

Abstract
Metabolism of the essential amino acid tryptophan is a key metabolic pathway that restricts antitumor immunity and is a drug development target for cancer immunotherapy. Tryptophan metabolism is active in brain tumors including gliomas and promotes a malignant phenotype and contributes to the immunosuppressive tumor microenvironment. In recent years, improved understanding of the regulation and downstream function of tryptophan metabolism has been significantly expanded beyond the initial in vitro observation that the enzyme indoleamine-2,3-dioxygenase 1 (IDO1) promotes the depletion of intracellular tryptophan. Here, we revisit the specific roles of tryptophan metabolites in regulating brain functioning and neuronal integrity as well as in the context of brain tumors. This review summarizes recent developments in identifying key regulators, as well as the cellular and molecular effects of tryptophan metabolism with a particular focus on potential therapeutic targets in glioma
Introduction
Metabolism of the essential amino acid tryptophan is increasingly recognized as an important endogenous metabolic feedback loop that contributes to the regulation of magnitude, duration and cellular composition of immune responses. Tryptophan metabolism by cancer cells and/or cancer-associated stromal cells contributes to the suppression of antitumor immune responses.
Tryptophan metabolism also mediates primary and adaptive resistance to immunotherapeutic treatment with immune checkpoint inhibitors [1••]. These observations have resulted in the development of a broad array of drugs that aim to modulate or inhibit tryptophan metabolism. The first molecular target of small molecule inhibitors was the enzyme indoleamine-2,3-dioxygenase 1 (IDO1) that catalyzes the rate-limiting conversion of tryptophan into kynurenine. Tryptophan-2,3-dioxygenase 2 (TDO2) is another drug target that catalyzes tryptophan into kynurenine that is upregulated in brain tumors and other types of cancers. Moreover, the expression of interleukin-4-induced-1 (IL4I1), an enzyme that metabolizes tryptophan into indole derivatives, is inversely associated with overall survival in patients with low grade glioma (LGG) and glioblastoma (GBM) [2••] and represents yet another therapeutic target for cancer [3]. Recognition that the immunosuppressive effects of tryptophan metabolism are mediated not only by local tryptophan depletion [4,5] but also through the activation of immunosuppressive tryptophan catabolites including kynurenine, kynurenic acid, cinnabarinic acid, indole-3-pyruvic acid, and indole-3-aldehyde binding to the transcription factor aryl hydrocarbon receptor (AHR) [2••,6••,7,8,9••,10•,11], has initiated the development of selective AHR inhibitors.
While drugs that interfere with tryptophan metabolism are moved to clinical trials for cancer immunotherapy — with initial phase III results associated with negative outcomes [12,13] — tryptophan metabolism in brain tumors has been a particular focus of research in the past for several reasons. First, brain tumors and in particular, malignant glioma, are characterized by a profoundly immunosuppressive microenvironment that diminishes immunotherapy-based therapeutic effects [14•]. Here, activation of tryptophan metabolism and downstream effector mechanisms, including AHR-mediated transcription, have been shown to be major contributors [2••,6••,9••]. Second, tryptophan metabolism in the central nervous system (CNS) is highly compartmentalized due to selective expression of pathway enzymes, cell type-specific metabolic vulnerability and selective permeability of the blood brain barrier (BBB) for tryptophan metabolites [15,16•]. Third, tryptophan metabolism has profound effects on brain function and contributes to neuropsychiatric and neurodegenerative disorders through perturbation of neuronal function [16•]. When reflecting on tryptophan metabolism in brain tumors it is imperative to recognize its specific role in the brain (Figure 1).
Figure 1.
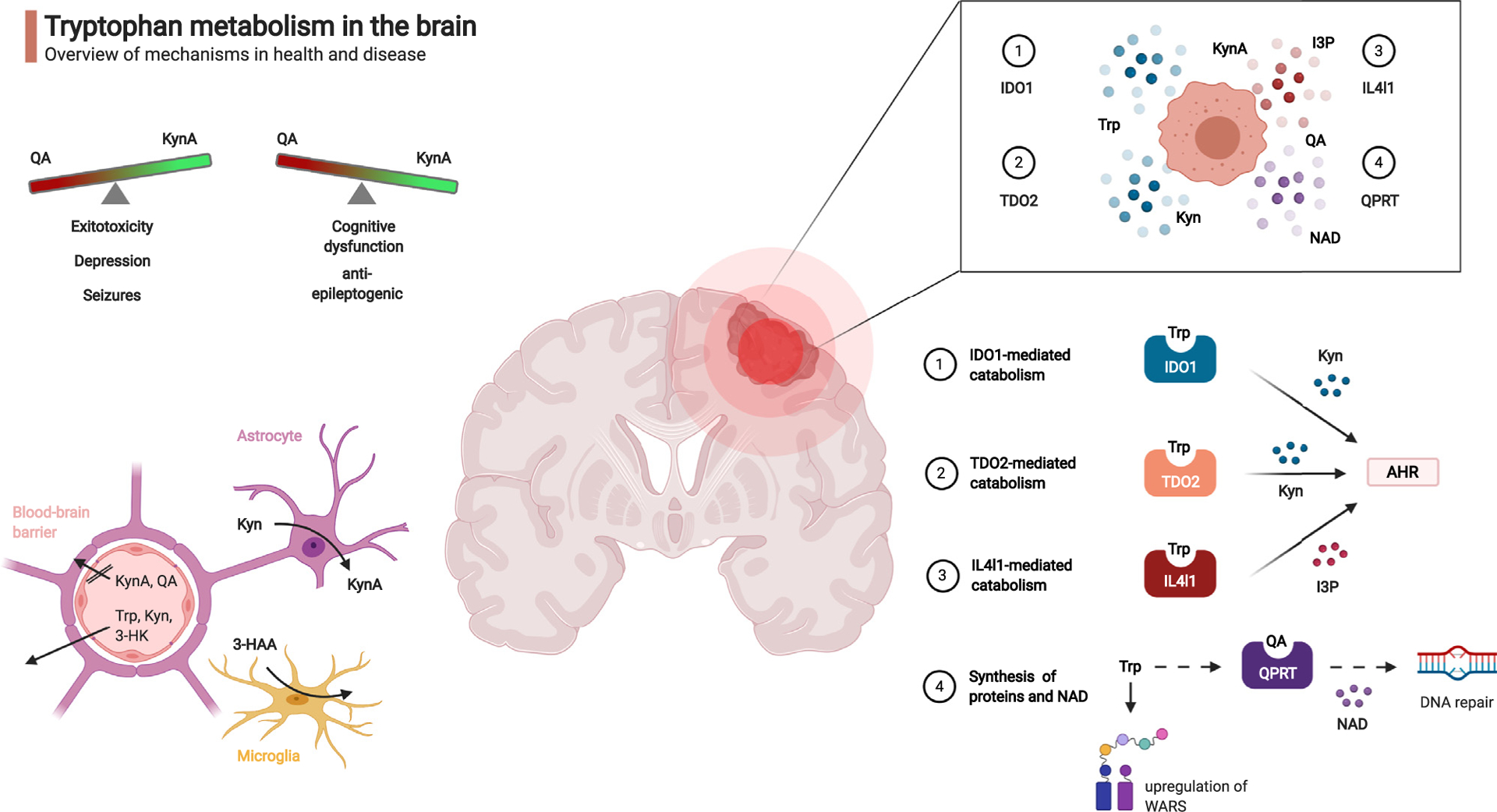
Tryptophan metabolism in the brain.
Tryptophan (Trp) catabolites regulate neurological and immunological functions in health and disease. Top left: overview of neuropsychiatric effects of Trp catabolites. Bottom left: Blood-brain barrier and glial cells determine local transport and metabolism of Trp and Trp-catabolites. Top right: Pleiotropic enzymatic digestion of exogenous Trp by brain tumor cells. Bottom right: Mechanisms for enzymatic digestion of Trp and biological effects of downstream catabolites. Kyn: kynurenine, KynA: kynurenic acid, QA: quinolinic acid, 3-HAA: 3-hydroxy-anthranilic acid, 3-HK: 3-hydroxykynurenine, IDO1: indoleamine-2,3-dioxygenase, IL4I1: Interleukin-4-Induced-1, I3P: indole-3-pyruvic acid, TDO2: tryptophan-2,3-dioxygenase, QPRT: quinolinic acid phosphoribosyltransferase, AHR: aryl hydrocarbon receptor, NAD: nicotinamide adenine dinucleotide, WARS: tryptophanyl-tRNA synthetase.
Tryptophan metabolism in the brain and its possible implications for brain tumors
Tryptophan metabolism in the brain is separated from systemic tryptophan metabolism by the BBB. In particular, tryptophan, kynurenine and 3-hydroxykynurenine are actively transported across the BBB via the large amino acid transporter whereas kynurenic acid and quinolinic acid are not [15]. In the brain, the production of tryptophan metabolitesishighly cell type-specific. Astrocytes primarily produce the neuroprotective metabolite kynurenic acid through kynurenine amino transferase enzyme activity, while microglial cells produce quinolinic acid from 3-hydroxy-anthranilic acid (3-HAA) through 3-HAA-3,4-dioxygenase (HAAO) activity [16•]. Unbalanced tryptophan levels or the dysregulation of downstream metabolites have been implicated in a broad range of CNS disorders including depression, schizophrenia and neurodegeneration [16•,17]. These pathologies are largely driven by the neuroactivity of specific tryptophan metabolites. Quinolinic acid mediates excitotoxicity and neurodegeneration by binding of the N-methyl-d-aspartate (NMDA) receptor. Conversely, kynurenic acid is an antagonist of ionotropic glutamate receptors and has been implicated in cognitive dysfunction in schizophrenia. For this reason, inhibitors of enzymes producing kynurenic acid and quinolinic acid, respectively, are being explored for the treatment of neuropsychiatric disorders [16•]. Importantly, both kynurenic acid and quinolinic acid are produced by gliomas [2••,6••,18,19] and may be involved in the frequent comorbidities seen in brain tumor patients. For instance, the high levels of kynurenic acid may contribute to the cognitive dysfunction in brain tumor patients. The control of neuronal excitability and seizures is another effect of tryptophan metabolite exposure.
Quinolinic acid administration causes seizures in animal models [20] and is antagonized by kynurenic acid [21]. Several lines of evidence suggest a role for quinolinic acid and kynurenic acid including: i) a nonsense mutation of the enzyme aminocarboxymuconate semialdehyde decarboxylase that likely results in channeling of tryptophan metabolism towards quinolinic acid and leads to familial cortical myoclonic tremor and epilepsy [22], ii) a ketogenic diet that reduces seizure frequency in pediatric epilepsy patients [23] and increases blood levels of kynurenic acid [24], as well as iii) the anticonvulsant levetiracetam that is widely used in brain tumor patients and inhibits the release of quinolinic acid but stimulates kynurenic acid production in interferon-gamma (IFN-γ)-treated rat cortical astrocytes [25]. Hence, the differential effects of kynurenine pathway metabolites on neuroactivity are important to consider as a potential cause of brain tumor-associated neuronal dysfunction resulting in the clinical manifestation of cognitive dysfunction and/or seizures. Depression, a comorbidity that is frequently observed among brain tumor patients [26] has also been linked with altered brain tryptophan metabolism; namely a shift away from the serotonergic, and instead, toward the kynurenine pathway [27,28]. Glioma patients suffering from depression have a higher level of tryptophan uptake in the frontal cortex and thalamus than patients without depression [29], which may reflect increased tryptophan metabolism in these brain areas.
Taken together, alterations in CNS tryptophan metabolism in patients with brain tumors may contribute to the multiple comorbidities of this disease (Figure 1).
In addition, the serotonergic branch of Trp metabolism can also modulate neuropsychiatric functions as it produces diverse neuroactive substances, including serotonin and melatonin [17]. Interestingly, several links have been discovered between the KP and the serotonergic pathway: The KP enzymes IDO1/2 participate in the serotonergic pathway as they are able to degrade both serotonin and melatonin [30]. Moreover, N-acetylserotonin acts as a positive allosteric modulator of IDO1 leading to increased kynurenine production [31•]. As serotonin and other monoamines are implicated in neurogenesis and development, a potential role of serotonin is discussed in gliomagenesis [32]. However, while treatment of glioblastoma patients with selective serotonin reuptake inhibitors (SSRI) benefits from a favorable safety profile and previous study results supporting their general use in cancer, it was not shown to improve overall survival [33].
IDO1 in brain tumors
IDO1 immunohistochemistry of primary brain tumors and brain metastases has revealed an expression pattern that is largely confined to tumor-infiltrating or resident myeloid cells and tumor-associated endothelial cells [6••,34]. Although it is tempting to speculate that endothelial IDO1 expression contributes to the immunosuppressive environment in the perivascular niche, studies in normal brain endothelial cells have demonstrated that IDO1 is involved in regulating vascular tone during inflammation through the production of kynurenine [35]. TCGA analyses and animal experiments have shown that IDO1 gene expression in the brain and in primary brain tumors increases with age and is associated with poor outcomes and resistance to immunotherapy with immune checkpoint inhibitor treatment [36•]. Considering preclinical evidence [37] a phase 1 clinical trial is underway to evaluate the safety of the IDO enzyme inhibitor BMS-986205, one of five IDO inhibitors currently under clinical evaluation [38] in patients with GBM and in combination with irradiation and the programmed cell death protein-1 (PD-1) antibody nivolumab (ClinicalTrials.gov identifier NCT04047706). Taking into account the high safety profile of this treatment combination, NRG Oncology is current in the final stages of approval for translating this into a phase II/III randomized multisite trial in the United States for patients with wildtype IDH GBM (Table 1).
Table 1.
Clinical trials with IDO1 inhibitors in brain cancer
| IDO1 inhibitor | Indication | Age | Combination partner | Trial ID | Development phase | Status |
|---|---|---|---|---|---|---|
| Indoximod | Glioblastoma multiforme, glioma, gliosarcoma, malignant brain tumor | 16 years and older | Temozolomide, Bevacizumab, Stereotactic Radiation | NCT02052648 | Phase I/II | Completed |
| Indoximod | Glioblastoma multiforme, glioma, gliosarcoma, malignant brain tumor, ependymoma, medulloblastoma, diffuse intrinsic pontine glioma, primary CNS tumor | 3 years to 21 years | Temozolomide, Conformal Radiation, Cyclophosphamide, Etoposide | NCT02502708 | Phase I | Completed |
| Indoximod | Glioblastoma, medulloblastoma, ependymoma, diffuse intrinsic pontine glioma | 3 years to 21 years | Partial Radiation, Full-dose Radiation, Temozolomide, Cyclophosphamide, Etoposide, Lomustine | NCT04049669 | Phase II | Recruiting |
| PF-06840003 | Oligodendroglioma, astrocytoma, malignant glioma | 18 years and older | Not annotated | NCT02764151 | Phase I | Terminated, Reardon et al. [81] |
| BMS-986205 | Glioblastoma | 18 years and older | Nivolumab, Radiation Therapy, Temozolomide | NCT04047706 | Phase I | Recruiting |
| Epacadostat | Glioma, Glioblastoma | 18 years and older | INCMGA00012, Bevacizumab, Radiation Therapy | NCT03532295 | Phase II | Recruiting |
| INCBO24360 (Epacadostat) | Glioblastoma | 18 years and older | Nivolumab | NCT03707457 | Phase I | Terminated |
TDO2 in brain tumors
For a long period of time, TDO2 was thought to be specifically expressed in the liver to regulate systemic tryptophan levels and energy homeostasis through the provision of nicotinamide adenine dinucleotide (NAD). However, it is now recognized that TDO2 is upregulated in several types of cancer including brain tumors [6••,39] and is inversely associated with survival outcomes in patients with glioma [6••,40,41] and meningioma [42,43]. Constitutive TDO2 expression in GBM is driven by a CCAAT/enhancer-binding protein β (C/EBPb)-binding site in the TDO2 promoter and sustained by interleukin-1b via C/EBPb in a mitogen-activated protein kinase (MAPK)-dependent fashion [6••,41]. Prostaglandins also upregulate TDO2 expression and enzyme activity in malignant glioma via activation of prostaglandin E receptor-4 (EP4) [40]. In contrast and unlike in liver cells, steroids limit TDO2 expression in glioma [44]. Moreover, hypoxia suppresses TDO2 expression both in the liver and in glioblastoma [45,46]. After discovering that TDO2 and IDO1 are both expressed by cancer cells and co-contribute to tumor-induced immunosuppression, developmental drug research has aimed to identify specific TDO2 and dual IDO1/TDO2 inhibitors for the application to patients with brain tumors. Similar to the inhibitors for IDO1, TDO2 inhibitors also increase the antitumor efficacy of immune checkpoint inhibitors in preclinical tumor models [47•]. Selective TDO2 and IDO1/TDO2 dual inhibitors have been developed [48]. However, the dual IDO1/TDO2 inhibitor M4112 that inhibits IDO1 ex vivo, does not significantly reduce plasma kynurenine levels [49].
Tryptophan metabolism and protein synthesis
IDO1-expressing and TDO2-expressing brain tumors have a high demand for tryptophan uptake to compensate for the IDO1/TDO2-mediated tryptophan consumption. Positron emission tomography (PET) imaging with α-[11C]-methyl-l-tryptophan (AMT) or new tracers containing fluorine-18 demonstrate increased tryptophan uptake and/or metabolism in patients’ brain tumors [29,50]. Importantly, this imaging strategy may guide the clinical evaluation of drugs targeting tryptophan metabolism [51]. If the demand cannot be met by increased import, depletion of intracellular tryptophan results in incapacitation of protein synthesis through activation of the integrated stress response via general control non-derepressible-2 (GCN2) kinase, leading to phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α) and induction of activating transcription factor 4 (ATF4). While GCN2 activation was initially implicated in the proliferation arrest of CD8+ T cells induced by IDO1-positive dendritic cells (DCs) [52], recent evidence in experimental glioma models suggests that GCN2 is required for CD8+ T cell survival under conditions of tryptophan deficiency [53]. Particularly in the brain, however, tryptophan levels are rapidly replenished through diet, thus limiting the physiological relevance of in vitro studies. In addition, free tryptophan is loaded on tRNA by tryptophanyl-tRNA synthetase (WARS) and tryptophan-dependent protein synthesis is maintained by upregulation of WARS via IFN-γ and/or GCN2-p-eIF2α-ATF4 signaling in glioblastoma [54]. Moreover, IFN- γ-induced tryptophan depletion has recently been demonstrated to lead to ribosomal frame shifting in melanoma, resulting in the generation of aberrant peptides that are presented on HLA-I molecules and can prime T cells, thereby possibly enhancing anti-tumor immunity [55••] (Figure 2).
Figure 2.
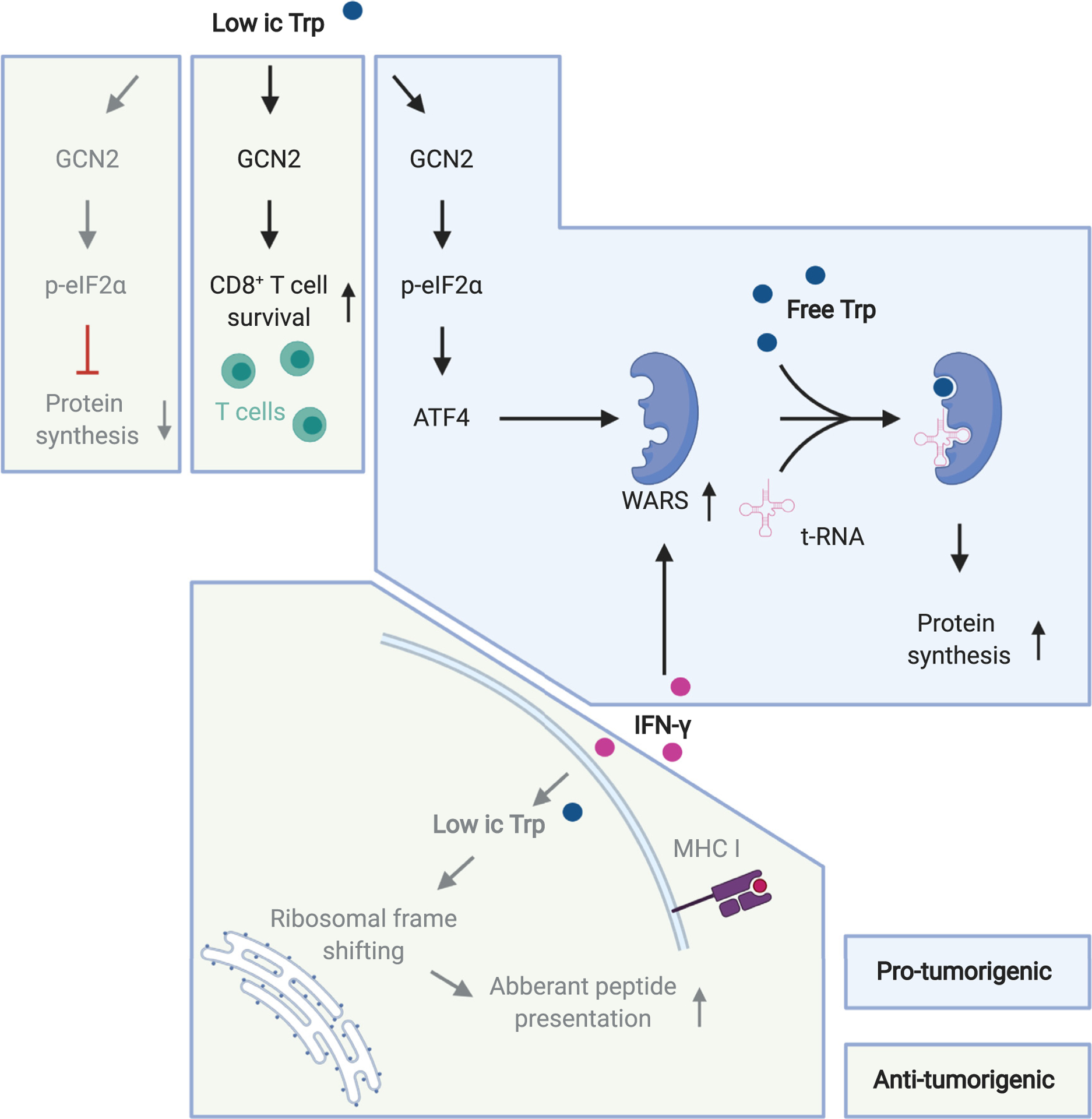
Effects of tryptophan deprivation.
Low intracellular (ic) tryptophan (Trp) levels can mediate pro-tumorigenic (blue background) and anti-tumorigenic effects (green background). Mechanisms described in glioma models are highlighted in black letters, mechanisms described in other systems are highlighted in gray. Abbreviations: GCN2: general control non-derepressible-2, p-eIF2α: phosphorylated eukaryotic translation initiation factor 2α, ATF4: activating transcription factor 4, WARS: tryptophanyl-tRNA synthetase, IFN-γ: interferon-gamma.
Tryptophan as a precursor for NAD
If all enzymes of the kynurenine pathway are present, tryptophan is ultimately metabolized into the energetic substrate NAD. NAD is an important cofactor for enzymes such as the DNA repair proteins poly(ADP-ribose) polymerases (PARPs). In brain tumors, quinolinic acid, a downstream tryptophan metabolite, is converted into NAD by glioma cells through the expression and activity of quinolinic acid phosphoribosyltransferase (QPRT). The quinolinic acid-producing enzyme HAAO is confined to tumor-infiltrating microglia indicating that gliomas exploit microglial-derived quinolinic acid as an alternative source of replenishing intracellular NAD pools as a resistance mechanism against DNA-damaging treatments such as alkylating chemotherapy and irradiation [19,56]. Interestingly, upregulation of QPRT and ensuing NAD production also mediates resistance to combined treatment with the histone deacetylase inhibitor panobinostat and the proteasome inhibitor bortezomib in glioma patients [57], further underscoring how tryptophan catabolism can contribute to higher NAD levels in glioma (Figure 3). Higher NAD levels could also contribute to a higher activity of one of the Trp-degrading enzymes, IDO1. The reduced form of NAD, NADH, has been shown to react with ferric IDO1 under aerobic conditions, fostering the formation of the active, ferrous dioxygen adduct form of IDO1 thus increasing its enzymatic activity [58]. This reaction in which IDO1 acts as an NADH oxidase links the downstream Trp-metabolic product NADH to an initial enzyme of the KP in a feed-forward loop.
Figure 3.
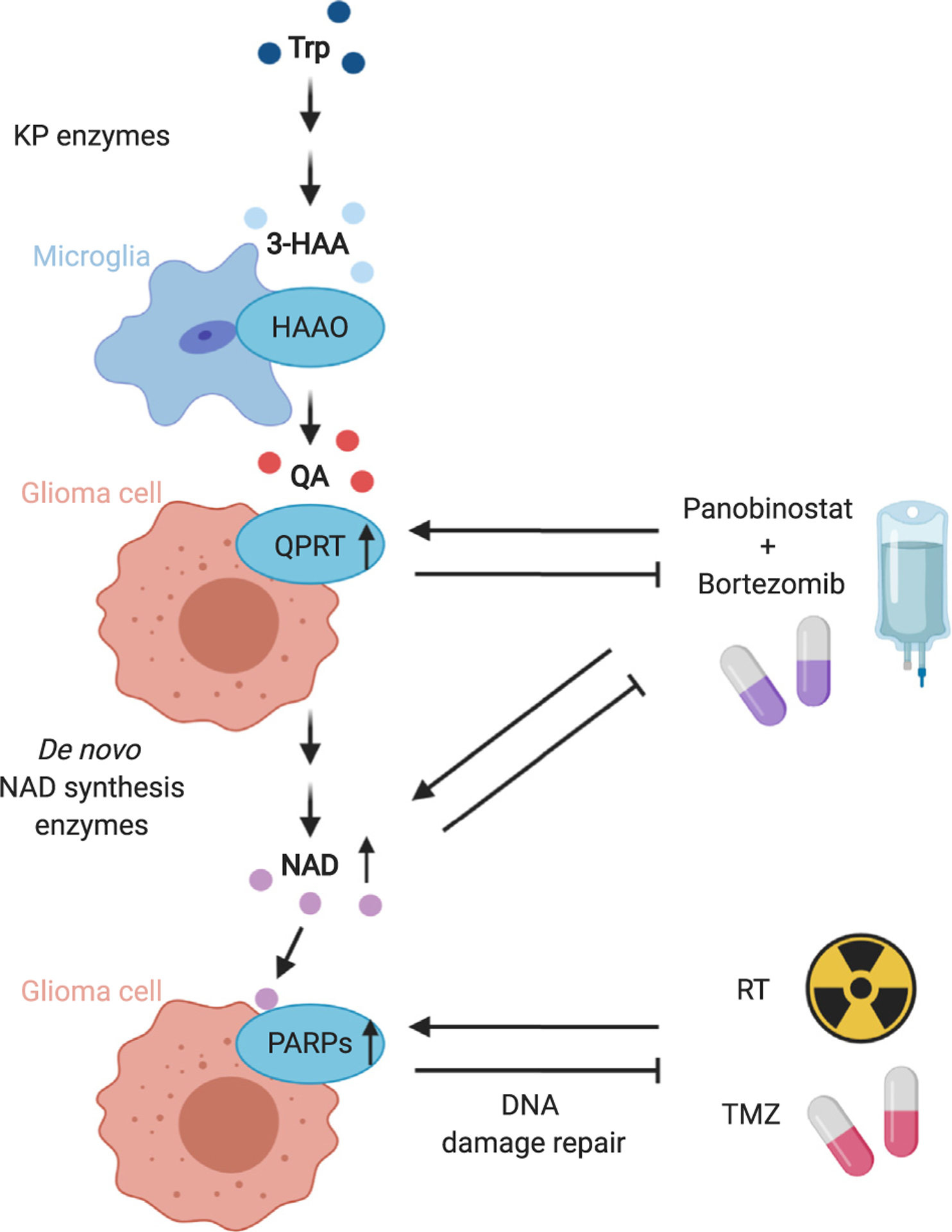
Tryptophan metabolism in glioma yields NAD that can mediate therapy resistance.
Collaborative degradation of tryptophan (Trp) in glioma by microglia and glioma cells yields nicotinamide adenine dinucleotide (NAD) that can mediate therapy resistance in glioma. Therapeutic agents induce quinolinic acid phosphoribosyltransferase (QPRT), NAD and poly(ADP-ribose) polymerases (PARPs) that consecutively mediate resistance to therapy, for example, the histone deacetylase inhibitor panobinostat and the proteasome inhibitor bortezomib. Abbreviations: KP: kynurenine pathway, 3-HAA: 3-hydroxy-anthranilic acid, HAAO: 3-HAA-3,4-dioxygenase, QA: quinolinic acid, RT: radiotherapy, TMZ: temozolomide.
AHR in brain tumors
There is increasing evidence that the immunosuppressive effects of tryptophan metabolism in infection, auto-immunity and tumor immunity are mediated by activating the AHR. The AHR is a cytosolic transcription factor that is highly expressed at environmental interfaces such as the skin and gut [59,60] and is responsible for the detoxification of polyaromatic hydrocarbons. Ligand binding causes AHR translocation from the cytoplasm into the nucleus resulting in heterodimerization with the AHR nuclear translocator (ARNT) [61]. The transcriptional complex binds dioxin or xenobiotic response elements (DRE or XRE) in the promoter regions of AHR target genes. In addition to the genes that encode detoxifying enzymes including cytochrome p450 oxidases, several additional AHR targets include immuno-regulatory genes. In brain tumors and other types of cancer, both AHR and AHR target genes are upregulated and correlate with the expression of IDO1 and TDO2 indicating that the production of kynurenine and its downstream metabolites are responsible for AHR activation [2••,6••]. More recently, IL4I1 was identified as an additional AHR-activating enzyme in cancer including gliomas. IL4I1 produces the AHR-activating ligands indole-3-pyruvic acid, kynurenic acid and indole-3-aldehyde and suppresses antitumor immune response [2••]. Being AHR target genes themselves, the three tryptophan-catabolic enzymes IDO1, TDO2 and IL4I1 are linked to AHR in a positive feedback loop [2••,62•,63] such that AHR induces or upregulates its own ligands.
In glioblastoma cells, AHR promotes motility mediated by kynurenine and indole-3-pyruvic acid as well as clonogenic survival [2••,6••]. AHR activation enhances transforming growth factor beta signaling in glioma cells [64], which may in part underlie the pro-migratory and immunosuppressive effects of AHR. Aside from its direct effects on cancer cells, AHR also influences diverse immune cell functions. AHR functions as a master regulator [65••] that controls differentiation and effector functions of T cells [66,67], macrophages [10•,68], DCs [68], natural killer cells [69] and B cells [70–72]. In glioblastoma, AHR contributes to the regulation of tumor-associated macrophage (TAM) recruitment and functioning [9••]. In these cells, AHR activation enhances the expression of C-C motif chemokine receptor 2 (CCR2), the receptor for glioma cell-derived C-C motif chemokine ligand 2 (CCL2) and upregulates the ectonucleotidase CD39. Together with CD73 [73], CD39 generates adenosine that causes CD8+ T cell dysfunction [9••]. Also, immune checkpoint molecules such as programmed cell death protein–ligand 1 (PD-L1) on TAMs [9••] and PD-1 on T cells [8] are induced by AHR activity.
Moreover, AHR activation reduces the viability of CD8+ T cells [74]. AHR has been implicated in the differentiation of naïve CD4+FoxP3− T cells into inducible CD4+FoxP3+ regulatory T cells (Tregs) [75,76] and is a potent transcriptional driver of interleukin (IL)-10 expression in diverse immune cell subsets [70,77–79]. In DCs, AHR dampens the induction of pro-inflammatory cytokines resulting in tolerogenic DCs that foster Treg differentiation [76]. Moreover, AHR reduces MHC class II expression and limits CD4+ T cell priming [10•]. Depending on its duration [66], AHR activation drives either T helper (TH) 17 cell or Treg differentiation [80]. Through its effects on tumor cells as well as select other immune cell subsets, AHR activation may importantly contribute to immune escape and malignancy in brain cancer (Figure 4).
Figure 4.
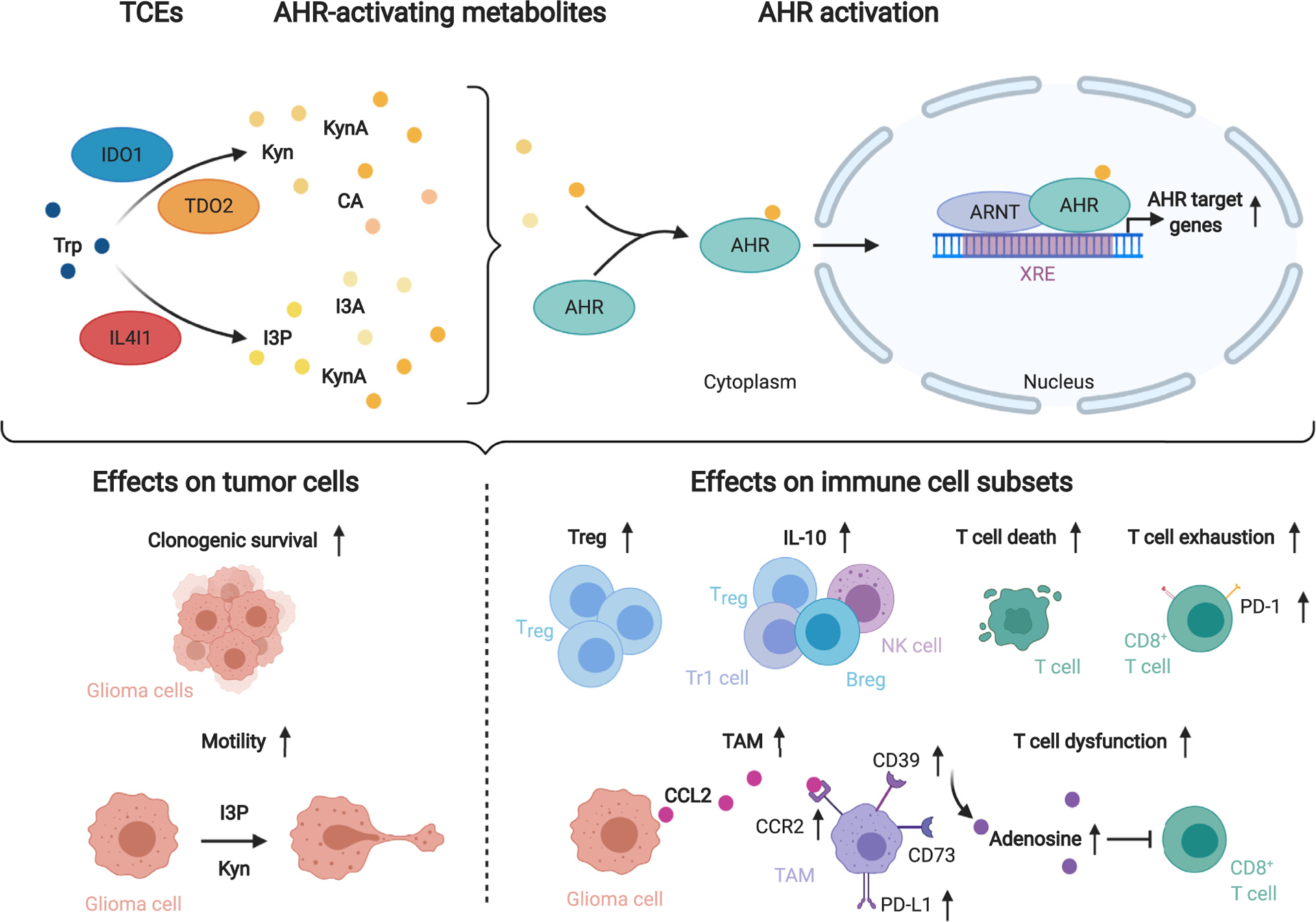
Tryptophan catabolism promotes tumor cell malignancy and immune suppression via activation of the aryl hydrocarbon receptor (AHR).
The tryptophan (Trp)-catabolic enzymes (TCEs) indoleamine-2,3-dioxygenase 1 (IDO1) and tryptophan-2,3-dioxygenase 2 (TDO2) initiate Trp degradation to the AHR-activating metabolites kynurenine (Kyn), kynurenic acid (KynA) and cinnabarinic acid (CA), while interleukin-4-induced-1 (IL4I1) generates the AHR agonists indole-3-pyruvic acid (I3P), indole-3-aldehyde (I3A) and KynA. Upon ligand binding the AHR translocates to the nucleus and binds to AHR nuclear translocator (ARNT). The heterodimer binds to xenobiotic response elements (XRE) in the promoters of AHR target genes increasing their expression. AHR activation enhances glioma cell malignancy. Furthermore, several immune cell subsets, including CD8+ T cells, regulatory T cells (Treg), Type 1 regulatory T cells (Tr1), regulatory B cells (Breg), natural killer (NK) cells and tumor-associated macrophages (TAM) are influenced by AHR activation. TAMs express the ectonucleotidases CD39 and CD73 that collaboratively produce adenosine which limit T cell function. Abbreviations: Interleukin (IL)-10, Programmed cell death protein-1 (PD-1), PD-ligand 1 (PD-L1), C-C motif chemokine receptor 2 (CCR2), C-C motif chemokine ligand 2 (CCL2).
Conclusion
Tryptophan metabolism is active in brain tumors and promotes their malignant phenotype through mechanisms beyond IDO1-mediated tryptophan degradation. Therapeutic development of strategies interfering with tryptophan metabolism in brain tumors needs to take into account the specific regulation and function of tryptophan metabolites in controlling neuronal function and integrity. The identification of novel key regulators and downstream mediators of tryptophan metabolism as well as repurposing of drugs targeting tryptophan metabolism in neuropsychiatric disorders offers novel targets and opportunities for therapeutic intervention.
Acknowledgements
This work was supported by grants from the German Cancer Aid (70113515) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (406052676; PL-315/5-1) to M.P. This work was supported in part by National Institutes of Health (NIH) P50 CA221747 (D.A.W.), NIH R01 NS097851 (D.A.W.), NIH K02 AG068617 (D.A.W.), BrainUp grant 2136 (D.A.W.), the Gail Boyter Magness (GBM) Foundation (D.A.W.), the Grace Giving Foundation (D.A.W.), the Stephen Coffman Charitable Trust (D.A.W.), and the 5 for the Fight organization (D.A.W.). V.P. was supported by the DKFZ Clinician Scientist Program and the Dieter Morszeck Foundation. M.F. is member of the MD/PhD program at Heidelberg University. M.F. received fellowships by the Heidelberg Biosciences International Graduate School (HBIGS), the German Academic Exchange Service (DAAD) and the Excellence Initiative of the German Council of Science and Humanities and the German Research Foundation (DFG). M. P. and C.A.O. were supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)-Project-ID 404521405, SFB 1389-UNITE Glioblastoma.
Footnotes
Conflict of interest statement
M.P. and C.A.O. are listed as inventors on patents on diagnostic tools for tryptophan metabolism (WO2008108994 (M. P.) and WO2017072368 (M.P. and C.A.O.). M.P. is inventor on patents on therapeutic targeting of the aryl hydrocarbon receptor in brain tumors and other types of cancer (WO2018146010A1, WO2019101643A1, WO20191016 47A1, WO2019101641A1, WO2019101641A1, WO2019101 642A1). C.A.O. is inventor on patents on AHR activity (WO2013034685 and WO20203715471) as well as tryptophan metabolism as a biomarker (WO2020208190 and PCT/EP2020/085647).
Figures were created using BioRender.com.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.••.Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bosse D, Lalani AA, Gopal S, Jin C, Horak C et al. : Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun 2019, 10:4346. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that an increase in the Kyn/Trp ratio under nivolumab therapy associates with reduced overall survival in melanoma and renal cell carcinoma patients.
- 2.••.Sadik A, Somarribas Patterson LF, Ozturk S, Mohapatra SR, Panitz V, Secker PF, Pfander P, Loth S, Salem H, Prentzell MT et al. : IL4I1 is a metabolic immune checkpoint that activates the AHR and promotes tumor progression. Cell 2020, 182:1252–1270.e34 [DOI] [PubMed] [Google Scholar]; This paper identifies AHR activation through the tryptophan-catabolic enzyme interleukin-4-induced-1 (IL4I1), which drives tumor cell-intrinsic malignant properties and suppresses adaptive immunity.
- 3.MacKinnon A, Bhupathi D, Chen J, Huang T, Li W, Ma Y, Sotirovska N, Steggerda S, Zhang W, Parlati F: 705 anti-tumor activity of CB-668, a potent, selective and orally bioavailable small-molecule inhibitor of the immuno-suppressive enzyme Interleukin 4 (IL-4)-Induced Gene 1 (IL4I1). J Immunother Cancer 2020, 8(Suppl. 3):A423–A424. [Google Scholar]
- 4.Sonner JK, Deumelandt K, Ott M, Thome CM, Rauschenbach KJ, Schulz S, Munteanu B, Mohapatra S, Adam I, Hofer AC et al. : The stress kinase GCN2 does not mediate suppression of antitumor T cell responses by tryptophan catabolism in experimental melanomas. Oncoimmunology 2016, 5:e1240858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van de Velde LA, Guo XJ, Barbaric L, Smith AM, Oguin TH 3rd, Thomas PG, Murray PJ: Stress kinase GCN2 controls the proliferative fitness and trafficking of cytotoxic T cells independent of environmental amino acid sensing. Cell Rep 2016, 17:2247–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.••.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M et al. : An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478:197–203 [DOI] [PubMed] [Google Scholar]; This paper establishes the role of TDO2-derived kynurenine as an immunosuppressive AHR ligand in gliomas.
- 7.Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, Ji T, Zhang H, Dong W, Jin X et al. : Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat Commun 2017, 8:15207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, Fiskesund R, Xie J, Liu J, Yin X et al. : Tumor-repopulating cells induce PD-1 expression in CD8(+) T cells by transferring kynurenine and AhR activation. Cancer Cell 2018, 33:480–494 e7. [DOI] [PubMed] [Google Scholar]
- 9.••.Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, Gutierrez-Vazquez C, Kenison J, Tjon EC, Barroso A et al. : Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 2019, 22:729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper highlights the effects of AHR activation on the recruitment and immunosuppressive properties of tumor-associated macrophages in glioblastoma.
- 10.•.Campesato LF, Budhu S, Tchaicha J, Weng CH, Gigoux M, Cohen IJ, Redmond D, Mangarin L, Pourpe S, Liu C et al. : Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun 2020, 11:4011. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that pharmacological inhibition of AHR restricts tumor growth in IDO1-overexpressing or TDO2-overexpressing murine tumor models which is enhanced in combination with anti-PD-1 therapy.
- 11.Lowe MM, Mold JE, Kanwar B, Huang Y, Louie A, Pollastri MP, Wang C, Patel G, Franks DG, Schlezinger J et al. : Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS One 2014, 9: e87877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC: Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol 2019, 41:41–48. [DOI] [PubMed] [Google Scholar]
- 13.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, Arance A, Carlino MS, Grob JJ, Kim TM et al. : Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol 2019, 20:1083–1097. [DOI] [PubMed] [Google Scholar]
- 14.•.Sampson JH, Gunn MD, Fecci PE, Ashley DM: Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer 2020, 20:12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]; This review summarizes immunological characteristics of the brain and their interplay with immunotherapeutic approaches in brain tumors.
- 15.Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR: Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem 1991, 56:2007–2017. [DOI] [PubMed] [Google Scholar]
- 16.•.Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA: Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 2019, 18:379–401 [DOI] [PubMed] [Google Scholar]; This review gives an overview of the diverse roles of Trp metabolism in health and disease.
- 17.Cervenka I, Agudelo LZ, Ruas JL: Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357. [DOI] [PubMed] [Google Scholar]
- 18.Vezzani A, Gramsbergen JB, Speciale C, Schwarcz R: Production of quinolinic acid and kynurenic acid by human glioma. Adv Exp Med Biol 1991, 294:691–695. [DOI] [PubMed] [Google Scholar]
- 19.Sahm F, Oezen I, Opitz CA, Radlwimmer B, von Deimling A, Ahrendt T, Adams S, Bode HB, Guillemin GJ, Wick W, Platten M: The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res 2020, 73:3225–3234. [DOI] [PubMed] [Google Scholar]
- 20.Schwarcz R, Speciale C, French ED: Hippocampal kynurenines as etiological factors in seizure disorders. Pol J Pharmacol Pharm 1987, 39:485–494. [PubMed] [Google Scholar]
- 21.Foster AC, Vezzani A, French ED, Schwarcz R: Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci Lett 1984, 48:273–278. [DOI] [PubMed] [Google Scholar]
- 22.Marti-Masso JF, Bergareche A, Makarov V, Ruiz-Martinez J, Gorostidi A, Lopez de Munain A, Poza JJ, Striano P, Buxbaum JD, Paisan-Ruiz C: The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and parkinsonism. J Mol Med (Berl) 2013, 91:1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-McGill KJ, Bresnahan R, Levy RG, Cooper PN: Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev 2020, 6:CD001903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarnowska I, Wrobel-Dudzinska D, Tulidowicz-Bielak M, Kocki T, Mitosek-Szewczyk K, Gasior M, Turski WA: Changes in tryptophan and kynurenine pathway metabolites in the blood of children treated with ketogenic diet for refractory epilepsy. Seizure 2019, 69:265–272. [DOI] [PubMed] [Google Scholar]
- 25.Fukuyama K, Okada M: Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br J Pharmacol 2018, 175:4253–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto-Meyer S, Lumibao J, Kim E, Ladomersky E, Zhai L, Lauing KL, Scholtens DM, Penedo F, Amidei C, Lukas RV, Wainwright DA: The interplay among psychological distress, the immune system, and brain tumor patient outcomes. Curr Opin Behav Sci 2019, 28:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marx W, McGuinness AJ, Rocks T, Ruusunen A, Cleminson J, Walker AJ, Gomes-da-Costa S, Lane M, Sanches M, Diaz AP et al. : The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: a meta-analysis of 101 studies. Mol Psychiatry 2020. [DOI] [PubMed] [Google Scholar]
- 28.Savitz J: The kynurenine pathway: a finger in every pie. Mol Psychiatry 2020, 25:131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.John F, Michelhaugh SK, Barger GR, Mittal S, Juhasz C: Depression and tryptophan metabolism in patients with primary brain tumors: clinical and molecular imaging correlates. Brain Imaging Behav 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mondanelli G, Volpi C: The double life of serotonin metabolites: in the mood for joining neuronal and immune systems. Curr Opin Immunol 2020, 70: 1–6. [DOI] [PubMed] [Google Scholar]
- 31.•.Mondanelli G, Coletti A, Greco FA, Pallotta MT, Orabona C, Iacono A, Belladonna ML, Albini E, Panfili E, Fallarino F et al. : Positive allosteric modulation of indoleamine 2,3-dioxygenase 1 restrains neuroinflammation. Proc Natl Acad Sci U S A 2020, 117:3848–3857 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that the serotonergic pathway metabolite N-acetlyserotonin increases IDO1 activity and consequently enhances kynurenine production.
- 32.Caragher SP, Hall RR, Ahsan R, Ahmed AU: Monoamines in glioblastoma: complex biology with therapeutic potential. Neuro Oncol 2018, 20:1014–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto-Meyer S, DeFaccio R, Dussold C, Ladomersky E, Zhai L, Lauing KL, Bollu LR, Amidei C, Lukas RV, Scholtens DM, Wainwright DA: A retrospective survival analysis of Glioblastoma patients treated with selective serotonin reuptake inhibitors. Brain Behav Immun Health 2020, 2:100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrera-Rios D, Mughal SS, Teuber-Hanselmann S, Pierscianek D, Sucker A, Jansen P, Schimming T, Klode J, Reifenberger J, Felsberg J et al. : Macrophages/microglia represent the major source of indolamine 2,3-dioxygenase expression in melanoma metastases of the brain. Front Immunol 2020, 11:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, Changsirivathanathamrong D, Wu BJ, Ball HJ, Thomas SR et al. : Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med 2010, 16:279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.•.Ladomersky E, Zhai L, Lauing KL, Bell A, Xu J, Kocherginsky M, Zhang B, Wu JD, Podojil JR, Platanias LC et al. : Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin Cancer Res 2020, 26:5232–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that Ido1 expression is increased in the brains of aged glioblastoma-bearing mice, which associates with worse survival under immunotherapy.
- 37.Ladomersky E, Zhai L, Lenzen A, Lauing KL, Qian J, Scholtens DM, Gritsina G, Sun X, Liu Y, Yu F et al. : IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clin Cancer Res 2018, 24:2559–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E: Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9:1777625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, De Plaen E, Uyttenhove C, Wouters J, Masereel B, Van den Eynde BJ: Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A 2012, 109:2497–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochs K, Ott M, Rauschenbach KJ, Deumelandt K, Sahm F, Opitz CA, von Deimling A, Wick W, Platten M: Tryptophan-2,3-dioxygenase is regulated by prostaglandin E2 in malignant glioma via a positive signaling loop involving prostaglandin E receptor-4. J Neurochem 2016, 136:1142–1154. [DOI] [PubMed] [Google Scholar]
- 41.Kudo T, Prentzell MT, Mohapatra SR, Sahm F, Zhao Z, Grummt I, Wick W, Opitz CA, Platten M, Green EW: Constitutive expression of the immunosuppressive tryptophan dioxygenase TDO2 in glioblastoma is driven by the transcription factor C/EBPbeta. Front Immunol 2020, 11:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guastella AR, Michelhaugh SK, Klinger NV, Fadel HA, Kiousis S, Ali-Fehmi R, Kupsky WJ, Juhasz C, Mittal S: Investigation of the aryl hydrocarbon receptor and the intrinsic tumoral component of the kynurenine pathway of tryptophan metabolism in primary brain tumors. J Neurooncol 2018, 139:239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hao S, Huang G, Feng J, Li D, Wang K, Wang L, Wu Z, Wan H, Zhang L, Zhang J: Non-NF2 mutations have a key effect on inhibitory immune checkpoints and tumor pathogenesis in skull base meningiomas. J Neurooncol 2019, 144:11–20. [DOI] [PubMed] [Google Scholar]
- 44.Ott M, Litzenburger UM, Rauschenbach KJ, Bunse L, Ochs K, Sahm F, Pusch S, Opitz CA, Blaes J, von Deimling A et al. : Suppression of TDO-mediated tryptophan catabolism in glioblastoma cells by a steroid-responsive FKBP52-dependent pathway. Glia 2015, 63:78–90. [DOI] [PubMed] [Google Scholar]
- 45.Mohapatra SR, Sadik A, Tykocinski LO, Dietze J, Poschet G, Heiland I, Opitz CA: Hypoxia inducible factor 1alpha inhibits the expression of immunosuppressive tryptophan-2,3-dioxygenase in glioblastoma. Front Immunol 2019, 10:2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohapatra SR, Sadik A, Sharma S, Poschet G, Gegner HM, Lanz TV, Lucarelli P, Klingmüller U, Platten M, Heiland I, Opitz CA: Front Immunol 2021, 12:590532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.•.Schramme F, Crosignani S, Frederix K, Hoffmann D, Pilotte L, Stroobant V, Preillon J, Driessens G, Van den Eynde BJ: Inhibition of tryptophan-dioxygenase activity increases the antitumor efficacy of immune checkpoint inhibitors. Cancer Immunol Res 2020, 8:32–45 [DOI] [PubMed] [Google Scholar]; This paper links the effectiveness of immune checkpoint therapy to TDO2 activity and tryptophan levels in the blood.
- 48.Winters M, DuHadaway JB, Pham KN, Lewis-Ballester A, Badir S, Wai J, Sheikh E, Yeh SR, Prendergast GC, Muller AJ, Malachowski WP: Diaryl hydroxylamines as pan or dual inhibitors of indoleamine 2,3-dioxygenase-1, indoleamine 2,3-dioxygenase-2 and tryptophan dioxygenase. Eur J Med Chem 2019, 162:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naing A, Eder JP, Piha-Paul SA, Gimmi C, Hussey E, Zhang S, Hildebrand V, Hosagrahara V, Habermehl C, Moisan J, Papadopoulos KP: Preclinical investigations and a first-in-human phase I trial of M4112, the first dual inhibitor of indoleamine 2,3-dioxygenase 1 and tryptophan 2,3-dioxygenase 2, in patients with advanced solid tumors. J Immunother Cancer 2020, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.John F, Bosnyak E, Robinette NL, Amit-Yousif AJ, Barger GR, Shah KD, Michelhaugh SK, Klinger NV, Mittal S, Juhasz C: Multimodal imaging-defined subregions in newly diagnosed glioblastoma: impact on overall survival. Neuro Oncol 2019, 21:264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukas RV, Juhasz C, Wainwright DA, James CD, Kennedy E, Stupp R, Lesniak MS: Imaging tryptophan uptake with positron emission tomography in glioblastoma patients treated with indoximod. J Neurooncol 2019, 141:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL: GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22:633–642. [DOI] [PubMed] [Google Scholar]
- 53.Rashidi A, Miska J, Lee-Chang C, Kanojia D, Panek WK, Lopez-Rosas A, Zhang P, Han Y, Xiao T, Pituch KC et al. : GCN2 is essential for CD8(+) T cell survival and function in murine models of malignant glioma. Cancer Immunol Immunother 2020, 69:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adam I, Dewi DL, Mooiweer J, Sadik A, Mohapatra SR, Berdel B, Keil M, Sonner JK, Thedieck K, Rose AJ et al. : Upregulation of tryptophanyl-tRNA synthethase adapts human cancer cells to nutritional stress caused by tryptophan degradation. Oncoimmunology 2018, 7:e1486353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.••.Bartok O, Pataskar A, Nagel R, Laos M, Goldfarb E, Hayoun D, Levy R, Korner PR, Kreuger IZM, Champagne J et al. : Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature 2021, 590:332–337 [DOI] [PubMed] [Google Scholar]; This paper shows that interferon-gamma-driven tryptophan depletion leads to ribosomal frame shifting and the presentation of aberrant peptides in melanoma.
- 56.Adams S, Teo C, McDonald KL, Zinger A, Bustamante S, Lim CK, Sundaram G, Braidy N, Brew BJ, Guillemin GJ: Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS One 2014, 9:e112945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jane EP, Premkumar DR, Thambireddy S, Golbourn B, Agnihotri S, Bertrand KC, Mack SC, Myers I, Chattopadhyay A, Taylor DL et al. : Targeting NAD(+) biosynthesis overcomes panobinostat and bortezomib-induced malignant glioma resistance. Mol Cancer Res 2020, 18:1004–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosell FI, Kuo HH, Mauk AG: NADH oxidase activity of indoleamine 2,3-dioxygenase. J Biol Chem 2011, 286:29273–29283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E, Li V, Maradana MR, Schiering C, Stockinger B: The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 2018, 49:353–362.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esser C, Rannug A: The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev 2015, 67:259–279. [DOI] [PubMed] [Google Scholar]
- 61.Schulte KW, Green E, Wilz A, Platten M, Daumke O: Structural basis for Aryl hydrocarbon receptor-mediated gene activation. Structure 2017, 25:1025–1033.e3. [DOI] [PubMed] [Google Scholar]
- 62.•.Opitz CA, Somarribas Patterson LF, Mohapatra SR, Dewi DL, Sadik A, Platten M, Trump S: The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer 2020, 122:30–44 [DOI] [PMC free article] [PubMed] [Google Scholar]; This review highlights therapies that modulate tumoral tryptophan metabolism.
- 63.Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X et al. : Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5:1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gramatzki D, Pantazis G, Schittenhelm J, Tabatabai G, Kohle C, Wick W, Schwarz M, Weller M, Tritschler I: Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 2009, 28:2593–2605. [DOI] [PubMed] [Google Scholar]
- 65.••.Rothhammer V, Quintana FJ: The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 2019, 19:184–197 [DOI] [PubMed] [Google Scholar]; This review gives an overview of the ligand-specificity, cell-type-specificity and context-specificity of AHR with a special focus on the immune system.
- 66.Ehrlich AK, Pennington JM, Bisson WH, Kolluri SK, Kerkvliet NI: TCDD, FICZ, and other high affinity AhR ligands dose-dependently determine the fate of CD4+ T cell differentiation. Toxicol Sci 2018, 161:310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prasad Singh N, Nagarkatti M, Nagarkatti P: From suppressor T cells to regulatory T cells: how the journey that began with the discovery of the toxic effects of TCDD led to better understanding of the role of AhR in immunoregulation. Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, Tang-Huau TL, Bohec M, Baulande S, Hacohen N et al. : Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity 2017, 47:582–596.e6. [DOI] [PubMed] [Google Scholar]
- 69.Shin JH, Zhang L, Murillo-Sauca O, Kim J, Kohrt HE, Bui JD, Sunwoo JB: Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A 2013, 110:12391–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piper CJM, Rosser EC, Oleinika K, Nistala K, Krausgruber T, Rendeiro AF, Banos A, Drozdov I, Villa M, Thomson S et al. : Aryl hydrocarbon receptor contributes to the transcriptional program of IL-10-producing regulatory B cells. Cell Rep 2019, 29:1878–1892.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vaidyanathan B, Chaudhry A, Yewdell WT, Angeletti D, Yen WF, Wheatley AK, Bradfield CA, McDermott AB, Yewdell JW, Rudensky AY, Chaudhuri J: The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J Exp Med 2017, 214:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Villa M, Gialitakis M, Tolaini M, Ahlfors H, Henderson CJ, Wolf CR, Brink R, Stockinger B: Aryl hydrocarbon receptor is required for optimal B-cell proliferation. EMBO J 2017, 36:116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B: Targeting CD73 to augment cancer immunotherapy. Curr Opin Pharmacol 2020, 53:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greene LI, Bruno TC, Christenson JL, D’Alessandro A, Culp-Hill R, Torkko K, Borges VF, Slansky JE, Richer JK: A role for tryptophan-2,3-dioxygenase in CD8 T-cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol Cancer Res 2019, 17:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA: An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 2010, 185:3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL: An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2010, 107:20768–20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK: The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol 2010, 11:854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ: Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol 2010, 11:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wagage S, John B, Krock BL, Hall AO, Randall LM, Karp CL, Simon MC, Hunter CA: The aryl hydrocarbon receptor promotes IL-10 production by NK cells. J Immunol 2014, 192:1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL: Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453:65–71. [DOI] [PubMed] [Google Scholar]
- 81.Reardon DA, Desjardins A, Rixe O, Cloughesy T, Alekar S, Williams JH, Li R, Taylor CT, Lassman AB: A phase 1 study of PF-06840003, an oral indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor in patients with recurrent malignant glioma. Invest New Drugs 2020, 38:1784–1795. [DOI] [PubMed] [Google Scholar]