

Abstract
Emerging Plasmodium parasite drug resistance is threatening progress towards malaria control and elimination. While recent efforts in cell-based, high-throughput drug screening have produced first-in-class drugs with promising activities against different Plasmodium life cycle stages, most of these antimalarial agents have elusive mechanisms of action. Though challenging to address, target identification can provide valuable information to facilitate lead optimization and preclinical drug prioritization. Recently, proteome-wide methods for direct assessment of drug-protein interactions have emerged as powerful tools in a number of systems, including Plasmodium. In this review, we will discuss current chemoproteomic strategies that have been adapted to antimalarial drug target discovery, including affinity- and activity-based protein profiling and the energetics-based techniques thermal proteome profiling and stability of proteins from rates of oxidation. The successful application of chemoproteomics to the Plasmodium blood stage highlights the potential of these methods to link inhibitors to their molecular targets in more elusive Plasmodium life stages and intracellular pathogens in the future.
Keywords: target identification, malaria, Plasmodium, mechanism of action, chemoproteomics
Graphical Abstract
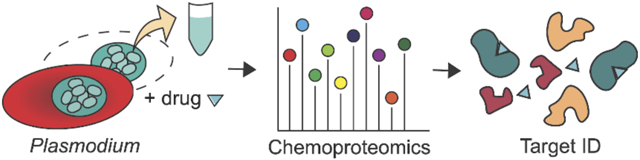
Chemoproteomics adapted to Plasmodium parasites enable proteome-wide assessment of drug-protein interactions. These methods assist in target deconvolution of novel antimalarial compounds, commonly discovered in phenotypic screens, that have unknown mechanisms of action. In this review, we detail current methods of label-based and energetics-based chemoproteomics and their application for Plasmodium parasite drug target discovery.
1. Introduction
Malaria continues to impose an enormous public health burden worldwide and has claimed more than two million human lives over the past five years.[1] Although substantial efforts and funding have been invested to control and eliminate malaria, progress has stalled since 2015. Expanding parasite resistance to the first-line antimalarial drugs, the dependence on insecticide-treated bed nets, and vector resistance to current insecticides are notable concerns to achieve disease eradication.[2] There are five protozoan parasite species of Plasmodium that cause malaria, but Plasmodium falciparum and Plasmodium vivax are the primary causative agents of this debilitating disease. Plasmodium parasites are transmitted by mosquito bites to the human host, where they first invade hepatocytes and undergo multiple rounds of DNA replication to form liver-stage schizonts. Tens of thousands of progeny cells are then released into the bloodstream where they invade red blood cells (RBCs). During the cyclical asexual stage, parasites transition through the ring (early), trophozoite (mid), and schizont (late) stages. The blood-stage schizonts produce parasites for further RBC invasion, but it is also during this period that a small proportion of parasites differentiate into sexual stage gametocytes that can be transmitted to mosquitoes to complete their life cycle. New first-in-class drugs with resolved antiparasitic mechanisms are critical to addressing this global health issue.
Advances in cell-based, high-throughput phenotypic screens have significantly improved the efficiency of antimalarial drug discovery.[3] Thousands to millions of compounds have been evaluated for their activities against different Plasmodium life cycle stages, and dozens of them are currently in preclinical and clinical studies.[4-8] Although drugs can be approved without a full understanding of their mechanisms of action, a well-characterized interaction between a candidate drug and its molecular target – possibly parasite or human biomolecules – can greatly facilitate the drug development process. Namely, identifying the direct target(s) of an inhibitor provides the molecular basis for optimizing structure–activity relationships and can also unveil novel druggable targets that in turn aid in target-based drug discovery. Complicating drug-target discovery efforts has been the increasing appreciation that many drugs, including antimalarial inhibitors, deliver their therapeutic effects by targeting multiple proteins.[9-11] This suggests that comprehensive profiling of drug targets is necessary to elucidate complicated mechanisms of drug action and prioritize compounds for further drug development.
Over the past decade, omics-based approaches, including genomics and proteomics, have been applied to investigate the therapeutic targets of many antimalarial agents.[12-15] In vitro evolution and subsequent identification of evolved genetic variants, such as by tiling microarrays or whole genome sequencing, is perhaps the most extensively used approach. It has determined the potential targets of many promising candidate drugs, including imidazopyrazines to PfPI4K and the compound DDD107498 to PfeEF2.[12, 16-18] This approach requires parasites to develop drug resistance mutations and the completion of subsequent validation studies to distinguish resistance-conferring mutations from background mutations in the Plasmodium genome (particularly important with the AT-rich genome of P. falciparum). This method has been critical to advancing many antimalarial drug leads, and it provides strong evidence of a potential target if replicate resistance-conferring variants map to a single gene. However, with this approach, if a lead fails to generate resistance, while an attractive feature, the mode of action cannot be determined. Further, indirect versus direct interactions need to be considered, as the method could identify mutations in parasite fitness or downstream pathway members instead of a binding protein. Comparative transcriptomic and metabolomic studies are similarly complicated by the inability to directly address drug targets through analysis and require further testing to validate a direct interaction.
Chemoproteomics offers an invaluable tool to directly probe the interactions between drugs and their protein targets. With advances in mass spectrometry (MS) technologies, such as increasing instrument sensitivity coupled with the development of isobaric mass tags for multiplex analyses, chemoproteomics has been increasingly implemented in academia and industry for drug target discovery in numerous model systems.[19-20] However, it was not until recent years that MS-based techniques were adapted to include intracellular Plasmodium parasites for target deconvolution.[21-22] This feat demonstrates the versatility of these approaches in interrogating challenging pathogenic systems, which will undoubtedly expand to more elusive organisms and life stages in the future. In this review, we discuss the utility of different chemoproteomic methods, ranging from affinity- and activity-based protein profiling to the energetics-based techniques termed thermal proteome profiling (TPP) and stability of proteins from rates of oxidation (SPROX) in antimalarial target discovery.
2. Affinity-based target profiling
Affinity-based protein profiling (AfBPP), also termed compound-centric chemical proteomics, has been employed to identify the protein targets of various antimalarial drugs. This approach enables target enrichment using an affinity matrix, where the drug is immobilized on a solid support, such as agarose beads, to pull down its binding proteins (Figure 1A).[23-24] The affinity-purified proteins are then eluted and analyzed by MS. For example, this method identified casein kinase 1 as the unique target of purvalanol B (a cyclin-dependent kinase inhibitor) in the P. falciparum blood stage and other protozoan parasites.[25] Specifically, casein kinase 1 was identified via protein sequencing after analyzing proteins pulled down by purvalanol B, but not its inactive methylated derivative.
Figure 1.

Label-based chemoproteomic methods for malaria drug target identification. A) Affinity-based protein profiling (AfBPP). Plasmodium parasite lysates are incubated with bead-immobilized compounds. After washes, the ligand-binding proteins can be eluted for MS analysis. B) Activity-based protein profiling (ABPP). Enriched Plasmodium-infected RBCs or the parasite lysates are incubated with an ABPP probe in the presence or absence of drugs. The RBCs are lysed (for intact cells), and a reporter tag (e.g., biotin or fluorescent tags) can be conjugated to the reactive head group via click chemistry. Proteins covalently modified by the probe are enriched using an avidin or streptavidin column for MS analysis. Fluorescently tagged proteins can be visualized by gel electrophoresis. Proteins targeted by the drug will show a deceased signal as the ABPP probe is competed out by the compound.
The 2-aminopyridine MMV390048 has been identified as a promising candidate drug against all life cycle stages of malaria parasites.[26] This compound, currently under phase IIa clinical trial, was found to target P. falciparum phosphatidylinositol 4-kinase (PI4K) using a combination of in vitro evolution and AfBPP.[26-27] In one study, free MMV390048 was pre-incubated in Plasmodium lysate before direct pull-down experiments with bead-immobilized drug. To further correlate the binding proteins with the drug’s antiparasitic activity, the lysate was pre-incubated with the inactive analog MMV034137 (low antimalarial activity) in parallel experiments. Importantly, MMV390048, but not the inactive analog, competed to prevent PfPI4K binding to the beads. Subsequent enzyme activity assays using purified Plasmodium PI4K confirmed a direct interaction between the protein and MMV390048.[26] The demonstrated utility of this competitive AfBPP approach offers a means to reduce false positives generated by compound immobilization for target identification in Plasmodium (Figure 1A).
An alternative AfBPP strategy is available for kinases, which uses Kinobeads with promiscuous kinase inhibitors covalently attached to a matrix.[28] This strategy was implemented as a complementary method to confirm the target of MMV390048.[26] The addition of MMV390048 to Plasmodium lysate selectively inhibited the binding of PfPI4K to the Kinobeads in a dose-response manner, consistent with the results obtained using the 2-aminopyridine-conjugated beads.[26] Kinobeads-based AfBPP has been applied to identify potential kinase targets of other antimalarial agents.[26, 29-31] Similarly, ATP-agarose beads can capture all ATP-binding proteins in Plasmodium, expanding the possible target class beyond kinases to purine-binding proteins.[32] Thus, all proteins containing a drug-targeted ATP-binding pocket can be elucidated by drug competition. These tools are particularly useful when chemical synthesis for compound immobilization is not amenable.
3. Activity-based protein profiling
Activity-based protein profiling (ABPP) was initially developed as a functional proteomics method to assess the active state of proteins in native conditions.[33] It has been successfully implemented in drug discovery and target identification workflows, with notable contributions to antimalarial drug studies. ABPP relies on chemical probes that specifically target proteins with conserved active sites (Figure 1B). These probes consist of a reactive head group linked to a reporter tag. The head groups are developed to covalently bind to protein active sites, particularly at the catalytic sites of an enzyme family. The reporter tags may contain a fluorophore or a biotin tag for in-gel visualization and pull-down assays, respectively. With these designs, proteins harboring the drug-targeted active sites can be studied.
Significant efforts have been dedicated to the development of ABPP probes to evaluate various enzymes in P. falciparum. A general cysteine protease activity-based probe DCG-04 has been shown to label Plasmodium falcipains.[34] Subsequent studies further described a range of ABPP probes that irreversibly labeled P. falciparum serine and cysteine proteases, including the multifunctional serine protease PfSUB1 and dipeptidyl peptidases (DPAPs).[35-36] These probes are useful for determining if an inhibitor targets these specific enzymes. For example, a nanomolar inhibitor of P. falciparum, symplostatin 4, induces a swollen digestive vacuole—a phenotype that could be linked to disrupted heme metabolism. Thus, it was hypothesized that symplostatin 4 targets Plasmodium digestive vacuole proteases, such as falcipains.[37] To test this hypothesis, fluorescently tagged DCG-04 and a DPAP ABPP probe, FY01, were added to P. falciparum lysate treated with symplostatin 4 or a less potent symplostatin 4 analog. The in-gel analysis then revealed the selective binding of symplostatin 4 to P. falciparum falcipains, as it competitively reduced the labeling of DCG-04 but not FY01 (i.e., symplostatin 4 did not inhibit the labeling of DPAPs that also reside in the digestive vacuole). Subsequent studies have largely expanded the scope of activity-based probes to target Plasmodium serine hydrolases[38], metallo-aminopeptidases[39] and the proteasome.[40-43] The expansion of ABPP probes to a larger number of diverse proteins could provide a platform to screen different enzymes as candidate targets of antimalarial inhibitors.[44]
ABPP is particularly useful when genetic approaches are not feasible. For example, the natural product salinipostin A was found to inhibit P. falciparum in a nanomolar range, but a resistance strain was unable to be established, preventing in vitro evolution target identification. Thus ABPP was a key tool to establishing its targets.[38, 45] Intriguingly, the chemical structure of this compound hinted that it covalently modified its protein targets, assisting in its design as an ABPP probe. Instead of directly linking salinipostin A to a reporter tag, an electrophilic alkyne was attached as a click chemistry handle.[38] Incorporating click chemistry as part of the ABPP probe design is advantageous as it allows in-cell studies given the smaller size and improved membrane permeability.[46-47] Importantly, the synthesized salinipostin A probe retained its antimalarial potency; thus, it could be used for subsequent target identification studies. Specifically, live parasites pretreated with and without salinipostin A were labeled with the alkyne probe, followed by cell lysis and click chemistry-mediated biotin conjugation.[38] Labeled proteins were then isolated by avidin enrichment, and multiple P. falciparum serine hydrolases were detected using MS analysis (with reduced signal upon compound pretreatment), suggesting the compound’s potential targets. Intriguingly, the salinipostin probe identified a previously uncharacterized hydrolase, PfMAGLLP, which is predicted to be essential in P. falciparum.[38] Subsequent biochemical studies suggested its role in lipid metabolism and highlighted the possibility to target this parasite-specific hydrolase.
In addition to studying drugs that target specific enzyme active sites, ABPP can be used to interrogate compounds that promiscuously target diverse protein classes. The first-line antimalarial artemisinin harbors an endoperoxide bridge that alkylates multiple Plasmodium cellular components and kills the parasites rapidly. To characterize the targets of artemisinin, two independent groups developed clickable artemisinin ABPP probes with a terminal azide or alkyne handle for in-cell labeling.[48-49] One group also included the inactive analogs (changing the endoperoxide bridge to a deoxyether) for both azide and alkyne probes to rule out non-covalent binders.[48] These studies led to the identification of several dozens of in situ labeled parasite proteins involved in essential metabolic pathways, translation, and redox defense. A similar strategy was employed to investigate the targets of the synthetic endoperoxide drug artefenomel (previously OZ439) that offers a single-dose cure for uncomplicated malaria (completed phase II clinical trial).[50-52] Not surprisingly, the majority of the artemisinin ABPP probe-labeled proteins were also labeled by the artefenomel probe with the same azide handle.[51] These findings highlight the pleiotropic effects of endoperoxides and underscore the ability of ABPP to uncover the targets of covalent inhibitors.
A major challenge of ABPP is the requirement to form a covalent bond between a probe and its protein target(s). This challenge can be addressed by incorporating a photoreactive moiety into the compound of interest. For example, the photoaffinity probes of two antimalarial agents, albitiazolium and diaminoquinazoline, were engineered for ABPP-based target identification.[53-54] The addition of diazirine or a phenyl azido group enabled crosslinking to proteins via UV irradiation, and the clickable moiety (which was also incorporated) was used for target isolation and MS analysis. However, interpreting data obtained from using these photoreactive probes is hampered by false positives that stem from probes crosslinking to non-specific binders. Thus, these experiments require careful controls and validation studies to unveil the true targets.[55] For example, photoaffinity labeling identified multiple albitiazolium-binding proteins, but a smaller subset was found to be selective after a competition study using unmodified albitiazolium. Among the selective proteins, a parasite choline/ethanolamine phosphotransferase exhibited the greatest decrease in labeling from albitiazolium competition, consistent with a proposed mechanism of action of disrupting phosphatidylcholine synthesis.
4. Thermal proteome profiling
Despite the utility of affinity- and activity-based protein profiling for target discovery, these techniques require compound modification or immobilization to achieve target protein enrichment. Such strategies may yield high false-positive and false-negative rates due to the altered chemical structures, which can affect protein affinity and selectivity as well as cause non-specific interactions. Additionally, the synthesis of ABPP probes can be challenging, as it demands pre-existing knowledge of the kinetics and molecular interactions between a ligand and its binding site.[56-57] These prerequisites are more difficult in malaria studies given that 75% of Plasmodium genes have unknown or putative functions.[58] Thus, detection methods that do not require ligand labeling and can allow proteome-wide screening would greatly benefit drug target deconvolution in Plasmodium, where the largely uncharted proteome has limited functional and structural annotation.
Thermal proteome profiling (TPP), also known as MS-based cellular thermal shift assay (MS-CETSA), provides an invaluable means to quantitatively determine ligand-induced protein thermostability changes on a proteome-wide scale. This approach is based on the biophysical properties of proteins where target engagement alters their structural stability, which can affect their thermal denaturation properties. This enables an ultracentrifugation step to separate soluble from insoluble protein fractions in thermal melts for MS quantification.[59] As such, TPP enables ligand–protein interaction profiling using unmodified compounds of interest. In a canonical experimental setup, equal aliquots of cell lysates (or intact cells) in the presence or absence of compounds are exposed to a temperature gradient, followed by precipitation of insoluble fractions by ultracentrifugation (Figure 2A). The soluble protein fraction is then digested with trypsin and labeled with isobaric mass tags for quantitative MS. The soluble fractions of each protein detected and quantified in a bottom-up proteomics readout can be plotted as a function of temperature to generate melting curves for every detected protein. From these curves, the melting temperature (Tm), at which half of the protein is denatured and precipitated, can be calculated in the presence and absence of ligand, and ultimately a ΔTm is determined. Therefore, ligand-induced protein thermal stability changes captured by TPP hint at possible inhibitor targets, and this methodology provides an avenue for deciphering mechanisms of action in various organisms, including intracellular malaria parasites.
Figure 2.

Energetics-based chemoproteomics to deconvolute malaria drug targets. A) Thermal proteome profiling (TPP). Enriched Plasmodium-infected RBCs or the parasite lysates are subjected to a temperature gradient in the presence or absence of drugs. The samples are lysed (for intact cells) and centrifuged to precipitate denatured protein aggregates. The soluble fractions (native proteins) are digested for quantitative bottom-up proteomics analysis and isobaric mass tag labeled for mass spectrometry. The abundance of each protein as a function of temperature can be plotted to create a melting curve, and the altered melting profile and melting temperature shift (ΔTm) indicates a drug-induced thermal stability change of the corresponding protein target. For the ITDR experiment, proteins are incubated at a fixed temperature with drugs at different concentrations. As such, the protein thermostability change can be measured in a dose-dependent manner. B) Stability of proteins from rates of oxidation (SPROX). The infected RBCs are permeabilized with saponin to remove host proteins. Enriched Plasmodium parasite lysate with and without drugs are incubated in denaturant-containing buffers (e.g., 0.5–6 M urea). After reaching the protein folding and unfolding equilibrium, the samples are treated with hydrogen peroxide, subsequently quenched, digested for quantitative bottom-up proteomics analysis and isobaric mass tag labeled for mass spectrometry. The abundance of wild-type as opposed to the oxidized methionine-containing peptide is then plotted against the denaturant concentration to generate the chemical denaturation curves and the transition midpoint shifts (ΔC1/2) upon ligand binding.
Recent studies have applied and adapted TPP to identify molecular targets of several antimalarial agents.[21-22] Dziekan et al. performed an elegant series of experiments to characterize the melting behavior of the P. falciparum proteome and demonstrated the feasibility of using TPP to identify antimalarial drug targets.[21] In addition to the canonical approach where the melting curves are generated using a saturating drug concentration, Dziekan et al. used an alternative TPP variant called isothermal dose-response CETSA (ITDR-CETSA) (Figure 2A).[21] The ITDR experiment measures protein thermal stability using various drug concentrations at constant temperatures. Typically, the median melting temperature is used to determine the dose-dependent thermal stability change, while a higher and a lower temperature can be chosen for proteins that are more or less thermostable.[21, 60-61] This ITDR strategy not only validated the known mechanisms of action of two antimalarial agents (pyrimethamine as a folic acid antagonist and E64d as a cysteine proteinase inhibitor) but also identified P. falciparum purine nucleoside phosphorylase (PfPNP) as a novel target of two quinoline drugs (quinine and mefloquine).[21] Subsequent biophysical, biochemical, and structural studies confirmed direct interactions between PfPNP and the quinoline drugs.[21] This study exemplifies the utility of ITDR-CETSA in resolving drug targets in malaria parasites. Furthermore, dose-response data derived from ITDR-CETSA was previously shown to correlate with target occupancy and may also be used to estimate relative affinities between compounds and different parasite proteins.[61-62]
To date, most chemoproteomic methods investigate ligand–protein interactions in cell lysates and have provided valuable information on basic biological processes, drug mechanisms and pharmaceutical toxicity. However, proteins in a non-physiological environment may display altered thermodynamic properties due to several factors such as dilution of cofactors or binding partners and disrupted biomolecular complexes during sample preparation.[63] In these scenarios, lysate experiments may not reflect the native state of proteins. By leveraging temperature perturbations to probe protein thermostability changes, TPP also allows direct measurement in living cells and intact tissues where the thermal denaturation properties of many proteins are conserved.[61-62] In-cell TPP has been used to profile various organisms ranging from bacteria to mammalian cells and has recently been adapted to the intracellular P. falciparum parasite.[11, 21, 62-65] Unlike the lysate experiments, this in-cell TPP method employs magnetic enrichment to isolate parasite-infected RBCs by leveraging the paramagnetic hemozoin crystals that are present in late trophozoites and schizonts. The enriched parasite-infected RBCs are then subjected to compound treatments and thermal challenges to assess possible protein thermostability changes. With this approach, the impact of changes in protein states in the cellular context is better accounted for in the target ligand's mode-of-action. This includes protein expression and conformational changes, altered post-translational modifications, changes in protein localization and interactions triggered by compound administration.[63-64] Thus, in-cell TPP not only identifies direct inhibitor targets but also captures the downstream effects of the compound, providing information on how antimalarial agents modulate the entire Plasmodium proteome. In-cell TPP could also allow the simultaneous interrogation of the compounds’ effects on the host erythrocyte proteome[21], which could reveal host-directed activities and/or off-target cytotoxicity.
The capability of TPP to unveil novel therapeutic targets in Plasmodium parasites has also been demonstrated. The P. falciparum TCP-1 ring complex (TRiC), also termed chaperonin containing TCP-1 (CCT), is an essential protein folding complex and may have a conserved function in cytoskeletal protein folding, as demonstrated in other eukaryotes.[66] Despite the essentiality of PfTRiC, it was not known to be druggable (i.e. no known Plasmodium TRiC inhibitors existed). By utilizing the TPP technique, PfTRiC was identified as a target of clemastine, an over-the-counter antihistamine that inhibits blood- and liver-stage Plasmodium parasites with no cytotoxicity detected in human cells.[22] Though PfTRiC shares a 74–86% amino acid sequence similarity to its human counterpart, clemastine exhibited a species selectivity for the Plasmodium protein. Subsequent cellular analyses confirmed that clemastine interferes with microtubule morphology and the biogenesis of the major TRiC substrate tubulin in P. falciparum blood-stage parasites.[22] Intriguingly, clemastine decreased the thermostability of all eight PfTRiC subunits, suggesting a unique mechanism of disruption of the protein folding nanomachine.
In addition to P. falciparum, TPP has identified molecular targets in other intracellular protozoan parasites. For example, an ATP analog ENH1 exhibited antiparasitic activities against both P. falciparum and the related apicomplexan parasite Toxoplasma gondii.[67] ENH1 modulates calcium homeostasis and blocks parasite egress, but its molecular targets were unknown.[68] TPP revealed that ENH1 directly binds to and inhibits T. gondii CDPK1, which contributes to parasite egress inhibition.[67] Additionally, TPP was used to validate the on-target activity of a pyrazolyl sulfonamide analog (DDD100097) to N-Myristoyltransferase in Leishmania donovani.[69] Collectively, TPP affords multiplexed, quantitative measurements of a compounds’ effects on proteome thermal stability, and it is widely applicable in many important human pathogens.
5. Stability of proteins from rates of oxidation
Stability of proteins from rates of oxidation (SPROX) is another energetics-based method for profiling drug–protein interactomes in complex biological samples. Unlike TPP, which measures relative soluble fractions as a proxy of protein thermal stability, SPROX quantifies the levels of methionine oxidation in proteins as a function of chemical denaturant to directly probe ligand-induced thermodynamic stability changes. Briefly, equal aliquots of cell lysates, with and without inhibitors, are subjected to a chemical denaturant, such as urea and guanidinium hydrochloride, in a concentration gradient, followed by a short hydrogen peroxide treatment that labels methionine residues in proteins as they become solvent exposed when the proteins unfold (Figure 2B).[70-71] After quenching the oxidation reactions, proteins are trypsinized, labeled with isobaric mass tags, enriched for methionine-containing peptides and then analyzed with LC/MS/MS. The relative amount of wild-type, as opposed to oxidized, methionine-containing peptides, can be plotted against the denaturant concentrations to produce chemical denaturation curves for the proteins to which the peptides map. Target proteins that are more or less resistant to chemical denaturation in the presence of ligand can be identified by the transition midpoint (C1/2) shift or ΔC1/2 value, which is the difference in the concentrations at which half of the methionine-containing peptide is oxidized.[70-71]
SPROX has been used to identify inhibitor targets in yeast and mammalian cells and can be broadly applied to more complex systems.[22, 71-73] In the aforementioned Plasmodium study on the clemastine mechanism of action, SPROX was also adapted to interrogate P. falciparum parasites.[22] Although TPP unveiled the interaction between clemastine and PfTRiC, the nature of the interaction was unclear, as the thermostability of all TRiC subunit(s) was decreased to a similar degree. This is because protein complexes tend to precipitate together under a thermal challenge, rendering similar melting behaviors for each subunit in a complex.[74] This phenomenon, known as thermal proximity coaggregation, has been observed in bacteria[63], human cells[74] and P. falciparum.[21] In contrast, SPROX probes the thermodynamic change using individual methionine-containing peptides, providing thermodynamic details on the molecular mechanism, including the peptide(s) involved in the ligand-induced structural stability change. Namely, compounds may bind to or allosterically regulate the regions in close structural proximity to peptides identified as hits. For example, clemastine stabilized a peptide that mapped to the apical loop of the PfTRiC delta subunit, hinting that this region, a known substrate binding site in human TRiC, may interact directly with clemastine.[22] Furthermore, the thermodynamic data derived from SPROX allows the estimation of binding affinities (Kd values), which can facilitate target ranking, drug discovery, and optimization.[19, 22, 75]
6. Summary and Outlook
To date, millions of compounds have been screened for the ability to inhibit Plasmodium, identifying thousands of compounds that exhibit submicromolar efficacy against the parasites.[6, 76-78] Compounds with superior antiparasitic activity and low cytotoxicity may be further optimized to generate drug leads and candidates for clinical trials. However, despite substantial efforts and progress, 84% of the candidate drugs entering preclinical development from the Medicines for Malaria Venture (MMV) portfolio did not reach FDA approval.[79] Toxicity from off-target effects and failure to bind to the intended target(s) under physiological conditions may contribute to this high attrition rate. A more comprehensive understanding of how a compound interacts with both the parasite and human proteomes could therefore facilitate drug development.
MS-based target identification studies have added to our knowledge of potential targets for numerous compounds. But many are carried out with cell lysates, which may not fully recapitulate the native states of proteins, often leading to false negatives. Thus far, TPP is the only chemoproteomic method that affords detection using unmodified compounds in unperturbed cells and tissues. Accordingly, TPP can examine downstream effects and stress responses induced by inhibitors in physiological settings.[80] This could be especially useful when the compounds bind to elusive or non-protein targets. Direct target identification for compounds that bind non-protein molecules, including RNA and DNA among others, is not feasible. However, shifts in melting temperature may be detected in downstream effectors that can hint at the overall impacted pathway(s). Conversely, it can be challenging to determine primary targets or to identify secondary phenotypes due to cell death and stress responses. In this regard, cross-referencing the datasets from in-cell TPP and the lysate experiment may facilitate data interpretation.
Likewise, each chemoproteomic strategy has its caveats, and by integrating different techniques, more in-depth insights into a compound’s mode of action may be obtained. Take ITDR-CETSA for example, although this technique provides a dose-response measurement that correlates with EC50 values, proteins that have altered melting behaviors, such as a change in melting curve slope, without melting temperature shifts may not be captured.[61] To this end, a two-dimensional TPP strategy was devised to measure thermostability changes at multiple compound concentrations throughout a temperature gradient.[81] This combination of ITDR and melting curve analyses increases the sensitivity of target detection as well as the estimation of the drug affinity. Additionally, smaller proteins may be less prone to precipitation during TPP while others may be more sensitive or recalcitrant to thermal challenges. The interpretation of ligand-induced thermostability changes can also be ambiguous, such as in the study with PfTRiC.[22] Thus, the use of alternative yet complementary chemoproteomic methods (e.g., SPROX) is advantageous not only to capture a more complete subset of proteins but also to provide a fuller understanding of a ligand's mode of action.
Like other omics-based approaches, subsequent target validation is required to link the proposed target to the drug’s activity. Genetic methods, such as the TetR-DOZI-RNA aptamer system and the selection-linked integration (SLI) system, are invaluable tools to validate targets that have been recently adapted to malaria parasites.[82-83] The TetR–aptamer system enables tunable protein expression at the posttranscriptional level, and can be used to examine the chemical–genetic interaction. Alternatively, the SLI method provides rapid selection for the intended genomic integration. Both methods can support the proposed target when compound sensitization is observed after conditional gene knockdown. Substantial structure–activity relationship (SAR) studies can also support a proposed target when essential moieties concerning the ligand-target interface are known. In fact, compounds with structure optimization and known SAR may produce better results in target deconvolution studies.
Many antimalarial agents have been proposed to target Plasmodium membrane proteins, such as cipargamin to PfATP4[84], atovaquone to the cytochrome bc1 complex[85] and DSM265 to PfDHODH.[86] Various transporters have also been considered potential therapeutic targets.[4, 12] Thus, the need remains to increase coverage of the membrane proteome to further enhance the utility of proteomics in target profiling. Though challenging, previous efforts using human cells demonstrated that low concentrations of Nonidet P-40 (NP-40) greatly improved TPP (and likely other chemoproteomic methods) membrane proteome coverage, without compromising overall melting behaviors. Specifically, 370–750 membrane proteins with good-quality melting curves were observed versus <80 membrane proteins without NP-40 treatment.[11, 87-88] Whether NP-40 (or other mild detergents) is compatible with the Plasmodium proteome and improves recovery of parasite membrane proteins remains to be determined, but such an achievement would greatly add to the ability to assess potential parasite targets.
Currently, most proteomics-based target identification studies that have been used with Plasmodium parasites were performed in the asexual P. falciparum blood stage, likely due to the relative ease of obtaining large amounts of enriched parasites during this stage. While targeting other Plasmodium species and life cycle stages is important for drug development, these parasites and developmental stages are less amenable to chemoproteomic analyses. For example, drugs targeting sexual gametocytes and the liver stage of parasite infection offer prophylactic and transmission-blocking interventions. Although the Plasmodium liver stage can be studied in vitro, proteomic studies are hampered by low parasite infection rates (<1% infected cells) and the need to breed and maintain parasite-infected mosquitoes.[89] Additionally, while sexual P. falciparum gametocytes can be produced in culture, low differentiation rates limit the amount of protein that can be obtained.[7, 90] Perhaps the most challenging stage in the parasite’s life cycle to interrogate with proteomics is the dormant P. vivax liver stage, termed hypnozoites, that causes relapsing malaria.[91] Despite these constraints, generating sufficient material for chemoproteomic studies may still be viable by establishing methods that enrich the specific life stages (gametocytes, liver-stage schizonts, and liver-stage hypnozoites).[90, 92] Further developments in Plasmodium culturing systems to obtain more materials coupled with the increased sensitivity and resolution of MS-based technologies could enable chemoproteomic studies in these elusive parasite stages in the future.[93-94]
Acknowledgments
Work was supported by the NIH (1DP2AI138239 to E.R.D., T32GM008555 to C.R.M.) for laboratory support and fellowship support.
References
- [1].WHO, World Malaria Report 2019.
- [2].Ranson H, N’guessan R, Lines J, Moiroux N, Nkuni Z, Corbel V, Trends in parasitology 2011, 27, 91–98. [DOI] [PubMed] [Google Scholar]
- [3].Hovlid ML, Winzeler EA, Trends in parasitology 2016, 32, 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Belete TM, Drug design, development and therapy 2020, 14, 3875–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ashley EA, Phyo AP, Drugs 2018, 78, 861–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Antonova-Koch Y, Meister S, Abraham M, Luth MR, Ottilie S, Lukens AK, Sakata-Kato T, Vanaerschot M, Owen E, Jado JC, Maher SP, Calla J, Plouffe D, Zhong Y, Chen K, Chaumeau V, Conway AJ, McNamara CW, Ibanez M, Gagaring K, Serrano FN, Eribez K, Taggard CM, Cheung AL, Lincoln C, Ambachew B, Rouillier M, Siegel D, Nosten F, Kyle DE, Gamo FJ, Zhou Y, Llinas M, Fidock DA, Wirth DF, Burrows J, Campo B, Winzeler EA, Science 2018, 362, eaat9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Delves MJ, Straschil U, Ruecker A, Miguel-Blanco C, Marques S, Dufour AC, Baum J, Sinden RE, Nature protocols 2016, 11, 1668–1680. [DOI] [PubMed] [Google Scholar]
- [8].Plouffe DM, Wree M, Du AY, Meister S, Li F, Patra K, Lubar A, Okitsu SL, Flannery EL, Kato N, Tanaseichuk O, Comer E, Zhou B, Kuhen K, Zhou Y, Leroy D, Schreiber SL, Scherer CA, Vinetz J, Winzeler EA, Cell host & microbe 2016, 19, 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Edwards RL, Odom John AR, F1000Research 2016, 5, 2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilkinson IVL, Terstappen GC, Russell AJ, Drug discovery today 2020, S1359-6446(20)30373-1. [DOI] [PubMed] [Google Scholar]
- [11].Dziekan JM, Wirjanata G, Dai L, Go KD, Yu H, Lim YT, Chen L, Wang LC, Puspita B, Prabhu N, Sobota RM, Nordlund P, Bozdech Z, Nature protocols 2020, 15, 1881–1921. [DOI] [PubMed] [Google Scholar]
- [12].Cowell AN, Winzeler EA, Genome medicine 2019, 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carolino K, Winzeler EA, Current opinion in microbiology 2020, 57, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Murithi JM, Owen ES, Istvan ES, Lee MCS, Ottilie S, Chibale K, Goldberg DE, Winzeler EA, Llinas M, Fidock DA, Vanaerschot M, Cell chemical biology 2020, 27, 158–171 e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cowell AN, Istvan ES, Lukens AK, Gomez-Lorenzo MG, Vanaerschot M, Sakata-Kato T, Flannery EL, Magistrado P, Owen E, Abraham M, LaMonte G, Painter HJ, Williams RM, Franco V, Linares M, Arriaga I, Bopp S, Corey VC, Gnadig NF, Coburn-Flynn O, Reimer C, Gupta P, Murithi JM, Moura PA, Fuchs O, Sasaki E, Kim SW, Teng CH, Wang LT, Akidil A, Adjalley S, Willis PA, Siegel D, Tanaseichuk O, Zhong Y, Zhou Y, Llinas M, Ottilie S, Gamo FJ, Lee MCS, Goldberg DE, Fidock DA, Wirth DF, Winzeler EA, Science 2018, 359, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luth MR, Gupta P, Ottilie S, Winzeler EA, ACS infectious diseases 2018, 4, 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Simon O, Yeung BK, Chatterjee AK, McCormack SL, Manary MJ, Zeeman AM, Dechering KJ, Kumar TS, Henrich PP, Gagaring K, Ibanez M, Kato N, Kuhen KL, Fischli C, Nagle A, Rottmann M, Plouffe DM, Bursulaya B, Meister S, Rameh L, Trappe J, Haasen D, Timmerman M, Sauerwein RW, Suwanarusk R, Russell B, Renia L, Nosten F, Tully DC, Kocken CH, Glynne RJ, Bodenreider C, Fidock DA, Diagana TT, Winzeler EA, Nature 2013, 504, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Luksch T, Martinez MS, Luksch T, Bolscher JM, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock PM, Churcher TS, Sala KA, Zakutansky SE, Jimenez-Diaz MB, Sanz LM, Riley J, Basak R, Campbell M, Avery VM, Sauerwein RW, Dechering KJ, Noviyanti R, Campo B, Frearson JA, Angulo-Barturen I, Ferrer-Bazaga S, Gamo FJ, Wyatt PG, Leroy D, Siegl P, Delves MJ, Kyle DE, Wittlin S, Marfurt J, Price RN, Sinden RE, Winzeler EA, Charman SA, Bebrevska L, Gray DW, Campbell S, Fairlamb AH, Willis PA, Rayner JC, Fidock DA, Read KD, Gilbert IH, Nature 2015, 522, 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu KY, Chembiochem : a European journal of chemical biology 2020, 21, 3189–3191. [DOI] [PubMed] [Google Scholar]
- [20].Friman T, Bioorganic & medicinal chemistry 2020, 28, 115174. [DOI] [PubMed] [Google Scholar]
- [21].Dziekan JM, Yu H, Chen D, Dai L, Wirjanata G, Larsson A, Prabhu N, Sobota RM, Bozdech Z, Nordlund P, Science translational medicine 2019, 11. eaau3174. [DOI] [PubMed] [Google Scholar]
- [22].Lu KY, Quan B, Sylvester K, Srivastava T, Fitzgerald MC, Derbyshire ER, Proceedings of the National Academy of Sciences of the United States of America 2020, 117, 5810–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rix U, Superti-Furga G, Nature chemical biology 2009, 5, 616–624. [DOI] [PubMed] [Google Scholar]
- [24].Medvedev A, Kopylov A, Buneeva O, Zgoda V, Archakov A, Proteomics 2012, 12, 621–637. [DOI] [PubMed] [Google Scholar]
- [25].Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, Fergusson D, Mottram J, Soete M, Dubremetz JF, Le Roch K, Doerig C, Schultz P, Meijer L, Chemistry & biology 2000, 7, 411–422. [DOI] [PubMed] [Google Scholar]
- [26].Paquet T, Le Manach C, Cabrera DG, Younis Y, Henrich PP, Abraham TS, Lee MCS, Basak R, Ghidelli-Disse S, Lafuente-Monasterio MJ, Bantscheff M, Ruecker A, Blagborough AM, Zakutansky SE, Zeeman AM, White KL, Shackleford DM, Mannila J, Morizzi J, Scheurer C, Angulo-Barturen I, Martinez MS, Ferrer S, Sanz LM, Gamo FJ, Reader J, Botha M, Dechering KJ, Sauerwein RW, Tungtaeng A, Vanachayangkul P, Lim CS, Burrows J, Witty MJ, Marsh KC, Bodenreider C, Rochford R, Solapure SM, Jimenez-Diaz MB, Wittlin S, Charman SA, Donini C, Campo B, Birkholtz LM, Hanson KK, Drewes G, Kocken CHM, Delves MJ, Leroy D, Fidock DA, Waterson D, Street LJ, Chibale K, Science translational medicine 2017, 9, eaad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sinxadi P, Donini C, Johnstone H, Langdon G, Wiesner L, Allen E, Duparc S, Chalon S, McCarthy JS, Lorch U, Chibale K, Mohrle J, Barnes KI, Antimicrobial agents and chemotherapy 2020, 64, e01896–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eberl HC, Werner T, Reinhard FB, Lehmann S, Thomson D, Chen P, Zhang C, Rau C, Muelbaier M, Drewes G, Drewry D, Bantscheff M, Scientific reports 2019, 9, 14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Penzo M, de Las Heras-Duena L, Mata-Cantero L, Diaz-Hernandez B, Vazquez-Muniz MJ, Ghidelli-Disse S, Drewes G, Fernandez-Alvaro E, Baker DA, Scientific reports 2019, 9, 7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Matralis AN, Malik A, Penzo M, Moreno I, Almela MJ, Camino I, Crespo B, Saadeddin A, Ghidelli-Disse S, Rueda L, Calderon F, Osborne SA, Drewes G, Boesche M, Fernandez-Alvaro E, Martin Hernando JI, Baker DA, Journal of medicinal chemistry 2019, 62, 9217–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Crowther GJ, Hillesland HK, Keyloun KR, Reid MC, Lafuente-Monasterio MJ, Ghidelli-Disse S, Leonard SE, He P, Jones JC, Krahn MM, Mo JS, Dasari KS, Fox AM, Boesche M, El Bakkouri M, Rivas KL, Leroy D, Hui R, Drewes G, Maly DJ, Van Voorhis WC, Ojo KK, PloS one 2016, 11, e0149996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, Foley M, Haystead TA, Molecular pharmacology 2002, 62, 1364–1372. [DOI] [PubMed] [Google Scholar]
- [33].Cravatt BF, Wright AT, Kozarich JW, Annual review of biochemistry 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- [34].Greenbaum DC, Baruch A, Grainger M, Bozdech Z, Medzihradszky KF, Engel J, DeRisi J, Holder AA, Bogyo M, Science 2002, 298, 2002–2006. [DOI] [PubMed] [Google Scholar]
- [35].Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, Bogyo M, Nature chemical biology 2008, 4, 203–213. [DOI] [PubMed] [Google Scholar]
- [36].Deu E, Leyva MJ, Albrow VE, Rice MJ, Ellman JA, Bogyo M, Chemistry & biology 2010, 17, 808–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stolze SC, Deu E, Kaschani F, Li N, Florea BI, Richau KH, Colby T, van der Hoorn RA, Overkleeft HS, Bogyo M, Kaiser M, Chemistry & biology 2012, 19, 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yoo E, Schulze CJ, Stokes BH, Onguka O, Yeo T, Mok S, Gnadig NF, Zhou Y, Kurita K, Foe IT, Terrell SM, Boucher MJ, Cieplak P, Kumpornsin K, Lee MCS, Linington RG, Long JZ, Uhlemann AC, Weerapana E, Fidock DA, Bogyo M, Cell chemical biology 2020, 27, 143–157 e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Harbut MB, Velmourougane G, Dalal S, Reiss G, Whisstock JC, Onder O, Brisson D, McGowan S, Klemba M, Greenbaum DC, Proceedings of the National Academy of Sciences of the United States of America 2011, 108, E526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yoo E, Stokes BH, de Jong H, Vanaerschot M, Kumar T, Lawrence N, Njoroge M, Garcia A, Van der Westhuyzen R, Momper JD, Ng CL, Fidock DA, Bogyo M, Journal of the American Chemical Society 2018, 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stokes BH, Yoo E, Murithi JM, Luth MR, Afanasyev P, da Fonseca PCA, Winzeler EA, Ng CL, Bogyo M, Fidock DA, PLoS pathogens 2019, 15, e1007722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li H, O'Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PC, Bogyo M, Nature 2016, 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li H, Ponder EL, Verdoes M, Asbjornsdottir KH, Deu E, Edgington LE, Lee JT, Kirk CJ, Demo SD, Williamson KC, Bogyo M, Chemistry & biology 2012, 19, 1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tan MSY, Davison D, Sanchez MI, Anderson BM, Howell S, Snijders A, Edgington-Mitchell LE, Deu E, PloS one 2020, 15, e0227341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schulze CJ, Navarro G, Ebert D, DeRisi J, Linington RG, The Journal of organic chemistry 2015, 80, 1312–1320. [DOI] [PubMed] [Google Scholar]
- [46].Speers AE, Adam GC, Cravatt BF, Journal of the American Chemical Society 2003, 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- [47].Speers AE, Cravatt BF, Chemistry & biology 2004, 11, 535–546. [DOI] [PubMed] [Google Scholar]
- [48].Ismail HM, Barton V, Phanchana M, Charoensutthivarakul S, Wong MH, Hemingway J, Biagini GA, O'Neill PM, Ward SA, Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 2080–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang J, Zhang CJ, Chia WN, Loh CC, Li Z, Lee YM, He Y, Yuan LX, Lim TK, Liu M, Liew CX, Lee YQ, Zhang J, Lu N, Lim CT, Hua ZC, Liu B, Shen HM, Tan KS, Lin Q, Nature communications 2015, 6, 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Collins KA, Abd-Rahman AN, Marquart L, Ballard E, Gobeau N, Griffin P, Chalon S, Mohrle JJ, McCarthy JS, The Journal of infectious diseases 2020, jiaa287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ismail HM, Barton VE, Panchana M, Charoensutthivarakul S, Biagini GA, Ward SA, O'Neill PM, Angewandte Chemie 2016, 128, 6511–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Phyo AP, Jittamala P, Nosten FH, Pukrittayakamee S, Imwong M, White NJ, Duparc S, Macintyre F, Baker M, Mohrle JJ, The Lancet. Infectious diseases 2016, 16, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Penarete-Vargas DM, Boisson A, Urbach S, Chantelauze H, Peyrottes S, Fraisse L, Vial HJ, PloS one 2014, 9, e113918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lubin AS, Rueda-Zubiaurre A, Matthews H, Baumann H, Fisher FR, Morales-Sanfrutos J, Hadavizadeh KS, Nardella F, Tate EW, Baum J, Scherf A, Fuchter MJ, ACS infectious diseases 2018, 4, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Smith E, Collins I, Future medicinal chemistry 2015, 7, 159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Speers AE, Cravatt BF, Chembiochem : a European journal of chemical biology 2004, 5, 41–47. [DOI] [PubMed] [Google Scholar]
- [57].Nodwell MB, Sieber SA, Topics in current chemistry 2012, 324, 1–41. [DOI] [PubMed] [Google Scholar]
- [58].Bohme U, Otto TD, Sanders M, Newbold CI, Berriman M, Wellcome open research 2019, 4, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mateus A, Maatta TA, Savitski MM, Proteome science 2016, 15, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lim YT, Prabhu N, Dai L, Go KD, Chen D, Sreekumar L, Egeblad L, Eriksson S, Chen L, Veerappan S, Teo HL, Tan CSH, Lengqvist J, Larsson A, Sobota RM, Nordlund P, PloS one 2018, 13, e0208273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dai L, Prabhu N, Yu LY, Bacanu S, Ramos AD, Nordlund P, Annual review of biochemistry 2019, 88, 383–408. [DOI] [PubMed] [Google Scholar]
- [62].Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, Kuster B, Nordlund P, Bantscheff M, Drewes G, Science 2014, 346, 1255784. [DOI] [PubMed] [Google Scholar]
- [63].Mateus A, Bobonis J, Kurzawa N, Stein F, Helm D, Hevler J, Typas A, Savitski MM, Molecular systems biology 2018, 14, e8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mateus A, Kurzawa N, Becher I, Sridharan S, Helm D, Stein F, Typas A, Savitski MM, Molecular systems biology 2020, 16, e9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sun W, Dai L, Yu H, Puspita B, Zhao T, Li F, Tan JL, Lim YT, Chen MW, Sobota RM, Tenen DG, Prabhu N, Nordlund P, Redox biology 2019, 24, 101168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Spillman NJ, Beck JR, Ganesan SM, Niles JC, Goldberg DE, Cellular microbiology 2017, 19, 10.1111/cmi.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Herneisen AL, Sidik SM, Markus BM, Drewry DH, Zuercher WJ, Lourido S, ACS chemical biology 2020, 15, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sidik SM, Hortua Triana MA, Paul AS, El Bakkouri M, Hackett CG, Tran F, Westwood NJ, Hui R, Zuercher WJ, Duraisingh MT, Moreno SN, Lourido S, The Journal of biological chemistry 2016, 291, 9566–9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Corpas-Lopez V, Moniz S, Thomas M, Wall RJ, Torrie LS, Zander-Dinse D, Tinti M, Brand S, Stojanovski L, Manthri S, Hallyburton I, Zuccotto F, Wyatt PG, De Rycker M, Horn D, Ferguson MAJ, Clos J, Read KD, Fairlamb AH, Gilbert IH, Wyllie S, ACS infectious diseases 2019, 5, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Strickland EC, Geer MA, Tran DT, Adhikari J, West GM, DeArmond PD, Xu Y, Fitzgerald MC, Nature protocols 2013, 8, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].West GM, Tucker CL, Xu T, Park SK, Han X, Yates JR 3rd, Fitzgerald MC, Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 9078–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Geer Wallace MA, Kwon DY, Weitzel DH, Lee CT, Stephenson TN, Chi JT, Mook RA Jr., Dewhirst MW, Hong J, Fitzgerald MC, Journal of proteome research 2016, 15, 2688–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ogburn RN, Jin L, Meng H, Fitzgerald MC, Journal of proteome research 2017, 16, 4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tan CSH, Go KD, Bisteau X, Dai L, Yong CH, Prabhu N, Ozturk MB, Lim YT, Sreekumar L, Lengqvist J, Tergaonkar V, Kaldis P, Sobota RM, Nordlund P, Science 2018, 359, 1170–1177. [DOI] [PubMed] [Google Scholar]
- [75].West GM, Tang L, Fitzgerald MC, Analytical chemistry 2008, 80, 4175–4185. [DOI] [PubMed] [Google Scholar]
- [76].Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF, Nature 2010, 465, 305–310. [DOI] [PubMed] [Google Scholar]
- [77].Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK, Nature 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA, Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 9059–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Burrows JN, Duparc S, Gutteridge WE, Hooft van Huijsduijnen R, Kaszubska W, Macintyre F, Mazzuri S, Mohrle JJ, Wells TNC, Malaria journal 2017, 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Prabhu N, Dai L, Nordlund P, Current opinion in chemical biology 2020, 54, 54–62. [DOI] [PubMed] [Google Scholar]
- [81].Becher I, Werner T, Doce C, Zaal EA, Togel I, Khan CA, Rueger A, Muelbaier M, Salzer E, Berkers CR, Fitzpatrick PF, Bantscheff M, Savitski MM, Nature chemical biology 2016, 12, 908–910. [DOI] [PubMed] [Google Scholar]
- [82].Birnbaum J, Flemming S, Reichard N, Soares AB, Mesen-Ramirez P, Jonscher E, Bergmann B, Spielmann T, Nature methods 2017, 14, 450–456. [DOI] [PubMed] [Google Scholar]
- [83].Goldfless SJ, Wagner JC, Niles JC, Nature communications, 2014, 5, 5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Spillman NJ, Kirk K, International journal for parasitology. Drugs and drug resistance 2015, 5, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Siregar JE, Kurisu G, Kobayashi T, Matsuzaki M, Sakamoto K, Mi-ichi F, Watanabe Y, Hirai M, Matsuoka H, Syafruddin D, Marzuki S, Kita K, Parasitology international 2015, 64, 295–300. [DOI] [PubMed] [Google Scholar]
- [86].Mandt REK, Lafuente-Monasterio MJ, Sakata-Kato T, Luth MR, Segura D, Pablos-Tanarro A, Viera S, Magan N, Ottilie S, Winzeler EA, Lukens AK, Gamo FJ, Wirth DF, Science translational medicine 2019, 11, eaav1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Reinhard FB, Eberhard D, Werner T, Franken H, Childs D, Doce C, Savitski MF, Huber W, Bantscheff M, Savitski MM, Drewes G, Nature methods 2015, 12, 1129–1131. [DOI] [PubMed] [Google Scholar]
- [88].Kawatkar A, Schefter M, Hermansson NO, Snijder A, Dekker N, Brown DG, Lundback T, Zhang AX, Castaldi MP, ACS chemical biology 2019, 14, 1913–1920. [DOI] [PubMed] [Google Scholar]
- [89].Raphemot R, Toro-Moreno M, Lu KY, Posfai D, Derbyshire ER, Cell chemical biology 2019, 26, 1253–1262 e1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Saliba KS, Jacobs-Lorena M, Methods in molecular biology 2013, 923, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lu KY, Derbyshire ER, Biochemistry 2020, 59, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Prudencio M, Rodrigues CD, Ataide R, Mota MM, Cellular microbiology 2008, 10, 218–224. [DOI] [PubMed] [Google Scholar]
- [93].McClure RA, Williams JD, ACS medicinal chemistry letters 2018, 9, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Couvillion SP, Zhu Y, Nagy G, Adkins JN, Ansong C, Renslow RS, Piehowski PD, Ibrahim YM, Kelly RT, Metz TO, The Analyst 2019, 144, 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]