

Abstract
A series of novel acyclic nucleoside phosphonates (ANPs) was synthesized as potential adenylate cyclase inhibitors, where the adenine nucleobase of adefovir (PMEA) was replaced with a 5-substituted 2-aminothiazole moiety. The design was based on the structure of MB05032, a potent and selective inhibitor of fructose 1,6-bisphosphatase and a good mimic of adenosine monophosphate (AMP). From the series of eighteen novel ANPs, which were prepared as phosphoroamidate prodrugs, fourteen compounds were potent (single digit micromolar or submicromolar) inhibitors of Bordetella pertussis adenylate cyclase toxin (ACT), mostly without observed cytotoxicity in J774A.1 macrophage cells. Selected phosphono diphosphates (nucleoside triphosphate analogues) were potent inhibitors of ACT (IC50 as low as 37 nM) and B. anthracis edema factor (IC50 as low as 235 nM) in enzymatic assays. Furthermore, several ANPs were found to be selective mammalian AC1 inhibitors in HEK293 cell-based assays (although with some associated cytotoxicity) and one compound exhibited selective inhibition of mammalian AC2 (only 12% of remaining adenylate cyclase activity) but no observed cytotoxicity. The mammalian AC1 inhibitors may represent potential leads in development of agents for treatment of human inflammatory and neuropathic pain.
Keywords: acyclic nucleoside phosphonates, adefovir, adenylate cyclase, Bacillus anthracis, Bordetella pertussis, inhibitors, prodrugs
Graphical Abstract
1. Introduction
Adenylate cyclases (ACs) are enzymes with key regulatory roles that are essential to all cells. Several distinct classes and numerous isoforms of ACs have been described, all catalyzing the same reaction: conversion of adenosine triphosphate (ATP) into 3’,5’-cyclic adenosine monophosphate (cAMP) and pyrophosphate. [1–3] In eukaryotes, cAMP acts as a ubiquitous second messenger, produced in response to extracellular stimulus, triggering a variety of intracellular processes.[1,2] The cytosolic concentration of cAMP is stringently regulated by a diverse set of ACs (cAMP formation) and phosphodiesterases (PDEs, cAMP degradation) isoforms and any alteration in the intracellular cAMP concentration has a profound effect on essential cellular processes such as metabolism, gene transcription, enzyme regulation, hormone secretion, and sensory transduction[4,5]
Numerous pathogenic bacteria produce, among other virulence factors, adenylate cyclase toxins that alter the cAMP concentration in cells, thus, disrupting key cellular processes such as signaling pathways.[6] These toxins facilitate the invasion and survival of the pathogen in the host and contribute to pathogenesis of infectious diseases.[6] The most extensively studied bacterial adenylate cyclase toxins are adenylate cyclase toxin from Bordetella pertussis (ACT) and edema factor from Bacillus anthracis (EF).[6–8] It has been speculated, that inhibition of bacterial adenylate cyclase toxins could be exploited as potential targets for prophylactic or therapeutic strategies to combat infectious diseases.[9,10] Furthermore, selective modulation of mammalian ACs’ activity represents also a promising strategy for the development of new agents to treat various human neurological and inflammatory diseases.[11,12]
Several inhibitors of B. pertussis ACT and B. anthracis EF have been previously reported. Seifert and co-workers discovered (N-methyl)anthraniloyl-substituted nucleoside triphosphates as potent competitive inhibitors of bacterial and mammalian ACs.[10,13–16] Jiao et al. have also reported covalent small-molecule inhibitors of EF which bear an electrophilic fluorosulfonylbenzene group.[17] Moreover, acyclic nucleoside phosphonates (ANPs),[18] a large group of nucleotide analogues with a broad spectrum of biological activities, have been reported to inhibit activity of bacterial ACs. It was described that the approved drug for HBV infection, adefovir dipivoxil (bis(POM)PMEA), upon conversion to its active metabolite adefovir diphosphate (PMEApp, 1, Fig. 1), effectively inhibited B. pertussis ACT[19] and B. anthracis EF,[20] In addition, it has been shown that various phosphoroamidate (bisamide) prodrugs of PMEA, such as bis(l-phenylalanine isopropyl ester) PMEA (2), possess enhanced plasma stability and lower toxicity compared to adefovir dipivoxil, but exhibited somewhat lower efficacy.[21] Subsequently, a large structure-activity relationship (SAR) study was also performed, where several series of ANPs were prepared, modified either at the heterocyclic base[22,23] or at the acyclic side chain.[24,25] Many of these compounds were potent inhibitor of ACT/EF and/or human ACs.
Fig. 1.
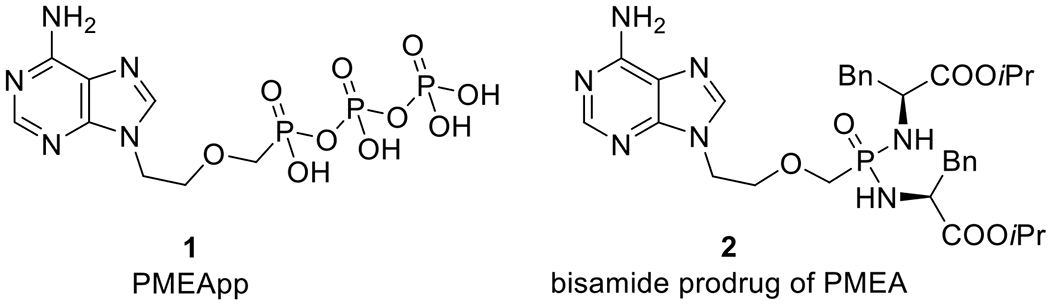
Structures of PMEApp (1) and bis(l-phenylalanine isopropyl ester) PMEA (2).
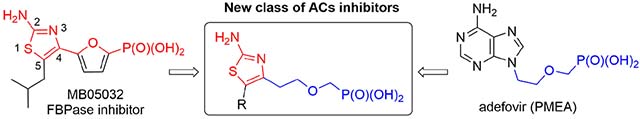
Based on the extensive SAR studies (summarized in the previous work[23]), which dealt with modifications of the promising adefovir (PMEA, 3, Fig. 2) scaffold, we decided to focus on non-purine mimics of the adenine nucleobase. 2-Aminothiazole derivative MB05032 (Fig. 2),[26–29] a potent and selective inhibitor of fructose 1,6-bisphosphatase (FBPase), was selected as a good AMP mimic. Thus, a novel series of ANPs were designed, which were derived from 5-substituted 2-aminothiazole and bearing either a phosphonomethoxymethyl (compounds I, Fig. 2) or a phosphonomethoxyethyl (compounds II, Fig. 2) side chain. In order to test the ability to inhibit ACT and mACs in cell-based assays, all ANPs were prepared in the form of cell-permeable phosphoroamidate (bisamide) prodrugs bearing non-toxic l-phenylalanine isopropyl ester moieties (as in compound 2, Fig. 1) that had been successfully used in the previous SAR studies.[21,23]
Fig. 2.
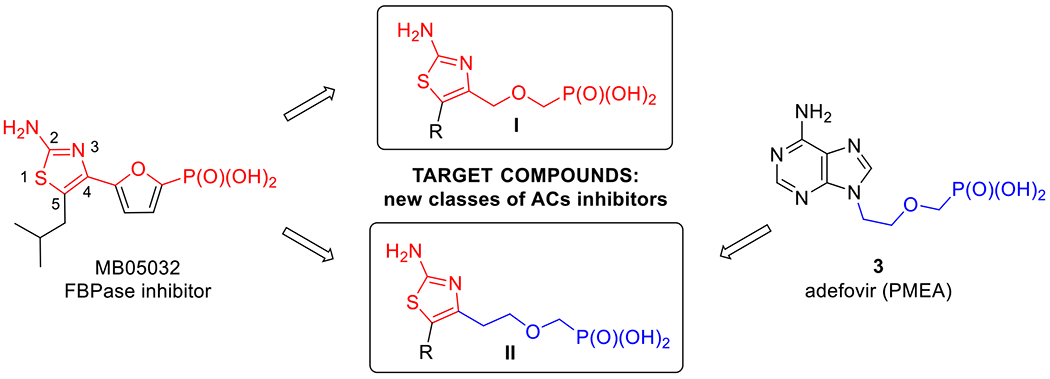
Drug design strategy combining FBPase inhibitor MB05032 and adefovir (3).
For direct evaluation of the inhibitory effects on bacterial enzymes (ACT and EF), selected candidates were subsequently prepared as free phosphonates (nucleotide analogues) and as phosphono diphosphates (nucleoside triphosphate analogues).
2. Results and discussion
2.1. Synthesis
First, as a proof of concept, commercially available MB05032 (CAS: 261365-11-1) was converted by the standard procedure[30] into bisamide prodrug 4 (48%), where also the corresponding monoamidate 5 (24%) was isolated from the reaction mixture (Scheme 1). Using the modified morpholidate methodology reported by Holý and Rosenberg,[31] we also prepared phosphono diphosphate derivative 6 in a 48% yield. The prepared compounds were evaluated as potential inhibitors of ACT enzyme. Desired phosphoroamidate 4 was a low-micromolar ACT inhibitor (IC50 = 2.12 μM, Table 2) in the cell-based assay and the triphosphate analogue 6 inhibited the ACT enzyme with an IC50 = 10.7 μM (Table 3). The ability of the compounds to inhibit ACT was our proof of concept that 2-aminothiazole derivatives represent a good mimic of the adenine-based analogues. Preliminary docking of target compounds (structure II, Fig. 2) into the re-interpreted[25] crystal structure of ACT with bound PMEApp (PDB: 1ZOT, resolution 2.2 Å)[19] suggested that aryl substituents at C-5 position of the 2-aminothiazole moiety may be superior to the 2-methylpropyl substituent of MB05032. Thus, series of 5-(het)aryl substituted 2-aminothiazole analogues derived from PMEA were designed and synthesized.
Scheme 1.
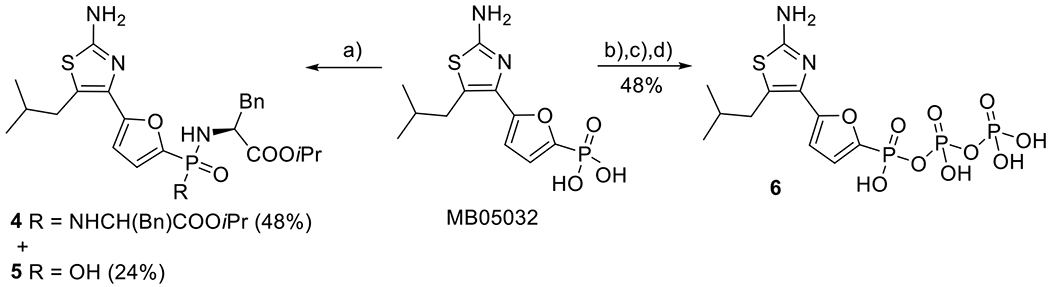
Synthesis of prodrugs 4 and 5 and phosphono diphosphate 6 derived from MB05032: a) TMSBr, pyridine, RT then (l)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C. b) TMSBr/CH3CN, RT; c) DCC, morpholine, tBuOH, H2O, reflux; d) (Bu3N)2P2O7, Bu3N, DMF.
Table 2.
The inhibition effect of prepared bisamides on bacterial adenylyl cyclase (ACT) and cell viability in J744A.1 murine macrophages and their effects on mammalian ACs isoforms in HEK293 cells stably transfected with AC1, AC2, and AC5.
| Compound | ACT inhibition | Viability | Response of Control [%]c | ||
|---|---|---|---|---|---|
| IC50 [μM]a | [%]b | AC1 | AC2 | AC5 | |
| 4 | 2.12 ± 0.58 | 88 | ND | ND | ND |
| 5 | 14.2 ± 4.3 | 86 | ND | ND | ND |
| 10 | >10 | 83 | 74 ± 3 | 143 ± 11 | 113 ± 1 |
| 13 * | 4.68 ± 1.56 | 54 | 61 ± 9 | 101 ± 10 | 147 ± 9 |
| 18 | >10 | 72 | 73 ± 18 | 185 ± 32 | 108 ± 13 |
| 21a * | 1.82 ± 0.58 | 112 | 48 ± 3 | 82 ± 21 | 177 ± 18 |
| 21b | 2.62 ± 0.62 | 99 | 37 ± 8 | 136 ± 25 | 99 ± 13 |
| 21c * | 0.45 ± 0.17 | 99 | 36 ± 10 | 62 ± 13 | 129 ± 4 |
| 21d | 0.68 ± 0.08 | 81 | 54 ± 7 | 156 ± 2 | 197 ± 14 |
| 21e | 1.72 ± 0.38 | 44 | 38 ± 9 | 150 ± 26 | 169 ± 26 |
| 21f * | 1.16 ± 0.01 | 57 | 29 ± 4 | 160 ± 31 | 159 ± 7 |
| 21g | 1.43 ± 0.04 | 86 | 32 ± 6 | 119 ± 13 | 172 ± 16 |
| 21h | 1.62 ±0.36 | 35 | 25 ±7 | 110 ± 9 | 140 ± 29 |
| 21i | >10 | 115 | 39 ± 3 | 134 ± 26 | 117 ± 4 |
| 21j | 0.26 ± 0.05 | 72 | 38 ± 9 | 104 ± 7 | 106 ± 8 |
| 21k | 8.45 ± 0.81 | 106 | 57 ± 3 | 109 ± 5 | 78 ± 4 |
| 21l | 0.66 ± 0.18 | 99 | 77 ± 6 | 12 ± 27 | 73 ± 5 |
| 21m | 4.30 ± 0.29 | 99 | 75 ± 12 | 122 ± 41 | 101 ± 13 |
| 21n | >10 | 94 | 64 ± 16 | 97 ± 28 | 77 ± 3 |
| 23 | 0.93 ± 0.19 | 36 | 23 ± 4 | 165 ± 41 | 131 ± 4 |
|
| |||||
| 2 | 0.15 ± 0.03 | 93 | 64 ± 9 | 200 ± 45 | 99 ± 8 |
| ST034307d | NDg | ND | 14 ± 13 | 398 ± 96 | 196 ± 31 |
| SKF83566e | ND | ND | 78 ± 8 | (−)7 ± 11 | 94 ± 5 |
| SQ22536f | ND | ND | 46 ± 2 | 27 ± 12 | 45 ± 15 |
Compound concentration that causes a 50% decrease in ACT-induced cAMP accumulation in J774A.1 macrophages; data are the mean ± SD of at least three independent experiments.
Percent cell viability in J774A.1 cells (n = 3) at a fixed prodrug concentration (10 μM) versus untreated control.
Data are the mean ± SEM relative to the control response (100%) of at least two independent experiments at 30 μM concetration.
ST034307 is a selective inhibitor of AC1.
SKF83566 is a selective inhibitor of AC2.
SQ22536 is a nonselective P-site inhibitor.
ND: not determined.
<60% cell viability in HEK-AC1 cells (n = 3).
Table 3.
Direct inhibition of bacterial adenylyl cyclases – ACT from Bordetella pertussis and EF from Bacillus anthracis – by selected ANP-diphosphates in a cell-free assay.
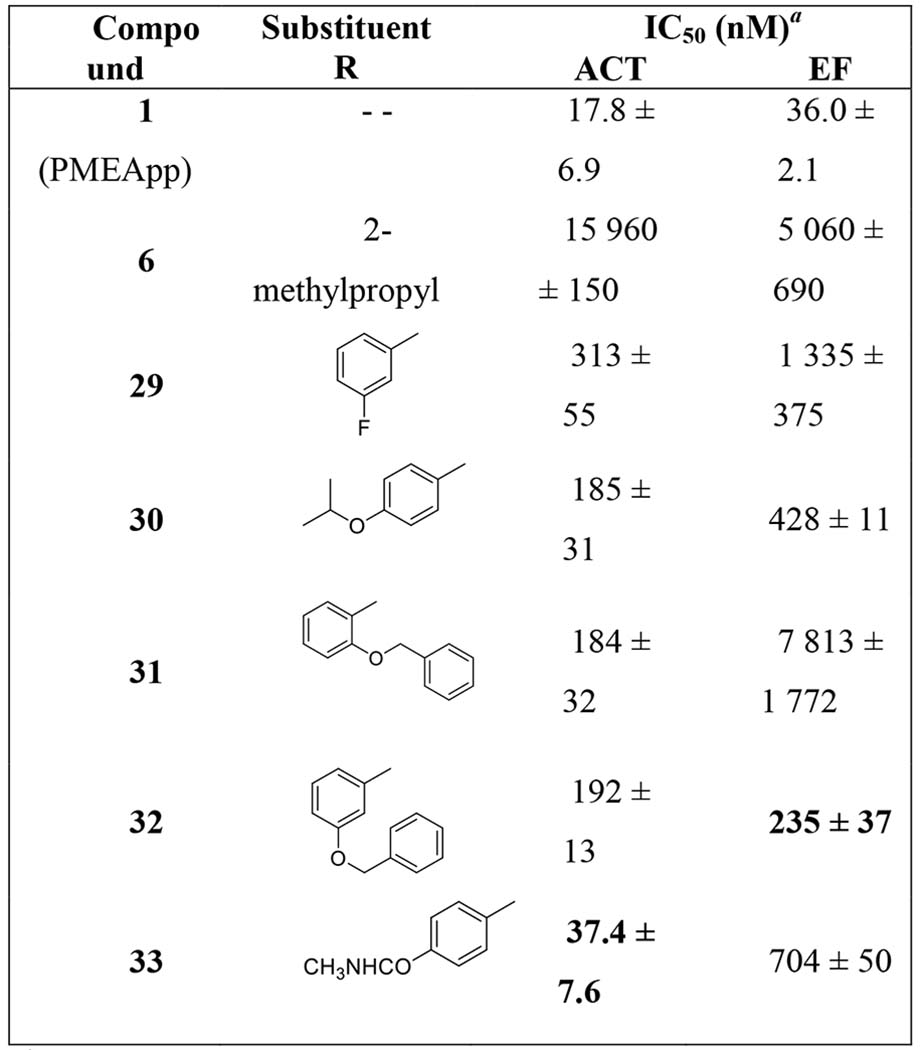
|
Data represent the mean IC50 values ± SD of at least three independent experiments, calculated by GraphPad Prism software from dose-response curves.
2-Aminothiazole derivatives with phosphonomethoxymethyl side chain (Scheme 2) were prepared from the N-Boc protected 4-hydroxymethyl derivative 7.[32] The hydroxymethyl group of compound 7 was alkylated using bromomethyl phosphonate and sodium hydride and phosphonate 8 was obtained in a 54% yield. Unsubstituted derivative 10 was prepared by removal of the Boc group with trifluoroacetic acid (to yield compound 9, 91%) and subsequent conversion of the phosphonate diester into the bisamide prodrug using l-phenylalanine isopropyl ester under the previously reported conditions.[30] Bromination of compound 8 (to yield 5-bromothiazole derivative 11, 63%), followed by Suzuki reaction[33] with phenyl boronic acid and Boc group removal, afforded 5-phenylthiazolo derivative 12 in a 74% yield (from 11). Interestingly, Suzuki reaction of 5-bromothiazole with unprotected 2-amino group did not proceed under the identical conditions (data not shown). Final bisamidate 13 was then obtained from compound 12 in a 42% yield.
Scheme 2.
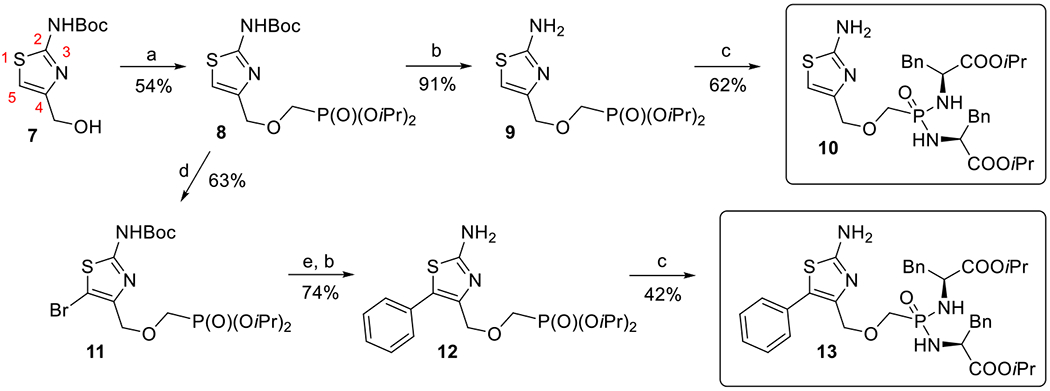
Synthesis of 2-aminothiazole derivatives with phosphonomethoxymethyl linker: a) NaH, BrCH2P(O)(OiPr)2, RT; b) TFA, RT; c) TMSBr, pyridine, RT then (l)-NH2CH(Bn)COOiPr·HCl, Et3N, Aldrithiol-2, Ph3P, pyridine, 65 °C; d) Br2, CCl4, RT; e) PhB(OH)2, Pd(PPh3)4, Cs2CO3, dioxane, H2O, 80 °C.
2-Aminothiazole derivatives bearing the phosphonomethoxyethyl side chain (Scheme 3) were prepared in a similar manner as the phosphonomethoxymethyl derivatives (Scheme 2). The amino group of commercially available ethyl (2-aminothiazol-4-yl)acetate (14) was first protected using Boc group, and then the ester group was reduced with an excess of NaBH4 to afford alcohol 15.[34] Compound 15 was converted by alkylation with bromomethanephosphonate into phosphonate 16 (81%), from which the Boc group was removed to yield 2-aminothiazole derivative 17 in a 93% yield. Final bisamidate 18 was prepared in a 48% yield by treatment of compound 17 using the standard procedure.[30]
Scheme 3.
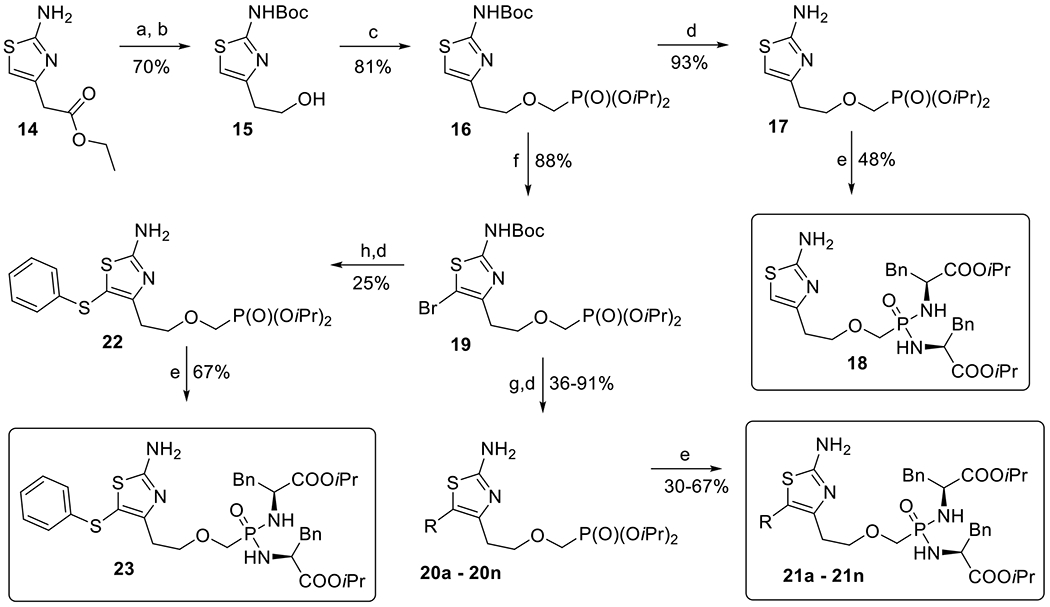
Synthesis of 2-aminothiazole derivatives with phosphomethoxyethyl linker: a) Boc2O, TEA, THF, 50 °C; b) NaBH4, EtOH, RT; c) NaH, BrCH2P(O)(OiPr)2, RT; d) TFA, RT; e) TMSBr, pyridine, RT then (L)-NH2CH(Bn)COOiPr·HCl, Et3N, Aldrithiol-2, Ph3P, pyridine, 65 °C; f) Br2, CCl4, RT; g) RB(OH)2, Pd(PPh3)4, Cs2CO3, dioxane, H2O, 80 °C; h) thiophenol, CuI, phenantroline, Et3N, toluene, 110 °C.
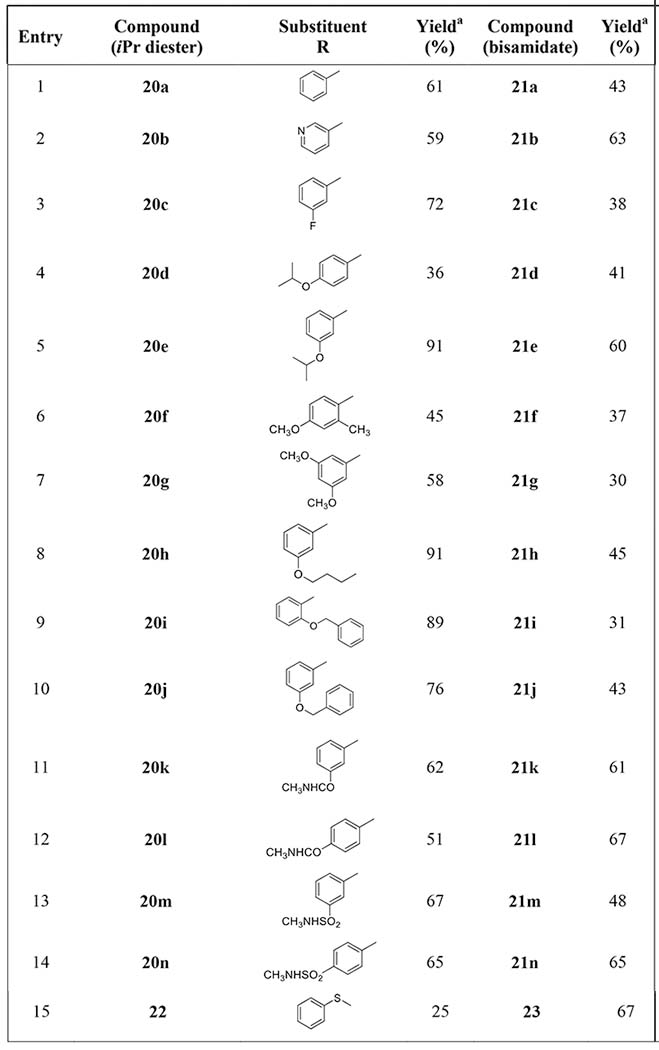
Bromination of 2-aminothiazole 16 with bromine in CCl4 afforded 5-bromo-2-aminothiazole 19 in a 88% yield (Scheme 3), which was used as a key intermediate for the introduction of various aryl substituents into the position C-5, namely derivatives 20a–20n and 22 (Scheme 3, Table 1). Compounds 20a–20n were prepared in moderate to high yields (36–91%, Table 1) by Suzuki coupling[33] of 5-bromo-2-aminothiazole 19 with commercially available boronic acids and by subsequent Boc group removal with TFA. Ullmann reaction of bromo derivative 19 with thiophenol, CuI and phenanthroline as a ligand,[35] followed by the standard Boc group removal, afforded phenylsulfanyl derivative 22 in a 25% yield (two steps). All phosphonate diesters 20a–20n and 22 were converted by the above described procedure[30] to the corresponding bisamides 21a–21n and 23, respectively, in good to moderate yields (30–67%, Scheme 3, Table 1).
Table 1.
Compounds prepared from 5-bromo-2-aminothiazole 19. Yields of Suzuki crosscoupling reaction (compounds 20a–20n), Ullmann reaction (product 22) and prodrug synthesis (compounds 21a–21n and 23).
|
Isolated yields.
Finally, in order to evaluate the most promising analogues in enzymatic assays using bacterial ACs, selected phosphonate diesters (20c, 20d, 20i, 20j, 20l) were converted into the corresponding free phosphonates 24–28, using a modified procedure of McKenna and Schmidhauser,[36] or the phosphono diphosphates 29–33 (Scheme 4), following the literature procedure using 1,1′-carbonyldiimidazole.[37]
Scheme 4.
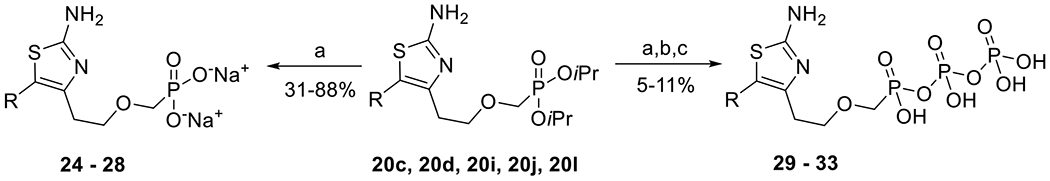
Synthesis of phosphonates 24–28 and phosphono diphosphates 29–33: a) TMSBr, pyridine, RT then DOWEX (50WX8 Na+); b) TBAH, 2 M aq. TEAB, MeOH, dioxane, RT; c) 1,1′-carbonyldiimidazole, DMSO, RT then (Bu3N)2P2O7, DMSO, RT.
2.2. Inhibition of ACT in the cell-based assay
All final 2-aminothiazoles bearing the phosphonate moiety in the form of bisamide prodrugs (compounds 4, 5, 10, 13, 18, 21a-21n, and 23) were tested for their ability to inhibit adenylate cyclase toxin’s (ACT) activity in J774A.1 macrophage cells (Table 2).[23] Murine macrophages J774.1 were pre-incubated with various concentrations of the tested compound and subsequently exposed to Bordetella pertussis ACT. The cells were lysed, and the amount of cAMP produced was determined. The compounds were also evaluated for their effects on the viability of the J774A.1 cells (Table 2).
Interestingly, compounds 10 and 18, 2-aminothiazole derivatives not substituted at C-5, did not inhibit ACT. All other compounds tested, except for 21i and 21n, which were neither potent nor cytotoxic, were able to inhibit cAMP production with a single digit micromolar or submicromolar IC50 value range (Table 2). Furthermore, the length of the acyclic moiety seemed to be crucial for potential activity, as observed for 5-phenyl substituted 2-aminothiazoles 13 and 21a: compound 21a bearing the longer phosphonomethoxyethyl moiety was about 2.6 times more potent and less cytotoxic in J774A.1 cells, compared to analogue 13 with the shorter phosphonomethoxymethyl side chain (Table 2). The most potent (submicromolar) inhibitors of cAMP production in the series were compounds 21c, 21d, 21j, 211 and 23, however, some of them exhibited an increased cytotoxity to J774A.1 cells (compound 23) or to HEK-AC1 (compound 21c) cells, which were used for the screening of mammalian ACs’ activity. Compounds that showed no signs of cytotoxicity for either J774A.1 macrophage cells or HEK-AC1 cells but retained a potent inhibitory activity towards ACT were 3-pyridyl derivative 21b, 4-isopropoxyphenyl derivative 21d, 3,5-dimethoxyphenyl derivative 21g, 4-(N-methylaminocarbonyl)phenyl derivative 21l, and 3-(N-methylsulphonamido)phenyl derivative 21m. To sum up, the highest potency to inhibit cAMP production with no marked effect on J774A.1 cells’ viability was observed for compounds bearing a p-substituted phenyl ring in C-5 position of the 2-aminothiazole moiety, namely compounds 21d and 211, with IC50 values 680 nM and 660 nM, respectively (Table 2). All of the compounds tested, however, exhibited a lower potency to inhibit ACT compared to the previously reported data[23] for the phosphoroamidate prodrugs of adefovir (compound 2 had an IC50 = 150 nM).
2.3. Inhibition of mammalian ACs
Next, all bisamide prodrugs (i.e. compounds 10, 13, 18, 21a–21n, and 23) were evaluated as potential inhibitors of mammalian adenylate cyclases’ (mACs) activity. Specifically, the AC isoforms: AC1, AC2, and AC5 – representing the three major mAC subfamilies – were selected to explore the potential selectivity of the studied compounds between bacterial and mammalian ACs, but to also determine the selectivity among the mAC subfamilies (Table 2). Cyclic AMP (cAMP) values after treatment with compounds were normalized to the response (100%) observed in cells treated only with the respective adenylate cyclase isoform selective stimulant.
Many of the compounds showed evident to moderate selectivity to inhibit AC1 over AC2 and/or AC5 (Table 2). Several compounds (at 30 μM) even seemed to potentiate the activity of either AC2 (e.g. 18 and 23), AC5 (e.g. 21a and 21g) or both AC2/AC5 (21d–21f). Potentiation of AC2 was also observed for the previously described AC1 inhibitor, ST034307.[38]
Compound 21h was found to be the most efficacious selective AC1 inhibitor (25% of remaining activity) while being nontoxic in both J774A.1 and the HEK-AC1 cells. Other AC1 selective inhibitors were derivatives 21f and 23 (29% and 23% of remaining activity, respectively) but these compounds exhibited some level of cytotoxicity. Interestingly, compound 211 was the most selective AC2 inhibitor (only 12% of remaining activity) while exhibiting no cytotoxicity.
2.4. Direct inhibition of ACT and EF activity in cell-free assays
It is expected that the masking groups (l-phenylalanine isopropyl ester) on the phosphonate moiety are cleaved off in the cell by the putative mechanism reported for the bisamide prodrug cleavage,[39,40] followed by phosphorylation of free phosphonic acid to form phosphono diphosphate, a nucleoside triphosphate analogue, which represents the active species that is able to inhibit adenylate cyclases.
Thus, based on the results of the cell-based assay (Table 2), five compounds were selected for their direct evaluation in the enzymatic assays: the four most potent ACT inhibitors, bisamides 21c, 21d, 21j, and 211, and for comparison, one of the less active analogues, bisamide 21i. Corresponding free phosphonic acids 24–28 and phosphono diphosphates 29–33 were prepared (Scheme 4) and subsequently evaluated (in comparison with compound 1, PMEApp)[19,20] as inhibitors of commercially available B. pertussis ACT and B. anthracis EF (Table 3). While free phosphonates 24–28 (nucleotide analogues) did not exhibit any inhibitory activity (data not shown), as anticipated, all phosphono diphosphates 29–33 (nucleoside triphosphate analogues) were potent inhibitors of both enzymes with IC50 values ranging from 37 nM to 7.8 μM. Nevertheless, all novel compounds were less potent inhibitors of ACT and EF compared to the previously studied PMEApp (1, Table 3).
Compound 33 was the most potent inhibitor of ACT (IC50 = 37 nM) from the series (Table 3), only about twice less potent compared to PMEApp (1). Compound 32 was the most potent inhibitor of EF (IC50 = 235 nM), 6.5 times weaker than PMEApp (1). The results suggested that B. pertussis ACT may be more sensitive to inhibition by ANPs compared to B. anthracis EF. Compound 31, the congener derived from the inactive compound 21i (Table 2), was the least potent inhibitor of EF (IC50 = 7.8 μM), but its ACT inhibitory activity (IC50 = 184 nM) was comparable (30 and 32) or better (29, Table 3) than other derivatives.
3. Conclusions
A series of eighteen acyclic nucleoside phosphonates (ANPs), consisting of a substituted 2-aminothiazole ring and the 2-(phosphonomethoxy)methyl or 2-(phosphonomethoxy)ethyl moiety in the form of bisamide prodrug, were synthesized as potential inhibitors of bacterial and/or mammalian adenylate cyclases (ACs). The prepared compounds were structural analogues of bisamide prodrug of adefovir (PMEA), compound 2, where the adenine base was replaced with the 2-aminothiazole moiety inspired by compound MB05032. Bisamide prodrugs with l-phenylalanine isopropyl ester were used, based on their previously reported[21] improved stability in plasma and decreased cytotoxicity relative to the original adefovir dipivoxil (bis(POM)PMEA) compound.
Prepared compounds, with the exception of four derivatives (10, 18, 21i, and 21n), were potent inhibitors of adenylate cyclase toxin (ACT) from B. pertussis in the J774A.1 macrophage assay, with IC50 values in the low micromolar to submicromolar range. 4-Isopropoxyphenyl derivative 21d (IC50 = 680 nM) and 4-(N-methylaminocarbonyl)phenyl derivative 21l (IC50 = 660 nM) were the most potent ACT inhibitors in the cell-based assay with no apparent cytotoxicity for either J774A.1 macrophage cells or HEK-AC1 cells. Nevertheless, their potency was about 45 times lower, compared to that of the PMEA bisamide 2 compound.
Five ANP-diphosphates (nucleoside triphosphate analogues) were also prepared for their direct evaluation on the B. pertussis ACT and B. anthracis EF cell-free assays. Compounds 29, 30, 32, and 33 were structural analogues of the most potent bisamides 21c, 21d, 21j, and 21l, and compound 31 was derived from inactive bisamide 21i. ANP-diphosphates 29, 30, 32, and 33 were potent inhibitors of the two bacterial adenylate cyclases tested: ACT (IC50 values 37–312 nM) and EF (IC50 values 0.235–1.34 μM). Analogue 31, derived from the less active prodrug 21i, was the least potent inhibitor of EF (IC50 = 7.81 μM), but exhibited a similar inhibitory activity against ACT (IC50 = 184 nM) in comparison to some other compounds (namely 29, 30, and 32).
Finally, some of the prepared prodrugs were discovered to selectively inhibit certain mammalian ACs with considerable efficacy. The most robust AC1 inhibitors were compounds 21f, 21h and 23, however, their activity reflected pronounced cytotoxicity in J774A.1 and/or HEK-AC1 cells. Compounds 21b, 21g, and 21i were also selective and potent AC1 inhibitors (<40% of remaining activity) while being non-cytotoxic. Compound 21l, on the other hand, was identified as a selective AC2 inhibitor (only 12% of remaining activity) without noticeable cytotoxicity. The AC1 inhibitors identified within the current study may be promising lead structures for further optimization of their structure and for potential development of non-opioid alternatives for the treatment of inflammatory and neuropathic pain.[38,41] Additional genetic and preclinical studies are needed to identify therapeutic applications for AC2 inhibitors, however, it has been suggested that such molecules may have an utility in muscle and lung pathologies or certain cancers.[42]
4. Experimental Section
Experimental details are given in the Supporting information.
Supplementary Material
Synthesis of 18 novel acyclic nucleoside phosphonates described
14 prodrugs are potent ACT inhibitors (0.26 – 8.45 μM) in J744A. 1 cells
Several compounds exhibited evident to moderate selectivity to inhibit AC1 over AC2 and/or AC5 in HEK293 cells
Prepared phosphono disphosphates are potent inhibitors of ACT and EF
Acknowledgements
This research was funded by the Institute of Organic Chemistry and Biochemistry (RVO61388963), the Ministry of Education, Youth and Sports (MŠMT in Czech) in the program INTER-EXCELLENCE (project LTAUSA18086) and the National Institutes of Health MH101673.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have declared no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Khannpnavar B, Mehta V, Qi C, Korkhov V, Curr. Opin. Struct. Biol 2020, 63, 34–41 [DOI] [PubMed] [Google Scholar]
- [2].Sadana R, Dessauer CW, Neurosignals 2009, 17, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Linder JU, Cell. Mol. Life. Sci 2006, 63, 1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hanoune J, Defer N, Annu. Rev. Pharmacol. Toxicol 2001, 41, 145–174. [DOI] [PubMed] [Google Scholar]
- [5].Sunahara RK, Dessauer CW, Gilman AG, Annu. Rev. Pharmacol. Toxicol 1996, 36, 461–480. [DOI] [PubMed] [Google Scholar]
- [6].Ahuja N, Kumar P, Bhatnagar R, Crit. Rev. Microbiol 2004, 30, 187–196. [DOI] [PubMed] [Google Scholar]
- [7].Ladant D, Ullmann A, Trends Microbiol. 1999, 7, 172–176. [DOI] [PubMed] [Google Scholar]
- [8].Fedele G, Schiavoni I, Adkins I, Klimova N, Sebo P, Toxins 2017, 9, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tang W-J, Guo Q, Mol. Asp. Med 2009, 30, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Seifert R, Dove S, Trends Microbiol. 2012, 20, 343–351. [DOI] [PubMed] [Google Scholar]
- [11].Seifert R, Lushington GH, Mou T-C, Gille A, Sprang SR, Trends Pharmacol. Sci 2012, 33, 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brand CS, Hocker HJ, Gorfe AA, Cavasotto CN, Dessauer CW, J. Pharmacol. Exp. Ther 2013, 347, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gille A, Seifert R, J. Biol. Chem 2003, 278, 12672–12679. [DOI] [PubMed] [Google Scholar]
- [14].Gille A, Lushington GH, Mou TC, Doughty MB, Johnson RA, Seifert R, J. Biol. Chem 2004, 279, 19955–19969. [DOI] [PubMed] [Google Scholar]
- [15].Taha HM, Schmidt J, Göttle M, Suryanarayana S, Shen Y, Tang W-J, Gille A, Geduhn J, König B, Dove S, Seifert R, Mol. Pharmacol 2009, 75, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Taha H, Dove S, Geduhn J, König B, Shen Y, Tang W-J, Seifert R, Naunyn-Schmiedeberg’s Arch. Pharmacol 2012, 385, 57–68. [DOI] [PubMed] [Google Scholar]
- [17].Jiao G-S, Kim S, Moayeri M, Thai A, Cregar-Hernandez L, McKasson L, O’Malley S, Leppla SH, Johnson AT, Bioorg. Med. Chem. Lett 2018, 28, 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].De Clercq E, Holý A, Rosenberg I, Sakuma T, Balzarini J, Maudgal PC, Nature 1986, 323, 464–467. [DOI] [PubMed] [Google Scholar]
- [19].Guo Q, Shen Y, Lee Y-S, Gibbs CS, Mrksich M, Tang W-J, EMBO J. 2005, 24, 3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shen Y, Zhukovskaya NL, Zimmer MI, Soelaiman S, Bergson P, Wang C-R, Gibbs CS, Tang W-J, Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Šmídková M, Dvořáková A, Tloušt’ová E, Česnek M, Janeba Z, Mertlíková-Kaiserová H, Antimicrob. Agents Chemother 2014, 58, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Česnek M, Jansa P, Šmídková M Mertlíková-Kaiserová H, Dračínský M, Brust TF, Pávek P, Tretnar F, Watts VJ, Janeba Z, ChemMedChem 2015, 10, 1351–1364. [DOI] [PubMed] [Google Scholar]
- [23].Česnek M, Skácel J, Jansa P, Dračínský M, Šmídková M, Mertlíková-Kaiserová H, Soto-Velasquez MP, Watts VJ, Janeba Z, ChemMedChem 2018, 13, 1779–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Břehová P, Šmídková M, Skácel J, Dračínský M, Mertlíková-Kaiserová H, Soto-Velasquez MP, Watts VJ, Janeba Z, ChemMedChem 2016, 11, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Frydrych J, Skácel J, Šmídková M, Mertlíková-Kaiserová H, Dračínský M, Gnanasekaran R, Lepšík M, Soto-Velasquez M, Watts VJ, Janeba Z, ChemMedChem 2018, 13, 199–206. [DOI] [PubMed] [Google Scholar]
- [26].Erion MD, van Poelje PD, Dang Q, Kasibhatla SR, Potter SC, Reddy MR, Reddy KR, Jiang T, Lipscomb WN, PNAS, 2005, 102, 7970–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dang Q, Kasibhatla SR, Reddy KR, Jiang T, Reddy MR, Potter SC, Fujitaki JM, van Poelje PD, Huang J, Lipscomb WN, Erion MD, J. Am. Chem. Soc 2007, 129, 15491–15502. [DOI] [PubMed] [Google Scholar]
- [28].Dang Q, Kasibhatla SR, Jiang T, Fan K, Liu Y, Taplin F, Schulz W, Cashion DK, Reddy KR, van Poelje PD, Fujitaki JM, Potter SC, Erion MD, J. Med. Chem 2008, 51, 4331–4339. [DOI] [PubMed] [Google Scholar]
- [29].Dang Q, Liu Y, Cashion DK, Kasibhatla SR, Jiang T, Taplin F, Jacintho JD, Li H, Sun Z, Fan Y, Da Re J, Tian F, Li W, Gibson T, Lemus R, van Poelje PD, Potter SC, Erion MD, J. Med. Chem 2011, 54, 153–165. [DOI] [PubMed] [Google Scholar]
- [30].Jansa P, Baszczyňski O, Dračínský M, Votruba I, Zídek Z, Bahador G, Stepan G, Cihlar T, Mackman R, Holý A, Janeba Z, Eur. J. Med. Chem 2011, 46, 3748–3754. [DOI] [PubMed] [Google Scholar]
- [31].Holý A, Rosenberg I, Collect. Czech. Chem. Commun 1987, 52, 2801–2809. [Google Scholar]
- [32].Karuvalam RP, Haridas KR, Nayak SK, Guru Row TN, Rajeesh P, Rishikesan R, Kumari NS, Eur. J. Mech. Chem 2012, 49, 172–182. [DOI] [PubMed] [Google Scholar]
- [33].Pi Z, Sutton J, Lloyd J, Hua J, Price L, Wu Q, Chang M, Zheng J, Rehfuss R, Huang Ch. S., Wexler RR, Lam PYS, Bioorg. Med. Chem. Lett 2013, 23, 4206–4209. [DOI] [PubMed] [Google Scholar]
- [34].Getlik M, Grutter C, Simard JR, Nguyen HD, Robubi A, Aust B, van Otterlo WAL, Rauh D, Eur. J. Med. Chem 2012, 48, 1–15. [DOI] [PubMed] [Google Scholar]
- [35].Sperotto E, van Klink GPM, van Koten G, de Vries JG, Dalton Trans. 2010, 39, 10338–10351. [DOI] [PubMed] [Google Scholar]
- [36].McKenna CE, Schmidhauser J, J. Chem. Soc., Chem. Commun 1979, 739. [Google Scholar]
- [37].Mackman RL, Zhang L, Prasad V, Boojamra CG, Douglas J, Grant D, Hui H, Kim CU, Laflamme G, Parrish J, Stoycheva AD, Swaminathan S, Wang KY, Cihlar T, Bioorg. Med. Chem 2007, 15, 5519–5528. [DOI] [PubMed] [Google Scholar]
- [38].Brust TF, Alongkronrusmee D, Soto-Velasquez M, Baldwin TA, Ye Z, Dai M, Dessauer CW, van Rijn RM, Watts VJ, Sci. Signal 2017, 10, eaah5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McGuigan C, Madela K, Aljarah M, Bourdin C, Arrica M, Barrett E, Jones S, Kolykhalov A, Bleiman B, Bryant KD, Ganguly B, Gorovits E, Henson G, Hunley D, Hutchins J, Muhammad J, Obikhod A, Patti J, Walters CR, Wang J, Vernachio J, Ramamurty CVS, Battina SK, Chamberlain S, J. Med. Chem 2011, 54, 8632–8645. [DOI] [PubMed] [Google Scholar]
- [40].Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF, Chem. Rev 2014, 114, 9154–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Watts VJ, Neuropsychopharmacol. 2018, 43, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Conley JM, Brand CS, Bogard AS, Pratt EPS, Xu R, Hockerman GH, Ostrom RS, Dessauer CW, Watts VJ, J. Pharmacol. Exp. Ther 2013, 347, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.