

Abstract
Multiple signals are involved in the regulation of developmental myelination by Schwann cells and in the maintenance of a normal myelin homeostasis throughout adult life, preserving the integrity of the axons in the PNS. Recent studies suggest that Mek/ERK1/2-MAPK and PI3K/Akt/mTOR intracellular signaling pathways play important, often overlapping roles in the regulation of myelination in the PNS. In addition, hyperactivation of these signaling pathways in Schwann cells leads to a late onset of various pathological changes in the sciatic nerves. However, it remains poorly understood whether these pathways function independently or sequentially or converge using a common mechanism to facilitate Schwann cell differentiation and myelin growth during development and in causing pathological changes in the adult animals. To address these questions, we analyzed multiple genetically modified mice using simultaneous loss- and constitutive gain-of-function approaches. We found that during development, the Mek/ERK1/2-MAPK pathway plays a primary role in Schwann cell differentiation, distinct from mTOR. However, during active myelination, ERK1/2 is dependent on mTOR signaling to drive the growth of the myelin sheath and regulate its thickness. Finally, our data suggest that peripheral nerve pathology during adulthood caused by hyperactivation of Mek/ERK1/2-MAPK or PI3K is likely to be independent or dependent on mTOR-signaling in different contexts. Thus, this study highlights the complexities in the roles played by two major intracellular signaling pathways in Schwann cells that affect their differentiation, myelination, and later PNS pathology and predicts that potential therapeutic modulation of these pathways in PNS neuropathies could be a complex process.
Keywords: differentiation, myelin, myelination, Schwann cells
1 |. INTRODUCTION
Proper formation and maintenance of myelin and Remak bundles by myelinating and non-myelinating Schwann cells, respectively, are critically important for peripheral nerve fibers to perform their essential motor and sensory functions, including efficient nerve conduction, providing trophic support to axons, and maintaining overall homeostasis in the PNS (Salzer, 2015; Wilson et al., 2020). It is expected that multiple extracellular and intracellular regulatory mechanisms must be involved in orchestrating these complex processes, since several human hereditary peripheral neuropathies are associated with dysregulation of Schwann cell functions (Scherer & Wrabetz, 2008; Suter & Scherer, 2003). Therefore, a better understanding of the regulatory mechanisms that govern normal Schwann cell functions and those underlying their dysfunction would contribute in the long run to identify potential therapeutic targets for ameliorating the pathology in human neuropathies.
One of the primary extracellular signals that control almost every aspect of PNS myelination is axonal NRG1III (Michailov et al., 2004; Taveggia et al., 2005), which activates Schwann cell ErbB-receptors, leading to intracellular signal transduction, primarily by the classical phosphatidylinositol-4,5-bisphosphate-3-kinase (PI3K) and the mitogen-activated protein-kinase (MAPK) pathways, both of which have been implicated as essential for proper PNS myelination (Figlia et al., 2017a; Newbern & Birchmeier, 2010). Briefly, enhanced myelin growth and increased myelin thickness in the PNS are common features found in several mouse mutants with sustained hyperactivation of PI3K/Akt/mTOR pathways, which is attributed to mTORC1, a major downstream signaling complex comprising the mTOR core kinase, Raptor, and several other adaptor molecules (Beirowski et al., 2017; Domènech-Estévez et al., 2016; Figlia et al., 2017b; Goebbels et al., 2010). Conversely, thin myelin sheaths were observed in mice with Schwann cell-specific ablation of mTOR or Raptor (Norrmén et al., 2014; Sherman et al., 2012). Similarly, Schwann cell-specific constitutive activation of Mek1, upstream of ERK1/2 (extracellular-signal-regulated kinase-1 and −2), primary mediators of the MAPK pathway (Rubinfeld & Seger, 2005), enhanced myelin growth, increasing myelin thickness, and conversely, ablation of Erk1/2 led to hypomyelination in mutant mice (Ishii et al., 2013; Newbern et al., 2011; Sheean et al., 2014). Collectively, these studies argue for a beneficial role of both these pathways in developmental myelination. Paradoxically, sustained hyperactivation of either of these two pathways in Schwann cells can also be detrimental in adulthood, as late onset of pathological changes, including abnormal myelin structures, was observed in sciatic nerves of mutant mice, similar to the pathology seen in certain human hereditary peripheral neuropathies (Beirowski et al., 2017; Domènech-Estévez et al., 2016; Figlia et al., 2017b; Goebbels et al., 2012; Ishii et al., 2016).
Therefore, given the similarities in the outcomes of their perturbation, an important question that emerges is whether these two pathways work as independent parallel pathways or whether there are molecular overlaps in their downstream signaling. Secondly, since these two pathways are also known to regulate CNS myelination (Bercury et al., 2014; Gonsalvez et al., 2016; Ishii et al., 2019; Ishii et al., 2012; Lebrun-Julien et al., 2014; Wahl et al., 2014), another open question is whether they signal differently, affecting myelination in a Schwann cell- and/or oligodendrocyte-specific manner, or whether the signaling mechanisms are conserved between CNS and PNS.
To address these questions, we generated and analyzed a series of genetically modified mice and examined the roles of these two pathways in Schwann cells. Our loss-of-function data suggest a primary role of ERK1/2 in Schwann cell differentiation, in contrast to a primary role of mTOR in oligodendrocyte differentiation, as shown previously. However, ERK1/2 is dependent on mTOR signaling for regulating myelin thickness in the PNS, similar to that reported in the CNS. Finally, our data suggest that both mTOR-dependent and mTOR-independent mechanisms are likely to act downstream of hyperactivated Mek/ERK1/2-MAPK and PI3K in a context-dependent manner, to cause peripheral nerve pathology during adulthood. Thus, Mek/ERK1/2-MAPK and the PI3K/Akt/mTOR signaling in Schwann cells play both independent and cooperative roles in PNS myelination and in the neuropathology of peripheral nerves caused by their dysregulation.
2 |. MATERIAL AND METHODS
2.1 |. Mouse lines
We generated mouse lines CnpCre/+;ERK1−/−;ERK2flox/flox and CnpCre/+;mTORflox/flox referred to here as CnpCre;ERK1/2-dKO and CnpCre; mTOR-KO where ERK1/2 or mTOR is specifically ablated in CnpCre expressing Schwann cells (2ʹ,3ʹ-cyclic nucleotide 3ʹ-phosphohydrolase; (Gravel et al., 1998; Lappe-Siefke et al., 2003)). This was done by appropriate mating of ERK1−/−;ERK2flox/flox or mTORflox/flox line (Jackson Laboratory, Jackson Labs: Stock#:011009; (Risson et al., 2009)) with the CnpCre/+ (Lappe-Siefke et al., 2003) mice. Furthermore, to simultaneously elevate ERK1/2 activity in mTOR-deficient Schwann cell, we generated transgenic mouse lines CnpCre/+; mTORflox/flox;Rosa26Sto pFlMek1DD (referred to here as CnpCre;mTOR-KO;MekDD) by mating the mTORflox/flox mice with our heterozygous CnpCre/+;Rosa26StopFlMek1DD (CnpCre;Mek1DD) mice described previously (Ishii et al., 2013; Ishii et al., 2016; Srinivasan et al., 2009) to produce progeny where Cre-mediated excision of floxed STOP cassette leads to the expression of constitutively active Mek1 transgene (Mek1DD) and simultaneous loss of mTOR gene.
For some experiments, we also generated mice in which mTOR was conditionally ablated in PLP-expressing mature Schwann cells upon intraperitoneal injection of Tamoxifen (Tm) to adult mice. These mice were developed by appropriate mating of PlpCreERT;mTORflox/flox mice (proteolipid protein; Jackson Laboratory; (Doerflinger et al., 2003; Leone et al., 2003)) with either homozygous Rosa26StopFl-Mek1DD line (Mek1DD/Mek1DD) or Rosa26StopFl-P110* line (PikDD/PikDD) ((Srinivasan et al., 2009); Jackson Laboratory, Stock#:012343), to produce Tm-inducible PlpCreERT;mTORflox/flox;Rosa26StopFl-Mek1DD (referred to as PlpCreERT;mTOR-KO;MekDD) and PlpCreERT;mTORflox/flox; Rosa26StopFl-P110* lines (referred to as PlpCreERT;mTOR-KO;PikDD). Mice heterozygous for mTOR (mTORflox/+) were produced in the same litter and referred to as CnpCre;mTOR-het;MekDD and PlpCreERT;mTOR-het;PikDD. In these mice, Cre-mediated excision of floxed STOP cassette leads to the expression of constitutively active P110*-transgene which is a mutant form of P110α (Pik3ca; the catalytic subunit of PI3K) that was made constitutively active by the addition of the p85 iSH2 domain (region between the two Src homology two domains of p85 that is required for the enzymatic activity of p110) to the p110 N-terminus. For all the lines of transgenic mice generated, littermates lacking Cre are referred to as “controls,” facilitating comparison among the genotypes. The genetic background of all the mouse lines used in this study was C57BL/6. Genotyping of different lines of mice was performed by PCR analysis using the appropriate primers as described previously (Ishii et al., 2012). Both male and female mice were used for the study.
2.2 |. Electron microscopy
Transgenic and littermate control mice of both sexes were perfused with 4% paraformaldehyde, 2% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4 (Electron Microscopy Sciences, Hatfield, PA). Sciatic nerves of mice were postfixed in 1% OsO4. Samples were dehydrated through graded ethanol, stained en bloc with uranyl acetate, and embedded in Poly/Bed812 resin (Polysciences Inc., Warrington, PA). Semithin (1 μm) sections were stained with toluidine blue. Ultrathin (0.1 μm) sections from matching areas of experimental and control tissue blocks were cut and visualized using an electron microscope (JEOL1200CX) at 80 kV. Digitized images (magnification 1000×) were used to determine the g-ratios of randomly selected myelinated axons. Approximately 50–100 axons were measured per genotype. Statistical analysis was performed on average g-ratios of all axons using ANOVA. For comparison of control and mutant mice, note that higher g-ratios indicate thinner myelin sheath.
2.3 |. Immunohistochemistry
As described previously (Furusho et al., 2012; Ishii et al., 2012), sciatic nerves from control and mutant mice of both sexes, perfused with PBS or 4% paraformaldehyde/PBS, were subjected to overnight post-fixation in 4% paraformaldehyde/PBS, and then another overnight in 20% sucrose/PBS. Frozen transverse and longitudinal sections (10 μm) of sciatic nerve were cut. Specimen were rehydrated and blocked (1 h) in PBS, 10% NGS, 0.3% TritonX100 and incubated overnight (4°C) in myelin protein zero (MPZ, 1:1000, Dr. Bruce Trapp, Cleveland Clinic) and Neurofilament M (Nf-m; 1:200, Millipore, MA). Prior to immunolabeling Krox-20 (1/200; Dr. Dies Meijer, The University of Edinburgh) and Oct-6 (1/200; Dr. Dies Meijer, The University of Edinburgh) sciatic nerve sections were subjected to antigen retrieval by 5 min of incubation at 95°C in citrate buffer, pH 6.0 followed by washes with PBS (3 times, 10 min). After washing the primary antibodies, the specimens were incubated in secondary antibodies conjugated to Alexa 488 (1:500; Molecular Probes, CA) and Cy3 (1:500; Jackson Immuno-Research, PA), and nuclei were counter-stained with Hoechst blue dye 33,342 (1 μg/ml; Sigma, MO).
2.4 |. In situ hybridization
Transverse sections of the sciatic nerve from mice of both sexes were prepared as above, and in situ hybridization (ISH) was performed as previously described (Furusho et al., 2012; Furusho et al., 2011; Ishii et al., 2012), using riboprobes specific for MBP mRNA (Dr. M. Qiu, Hangzhou Normal University, China). Briefly, after incubation in 1 μg/ml proteinase K at 37°C for 30 min, sections were hybridized over night at 65 °C with digoxigenin-labeled antisense cRNA probe, and washed in 50% formamide, 2× SSC, and 1% SDS at 65°C for 2–3 h, followed by rinses in 2× SSC and 0.2× SSC at room temperature, and 0.1× SSC at 60 °C. After blocking in 1% Tween20 and 1% normal goat serum (1 h), sections were incubated (overnight) in alkaline phosphatase-conjugated anti-digoxigenin antibody (1:5000; Roche Diagnostics, Penzberg, Germany). The color was developed with 4-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate.
2.5 |. Immunoblotting
Immunoblotting was performed as described previously (Fortin et al., 2005). Briefly, equal amounts of total proteins from lysates of sciatic nerves from mice of both sexes were loaded onto SDS-PAGE, transferred to PVDF membranes, and immunolabeled for MPZ (1:10,000; Dr. Bruce Trapp), Krox20 (1:1000; Dr. Dies Meijer), Oct 6 (1:1000; Dr. Dies Meijer), phospho-Erk1/2 (1:10,000, Cell Signaling Technology), phospho-mTOR2448 (1:1000, Cell Signaling Technology) phospho-p70S6KT389 (Thermo Fisher Scientific, Inc., MA), phospho-S6RPS235,S236 (1:1000, Cell Signaling Technology) and GAPDH (1:60,000; Biodesign International, Saco, ME) or β-actin (1:400,000, Sigma Aldrich, MO) as a loading control. Quantification of the bands was done by Image-J software. Statistical analysis was done by one-way ANOVA test.
2.6 |. Giemsa staining for mast cells
Giemsa stock solution in glycerol (50%) and methanol (50%) was diluted in PBS. Longitudinal sections of sciatic nerves were incubated in the diluted solution (0.1% Giemsa/2.5% glycerol/2.5% methanol/PBS) for 10 min at room temperature. After a 5–10 min wash in PBS, the sections were differentiated in 0.25% acetic acid.
2.7 |. Teased fibers preparation
Teased fibers were prepared from sciatic nerves dissected from gluteraldehyde perfused animals according to the procedure described by Viader et al. (Viader et al., 2011) with slight modifications. Specifically, sciatic nerves were washed with 0.1 M cacodylate buffer and then incubated in 1% osmiumtetroxide and 1.5% potassium ferricyanide in 0.1 M cacodylate buffer for 1 h, followed by washes in PBS and incubation in 33%, 66%, and 100% glycerol/PBS for 6 h each. Nerves were then treated with 0.6% Sudan black dissolved in 70% ethanol at room temperature for 30 min, rinsed with 70% ethanol and water, and then placed back in 100% glycerol. Finally, nerves were teased in 100% glycerol and cover slipped for imaging. For phospho-ERK1/2 immunolabeling, unfixed nerves are teased into single individual fibers and arranged onto slides. Fibers were subjected to antigen retrieval by incubation in citrate buffer (pH 6.0, 5 min, 95°C). After three washes in PBS (10 min each), the slides were blocked in 10% NGS for 1 h and incubated overnight at 4°C in anti-phospho-ERK1/2 (1:400; Cell Signaling Technology), which was diluted in 10% NGS/0.3% Triton X-100. The ABC system (Vector Laboratories) and DAB (Sigma) were used to develop the color.
3 |. RESULTS
3.1 |. Differentiation of Schwann cell precursors is differentially affected by conditional ablation of ERK1/2 or mTOR
In the PNS, CNP is first expressed embryonically by Schwann cell precursors that give rise to both myelinating and non-myelinating Schwann cells (Gravel et al., 1998; Jessen & Mirsky, 2005). We utilized CnpCre to conditionally ablate ERK2 or mTOR from Schwann cell precursors to compare in parallel the effects of their loss on Schwann cell differentiation. Specifically, ERK1−/−;ERK2flox/flox mice (Fan et al., 2009) were bred with CnpCre+/− mice (Lappe-Siefke et al., 2003) to generate CnpCre+/−; ERK1−/−;ERK2flox/flox mice (referred to here as CnpCre;ERK1/2-dKO). Similarly, mTORflox/flox mice (Jackson Labs) were bred with CnpCre+/− mice to generate CnpCre+/−;mTORflox/flox mice (referred to here as CnpCre;mTOR-KO). Littermate mice with no Cre were used as controls. Mice heterozygous for mTOR (CnpCre;mTOR-het), showed no phenotype and were indistinguishable from control.
We first examined the ultrastructure of sciatic nerves by electron microscopy (EM) analysis (Figure 1(a)) and found that in the CnpCre; ERK1/2-dKO mice, the majority of axons remained unmyelinated, even at P30. In contrast, in the CnpCre;mTOR-KO, the majority of axons were wrapped by compact myelin sheaths. Consistent with previous reports, the myelin in our CnpCre;mTOR-KO mice appeared thinner than control (Sherman et al., 2012). A similar phenotype was reported when Raptor, major component of mTORC1, was ablated using DhhCre line (Norrmén et al., 2014). Quantification of the numbers of myelinated and unmyelinated axons confirmed these observations and showed a statistically significant reduction in the percentage of axons that were myelinated in the CnpCre;ERK1/2-dKO compared to control and CnpCre;mTOR-KO mice (Figure 1(b)) [% of myelinated axons and p-values: Control (100%) vs. CnpCre;ERK1/2-dKO (17.4%): p = 8.0 ×10−6; Control (100%) vs. CnpCre;mTOR-KO (98.7%): p = .37]. Higher magnification EM images of CnpCre;Erk1/2-dKO sciatic nerves showed that Schwann cells were arrested at different stages of their maturation–that is, as immature Schwann cells engulfing numerous unsorted axons and as promyelinating Schwann cells that had undergone sorting and had established a one-to-one ratio with axons but failed to make compact myelin sheaths (Figure 1(c)).
FIGURE 1.
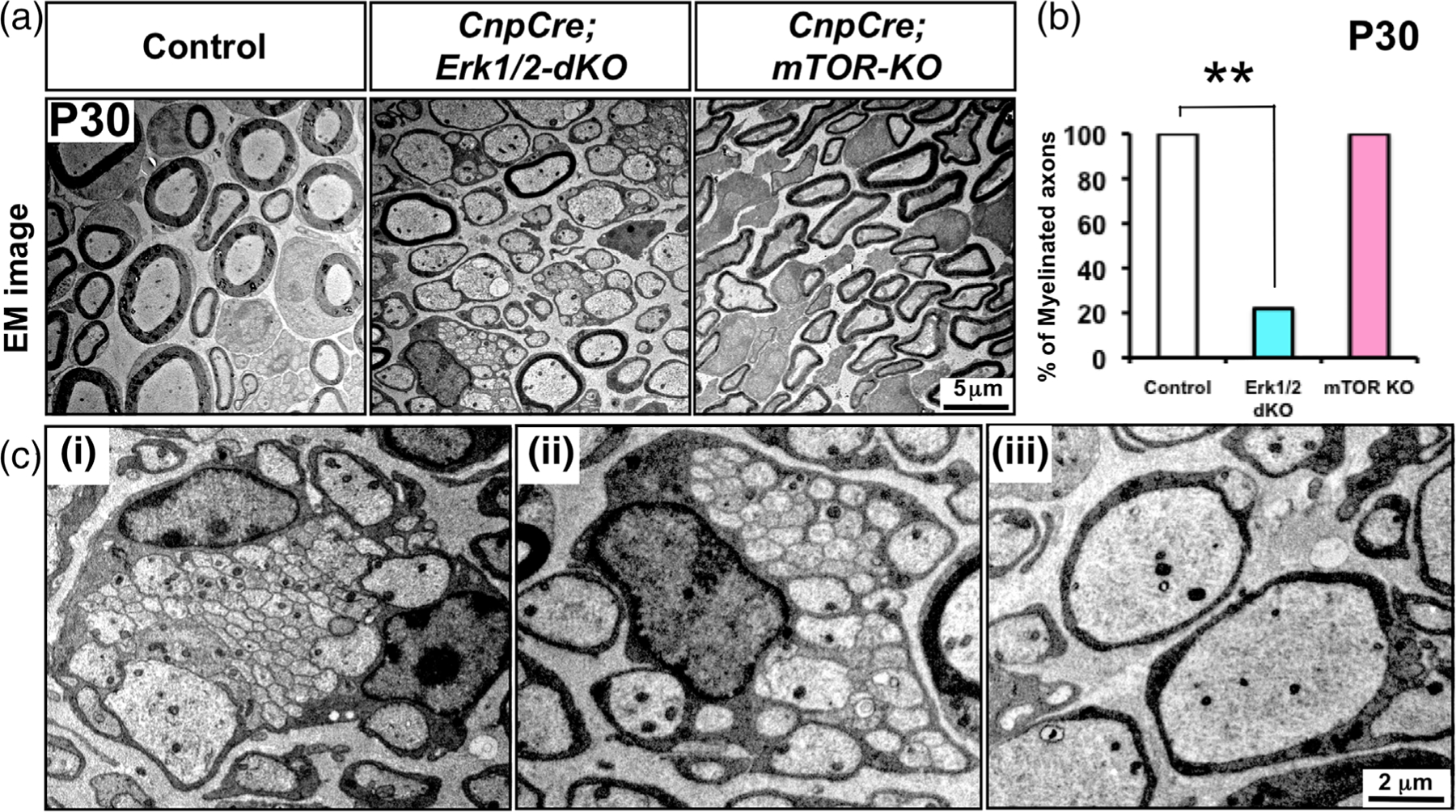
Ultrastructure of sciatic nerves show that the majority of axons remains unmyelinated in the conditional ERK1/2 dKO mice in contrast to mTOR KO mice and that chwann cell differentiation is arrested at different stages of maturation. (a) EM images of sciatic nerve cross-sections at postnatal day (P) 30 from control, CnpCre;ERK1/2-dKO, and CnpCre;mTOR-KO mice show that the majority of axons are unmyelinated in CnpCre;ERK1/2-dKO, whereas in the CnpCre;mTOR-KO mice, all axons are ensheathed by myelin sheaths, although they are thinner than controls, as expected. (b) Quantification confirms that the percentage of myelinated axons is significantly lower in CnpCre;ERK1/2-dKO but is comparable to control in the CnpCre;mTOR-KO mice. (c) Higher-magnification EM images of CnpCre;ERK1/2-dKO sciatic nerves show examples of Schwann cells arrested at different stages of maturation, (i,ii) as immature Schwann cells engulfing numerous small size unsorted axons, and (iii) promyelinating Schwann cells that have sorted and have established a one-to-one ratio with axons but have failed to make compact myelin sheath. Scale bars as indicated. Error bars indicate SEM, **p < .01, N = 3 [Color figure can be viewed at wileyonlinelibrary.com]
We next asked whether the expression of major myelin proteins and transcription factors is affected differently in the sciatic nerves of ERK1/2 dKO compared to mTOR-KO mice. But we first performed immunoblot analysis for p-Erk1/2 and pan-mTOR and confirmed that as expected p-Erk1/2 expression was indeed downregulated in the CnpCre;Erk1/2-dKO and pan-mTOR in the CnpCre;mTOR-KO mice compared to their respective controls (Figure 2(a)). We next examined the sciatic nerves from CnpCre;ERK1/2-dKO and CnpCre;mTOR-KO mice by immunolabeling, in situ hybridization, and western blotting for the expression of major myelin proteins, myelin protein zero (MPZ) and myelin basic protein (MBP), and key transcription factors, Krox-20 (marker of myelinating Schwann cells) and Oct-6 (marker of promyelinating Schwann cells), which normally are reciprocally up- and down-regulated, respectively, at the onset of myelination. Consistent with the arrest of differentiation observed in the CnpCre; ERK1/2-dKO mice by ultrastructural analysis, we found that compared to control, these mice showed a highly significant reduction in the levels of MPZ protein and MBP mRNA (Figure 2(b,c)). Furthermore, as expected at P30, control sciatic nerves showed a low expression of Oct-6 and high expression of Krox-20 in Schwann cells (Figure 2(b)). However, in ERK1/2-deficient Schwann cells, Krox-20 failed to be upregulated, and Oct-6 continued its expression, even at P30. In contrast, CnpCre;mTOR-KO mice showed only a partial reduction in MPZ protein and MBP mRNA signals, consistent with the observed hypomyelination of axons in these mice. Interestingly, the signal intensity of Krox-20 protein was found to be considerably increased in these mutants compared to controls, as was reported previously in Raptor KO mice (Figlia et al., 2017b). Oct-6 expression was retained in some mTOR-deficient Schwann cells, presumably due to a transient delay in the onset of myelination, as was reported previously in Raptor KO mice (Norrmén et al., 2014) (Figure 2(b)). Quantification of MPZ, Krox-20, and Oct-6 protein levels by immunoblot analysis of sciatic nerves confirmed the statistically significant changes in the levels of these proteins in mutant mice, as was indicated by immunolabeling of sciatic nerve sections (Figure 2(d)).
FIGURE 2.
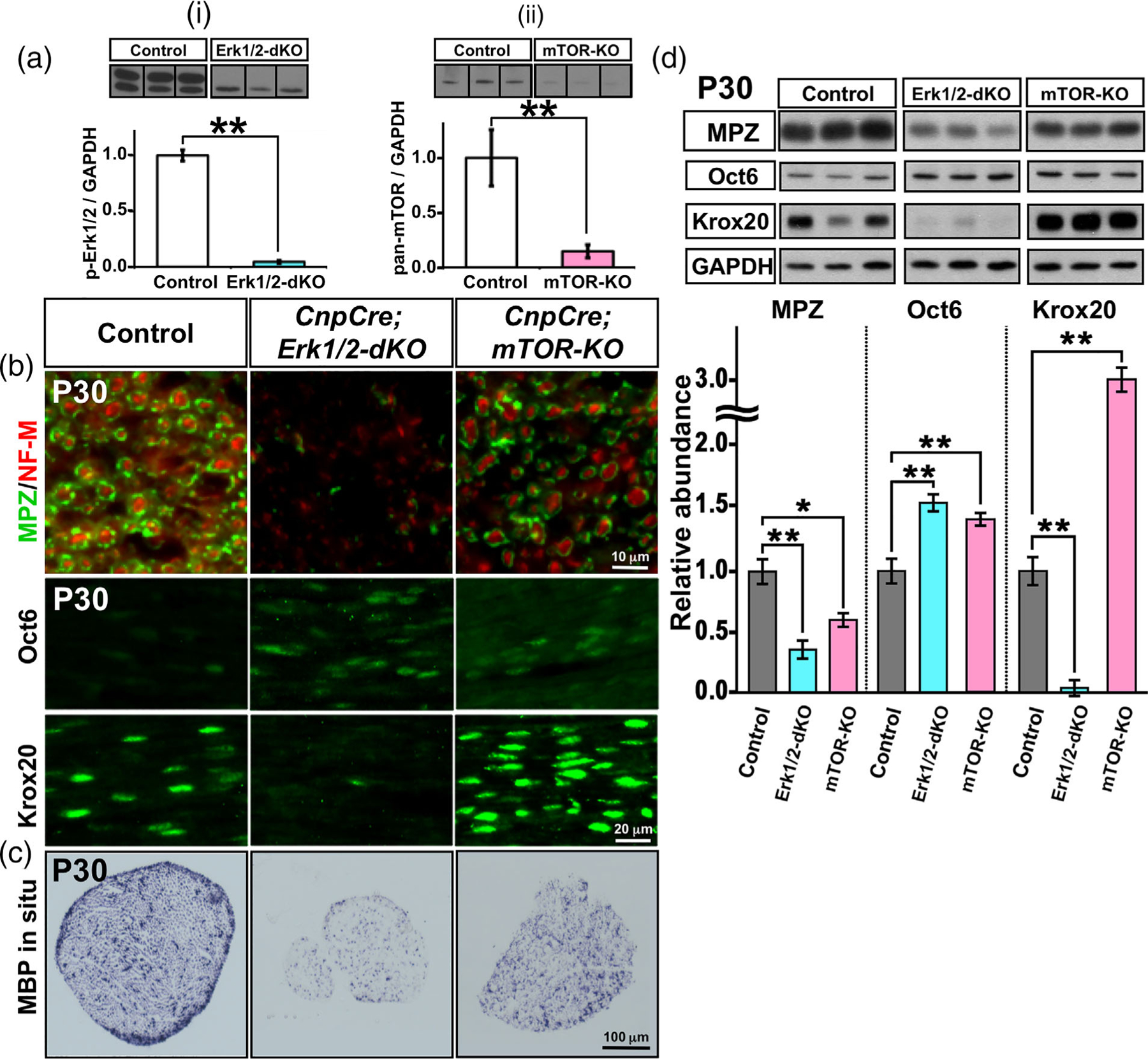
Expression of major myelin proteins and transcription factors is affected differently in ERK1/2 dKO compared to mTOR KO mice. (a) Immunoblot analysis and quantification for p-Erk1/2 and pan-mTOR levels in total proteins homogenates from sciatic nerves at postnatal day 30 (P30) shows a significant reduction of p-Erk1/2 expression in the CnpCre;ERK1/2-dKO (i) and of pan-mTOR expression in the CnpCre;mTOR-KO (ii) mice compared to their respective controls. GAPDH was used as a loading control. N = 3 for each condition. Error bars indicate SEM. **p < .01, Student’s t-test. Immunolabeling (b) and in situ hybridization (c) of sciatic nerve sections at P30 from control, CnpCre;ERK1/2-dKO, and CnpCre;mTOR-KO mice show a dramatic reduction in the signals of myelin protein zero (MPZ), Krox-20 proteins, and MBP mRNA and an increase in Oct-6 protein in Schwann cells of CnpCre;ERK1/2-dKO compared to control mice. In contrast, CnpCre;mTOR-KO mice showed only a partial reduction in MPZ protein, MBP mRNA, and increase in Oct-6 signals but a strong increase in the intensity of Krox-20 signal compared to controls. (d) Immunoblot analysis of total proteins homogenates and quantification of the band intensity from sciatic nerve of CnpCre; ERK1/2-dKO, CnpCre;mTOR-KO, and littermate control mice at P30 shows a highly significant reduction in MPZ and Krox-20 and increase in Oct-6 protein levels in the CnpCre;ERK1/2-dKO compared to control mice. In the CnpCre;mTOR-KO mice, there is a reduction in MPZ and a highly significant increase in Krox-20 and Oct-6 compared to controls. GAPDH was used as a loading control. Nf-m, neurofilament-m. scale bars, as indicated. Error bars indicate SEM. *p < .05, **p < .01, one-way ANOVA. N = 3 for each condition [Color figure can be viewed at wileyonlinelibrary.com]
We conclude that the ablation of ERK1/2 in Schwann cells leads to a highly significant downregulation of major myelin gene/protein and the key transcription factor Krox-20, required for onset of PNS myelination, correlating with arrest of Schwann cell differentiation and myelination. In contrast, mice lacking mTOR in Schwann cells were able to myelinate axons but with thinner myelin sheath than control, indicating that the absence of ERK1/2 affects Schwann cell differentiation in a different and more dramatic manner than the absence of mTOR.
3.2 |. Sustained elevation of Mek/ERK1/2 activity in Schwann cells failed to rescue the deficits in myelin thickness caused by the loss of mTOR
Previous gain- and loss-of-function genetic studies have suggested that both the Mek/ERK1/2-MAPK and the PI3K/Akt/mTOR signaling pathways are involved in regulating the thickness of PNS myelin (Beirowski et al., 2017; Cotter et al., 2010; Domènech-Estévez et al., 2016; Figlia et al., 2017b; Goebbels et al., 2010; Ishii et al., 2013; Norrmén et al., 2014; Sheean et al., 2014; Sherman et al., 2012). However, it was unclear whether these two major signaling pathways play independent parallel roles in vivo or cooperate with each other using a common downstream mechanism to regulate myelin thickness. To address this question, we asked whether sustained elevation of ERK1/2 activity (by constitutive activation of Mek1) in mTOR-deficient Schwann cells could rescue the deficits in myelin thickness observed in the CnpCre; mTOR-KO mice (Sherman et al., 2012). We therefore generated and analyzed a transgenic mouse line (referred to as CnpCre;mTOR-KO;MekDD) where mTOR was deleted (mTOR-KO) and simultaneously Mek1 was constitutively activated (MekDD) specifically in Cnp-expressing Schwann cells and compared it to the CnpCre;mTOR-KO mice (Figure 3(a)).
FIGURE 3.
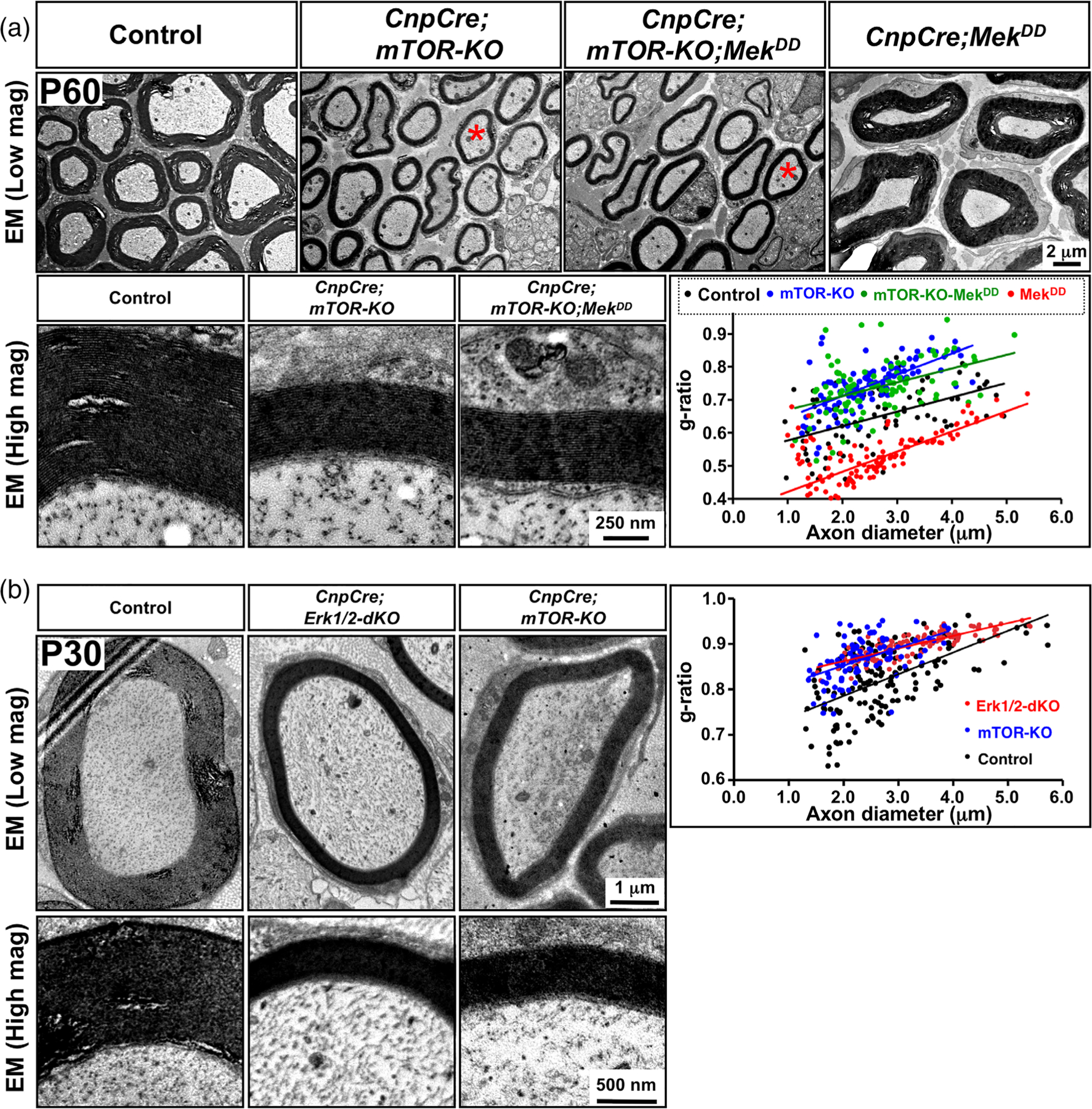
Sustained elevation of ERK1/2 activity in Schwann cells failed to rescue the deficits in myelin thickness in the mTOR KO mice. (a) EM images of sciatic nerve cross-sections at low and high magnification show reduced myelin thickness in the CnpCre;mTOR-KO compared to control, which was not rescued by ERK1/2 elevation in the CnpCre;mTOR-KO;MekDD mice, even though its elevation increases myelin thickness in CnpCre;MekDD compared to control mice. Quantification of myelin thickness by g-ratio analysis, presented as scatter plots relative to axon diameters, indicates that compared to control (black dots), both CnpCre;mTOR-KO (blue dots) and CnpCre;mTOR-KO;MekDD (green dots) show reduced myelin thickness (higher g-ratios), while CnpCre;MekDD mice (red dots) show thicker myelin sheaths compared to controls (lower g-ratios). (b) EM images of sciatic nerve cross-sections at low and high magnification at P30 also show that the thickness of myelin in the CnpCre; ERK1/2-dKO mice is thinner than in the control, but is similar to the CnpCre;mTOR-KO mice. Approximately 100 axons from two mice of each genotype were analyzed. Scale bar as indicated. Red asterisks in (a), shows axons of similar diameter [Color figure can be viewed at wileyonlinelibrary.com]
Ultrastructural examination of sciatic nerves and quantification of myelin thickness by g-ratio analysis showed that consistent with previous reports (Sherman et al., 2012), the majority of the axons in the sciatic nerves of the CnpCre;mTOR-KO mice were myelinated by myelin sheaths that were thinner than control even at P30 (Figure 3(a)). Importantly, we found that sustained over-activation of ERK1/2 could not abrogate this defect, and the myelin sheaths, wrapping similar diameter axons, remained thinner than control in the CnpCre;mTOR-KO;MekDD mice similar to that in the CnpCre;mTOR-KO mice (Figure 3 (a)) [average g-ratios and p-values: Control (0.66) vs. CnpCre;mTOR-KO (0.75): p = 1.1 ×10−12 vs. CnpCre;mTOR-KO;MekDD (0.74): p = 2.4X10−9].
Moreover, we have shown previously that sustained overactivation of Mek/ERK1/2 in Schwann cells led to the formation of significantly thicker myelin sheaths in the CnpCre;MekDD mice compared to controls (Ishii et al., 2013; Sheean et al., 2014). We first confirmed these findings and once again showed the formation of significantly thicker myelin sheaths compared to controls, in CnpCre;MekDD mice (Figure 3(a)). This Mek/ERK1/2-driven increase in myelin thickness was completely abrogated when mTOR was simultaneously ablated in these mice (CnpCre; mTOR-KO;MekDD). Quantification of myelin thickness by g-ratio analysis confirmed that compared to control, increased myelin thickness was observed in the CnpCre;MekDD mice but not in the CnpCre;mTOR-KO; MekDD mice (Figure 3(a)) [average g-ratios and p-values: Control (0.66) vs. MekDD (0.51): p = .2×10−24 vs. CnpCre;mTOR-KO;MekDD (0.74): p = 2.4X10−9]. These data together indicate a role of mTOR signaling in the ERK1/2-mediated increase of myelin thickness during PNS myelination.
Furthermore, as we have shown above (Figure 1(a,b)), in the CnpCre;Erk1/2-dKO mice, 80% of axons remained unmyelinated, even at P30 (last time point studies due to lethality beyond this). We found that the remaining 20% of axons of similar diameter appeared to be wrapped by thinner-than-normal myelin sheaths. We quantified myelin thickness of these myelinated axons by g-ratio analysis and found that similar to CnpCre;mTOR-KO, the myelin sheath was significantly thinner than control in these mice [average g-ratios and p-values: Control (0.82) vs. CnpCre;Erk1/2-dKO (0.90), p = .1 ×10−20 vs. CnpCre;mTOR-KO (0.87)] (Figure 3(b)). These loss- and gain-of-function studies together indicate that ERK1/2 is needed not only for differentiation of Schwann but also for the growth of the myelin sheath in the PNS.
Taken together, we conclude that the deficits in myelin growth caused by the loss of mTOR during developmental myelination in the PNS could not be abrogated by simultaneous elevation of ERK1/2 activity. Conversely, the increase of myelin thickness caused by the hyperactivation of ERK1/2 was abrogated when mTOR was simultaneously ablated in Schwann cell precursors, together suggesting that ERK1/2-mediated regulation of myelin thickness is dependent on mTOR signaling in the PNS.
3.3 |. Ablation of mTOR early during development abrogates pathological changes caused by the simultaneous hyperactivation of Mek/ERK1/2 in myelinating and non-myelinating Schwann cells
We have previously shown that increasing the level of ERK1/2 in Schwann cell precursors leads to a progressive late onset of pathological changes in the sciatic nerves of adult and aging mice, which is characterized by abnormal myelin growth and the formation of “tomacula-like” structures in the CnpCre;MekDD mice (Ishii et al., 2016). Similar abnormalities were reported when Egr2Cre line was used to constitutively express MekDD (Sheean et al., 2014). Here, we asked whether these ERK1/2-mediated pathological changes involved mTOR signaling in Schwann cells and therefore could potentially be abrogated by simultaneous deletion of mTOR in these mice. To address this question, we hyperactivated Mek/ERK1/2 and simultaneously ablated mTOR from Schwann cell precursors and analyzed the sciatic nerves of control, CnpCre;MekDD, CnpCre;mTOR-het;MekDD, and CnpCre;mTOR-KO;MekDD mice at 2–8 months of age (Figure 4(a)). As expected, the aberrant myelin structures and increased extracellular spaces were observed in the CnpCre;MekDD mice at 2 months of age, which became more pronounced by 6 months. Similar pathology was observed in the CnpCre;mTOR-het;MekDD mice compared to controls. However, these pathological changes were completely abrogated when both alleles of mTOR were deleted in Schwann cells in which Mek/ERK1/2 were also hyperactivated (CnpCre;mTOR-KO; MekDD). We also examined teased fiber preparations of sciatic nerves stained with Sudan black (Figure 4(b)). Abnormal myelin with focal thickening at the paranodes (tomacula) was clearly identified in the CnpCre;MekDD and CnpCre;mTOR-het;MekDD mice but not in the CnpCre;mTOR-KO;MekDD mice. To quantify the presence of pathological structures in the mutant mice we examined semithin cross sections and found that about 25%–30% of myelinated axons showed abnormal myelin structures in the CnpCre;MekDD and CnpCre;mTOR-het; MekDD mice, whereas the CnpCre;mTOR-KO;MekDD and control mice did not show these abnormalities (Figure 4(e)).
FIGURE 4.
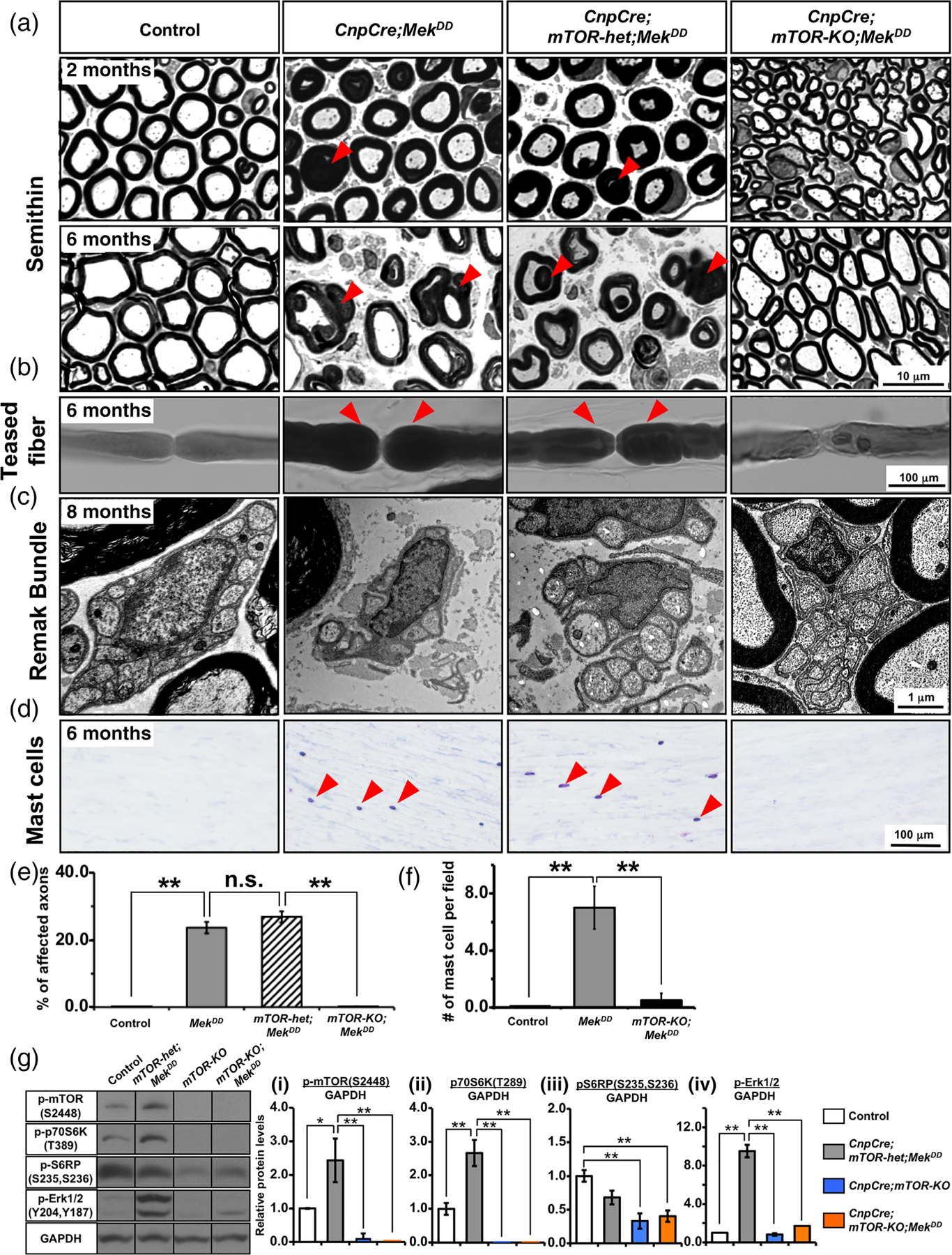
Ablation of mTOR in Schwann cell precursors early during development abrogates pathological changes in myelinating and non-myelinating caused by the simultaneous hyperactivation of Mek/ERK1/2. (a) Semithin sections of sciatic nerves from control, CnpCre;MekDD, CnpCre;mTOR-het;MekDD, and CnpCre;mTOR-KO;MekDD mice at 2 and 6 months show abnormal myelin figures (examples, red arrowheads) and enlarged extracellular space already at 2 months in the CnpCre;MekDD and CnpCre;mTOR-het;MekDD mice but not in the CnpCre;mTOR-KO;MekDD mice. By 6 months of age, abnormal myelin figures and the enlargement of extracellular space are more pronounced in both the CnpCre;MekDD and CnpCre;mTOR-het;MekDD mice but are not seen in the CnpCre;mTOR-KO;MekDD or control mice. (b) Sudan black staining of teased fiber preparation of sciatic nerve shows thickening of myelin at the paranodes (tomacula, red arrowheads) in the CnpCre;MekDD and CnpCre;mTOR-het; MekDD mice but not in the control or CnpCre;mTOR-KO;MekDD mice. (c) High-magnification EM images of sciatic nerve cross-sections show normal Remak bundles in control, but its structure appears disrupted in the CnpCre;MekDD and CnpCre;mTOR-het;MekDD mice. In contrast, CnpCre; mTOR-KO;MekDD mice show normal Remak bundles. (d) Longitudinal sections of sciatic nerves, stained at 6 months with Giemsa stain (blue) to identify mast cells (arrowheads), show increased infiltration in the CnpCre;MekDD and CnpCre;mTOR-het;MekDD but not in the CnpCre;mTOR-KO; MekDD mice. (e) Quantification of abnormal myelin/axon structures in seminthin sections of sciatic nerves at 6 months show a statistically significant increase in the percentage of affected axons in the CnpCre;MekDD and CnpCre;mTOR-het;MekDD mice compared with control mice. This increase was completely abrogated in the CnpCre;mTOR-KO;MekDD mice. 10 fields at 100x per genotype were analyzed from 2 mice per group. (f) Quantification of mast cell infiltration in the longitudinal section of sciatic nerves show a statistically significant increased at 6 months in the CnpCre;MekDD compared with the control mice which was abrogated in the CnpCre;mTOR-KO;MekDD mice. 2–3 fields at 20x per genotype were analyzed from at least 2–3 mice per group. (g) Immunoblot analysis and quantification for p-mTORS2448, p-p70S6K T389, p-S6RPS235,S236 and p-Erk1/2 levels in total proteins homogenates from sciatic nerve at 2 months shows a significant increase in p-mTORS2448, p-p70S6KT389 and p-Erk1/2 expression in the CnpCre;mTOR-het;MekDD compared to control. These increases were abrogated when mTOR was ablated in CnpCre; mTOR-KO and CnpCre;mTOR-KO;MekDD mice and these mice showed dramatically reduced levels of p-mTORS2448, p-p70S6KT389 p-S6RPS235,S236 and p-Erk1/2 compared to controls. GAPDH was used as a loading control. N = 3 for each condition. Error bars indicate SEM. *p < .05, **p < .01, one-way ANOVA. Scale bar as indicated. Representative images of sciatic nerves are shown [Color figure can be viewed at wileyonlinelibrary.com]
We next examined the effects of direct hyperactivation of MAPK/ERK1/2 in non-myelinating Schwann cells that engulf small diameter axons to form structures called Remak bundle (Jessen & Mirsky, 2005). Remak bundle disruption and Mek activation have been previously reported in mice with Nf1 mutation (Mayes et al., 2011; Wu et al., 2008). We found that while sciatic nerve cross-sections from 6–8-month-old control mice showed normal Remak bundles, they appeared disrupted and disorganized in the CnpCre; MekDD and CnpCre;mTOR-het;MekDD mice. In contrast, CnpCre;mTOR-KO;MekDD mice showed normal Remak bundles, indicating complete rescue of the defect by mTOR ablation in these mice (Figure 4(c)).
Inflammatory reactions, such as mast cell infiltration plays a role in Schwann cell-mediated nerve pathology (Monk et al., 2007). We had previously shown an increase in mast cell infiltration in the sciatic nerves of CnpCre;MekDD mice (Ishii et al., 2016). Here we examined the affect of mTOR ablation on mast cell infiltration by Giemsa staining of longitudinal sections of sciatic nerves (Figure 4(d)). We found that the increased infiltration of mast cells was observed not only in the sciatic nerves of the CnpCre;MekDD but also in CnpCre; mTOR-het;MekDD mice. However, this increase did not occur when mTOR was ablated in the CnpCre;mTOR-KO;MekDD mice. Quantification of the numbers of mast cells confirmed these findings and showed that while the numbers of mast cells were significantly increased in CnpCre;MekDD sciatic nerves compared to controls, there was no such increase in CnpCre;mTOR-KO;MekDD mice (Figure 4(f)).
Finally, in order to get a better understanding of the status of signaling downstream of mTOR, in Schwann cell of mice where mTOR was ablated and simultaneously Erk1/2 was activated during Schwann cell development, we performed immunoblot analysis for p-p70S6KT389 and p-S6RPS235,S236, classical mTOR targets, in sciatic nerve protein homogenates from the CnpCre;mTOR-het;MekDD, CnpCre;mTOR-KO, CnpCre;mTOR-KO;MekDD and control mice (Figure 4(g)). But first we examined the level of p-mTOR2448 and demonstrated that stimulation of Erk1/2 activity in CnpCre;mTOR-het; MekDD led to a significant elevation of p-mTOR2448 in sciatic nerves, similar to that we showed previously in the spinal cord of these mice (Furusho et al., 2017). We also demonstrated that p-mTOR2448 was dramatically downregulated in both the CnpCre;mTOR-KO and CnpCre; mTOR-KO;MekDD mice compared to control and CnpCre;mTOR-het; MekDD, as expected (Figure 4(g-i)). Similarly, p-p70S6KT389 and p-S6RPS235,S236 were also downregulated in these mice with mTOR ablation (Figure 4(g-ii,iii)).
In order to determine if Erk1/2 activation was affected by mTOR ablation in Schwann cells, we examined p-Erk1/2 levels by immunoblotting in sciatic nerve homogenates (Figure 4(g-iv)). We found that while p-Erk1/2 levels were significantly elevated in CnpCre;mTOR-KO;MekDD compared to control mice, as expected, this upregulation was abrogated in both CnpCre; mTOR-KO and CnpCre;mTOR-KO;MekDD mice where mTOR was ablated.
We conclude that the late onset of sciatic nerve pathology caused by the sustained elevation of Mek/ERK1/2 activity was completely abrogated when mTOR was simultaneously deleted from Cnp-expressing Schwann cells precursors early during development. Furthermore, the downregulation of p-p70S6KT389, p-S6RPS235,S236 and p-Erk1/2 activities correlated with the loss of mTOR in the CnpCre;mTOR-KO;MekDD and with the abrogation of the pathology in these mice.
3.4 |. Mek/ERK1/2-mediated myelin pathology was not abrogated when mTOR was simultaneously ablated from myelinating Schwann cells during adulthood
We have previously shown that hyperactivation of Mek/ERK1/2, when induced in myelinating Schwann cells during adulthood, leads to a similar late onset of myelin pathology as was observed when it was hyperactivated early during development (Ishii et al., 2016). We therefore asked whether hyperactivated Mek/ERK1/2 in this scenario also uses mTOR activation as a downstream signal to manifest pathological changes. To test this, we generated transgenic mice where Mek was constitutively activated (MekDD), and simultaneously, mTOR was ablated in the adult mice using tamoxifen-inducible PlpCreERT mice (referred to as PlpCreERT;mTOR-KO;MekDD) (Figure 5). We examined semithin sections of sciatic nerves from control, PlpCreERT;MekDD and PlpCreERT;mTOR-KO;MekDD mice injected with Tm at 1 month of age and analyzed at 12 months post-injection (MPI). As expected, we found numerous abnormal myelin figures in the PlpCreERT;MekDD but not in the controls, as we had shown previously (Ishii et al., 2016). Interestingly, the ablation of mTOR was unable to abrogate this myelin pathology and the PlpCreERT;MekDD and PlpCreERT;mTOR-KO;MekDD looked very similar (Figure 5(a)). Higher-magnification EM images of sciatic nerve cross-sections showed abnormal myelin structures in detail, including out-foldings with invaginating recurrent loops that appear as concentric rings of myelin, Wallerian-type degeneration with axons in different stages of degeneration. And aberrant hypermyelination with axonal compression, which were present in both PlpCreERT;MekDD (Figure 5(b-i–iii)) and PlpCreERT;mTOR-KO;MekDD (Figure 5(b-iv–vi)) mice. This was further confirmed by quantification of these abnormal structures on semithin cross sections of sciatic nerves from control and mutant mice (Figure 5(d)). We found a highly significant and similar increase in the percentage of affected axons in the PlpCreERT;MekDD and PlpCreERT;mTOR-KO;MekDD mice compared to controls.
FIGURE 5.
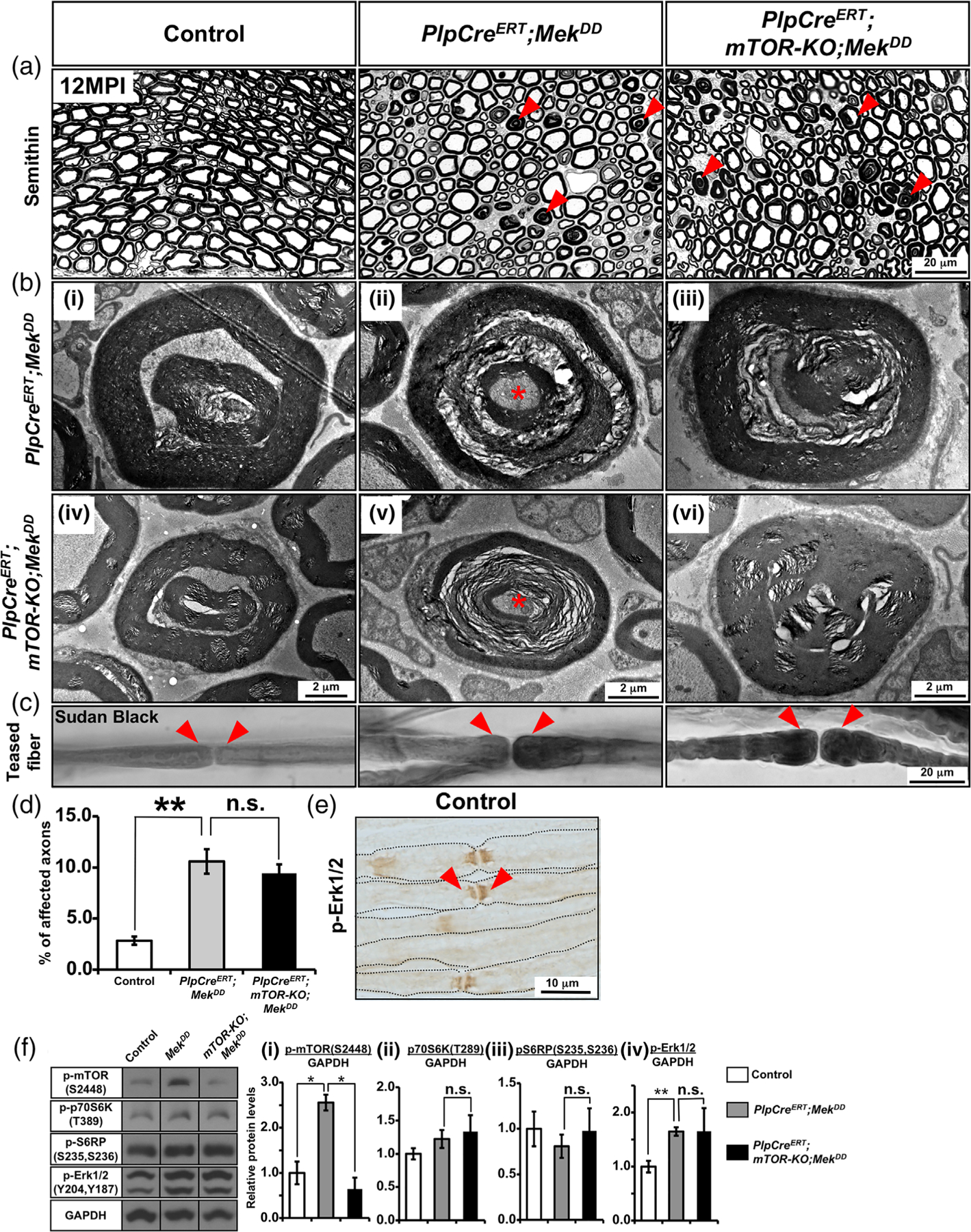
Mek/ERK1/2-mediated myelin pathology was not abrogated when mTOR was simultaneously ablated during adulthood from myelinating Schwann cells. (a) Semithin sections of sciatic nerves from control, PlpCreERT;MekDD, and PlpCreERT;mTOR-KO;MekDD mice injected with Tm at 1 month of age and analyzed at 12 months post-injection (MPI) show numerous abnormal myelin figures (red arrowheads) in both the PlpCreERT;MekDD and PlpCreERT;mTOR-KO;MekDD mice but not in the controls. (b) EM images at higher magnification show examples of abnormal myelin structures, including out-foldings with invaginating recurrent loops that appear as concentric rings of myelin (i,iv), Wallerian-type degeneration with axons in different stages of degeneration (ii,v), and aberrant hyper-myelination with axon compression (iii,vi) in the PlpCreERT; mTOR-KO;MekDD mice (iv–vi), which is similar to that in PlpCreERT;MekDD mice (i–iii). (c) Teased fiber preparation from sciatic nerves stained with Sudan black shows focal myelin thickening at the paranodes forming tomacula-like structures (red arrowheads) in both the PlpCreERT;MekDD mice and PlpCreERT;mTOR-KO;MekDD mice but not in the control. (d) Quantification of abnormal myelin/axon figures in seminthin sections of sciatic nerves from control and mutant mice at 12 MPI show a statistically significant increase in the percentage of affected axons in the PlpCreERT; MekDD mice compared with controls which was not abrogated in the PlpCreERT;mTOR-KO;MekDD mice. Ten fields at 100x per genotype were analyzed from 2 mice per group. (e) Immunolabeling of teased fibers from sciatic nerves of 2-month-old control mice show intense staining for phospho-ERK1/2 at the paranodal region of the myelinated fibers (arrowheads). Scale bar as indicated. Asterisks, degenerating axons. Representative images of sciatic nerves from 2–3 mice per genotype are shown. (f) Immunoblot analysis and quantification for (i) p-mTORS2448, (ii) p-p70S6KT389, (iii) p-S6RPS235,S236, and (iv) p-Erk1/2 levels in total protein homogenates from sciatic nerve at approximately 11 MPI shows a statistically significant increase in p-mTORS2448 and p-Erk1/2 expression levels in the PlpCreERT;MekDD compared to control mice. While ablation of mTOR led to significant decrease of p-mTORS2448 in the PlpCreERT;mTOR-KO;MekDD mice compared to PlpCreERT;MekDD, the levels of p-p70S6KT389, p-S6RPS235,S236 and p-ERK did not show such decrease. GAPDH was used as a loading control. N = 3 for each condition. Error bars indicate SEM. *p < .05, **p < .01, one-way ANOVA [Color figure can be viewed at wileyonlinelibrary.com]
Teased sciatic nerve fibers from PlpCreERT;MekDD mice at 12 MPI showed the presence of focal thickening of the myelin sheath at the paranodal regions (tomacula). However, this myelin pathology could not be abrogated after ablation of mTOR in the PlpCreERT;mTOR-KO;MekDD mice (Figure 5(c)). Since the myelin pathology appeared to originate from paranodes, we examined the expression pattern of activated ERK1/2 by immunolabeling teased fiber preparations of control sciatic nerves with anti-phospho-Erk1/2 and found a high level of p-Erk1/2 localized at the paranodes (Figure 5(e)).
Lastly, we performed immunoblot analysis for p-mTOR2448, p-p70S6KT389, p-S6RPS235,S236 and p-Erk1/2 on sciatic nerve homogenates from the PlpCreERT;MekDD, PlpCreERT;mTOR-KO;MekDD and control mice to determine how the ablation of mTOR and simultaneous activation of Erk1/2 in Schwann cells during adulthood would affect their activity (Figure 5(f)). We found that compared to controls, the level of p-mTOR2448 was significantly elevated in adult sciatic nerves when Erk1/2 activity was elevated in PlpCreERT;MekDD mice. Furthermore, as expected, a significant reduction of p-mTOR2448 was observed in the PlpCreERT;mTOR-KO;MekDD mice compared to PlpCreERT;MekDD which demonstrated that mTOR was indeed ablated in the adult sciatic nerves of these mice (Figure 5(f-i)). However, in contrast to p-mTOR2448, its downstream targets, p-p70S6KT389 and p-S6RPS235,S236, were neither elevated nor downregulated in PlpCreERT;MekDD or PlpCreERT;mTOR-KO;MekDD mice (Figure 5(f-ii,iii)). As expected, p-Erk1/2 levels were significantly elevated in PlpCreERT; MekDD compared to control mice. However, unlike CnpCre;mTOR-KO;MekDD the p-Erk1/2 activity remained elevated in the PlpCreERT; mTOR;MekDD mice at a level comparable to PlpCreERT;MekDD (Figure 5(f-iv)).
We conclude that the ERK1/2-mediated pathology cannot be abrogated when its hyperactivation and simultaneous ablation of mTOR occurs in adult mice. Furthermore, p-p70S6KT389 and p-S6RPS235,S236 and p-Erk1/2 activities were not downregulated upon mTOR ablation, together indicating that the Mek/ERK1/2-mediated manifestation of sciatic nerve pathology becomes mTOR-independent during adulthood.
3.5 |. PI3K-mediated pathology was also not abrogated when mTOR was simultaneously ablated during adulthood from myelinating and non-myelinating Schwann cells
It has been reported that ablation of PTEN in Schwann cells, which indirectly results in hyperactivation of the PI3K pathway, leads to a late onset of abnormal myelin pathology in the sciatic nerves of these mice (Goebbels et al., 2012). In this study, we generated and analyzed transgenic mice, where hyperactivation of PI3K was induced in Schwann cells of adult mice directly by constitutive activation of the Pik3ca (PikDD), which encodes the catalytic subunit of PI3K. We found that similar to the PTEN-KO mice, sciatic nerves from PlpCreERT;PikDD and PlpCreERT;mTOR-het;PikDD mice showed a late onset of pathological changes (Figure 6). Given the similarities in the pathological outcomes observed in the PlpCreERT;mTOR-het;PikDD and the PlpCreERT;mTOR-het;MekDD mice, we asked whether simultaneous ablation of mTOR induced during adulthood would have an impact on the PI3K-mediated pathology. We therefore generated tamoxifen-inducible conditional PlpCreERT;mTOR-KO;PikDD mice and compared them with control and PlpCreERT;mTOR-het;PikDD for potential sciatic nerve pathology at 5 MPI, following Tm injection at 1 month of age (Figure 6). Toluidine blue-stained semithin cross-sections and EM images from the PlpCreERT;mTOR-het;PikDD mice showed examples of abnormal myelin overgrowths, appearing as darkly stained myelin figures in semithin sections (Figure 6(a)), and in high-magnification EM images, as myelin out-foldings with evaginating recurrent loops, redundant myelin, constricting and displacing the axon, and multiple evaginating recurrent loops that appeared as if one Schwann cell was ensheathing multiple small diameter axons (Figure 6(b-i–iii)). Interestingly, ablation of mTOR failed to abrogate these myelin abnormalities in the PlpCreERT;mTOR-KO;PikDD mice (Figure 6(a,b-iv–vi)). Furthermore, examination of Sudan black-stained teased sciatic nerve fibers from the PlpCreERT;mTOR-het;PikDD mice showed abnormal myelin structures, which were also not abrogated in mice where mTOR was ablated (Figure 6(c)). These observations were confirmed by quantification of the abnormal myelin figures in semithin cross sections from sciatic nerves of control, PlpCreERT;PikDD and PlpCreERT;mTOR-KO; PikDD mice which showed that compared to control there was a statistically significant and similar increase in the percentage of affected axons in both the PlpCreERT;PikDD and PlpCreERT;mTOR-KO;PikDD mice (Figure 6(d)), indicating that like Mek, PI3K-mediated pathology could not be abrogated when mTOR was simultaneously ablated in the adult animals.
FIGURE 6.
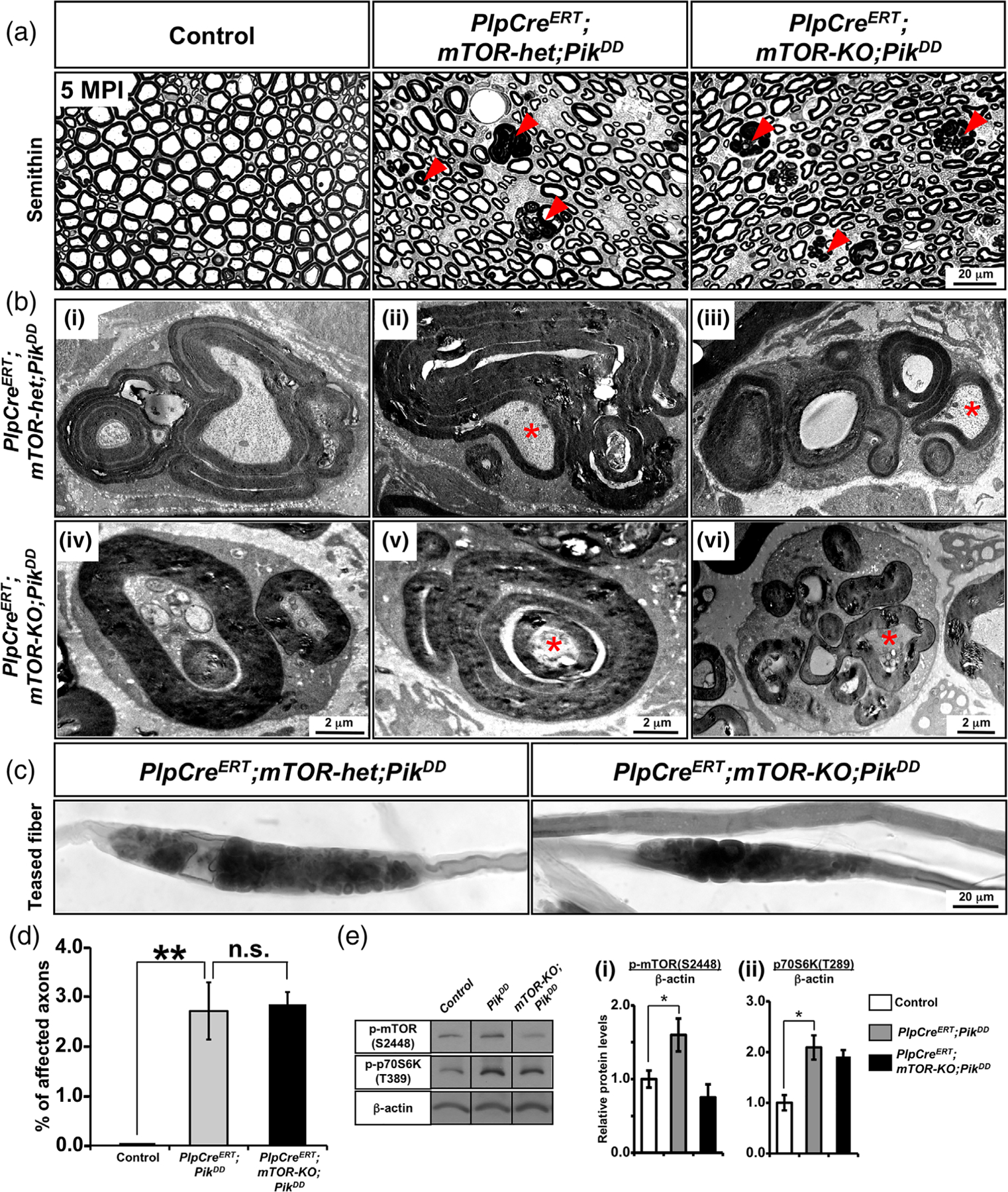
PI3K-mediated pathology was also not abrogated when mTOR was simultaneously ablated during adulthood from myelinating Schwann cells. (a) Semithin sections of sciatic nerves from control, PlpCreERT;mTOR-het;PikDD, and PlpCreERT;mTOR-KO;PikDD mice injected with Tm at 1 month of age and analyzed at 5 months postinjection (MPI) show numerous abnormal myelin figures (red arrowheads) in both the PlpCreERT;mTOR-het;PikDD and PlpCreERT;mTOR-KO;PikDD mice but not in the controls. (b) EM images at higher magnification show presence of abnormalities in both the PlpCreERT;mTOR-het;PikDD (i–iii) and PlpCreERT;mTOR-KO;PikDD (iv–vi), which includes (i,iv) myelin out-foldings with evaginating recurrent loops, (ii,v) redundant myelin, constricting and displacing the axon, and (iii,vi) multiple evaginating recurrent loops that appear as Schwann cell ensheathing multiple small-diameter axons. (c) Teased fiber preparation from sciatic nerve shows focal myelin thickening with abnormal structures in both the PlpCreERT;mTOR-het;PikDD and PlpCreERT;mTOR-KO;PikDD mice. Scale bar as indicated. Asterisks, axonal abnormalities. Representative images of sciatic nerves from 2–3 mice per genotype are shown. (d) Quantification of abnormal myelin figures in seminthin sections of sciatic nerves from mice at 5 MPI show a statistically significant increase in the percentage of affected axons in the PlpCreERT;mTOR-het;PikDD mice compared to control mice which failed to be rescue in the PlpCreERT;mTOR-KO;PikDD mice. Ten fields at 100x per genotype were analyzed from at least 2–3 mice per group. Error bars indicate SEM. **p < .01, one-way ANOVA. (e) Immunoblot analysis and quantification for p-mTORS2448 and p-p70S6KT389 levels in total protein homogenates from sciatic nerve at 2MPI shows a significant increase in p-mTORS2448 and p-p70S6KT389 levels in the PlpCreERT;PikDD mice compared to controls. While ablation of mTOR showed reduction trend of p-mTORS2448 levels in the PlpCreERT;mTOR-KO;PikDD compared to PlpCreERT;PikDD mice, the levels of p-p70S6KT389 did not show such a trend. β-Actin was used as a loading control. N = 3 for control and PlpCreERT;PikDD and N = 2 for PlpCreERT;mTOR-KO;PikDD. Error bars indicate SEM. *p < .05, Student’s t-test [Color figure can be viewed at wileyonlinelibrary.com]
Lastly, since Pik3ca could directly activate mTOR in the absence of PI3K activation (Sawade et al., 2020), we first wanted to confirm that mTOR was indeed activated in these mice. We therefore performed immunoblot analysis for p-mTOR2448 on sciatic nerve homogenates from the PlpCreERT;PikDD, PlpCreERT;mTOR-KO;PikDD and control mice (Figure 6(e)). As expected, we found that p-mTOR2448 was upregulated in the PlpCreERT;PikDD mice compared to controls and that ablation of mTOR abolished this increase in the PlpCreERT;mTOR-KO;PikDD mice (Figure 6(e-i)). We next examined the status of p70S6K activity and found that p-p70S6KT389 levels were also elevated in the PlpCreERT;PikDD mice and that they remained elevated even in the absence of mTOR in PlpCreERT;mTOR-KO;PikDD mice (Figure 6(e-ii)).
We next examined the structure of Remak bundles in control and mutant mice. While the Remak bundles in the control mice were well formed, we found that they were disrupted in the PlpCreERT;mTOR-het;PikDD mice (Figure 7(a)). Ablation of mTOR was unable to prevent this disruption of Remak bundles in the PlpCreERT;mTOR-KO;PikDD mice. EM images at high magnification showed other abnormalities in the PlpCreERT;mTOR-het;PikDD mice, which include multiple redundant membranous structures, often appearing to nonrandomly hyper-wrap small-diameter axons, and increased accumulation of extracellular collagen fibers, often surrounded by abnormal hyper-wrapped membranes (Figure 7(b-i–iii)). These abnormalities also did not get abrogated by mTOR ablation in the PlpCreERT;mTOR-KO;PikDD mice (Figure 7(b-iv–vi)).
FIGURE 7.
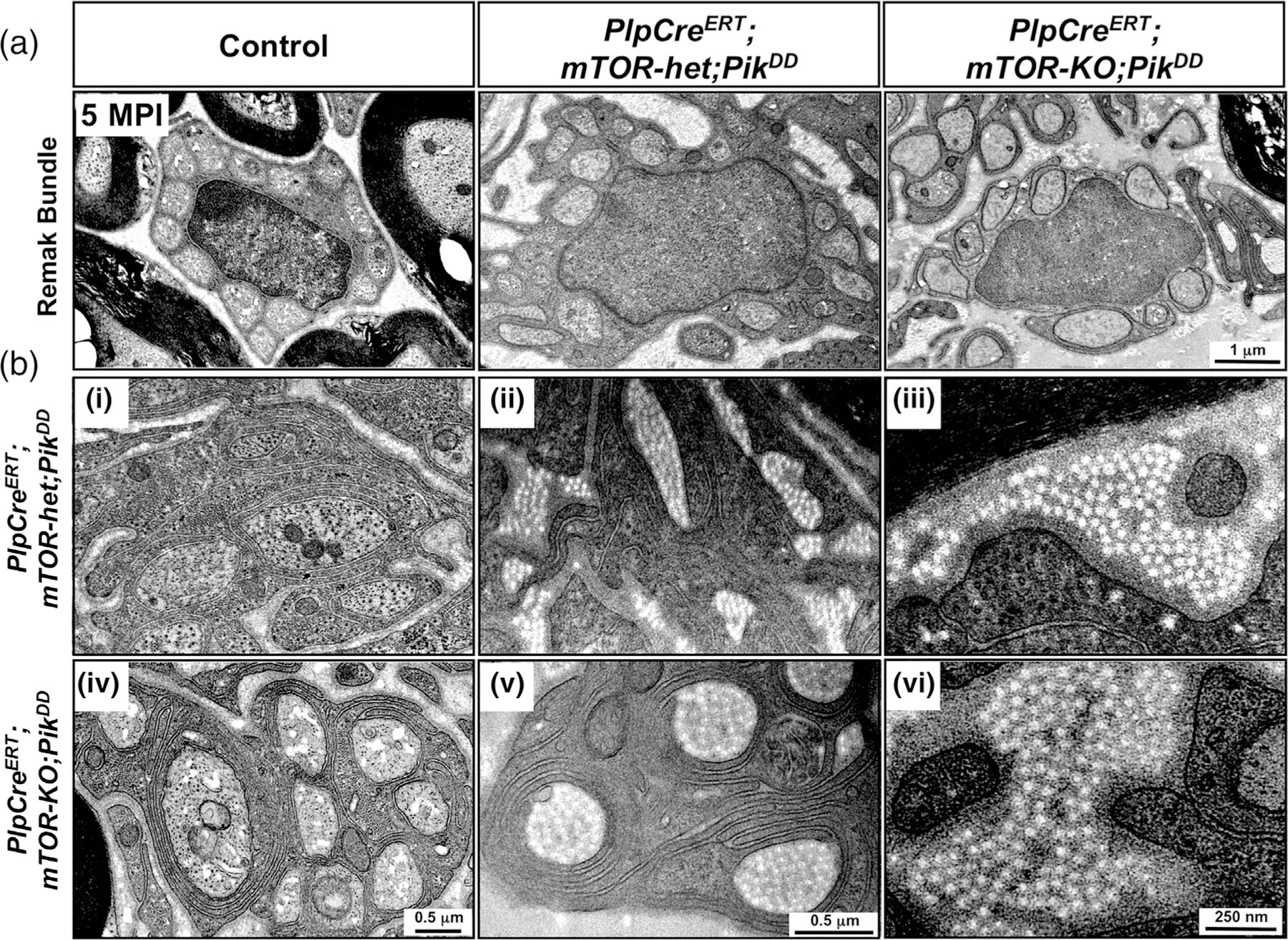
PI3K-mediated pathology was not abrogated when mTOR was simultaneously ablated during adulthood from non-myelinating Schwann cells. (a) High-magnification EM images of sciatic nerves from control, PlpCreERT;mTOR-het;PikDD, and PlpCreERT;mTOR-KO;PikDD mice injected with Tm at 1 month of age and analyzed at 5 months postinjection (MPI) show normal Remak bundle in control but a disruption of its structure in both the PlpCreERT;mTOR-het;PikDD and PlpCreERT;mTOR-KO;PikDD mice. (b) EM images at high magnification show other abnormalities that are present in both PlpCreERT;mTOR-het;PikDD (i–iii) and PlpCreERT;mTOR-KO;PikDD (iv–vi) mice, including (i,iv) multiple redundant membranous structures nonrandomly hyper-wrapping small-diameter axons, (ii,v) abnormal membranous hyper-wrapping of collagen fibrils, and (iii,vi) increased accumulation of extracellular collagen fibers. Scale bar as indicated. Representative images of sciatic nerves from 2–3 mice per genotype are shown
Taken together, we conclude that the ablation of mTOR in Schwann cells during adulthood was unable to abrogate any of the pathological changes induced by simultaneous elevation of PI3K activity in these cells.
4 |. DISCUSSION
This in vivo study investigated the relative involvement and interaction of the PI3K/Akt/mTOR and Mek/ERK1/2-MAPK pathways in Schwann cell differentiation, myelin formation, and maintenance of normal myelin homeostasis during adulthood, summarized in Figure 8. The data suggest that while the two pathways differentially regulate Schwann cell differentiation, they work together to enhance myelin thickness, converging at the level of mTORC1. Dysregulation of these pathways leads to a late onset of sciatic nerve pathology that is likely to utilize both mTOR-dependent and mTOR-independent mechanisms in a context-dependent manner.
FIGURE 8.
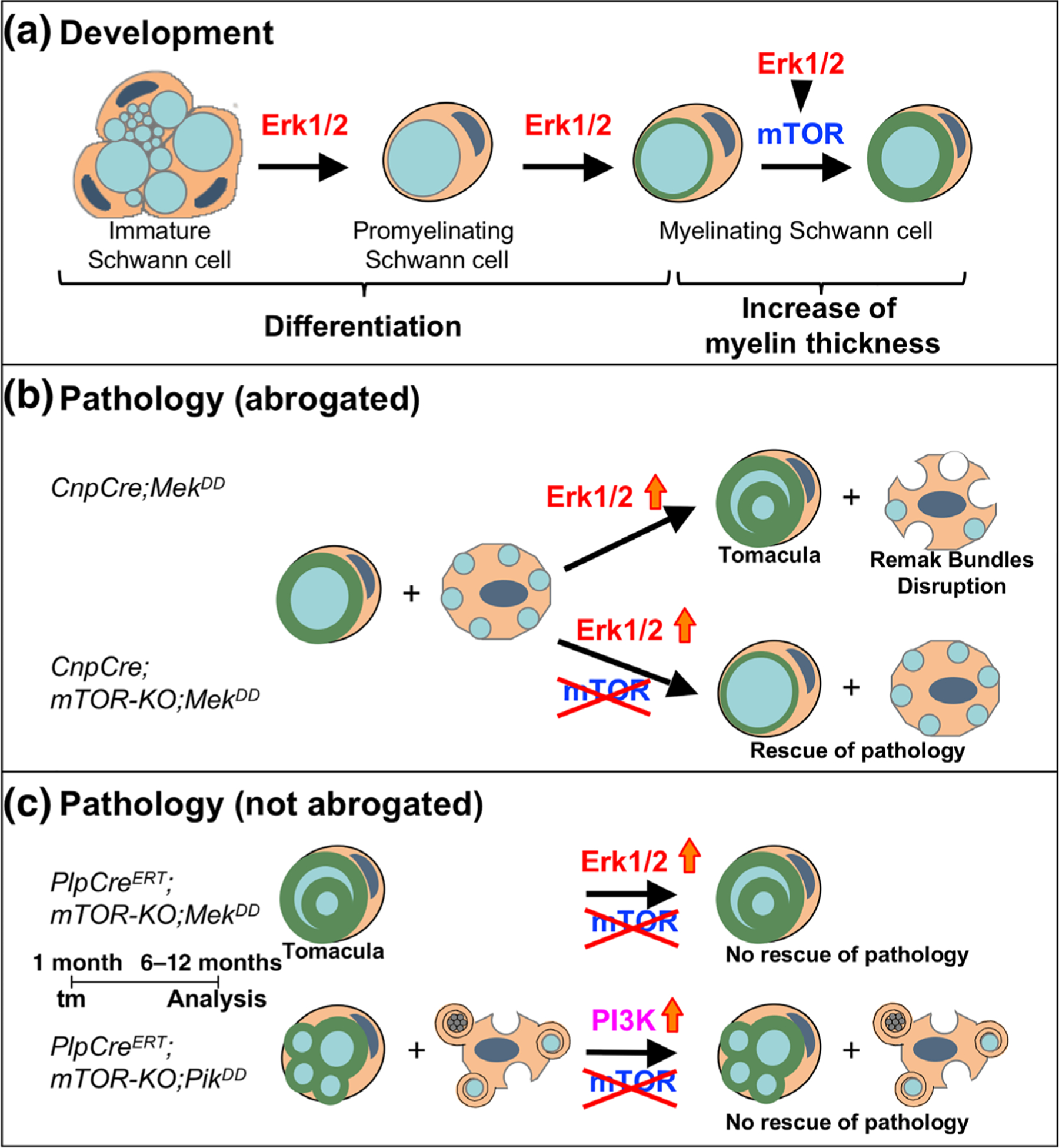
Working model for the role of Mek/ERK1/2-MAPK and PI3K/Akt/mTOR signaling pathways in the regulation of Schwann cell differentiation/myelination and in the manifestation of pathological changes in sciatic nerves during adulthood. (a) Loss-of-function studies show that during development, Schwann cell differentiation is primary regulated by the Mek/ERK1/2-MAPK pathway, independent of mTOR. However, during active myelination, ERK1/2 is dependent on mTOR signaling to drive the growth of the myelin sheath and regulate its thickness. (b) Sustained hyperactivation of Mek/ERK1/2 in immature Schwann cell precursors (CnpCre;MekDD) leads to a late onset of tomacula-like myelin pathology and disruption of Remak bundles. This was abrogated when mTOR was simultaneously ablated early during development (CnpCre; mTOR-KO;MekDD). (c) However, this Mek/ERK1/2-mediated pathology could not be abrogated when mTOR ablation was simultaneously induced later during adulthood (PlpCreERT;mTOR-KO;MekDD). Similarly, pathological changes induced by hyperactivation of PI3K (PlpCreERT;mTOR-KO;PikDD) could not be abrogated when mTOR ablation was simultaneously induced later during adulthood (PlpCreERT;mTOR-KO;PikDD) [Color figure can be viewed at wileyonlinelibrary.com]
Loss- and gain-of-function studies have shown that modulating Mek/ERK1/2 or mTOR signaling in immature Schwann cells affects their differentiation in different ways. Specifically, loss of ERK1/2 led to arrest of Schwann cell differentiation, and as a result, the majority of axons remained unmyelinated, even in adult mice ((Newbern et al., 2011); Figure 1), whereas ablation of mTOR or Raptor did not affect differentiation of Schwann cells to the promyelinating state in major ways and all axons became myelinated, although after a transient delay ((Norrmén et al., 2014; Sherman et al., 2012); Figure 1). Paradoxically, transient delay of promyelinating to myelinating transition was also observed when mTORC1 was hyperactivated by Schwann cell-specific perinatal deletion of TSC1 or PTEN, negative regulators of the PI3K/Akt/mTOR pathway (Beirowski et al., 2017; Figlia et al., 2017b). In contrast, constitutive hyperactivation of Mek/ERK1/2 in immature Schwann cells did not affect their differentiation (Ishii et al., 2013). Furthermore, Krox-20, the master transcription factor, was also differentially regulated by ERK1/2 and mTOR during Schwann cell differentiation. Specifically, we found that loss of ERK1/2 decreased Krox-20 expression, consistent with an arrest of Schwann cell differentiation. In contrast, loss of Schwann cell-specific Raptor (Figlia et al., 2017b) or mTOR (Figure 2) upregulated Krox-20 expression, and conversely, hyperactivation of mTORC1 signaling led to its downregulation (Figlia et al., 2017b). Collectively, these studies support the notion that Schwann cell differentiation is differentially regulated by the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways, with ERK1/2 playing a primary role in the PNS, in contrast to CNS, where mTORC1, not ERK1/2 signaling, plays a leading role in oligodendrocyte differentiation (Bercury et al., 2014; Gonsalvez et al., 2016; Ishii et al., 2019; Ishii et al., 2012; Lebrun-Julien et al., 2014; Wahl et al., 2014).
Following Schwann cell differentiation and initiation of myelin wrapping, the radial growth of the myelin sheath continues rapidly to match axon caliber to myelin thickness. Gain- and loss-of-function studies have shown that both Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways are important regulators of myelin thickness and that modulation of these pathways leads to very similar outcomes. Specifically, hyperactivation of mTORC1 in Schwann cell of transgenic mice by deletion of PTEN or TSC1 or by expression of membrane-targeted activated Akt (MyrAkt), an upstream activator of mTORC1, led to the formation of thicker-than-normal myelin sheaths (Beirowski et al., 2017; Domènech-Estévez et al., 2016; Figlia et al., 2017a; Figlia et al., 2017b; Goebbels et al., 2010). Similarly, hyperactivation of Mek/ERK1/2 in Schwann cells also resulted in increased myelin thickness (Ishii et al., 2013; Sheean et al., 2014). Conversely, ablation of mTOR or Raptor in Schwann cells led to the formation of thinner-than-normal myelin sheaths ((Norrmén et al., 2014; Sherman et al., 2012); Figure 1). Similarly, here we show that in ERK1/2-dKO, the 20% of axons that were myelinated had thin myelin sheaths. Given these similarities, the key question that we addressed here was whether Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways play independent, parallel roles or converge at a common downstream target to regulate myelin thickness in the PNS. Since we found that sustained elevation of Mek/ERK1/2 activity in Schwann cells failed to rescue the deficit in myelin thickness caused by the loss of mTOR and that the increase of myelin thickness driven by Mek/ERK1/2 hyperactivation was completely abrogated when mTOR was simultaneously ablated, we concluded that the two major signaling pathways work together, converging at the level of mTORC1 to regulate myelin thickness in the PNS. It is interesting to note that this unique mechanism of regulating myelin growth is highly conserved between the CNS and PNS, since we recently showed that ERK1/2 in oligodendrocytes also signal through mTOR (Ishii et al., 2019) to regulate myelin thickness.
Although a key role of myelinating Schwann cells is the formation of the myelin sheaths of the correct thickness during developmental myelination, it is equally important that Schwann cells maintain normal myelin homeostasis throughout life. This is because several human hereditary peripheral neuropathies are known to be associated with dysregulation of Schwann cell functions, including myelin abnormalities and axonal degeneration. (Scherer & Wrabetz, 2008; Suter & Scherer, 2003). Therefore, a better understanding of the regulatory mechanisms underlying their dysfunction would contribute in the long run to identify potential therapeutic targets for ameliorating the pathology in human neuropathies. Hyperactivation of mTORC1 activity is believed to be responsible for these abnormalities, since they could be corrected by rapamycin injection to postnatal or adult mice where the PI3K/Akt/mTOR pathway was hyperactivated in Schwann cells (Domènech-Estévez et al., 2016; Goebbels et al., 2012). Interestingly, we observed very similar myelin abnormalities in mice with hyperactivation of the Mek/ERK1/2-MAPK pathway in Schwann cells ((Ishii et al., 2016), present study). In both cases, myelin pathology appeared to originate from paranodal loops, where high levels of both phospho-ERK1/2 (present data) and phospho-Akt are preferentially localized (Goebbels et al., 2012). Hyperactivation of PI3K/Akt/mTOR or Mek/ERK1/2-MAPK pathways also led to similar Remak bundle pathology, in these mutant mice ((Domènech-Estévez et al., 2016; Figlia et al., 2017b; Goebbels et al., 2010); present study). Given these similarities, here, we addressed the question of whether the neuropathology was caused by independent parallel mechanisms or involved downstream convergence of these two pathways, as was seen during development. Our analysis of CnpCre;mTOR-KO;MekDD mice clearly showed that when hyperactivation of Mek/ERK1/2 and simultaneous deletion of mTOR occurred early in development, then mTORC1 targets, p-p70S6K and p-S6RP activities were downregulated and the late onset of sciatic nerve pathology was abrogated, suggesting that Mek/ERK1/2-induced dysfunction of Schwann cells was dependent on downstream mTOR signaling and supporting a model for the convergence of the two pathways at the level of mTORC1 in this scenario.
However, when we examined tamoxifen-inducible PlpCreERT; mTOR-KO;MekDD mice, where Mek/ERK1/2 was hyperactivated and simultaneously mTOR was conditionally ablated in the adult mice after myelination is largely terminated, we were surprised to see that the Mek/ERK1/2-induced sciatic nerve pathology was not abrogated. Furthermore, in a parallel study, we found that in the PlpCreERT;mTOR-KO;PikDD mice, where ablation of mTOR and simultaneous hyper-activation of PI3K were induced conditionally in the adult mice, the PI3K-mediated sciatic nerve pathology was also not abrogated. In both these cases, p-p70S6K and p-S6RP activity was not down-regulated in spite of mTOR ablation. Thus, it is plausible that in adult mice, manifestation of pathological changes by hyperactivated Mek/ERK1/2 or PI3K could potentially occur by mechanisms independent of mTOR.
Here we have hypothesized that the differences in the observed outcomes are due to difference in the timing of mTOR ablation/Mek activation that is, during development versus adulthood. An alternate interpretation could be that since Cnp has been reported to be also expressed by certain other cell types (Ballestero et al., 1999; Genoud et al., 2002; Heath & Hindman, 1986; Tognatta et al., 2017), the observed differences could be due to off target effects of CnpCre. Furthermore, since in the CnpCre/+ mice the Cre recombinase is knocked-in in one of the Cnp alleles (Lappe-Siefke et al., 2003), the haploinsufficiency for Cnp in the mutant mice combined with other gene perturbations (like mTOR and ERK1/2 perturbation), could reveal a pathological phenotype due to CNPase haploinsufficiency itself as was suggested previously (Della-Flora Nunes et al., 2017). However, it is to be noted that two other Cre line, DhhCre and Egr2Cre, whose expression begins embryonically similar to CnpCre and used widely to recombine in a Schwann cell-specific manner, showed the same phenotypes as the CnpCre line with regards to Raptor/mTOR ablation and MekDD activation (Ishii et al., 2016; Norrmén et al., 2014; Sheean et al., 2014; Sherman et al., 2012). Therefore, while not completely ruled out, we believe that the observed phenotypes of the mutant mice are unlikely to be meaningfully contributed by the CnpCre line itself but are most likely due to the different timing of ablation/stimulation during development versus adulthood.
Interestingly, previous studies that used rapamycin, which disrupts mTOR signaling in all cell types suggested that mTOR, presumably in Schwann cells, was responsible for some but not all of the PNS phenotypes driven by hyperactivation of the PI3K pathway. For example, while rapamycin injected for 8 weeks to 3-month-old mice abrogated the myelin out-foldings and tomacula phenotypes, no improvement of hyper-wrapping in the Schwann cell/Remak bundle units was detected in the rapamycin-treated transgenic mice (Domènech-Estévez et al., 2016). More recently, overgrowth of myelin observed after ablation of Fbxw7 in Schwann cells was reported to be due to the hyperactivation of mTOR; however, the ensheathment of multiple axons by mutant Schwann cells was found to be independent of mTOR signaling (Harty et al., 2019). Thus, evidence from previous studies, together with our present findings, supports the hypothesis that abnormal membrane growth in the PNS, by hyper-activation of PI3K or Mek/ERK1/2, is likely to utilize both mTOR-dependent and mTOR-independent mechanisms.
The identity of these mTOR-independent mechanisms that may operate in Schwann cells is currently poorly understood. However, it is known that both Mek/ERK1/2-MAPK and PI3K signaling controls numerous cytosolic targets and various transcription factors (Anjum & Blenis, 2008). Therefore, it is possible that one or more of these targets may act independently of mTOR and significantly influence Schwann cell intracellular signaling. For example, one plausible mTOR-independent mechanism could be the sustained increase in Krox-20 expression through Mek-dependent activation of YY1 (He et al., 2010), which could lead to increased myelin gene expression and uncontrolled membrane growth. Other likely candidates, Rac1 and Jun, previously proposed to perform the mTOR-independent roles of PI3K (Domènech-Estévez et al., 2016; Harty et al., 2019), could potentially, indirectly or directly, be targeted by the MAPK pathway, as well.
In summary, we show that while the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways play very different roles during Schwann cell differentiation, they cooperate with each other to regulate myelin thickness during developmental myelination, by using mTOR signaling as a common downstream mechanism. Finally, these studies have revealed that mTOR-independent, in addition to mTOR-dependent, mechanisms are likely to be involved in causing PNS neuropathology observed during adulthood in mice with sustained hyperactivation of Mek or PI3K, which mimics neuropathology in certain hereditary human neuropathies. Thus, given these complexities the therapeutic modulation of these pathways may prove to be a challenging task for ameliorating neuropathology in human neuropathies.
ACKNOWLEDGMENTS
This work was supported by the NIH, Grant NS38878. We would like to thank Dr. J.S. Richards (Baylor College of Medicine, Houston, TX) for the Erk1−/−;Erk2 floxed mice and Dr. K-A Nave (Max Planck Institute of Experimental Medicine, Goettingen, Germany) for the Cnp-Cre mice.
Funding information
NIH
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Anjum R, & Blenis J. (2008). The RSK family of kinases: Emerging roles in cellular signalling. Nature Reviews. Molecular Cell Biology, 29, 747–758. 10.1038/nrm2509 [DOI] [PubMed] [Google Scholar]
- Ballestero RP, Dybowski JA, Levy G, Agranoff BW, & Uhler MD (1999). Cloning and characterization of zRICH, a 20,30-cyclic-nucleotide 30-phosphodiesterase induced during zebrafish optic nerve regeneration. Journal of Neurochemistry, 72, 1362–1371. 10.1046/j.1471-4159.1999.721362.x [DOI] [PubMed] [Google Scholar]
- Beirowski B, Wong KM, Babetto E, & Milbrandt J. (2017). mTORC1 promotes proliferation of immature Schwann cells and myelin growth of differentiated Schwann cells. Proceedings of the National Academy of Sciences of the United States of America, 114, E4261–E4270. 10.1073/pnas.1620761114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL, & Macklin WB (2014). Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. The Journal of Neuroscience, 34, 4466–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter L, Ozçelik M, Jacob C, Pereira JA, Locher V, Baumann R, Relvas JB, Suter U, & Tricaud N. (2010). Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve over-myelination. Science, 328, 1415–1418. [DOI] [PubMed] [Google Scholar]
- Della-Flora Nunes G, Mueller L, Silvestri N, Patel MS, Wrabetz L, Feltri ML, & Poitelon Y. (2017). Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. Glia, 65, 1626–1639. 10.1002/glia.23184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, & Popko B. (2003). Inducible site-specific recombination in myelinating cells. Genesis, 35, 63–72. [DOI] [PubMed] [Google Scholar]
- Domènech-Estévez E, Baloui H, Meng X, Zhang Y, Deinhardt K, Dupree JL, Einheber S, Chrast R, & Salzer JL (2016). Akt regulates axon wrapping and myelin sheath thickness in the PNS. The Journal of Neuroscience, 36, 4506–4521. 10.1523/JNEUROSCI.3521-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HY, Liu Z, Shimada M, Sterneck E, Johnson PF, Hedrick SM, & Richards JS (2009). MAPK3/1 (ERK1/2) in ovarian granulosa cells are essential for female fertility. Science, 324, 938–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlia G, Gerber D, & Suter U. (2017a). Myelination and mTOR. Glia, 66, 693–707. 10.1002/glia.23273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlia G, Norrmén C, Pereira JA, Gerber D, & Suter U. (2017b). Dual function of the PI3K-Akt-mTORC1 axis in myelination of the peripheral nervous system. eLife, 6, e29241. 10.7554/eLife.29241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, & Bansal R. (2005). Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. The Journal of Neuroscience, 25, 7470–7479. 10.1523/JNEUROSCI.2120-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Nave KA, & Bansal R. (2012). Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. The Journal of Neuroscience, 32, 6631–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Ishii A, & Bansal R. (2017). Signaling by FGF receptor 2, not FGF receptor 1, regulates myelin thickness through activation of ERK1/2-MAPK, which promotes mTORC1 activity in an Akt-independent manner. The Journal of Neuroscience, 37, 2931–2946. 10.1523/JNEUROSCI.3316-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Kaga Y, Ishii A, Hébert JM, & Bansal R. (2011). Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. The Journal of Neuroscience, 31, 5055–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genoud S, Lappe-Siefke C, Goebbels S, Radtke F, Aguet M, Scherer SS, Suter U, Nave KA, & Mantei N. (2002). Notch1 control of oligodendrocyte differentiation in the spinal cord. The Journal of Cell Biology, 158, 709–718. 10.1083/jcb.200202002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Möbius W, Liu X, Lappe-Siefke C, Rossner MJ, Groszer M, Suter U, Frahm J, Boretius S, & Nave KA (2010). Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. The Journal of Neuroscience, 30, 8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Wolfer S, Wieser GL, Nientiedt T, Pieper A, Ruhwedel T, Groszer M, Sereda MW, & Nave KA (2012). Genetic disruption of PTEN in a novel mouse model of tomaculous neuropathy. EMBO Molecular Medicine, 4, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalvez D, Ferner AH, Peckham H, Murray SS, & Xiao J. (2016). The roles of extracellular related-kinases 1 and 2 signaling in CNS myelination. Neuropharmacology, 110, 586–593. 10.1016/j.neuropharm.2015.04.024 [DOI] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, & Braun PE (1998). Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of LacZ in transgenic mice. Journal of Neuroscience Research, 53, 393–404. 10.1002/(SICI)1097-4547(19980815)53 [DOI] [PubMed] [Google Scholar]
- Harty BL, Coelho F, Pease-Raissi SE, Mogha A, Ackerman SD, Herbert AL, Gereau RW 4th, Golden JP, Lyons DA, Chan JR, & Monk KR (2019). Myelinating Schwann cells Ensheath multiple axons in the absence of E3 ligase component Fbxw7. Nature Communications, 10(1), 2976. 10.1038/s41467-019-10881-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Kim JY, Dupree J, Tewari A, Melendez-Vasquez C, Svaren J, & Casaccia P. (2010). Yy1 as a molecular link between neuregulin and transcriptional modulation of peripheral myelination. Nature Neuroscience, 13, 1472–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AR, & Hindman HM (1986). CNPase activity in the vertebrate retina, retinal pigmented epithelium, and choroid. The Journal of Experimental Zoology, 238, 183–191. 10.1002/jez.1402380208 [DOI] [PubMed] [Google Scholar]
- Ishii A, Furusho M, & Bansal R. (2013). Sustained activation of ERK1/2 MAPK in oligodendrocytes and Schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. The Journal of Neuroscience, 33, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Dupree JL, & Bansal R. (2016). Strength of ERK1/2 MAPK activation determines its effect on myelin and axonal integrity in the adult CNS. The Journal of Neuroscience, 36, 6471–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Macklin W, & Bansal R. (2019). Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia, 67, 1277–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, & Bansal R. (2012). ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. The Journal of Neuroscience, 32, 8855–8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, & Mirsky R. (2005). The origin and development of glial cells in peripheral nerves. Nature Reviews Neuroscience, 6, 671–682. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, & Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nature Genetics, 33, 366–374. [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F, Bachmann L, Norrmén C, Trötzmüller M, Köfeler H, Rüegg MA, Hall MN, & Suter U. (2014). Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. The Journal of Neuroscience, 34, 8432–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone DP, Genoud S, Atanasoski S, Grausenburger R, Berger P, Metzger D, Macklin WB, Chambon P, & Suter U. (2003). Tamoxifen-inducible glia-specific Cre mice for somatic mutagenesis in oligodendrocytes and Schwann cells. Molecular and Cellular Neurosciences, 22, 430–440. [DOI] [PubMed] [Google Scholar]
- Mayes DA, Rizvi TA, Cancelas JA, Kolasinski NT, Ciraolo GM, Stemmer-Rachamimov AO, & Ratner N. (2011). Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Research, 71, 4675–4685. 10.1158/0008-5472.CAN-10-4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, & Nave KA (2004). Axonal neuregulin-1 regulates myelin sheath thickness. Science, 304, 700–703. 10.1126/science.1095862 [DOI] [PubMed] [Google Scholar]
- Monk KR, Wu J, Williams JP, Finney BA, Fitzgerald ME, Filippi MD, & Ratner N. (2007). Mast cells can contribute to axon-glial dissociation and fibrosis in peripheral nerve. Neuron Glia Biology, 3, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern J, & Birchmeier C. (2010). Nrg1/ErbB signaling networks in Schwann cell development and myelination. Seminars in Cell & Developmental Biology, 21, 922–928. 10.1016/j.semcdb.2010.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, & Snider WD (2011). Specific functions for ERK/-MAPK signaling during PNS development. Neuron, 69, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrmén C, Figlia G, Lebrun-Julien F, Pereira JA, Trötzmüller M, Köfeler HC, Rantanen V, Wessig C, van Deijk AL, Smit AB, Verheijen MH, Rüegg MA, Hall MN, & Suter U. (2014). mTORC1 controls PNS myelination along the mTORC1-RXRγ-SREBP-lipid biosynthesis axis in Schwann cells. Cell Reports, 9, 646–660. [DOI] [PubMed] [Google Scholar]
- Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, Richard-Bulteau H, Vignaud A, Baas D, Defour A, Freyssenet D, Tanti JF, Le-Marchand-Brustel Y, Ferrier B, Conjard-Duplany A, Romanino K, Bauché S, Hantaï D, Mueller M, … Gangloff YG (2009). Muscle inactivation of mTOR causes meta-bolic and dystrophin defects leading to severe myopathy. The Journal of Cell Biology, 187, 859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld H, & Seger R. (2005). The ERK cascade: A prototype of MAPK signaling. Molecular Biotechnology, 31, 151–174. 10.1385/MB:31:2:151 [DOI] [PubMed] [Google Scholar]
- Salzer JL (2015). Schwann cell myelination. Cold Spring Harbor Perspectives in Biology, 7, a020529. 10.1101/cshperspect.a020529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawade L, Grandi F, Mignanelli M, Patiño-López G, Klinkert K, Langa-Vives F, Di Guardo R, Echard A, Bolino A, & Haucke V. (2020). Rab35-regulated lipid turnover by myotubularins represses mTORC1 activity and controls myelin growth. Nature Communications, 11, 2835. 10.1038/s41467-020-16696-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, & Wrabetz L. (2008). Molecular mechanisms of inherited demyelinating neuropathies. Glia, 56, 1578–1589. 10.1002/glia.20751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheean ME, McShane E, Cheret C, Walcher J, Müller T, Wulf-Goldenberg A, Hoelper S, Garratt AN, Krüger M, Rajewsky K, Meijer D, Birchmeier W, Lewin GR, Selbach M, & Birchmeier C. (2014). Activation of MAPK overrides the termination of myelin growth and replaces Nrg1/ErbB3 signals during Schwann cell development and myelination. Genes & Development, 28, 290–303. 10.1101/gad.230045.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Krols M, Wu LM, Grove M, Nave KA, Gangloff YG, & Brophy PJ (2012). Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. The Journal of Neuroscience, 32, 1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, & Rajewsky K. (2009). PI3 kinase signals BCR-dependent mature B cell survival. Cell, 139, 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U, & Scherer SS (2003). Disease mechanisms in inherited neuropathies. Nature Reviews. Neuroscience, 4, 714–726. 10.1038/nrn1196 [DOI] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, & Salzer JL (2005). Neuregulin-1 type III determines the ensheathment fate of axons. Neuron, 47, 681–694. 10.1016/j.neuron.2005.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognatta R, Sun W, Goebbels S, Nave KA, Nishiyama A, Schoch S, Dimou L, & Dietrich D. (2017). Transient Cnp expression by early progenitors causes Cre-lox-based reporter lines to map profoundly different fates. Glia, 65, 342–359. 10.1002/glia.23095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viader A, Golden JP, Baloh RH, Schmidt RE, Hunter DA, & Milbrandt J. (2011). Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. The Journal of Neuroscience, 31, 10128–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SE, McLane LE, Bercury KK, Macklin WB, & Wood TL (2014). Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. The Journal of Neuroscience, 34, 4453–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ER, Della-Flora Nunes G, Weaver MR, Frick LR, & Feltri ML (2020). Schwann cell interactions during the development of the peripheral nervous system. Dev Neurobiol. 2020. April 12: 10.1002/dneu.22744. 10.1002/dneu.22744. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, & Ratner N. (2008). Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell, 13, 105–116. 10.1016/j.ccr.2007.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.