) was added with 960 μL of transfection-grade linear polyethylenimine hydrochloride at 1 mg mL−1 (Polysciences, PA, USA) for transfecting 240 mL of Expi293F cells cultured at a density of 2.5 million cells per mL per manufacturers’ instructions. Cells were then incubated with shaking (125 rpm) in a 37°C incubator with 5% CO2. On day 5 post transfection, media containing target proteins were collected. Cells were removed by a 2-step serial centrifugation with centrifugal force at 100 ×g for 10 minutes first, followed by 4,000 ×g for 30 minutes.
The recombinant human CD38 and CLL-1 extracellular domains were purified by the same protocol as previously reported.23 In brief, media with target proteins were first dialyzed against storage buffer for overnight (25 mM HEPES, 250 mM NaCl, pH 7.5) before applying to the Ni-NTA resin-based affinity chromatography. Dialyzed media were gradually loaded onto a gravity-flow column with 1 mL of Ni-NTA resin and let passing through the resin twice, followed by washing with 15 column volumes of wash buffer (20 mM Tris-HCl, 200 mM NaCl, 30 mM imidazole, pH 8). Recombinant proteins were eluted in 15 column volumes of elution buffer (20 mM Tris-HCl, 200 mM NaCl, 400 mM imidazole, pH 8) and dialyzed into PBS buffer (pH 7.4) for overnight at 4°C and then in the same freshly prepared buffer for another 8 hours under the same condition. Finally, recombinant proteins were concentrated with 10 kDa-cutoff concentrators (MilliporeSigma, MA, USA). The purified proteins were examined by Coomassie blue stained SDS-PAGE gels. Protein concentrations were determined by UV absorbance at 280 nm with a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, MA, USA) corrected by calculated molar extinction coefficients.
All antibodies were purified by protein G affinity chromatography resins (GenScript, NJ, USA) per the manufacturer’s protocol. Antibodies were eluted by 15 column volumes of 100 mM glycine (pH 2.7) and then dialyzed against PBS buffer (pH 7.4). After being concentrated by 30 kDa-cut-off concentrators (MilliporeSigma, MA, USA), the purified antibodies were examined by Coomassie blue stained SDS-PAGE gels. Protein concentrations were determined based on UV absorbance at 280 nm (corrected by calculated molar extinction coefficients) by a NanoDrop 2000C spectrophotometer.
Preparation of Drug-Linker Conjugate (2′-Cl-araNAD+-MMAF).
2′-Cl-araNAD+-MMAF was synthesized as reported previously.23 To a stirred solution of 2′-Cl-NAD+-N3 (7.7 mg, 0.01 mmol), CuSO4·5H2O (10.0 mg, 0.04 mmol, 4 eq) in H2O (0.5 mL) were added a solution of alkynyl-MMAF (9.2 mg, 0.012 mmol, 1.2 eq) in DMSO (0.2 mL), THPTA (86.9 mg, 0.2 mmol, 20 eq) and sodium-L-ascorbate (63.4 mg, 0.32 mmol, 32 eq) at room temperature. Then the reaction mixture was stirred at the same temperature until the reaction completed (monitored by HPLC). The product was purified via preparative HPLC (C18-A column, 150×10.0 mm, 5 μm) (mobile phase A: 0.1% formic acid (aq), mobile B: 0.1% formic acid in acetonitrile; flow rate = 2.0 mL/min; 0–2 min: 0–4% B, 2–4 min: 4–10% B, 4–6 min: 10–20% B, 6–12 min: 20–50% B, 12–17 min: 50–100% B, 17–20 min: 100–0% B) with detection of UV absorbance at 260 nm. Fractions containing the desired product were concentrated and lyophilized to yield the compound 2’-Cl-NAD+-MMAF (8.4 mg, 55%) as a colorless solid. HRMS (ESI) for C66H97ClN16O20P2Na22+ (M+2Na-2H)2+: Calcd.: 789.3085 Da; Obs: 789.3092 Da.
Preparation of ADCs and ADC Surrogates.
DAR2-ARC-IgG and DAR4-ARC-IgG at above 10 μM stock concentrations were combined with the drug-linker conjugate (2′-Cl-araNAD+-MMAF) in Tris buffer (50 mM Tris, pH 8.5) at a final molar concentration ratio of 1:200 (antibody : drug-linker conjugate) on ice for 1 hour. Then, buffer exchange was performed using PBS and filters with 30 kDa cut-off (MilliporeSigma, MA, USA) to remove free drug-linker conjugate.
To generate ADC surrogates with Alexa Fluor 488 or FITC as the payload, the linker 2′-Cl-araNAD+-N3 was first conjugated to DAR2-ARC-IgG or DAR4-ARC-IgG with the same method described above. Then, acquired fusion antibody-linkers were incubated with Alexa Fluor 488 DBCO (Click Chemistry Tools, AZ, USA) or DBCO-PEG3-FITC (CONJU-PROBE, CA, USA) in PBS buffer (pH 7.4) at a molar ratio of 1:50 (antibody-linker : fluorescent dye) for 30 minutes on ice. Finally, buffer exchange was performed using PBS (pH 7.4) and filters with 30 kDa cut-off to remove free surrogate payloads. The concentrations of generated ADCs and ADCs surrogates were determined by Bradford assay kits (Thermo Fisher Scientific, MA, USA).
In Vitro Plasma Stability of Alexa Fluor 488-Conjugated Surrogate ADCs.
Exendin-4 (GenScript, NJ, USA) at a concentration of 3 mg mL−1 was labeled with 1 mM N-hydroxysuccinimide (NHS)–fluorescein (Thermo Fisher Scientific, MA, USA) on ice for 2 hours in PBS buffer (pH 7.4). Then, the reaction mixture was passed through Zeba spin desalting columns (Thermo Fisher Scientific, MA) with molecular weight cut-off at 7 kDa for removing free NHS-fluorescein.
Acquired fluorescein-labeled exendin-4 (0.75 mg mL−1) and Alexa Fluor 488-conjugated surrogate DAR2-ARC-ADC (3 μM) and DAR4-ARC-ADC (3 μM) were incubated in fresh CD-1 mouse plasma containing 100 μg mL−1 of penicillin-streptomycin (Thermo Fisher Scientific, MA) in a 37°C incubator with 5% CO2 for 14 days. During the incubation, 1 μL of mixture was periodically extracted and frozen at −20°C until the final SDS-PAGE gel analysis. Prior to Coomassie staining, SDS-PAGE gels were imaged by a ChemiDoc Touch Imager (Bio-Rad, CA, USA) for the presence of intact conjugated surrogate ADCs or fluorescein-labeled exendin-4 under the fluorescence mode. The intensity of fluorescent bands on SDS-PAGE gels were quantified by Image Lab (Bio-Rad, CA, USA).
Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) for IgG Modification Analysis.
DAR2-ARC-IgG, DAR2-ARC-ADC, DAR4-ARC-IgG, and DAR4-ARC-ADC (~2 mg/mL) were deglycosylated with PNGase F (450 U mL−1) in the presence of 10 mM DTT, and mixed with 2,6 dihydroxyacetophenone (DHAP) solution (20 mg mL−1 in 50% acetonitrile:0.1% formic acid) at a ratio of 1:5 (by volume). Half microliter of each solution was spotted on a 384-Big Anchor MALDI target and let dry at room temperature. Crystalized samples were then analyzed using Bruker Rapiflex MALDI-TOF MS equipped with a Smartbeam 3D, 10 kHz, 355 nm Nd:YAG laser. The laser parameters were optimized as follows: scan range = 26 μm; number of shots per sample = 1000; laser frequency = 5000 Hz. The mass spectrometer was calibrated for high-mass range using protein A and trypsinogen standards under a linear mode. Data were analyzed using FlexAnalysis software and plotted using GraphPad Prism (CA, USA).
ADP-Ribosyl Cyclase Activities.
Acquired recombinant human CD38 extracellular domain, DAR2-ARC-IgG, DAR4-ARC-IgG, and the anti-hCLL-1 antibody were all diluted to the final concentration of 20 nM for CD38 or 10 nM for antibody or ARC-IgGs in 100 μL of PBS buffer (pH 7.4) with 100 μM of nicotinamide guanine dinucleotide (NGD+). The rate of cyclic guanine dinucleotide phosphate-ribose (cGDPR) generation was monitored by a Synergy H1 plate reader (BioTek, VT, USA) for its signature fluorescence (excitation at 300 nm; emission at 410 nm) for 5 minutes.
Binding to Recombinant Human CLL-1.
Purified recombinant human CLL-1 extracellular domain was coated overnight on 96-well ELISA plates at room temperature (Greiner Bio-One, NC) in 80 μL of PBS buffer (pH 7.4) at a final concentration of 50 μg mL−1. Then, coated wells were blocked with PBS (pH 7.4) containing 3% bovine serum albumin (BSA) (MilliporeSigma, MA, USA) for 2 hours at room temperature followed by a three-time-wash with 200 μL of 0.05% Tween-20-containing PBS (pH 7.4) buffer (PBST). Next, anti-CLL-1 antibody, DAR2-ARC-IgG, and DAR-4-ARC-IgG in PBS buffer (pH 7.4) were added into wells at a gradient of concentrations and let incubated at room temperature for 1 hour. After a three-time-wash by 0.05% PBST buffer (pH 7.4), 80 μL of 1000-fold diluted anti-human kappa light chain antibody with conjugated horse reddish peroxidase (HRP) (Thermo Fisher Scientific, MA, USA) in PBS buffer (pH 7.4) were added and let incubated at room temperature for 1 hour followed by a three-time-wash by 0.05% PBST buffer (pH 7.4). Finally, 80 μL of QuantaBlu fluorogenic substrate (Thermo Fisher Scientific, MA, USA) was added into wells. Fluorescence intensities (excitation at 325 nm; emission at 420 nm) were recorded by a Synergy H1 Plate Reader after a 5-minute incubation at room temperature. The results were analyzed by GraphPad Prism software for calculating the EC50 for different antibody constructs binding to the recombinant human CLL-1 extracellular domain.
Binding to Human CD16a and C1q.
Anti-hCLL-1 antibody, DAR2-ARC-IgG, and DAR4-ARC-IgG in PBS (pH 7.4) at various concentrations were incubated in the 96-well ELISA plates at room temperature for overnight. Wells were then blocked with PBS (pH 7.4) with 3% BSA for 2 hours at room temperature, followed by a three-time-wash with 200 μL of 0.05% PBST buffer (pH 7.4). Human CD16a-Fc chimera with a 6×His-tag (150 nM in PBS (pH 7.4), R&D Systems, MN) or human C1q protein (50 nM in PBS (pH 7.4), Quidel Corporation, CA) was added into the wells and incubated at room temperature for 2 hours. Following a three-time-wash with 200 μL of 0.05% PBST buffer (pH 7.4), an anti-6×His-tag antibody-HRP conjugate (50-fold dilution, Santa Cruz Biotechnology, TX) for bound CD16a or an anti-human C1q antibody-HRP conjugate (200-fold dilution, Invitrogen, MA) for bound C1q was added and incubated at room temperature for 2 hours. After a three-time-wash with 200 μL of 0.05% PBST buffer (pH 7.4), QuantaBlu fluorogenic substrate was added into wells. Fluorescence intensities (excitation at 325 nm; emission at 420 nm) were recorded by a Synergy H1 Plate Reader after a 5-minute incubation at room temperature. The results were analyzed by GraphPad Prism software for calculating the EC50 for binding of recombinant human CD16a and human C1q to different antibody constructs.
In Vitro Cytotoxicity of ADCs.
U937 or KG1a cells in RPMI1640 medium (Corning, NY, USA) supplemented with 10% FBS (Thermo Fisher Scientific, MA, USA) and 100 μg mL−1 penicillin-streptomycin (Thermo Fisher Scientific, MA, USA) at passage 3 were placed into 96-well plates (5,000 cells per well) (Corning, NY, USA) in a total volume of 100 μL per well. For the positive control wells, paclitaxel (MilliporeSigma, MA) was added to the final concentration of 5 μM. For the treatment group wells, DAR2-ARC-ADC and DAR4-ARC-ADC were added in a gradient of concentrations. For other control groups wells, anti-CLL-1 antibody, DAR2-ARC-IgG, DAR4-ARC-IgG, or 2’-Cl-araNAD+-MMAF were added in a gradient of concentrations. After a 72-hour incubation in a 37°C incubator with 5% CO2, 10 μL MTT reagent (Thermo Fisher Scientific, MA) was added into each well and let incubated for 2 hours in a 37°C incubator with 5% CO2. Then, 100 μL of lysis buffer (20% SDS and 50% dimethylformamide dissolved in water, pH 4.7) was added and let incubated for 1 hour at 37°C. Finally, absorbance at 580 nm was measured with a Synergy H1 plate reader (BioTek, VT, USA). The average readings from the positive control wells were designated as 0% viability, while the average readings from wells containing only cells with culture medium and antibiotics were designated as 100% viability. The data were fitted by the sigmoidal function in GraphPad Prism to calculate EC50 values.
Generation of Luciferase-Expressing U937 Cell Line.
HEK293T cells were maintained to achieve 70–95% confluence in DMEM medium (Corning, NY, USA) with 10% FBS. The pCDH-FFluc-GFP plasmid was used to transfect HEK293T cells together with packaging plasmids (pRRE, pVSVG, and pREV) with the calcium-phosphate transfection method. The lentivirus was collected and concentrated using filters with the molecular weight cut-off at 100 kDa (Thermo Fisher Scientific, MA, USA). U937 cells (approximately 5 × 105 cells) were then loaded on a 24-well plate together with the concentrated lentivirus for centrifugation at 2,200 rpm for 90 minutes. The cells were washed twice into fresh RPMI1640 medium on the next day. After the transduced U937 cells cultured in RPMI1640 medium supplemented with 10% FBS reached a total number of 3 million, cells were harvested by centrifugation at 100 ×g for 5 minutes and resuspended in DPBS (Corning, NY, USA) containing 10% FBS for sorting based on intracellular green fluorescence protein (GFP) intensity on a BD FACSAria Fusion Cell Sorter (CA, USA). Data were analyzed by FACSDiva (CA, USA) and top 5% of U937 cells with the strongest GFP fluorescence intensity were selected for the in vivo study.
Pharmacokinetics of Surrogate ADCs with FITC as the Payload.
Female CD-1 mice (Jackson Laboratory, ME, USA) at age of 6 weeks were randomly grouped into 5 mice per group. Surrogate ADCs with FITC as the payload at a dose of 5 mg kg−1 were administered by tail vein intravenous (I.V.) injection. Blood samples were collected at various time points throughout the 14-day-long study by tail venipuncture and processed for plasma samples by Multivette 600 μL lithium heparin gel collection tubes (Sarstedt Inc., Germany) per manufacturer’s protocol. To detect the presence of intact surrogate ADCs by ELISA, anti-FITC polyclonal antibody (Thermo Fisher Scientific, MA, USA) at 8 μg mL−1 in PBS buffer (pH 7.4) was first coated on high-binding ELISA plates (Greiner Bio-One, Austria) at 80 μL per well for overnight at room temperature. After a 3-time-wash by 0.05% PBST buffer (pH 7.4), 100-fold diluted plasma samples in PBS buffer (pH 7.4) were applied into wells and let incubated at room temperature for 2 hours followed by a 3-time-wash by 0.05% PBST buffer (pH 7.4). Next, 80 μL of 1000-fold diluted goat anti-human kappa light chain antibody with HRP (Thermo Fisher Scientific, MA, USA) in PBS buffer (pH 7.4) was added and let incubated for 1 hour at room temperature followed by a 3-time-wash by 0.05% PBST buffer (pH 7.4). Finally, 80 μL of QuantaBlu fluorogenic substrate (Thermo Fisher Scientific, MA) was added. Fluorescence intensity of each well (excitation at 325 nm; emission at 420 nm) was recorded by a Synergy H1 plate reader (BioTek, VT, USA). Standards for different concentrations of surrogate ADCs were prepared by diluting surrogate ADCs into fresh CD-1 mice plasma, and fluorescence signals corresponding to each standard concentration were acquired by the same method mentioned above. Concentrations of surrogate ADCs in plasma samples were calculated based on a standard curve of fluorescence-concentration extrapolated from the standards. Noncompartmental analysis in MatLab SimBiology (MathWorks, MA, USA) was employed to analyze pharmacokinetic parameters of surrogate ADCs.
In Vivo Efficacy Study.
Female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice at age of 6 weeks were purchased from the Jackson Laboratory (ME, USA). Each mouse was inoculated with 1 million luciferase-expressing U937 cells in 200 μL DPBS by tail vein I.V. injection. On day 2 post U937 cells implantation, mice were randomized into 5 per group and given 150 mg kg−1 of D-luciferin (Syd Labs, MA, USA), followed by imaging using an IVIS Lumina III Imaging System (PerkinElmer, MA, USA) for luminescence generated by luciferase-expressing U937 cells. On days 4, 7, and 10 post U937 cells inoculation, different groups of mice were given one of the following treatment options: (1) 200 μL of DPBS (vehicle only); (2) DAR4-ARC-IgG at 5 mg kg−1; (3) DAR2-ARC-ADC at 1 mg kg−1; (4) DAR2-ARC-ADC at 5 mg kg−1; (5) DAR4-ARC-ADC at 1 mg kg−1; (6) DAR4-ARC-ADC at 5 mg kg−1. In order to conduct both the interim efficacy analysis (when conditions of control group mice reach the endpoint per IACUC protocol) based on tissue sample analysis by flow cytometry as well as study for survival difference between groups, 2 groups of mice were given treatment option (3), (4), (5) or (6) with one group for interim efficacy analysis while the other group for survival analysis. Since the completion of first IVIS imaging, all live mice were continued to be imaged by IVIS weekly.
When mice in the control group (treatment option (1)) reached the humane endpoint, they were euthanized together with other groups of mice given the treatment option (2)-(6). Blood, spleen, and bone marrow samples were collected from each group, and samples were prepared into single cell suspension with red blood cells lysed by red blood cell lysis buffer (Biolegend, CA, USA) in DPBS containing 2% FBS. Prepared tissue samples were stained with anti-mouse CD34 PE antibody (119308, Biolegend, CA, USA), anti-mouse CD45 Pacific Blue antibody (MCD4528, Thermo Fisher Scientific, MA), anti-human CLL-1 APC antibody (353606, Biolegend, CA, USA), and propidium iodide (Thermo Fisher Scientific, MA). All samples were acquired by a BD LSRFortessa X-20 Cell Analyzer (CA, USA) and analyzed by FACSDiva and FlowJo software. Data for blood samples were first gated based on morphology (FSC-A/SSC-A) for peripheral blood myeloid cell population and then all live cells (propidium iodide-staining negative) populations were analyzed on fluorescence intensities of different antibodies used for staining. Data for spleen and bone marrow samples were first gated based on morphology (FSC-A/SSC-A) to remove non-cell debris, and then all live cells (propidium iodide-staining negative) populations were analyzed on fluorescence intensities of different antibodies used for staining. Gating strategies for all samples were shown in Figures S6–S8.
For toxicology study, part of the blood samples collected during the interim efficacy analysis study were processed for plasma samples by Multivette 600 μL lithium heparin gel collection tubes (Sarstedt Inc., Germany) per manufacturer’s protocol and frozen at −80°C. To measure the concentration of creatine in plasma, thawed plasma samples were first deproteinized by mixing with equal volume of 1.2 M trichloroacetic acid (Oakwood Chemicals, SC, USA) and the supernatants after centrifugation at 10,000 ×g for 5 minutes were then mixed with twice the volume of working solution, containing 38 mM picric acid premixed with equal volume of 1.2 M NaOH. After incubation at room temperature for 20 minutes, absorbance at 510 nm were recorded by a Synergy H1 plate reader (BioTek, VT, USA). Creatine powder (MilliporeSigma, MA, USA) was prepared into a series of concentrations of standards and utilized in the assay mentioned above for extrapolating a standard curve.
To measure alanine aminotransferase (ALT) activity in plasma, 5 μL of thawed plasma samples were first mixed with 25 μL of substrate solution (0.2 M alanine and 2 mM 2-oxoglutarate in 0.1 M disodium phosphate, pH 7.4) for 1 hour at 37°C. Then, the mixture was added with 25 μL of 1 mM 2,4-dinitrophenylhydrazine in 1 M HCl for 20 minutes at room temperature, followed by addition of 250 μL of 0.5 M NaOH. Absorbance at 510 nm was then measured by a Synergy H1 plate reader (BioTek, VT, USA). Pyruvate powder (MilliporeSigma, MA, USA) was prepared into a series of concentrations of standards and used to replace plasma samples in the above protocol for extrapolating a standard curve.
Statistical Analysis.
Two-tailed unpaired t tests were performed for comparison between two groups. Tumor growth curves of the control and treatment groups were analyzed using two-tailed unpaired t tests. P < 0.05 was defined as statistically significant. Data are shown as mean ± SD. Kaplan-Meier method was adopted to compare survival time between two groups of mice. All statistical analyses were performed using GraphPad Prism.
Supplementary Material
Acknowledgements
This work was supported in part by Sharon L. Cockrell Cancer Research Fund, STOP CANCER Research Career Development Award (to Y. Z.), Department of Defense CDMRP Career Development Award W81XWH-19-1-0272 (to Y.Z.), National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) grant R35GM137901 (to Y. Z.), and Tobacco Related-Disease Research Program New Investigator Award T30KT1021 (to Y.Z.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- (1).Ford C, Newman C, Johnson J, Woodhouse C, Reeder T, Rowland G, and Simmonds R (1983) Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer, British journal of cancer 47, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Walter RB (2020) Brief overview of antibody–drug conjugate therapy for acute leukemia, Taylor & Francis. [DOI] [PubMed] [Google Scholar]
- (3).Li F, Sutherland MK, Yu C, Walter RB, Westendorf L, Valliere-Douglass J, Pan L, Cronkite A, Sussman D, and Klussman K (2018) Characterization of SGN-CD123A, a potent CD123-directed antibody–drug conjugate for acute myeloid leukemia, Molecular cancer therapeutics 17, 554–564. [DOI] [PubMed] [Google Scholar]
- (4).Jain N, Smith SW, Ghone S, and Tomczuk B (2015) Current ADC linker chemistry, Pharmaceutical research 32, 3526–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, and Zabinski RF (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate, Clinical cancer research 10, 7063–7070. [DOI] [PubMed] [Google Scholar]
- (6).Wakankar AA, Feeney MB, Rivera J, Chen Y, Kim M, Sharma VK, and Wang YJ (2010) Physicochemical stability of the antibody− drug conjugate trastuzumab-DM1: changes due to modification and conjugation processes, Bioconjugate chemistry 21, 1588–1595. [DOI] [PubMed] [Google Scholar]
- (7).Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, and Stafin K (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids, Proceedings of the National Academy of Sciences 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bruins JJ, Westphal AH, Albada B, Wagner K, Bartels L, Spits H, van Berkel WJ, and van Delft FL (2017) Inducible, site-specific protein labeling by tyrosine oxidation–strain-promoted (4+ 2) cycloaddition, Bioconjugate chemistry 28, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li X, Nelson CG, Nair RR, Hazlehurst L, Moroni T, Martinez-Acedo P, Nanna AR, Hymel D, Burke TR Jr, and Rader C (2017) Stable and potent selenomab-drug conjugates, Cell chemical biology 24, 433–442. e436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lin S, Yang X, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, and Toste FD (2017) Redox-based reagents for chemoselective methionine bioconjugation, Science 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Okeley NM, Toki BE, Zhang X, Jeffrey SC, Burke PJ, Alley SC, and Senter PD (2013) Metabolic engineering of monoclonal antibody carbohydrates for antibody–drug conjugation, Bioconjugate chemistry 24, 1650–1655. [DOI] [PubMed] [Google Scholar]
- (12).Tian F, Lu Y, Manibusan A, Sellers A, Tran H, Sun Y, Phuong T, Barnett R, Hehli B, and Song F (2014) A general approach to site-specific antibody drug conjugates, Proceedings of the National Academy of Sciences 111, 1766–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).VanBrunt MP, Shanebeck K, Caldwell Z, Johnson J, Thompson P, Martin T, Dong H, Li G, Xu H, and D’Hooge F (2015) Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody–drug conjugates using click cycloaddition chemistry, Bioconjugate chemistry 26, 2249–2260. [DOI] [PubMed] [Google Scholar]
- (14).Zhou Q, Stefano JE, Manning C, Kyazike J, Chen B, Gianolio DA, Park A, Busch M, Bird J, and Zheng X (2014) Site-specific antibody–drug conjugation through glycoengineering, Bioconjugate chemistry 25, 510–520. [DOI] [PubMed] [Google Scholar]
- (15).Zhu Z, Ramakrishnan B, Li J, Wang Y, Feng Y, Prabakaran P, Colantonio S, Dyba MA, Qasba PK, and Dimitrov DS (2014) Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar, In MAbs, pp 1190–1200, Taylor & Francis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dennler P, Chiotellis A, Fischer E, Brégeon D, Belmant C, Gauthier L, Lhospice F, Romagne F. o., and Schibli R (2014) Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates, Bioconjugate chemistry 25, 569–578. [DOI] [PubMed] [Google Scholar]
- (17).Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin RG, Tran T-T, Ho W-H, Farias S, Casas MG, and Abdiche Y (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates, Chemistry & biology 20, 161–167. [DOI] [PubMed] [Google Scholar]
- (18).Dorywalska M, Strop P, Melton-Witt JA, Hasa-Moreno A, Farias SE, Galindo Casas M, Delaria K, Lui V, Poulsen K, and Sutton J (2015) Site-dependent degradation of a non-cleavable auristatin-based linker-payload in rodent plasma and its effect on ADC efficacy, PLoS One 10, e0132282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stefan N, Gébleux R, Waldmeier L, Hell T, Escher M, Wolter FI, Grawunder U, and Beerli RR (2017) Highly potent, anthracycline-based antibody–drug conjugates generated by enzymatic, site-specific conjugation, Molecular cancer therapeutics 16, 879–892. [DOI] [PubMed] [Google Scholar]
- (20).Beerli RR, Hell T, Merkel AS, and Grawunder U (2015) Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency, PloS one 10, e0131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Woitok M, Klose D, Di Fiore S, Richter W, Stein C, Gresch G, Grieger E, Barth S, Fischer R, and Kolberg K (2017) Comparison of a mouse and a novel human scFv-SNAP-auristatin F drug conjugate with potent activity against EGFR-overexpressing human solid tumor cells, OncoTargets and therapy 10, 3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nanna AR, Li X, Walseng E, Pedzisa L, Goydel RS, Hymel D, Burke TR Jr, Roush WR, and Rader C (2017) Harnessing a catalytic lysine residue for the one-step preparation of homogeneous antibody-drug conjugates, Nature communications 8, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dai Z, Zhang X-N, Nasertorabi F, Cheng Q, Li J, Katz BB, Smbatyan G, Pei H, Louie SG, and Lenz H-J (2020) Synthesis of site-specific antibody-drug conjugates by ADP-ribosyl cyclases, Science Advances 6, eaba6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lowenberg B, Downing JR, and Burnett A (1999) Acute myeloid leukemia, New England Journal of Medicine 341, 1051–1062. [DOI] [PubMed] [Google Scholar]
- (25).Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, Jongeneelen MA, Visser TJ, Bijl N, and Geuijen CA (2004) C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia, Cancer research 64, 8443–8450. [DOI] [PubMed] [Google Scholar]
- (26).Lin T. -y., Zhu Y, Li Y, Zhang H, Ma A-H, Long Q, Keck J, Lam KS, Pan C. -x., and Jonas BA (2019) Daunorubicin-containing CLL1-targeting nanomicelles have anti-leukemia stem cell activity in acute myeloid leukemia, Nanomedicine: Nanotechnology, Biology and Medicine 20, 102004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lu H, Zhou Q, Deshmukh V, Phull H, Ma J, Tardif V, Naik RR, Bouvard C, Zhang Y, and Choi S (2014) Targeting human C‐type lectin‐like molecule‐1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia, Angewandte Chemie International Edition 53, 9841–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).De Togni E, Kim MY, Cooper ML, Ritchey J, O’Neal J, Niswonger J, and DiPersio JF (2018) Chimeric Antigen Receptor T Cells Specific for CLL-1 for Treatment of Acute Myeloid Leukemia, Blood 132, 2205–2205.30266775 [Google Scholar]
- (29).Jiang Y-P, Liu BY, Zheng Q, Panuganti S, Chen R, Zhu J, Mishra M, Huang J, Dao-Pick T, and Roy S (2018) CLT030, a leukemic stem cell–targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia, Blood advances 2, 1738–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zheng B, Yu S-F, Del Rosario G, Leong SR, Lee GY, Vij R, Chiu C, Liang W-C, Wu Y, and Chalouni C (2019) An Anti–CLL-1 Antibody–Drug Conjugate for the Treatment of Acute Myeloid Leukemia, Clinical Cancer Research 25, 1358–1368. [DOI] [PubMed] [Google Scholar]
- (31).Zhao X, Singh S, Pardoux C, Zhao J, Hsi ED, Abo A, and Korver W (2010) Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia, Haematologica 95, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.