

Abstract
Background -
The proliferation of genetic profiling has revealed many associations between genetic variations and disease. However, large-scale phenotyping efforts in largely healthy populations, coupled with DNA sequencing, suggest variants currently annotated as pathogenic are more common in healthy populations than previously thought. In addition, novel and rare variants are frequently observed in genes associated with disease both in healthy individuals and those under suspicion of disease. This raises the question of whether these variants can be useful predictors of disease. To answer this question, we assessed the degree to which the presence of a variant in the cardiac potassium channel gene KCNH2 was diagnostically predictive for the autosomal dominant long QT syndrome.
Methods -
We estimated the probability of a long QT diagnosis given the presence of each KCNH2 variant using Bayesian methods that incorporated variant features such as changes in variant function, protein structure, and in silico predictions. We call this estimate the post-test probability of disease. Our method was applied to over 4,000 individuals heterozygous for 871 missense or in-frame insertion/deletion variants in KCNH2 and validated against a separate international cohort of 933 individuals heterozygous for 266 missense or in-frame insertion/deletion variants.
Results -
Our method was well-calibrated for the observed fraction of heterozygotes diagnosed with Long QT. Heuristically, we found that the innate diagnostic information one learns about a variant from three-dimensional variant location, in vitro functional data, and in silico predictors is equivalent to the diagnostic information one learns about that same variant by clinically phenotyping 10 heterozygotes. Most importantly, these data can be obtained in the absence of any clinical observations.
Conclusions -
We show how variant-specific features can inform a prior probability of disease for rare variants even in the absence of clinically-phenotyped heterozygotes.
Keywords: ion channel, long QT syndrome, K-channel, genetic variation, genetics, bioinformatics, Ion Channels/Membrane Transport, Functional Genomics, Computational Biology, Meta Analysis
Introduction
Sequencing an individual’s full genome or exome now costs less than many routine medical procedures. One resulting vision is that our genomes could be sequenced early in life for individualized medical advice about disease prevention and drug selection. However, most discovered variants will never be observed in a sufficient number of heterozygotes for a definitive association with disease.1, 2 Furthermore, even when a variant is strongly associated with disease, the clinical implications can vary strikingly across individuals.3, 4
The American College of Medical Genetics and Genomics (ACMG) put forward an interpretation framework that integrates criteria such as variant prevalence, function, and computational predictions into a single annotation from “Benign” to “Pathogenic”.5, 6 Each additional satisfied criterion raises or lowers the probability the variant is classified “Pathogenic”.6 However, even definitively annotated “Pathogenic” variants have heterogeneous or asymptomatic clinical presentations7, 8 and variants annotated “Benign” may still increase risk (see Discussion). Thus, the predictive value of rare variants remains unclear.9, 10 Because a positive test for most variants cannot be applied to enough heterozygous individuals to achieve a definitive association with disease, a statistical approach is required to estimate the post-test probability of disease and validate those predictions in different groups and cohorts.
The observation of a single (or few) heterozygous carrier(s) does not adequately inform the probability of disease. Rather, individuals heterozygous for these variants benefit from a prior probability informed by knowledge about their clinical characteristics or the population they are drawn from. This prior, informed without knowledge of the variant, more reasonably reflects the true disease probability. In contrast, we propose to construct a prior probability of disease conditioned on variant-specific features and to modify this estimate using observations of heterozygous carriers. Our analysis yields a prior probability conditioned on variant-specific features known to be relevant to the association between the gene and disease. In practice, disease probability estimates are calibrated largely by how variant-features associate with disease probability in well-characterized variants. Our final estimate is effectively the post-test probability of disease given the presence of a variant or the positive predictive value (PPV) of rare genetic variants. We use “post-test probability” interchangeably with “penetrance”, in which the former recapitulates diagnostic thinking and reflects the Bayesian framework of this approach.
In past work, we described an algorithm for estimating the probability of a diagnosis of Brugada syndrome given the presence of a variant in the cardiac sodium channel gene SCN5A.11 While we incorporated variant-specific covariates (e.g. sequence conservation, functional perturbation, structural location, etc.), Brugada syndrome is likely oligogenic and the clinical phenotype is sometimes difficult to assess. In this manuscript, we develop a similar algorithm for estimating the probability of long QT syndrome type 2 (LQT2), a well-characterized and monogenic disorder induced by variants in the cardiac potassium channel gene KCNH2.
The KCNH2 gene (also called the human Ether-à-go-go-Related Gene, or hERG) encodes an ion channel subunit that assembles into the homotetrameric KV11.1 potassium channel. This channel produces the rapid delayed-rectifier repolarizing current, IKr, which sustains cardiomyocyte repolarization throughout the action potential plateau phase.12, 13 Loss-of-function KCNH2 variants that reduce IKr are associated with LQT2, a congenital heart arrhythmia defined by a prolongation of the QT interval on an electrocardiogram (ECG). Individuals with this ECG feature are at a greater risk for torsades de pointes, a life-threatening arrhythmia. With our method, we estimate the probability that an individual heterozygous for a missense or in-frame insertion/deletion (indel) variant in KCNH2 presents with LQT2 (for each variant). We validated our approach in an international cohort of 933 individuals ascertained under suspicion of LQTS, heterozygous for 266 unique missense and in-frame indel variants in KCNH2. Our results suggest the probability of disease can be estimated accurately before knowing the phenotype of a given heterozygous individual. Our result is a point estimate and 95% credible interval of disease probability for each variant which can be calculated before observing a single heterozygote. This prior—conditioned on variant-specific features—can be directly combined with observed heterozygotes for a posterior probability of disease. In this way, the prior is comparable with observations of heterozygotes, we estimate roughly 10 observations. All data resulting from this study are presented in web-accessible format at https://variantbrowser.org/KCNH2/ (Figure S1).
Methods
All data and materials are publicly available on GitHub (https://github.com/kroncke-lab/Bayes_KCNH2_LQT2_Penetrance). Additionally, a compiled and curated form of the data presented here are available in the KCNH2 Variant Browser (https://variantbrowser.org/KCNH2/; Figure S1). Internal Review Board (#191563) was evaluated at Vanderbilt University Medical Center and found to meet 45 CFR 46.104 (d) category (4) for Exempt Review. Detailed methods are available as supplemental data.
Results
KCNH2 variant heterozygote datasets
In total, the literature combined with gnomAD produced 871 unique missense or insertion/deletion (indel) KCNH2 variants; 4,810 individuals were heterozygous for these variants (< 0.001 minor allele frequency [MAF]), 1,041 of which were diagnosed with LQT2 according to our classification criteria (see Materials and Methods for details). From five arrhythmia centers in France, Italy, and Japan, we collected a cohort of patients heterozygous for KCNH2 variants. From these sites, we identified 266 unique missense or in-frame indel variants in KCNH2, 933 heterozygote carriers of these variants, 594 of which met our criteria for LQT2. From this cohort, the average age of diagnosis or ascertainment (if criteria for affected status were not met) within the participating sites was 24 years old (standard deviation of 19 years; available for 744 individuals in the cohort dataset). Gender was distributed 45% male.
Rates of LQT2, observed in the literature and our cohort, is associated with in vitro and in silico features.
To assess the association between in vitro and in silico characteristics of IKr/KV11.1/KCNH2 and the fraction of heterozygotes which present with LQT2, we calculated non-parametric Spearman rank order coefficients (Spearman ρ) between these features and the observed literature (Figure 1, black) or cohort (Figure 1, red) LQT2 probability (the fraction of heterozygotes diagnosed with LQT2 over total number of heterozygotes). We evaluated common in silico predictors, electrophysiological parameters for IKr, and a by-residue average observed LQT2 probability in a three-dimensional shell surrounding each residue (LQT2 probability density, see Materials and Methods for details). The variant-specific features LQT2 probability density, REVEL, and heterologous measurement of variant IKr peak tail currents had Spearman ρ absolute value point estimates around 0.6 and 0.7 in the literature and cohort datasets, the highest that reached a nominal p-value of 0.05.
Figure 1.
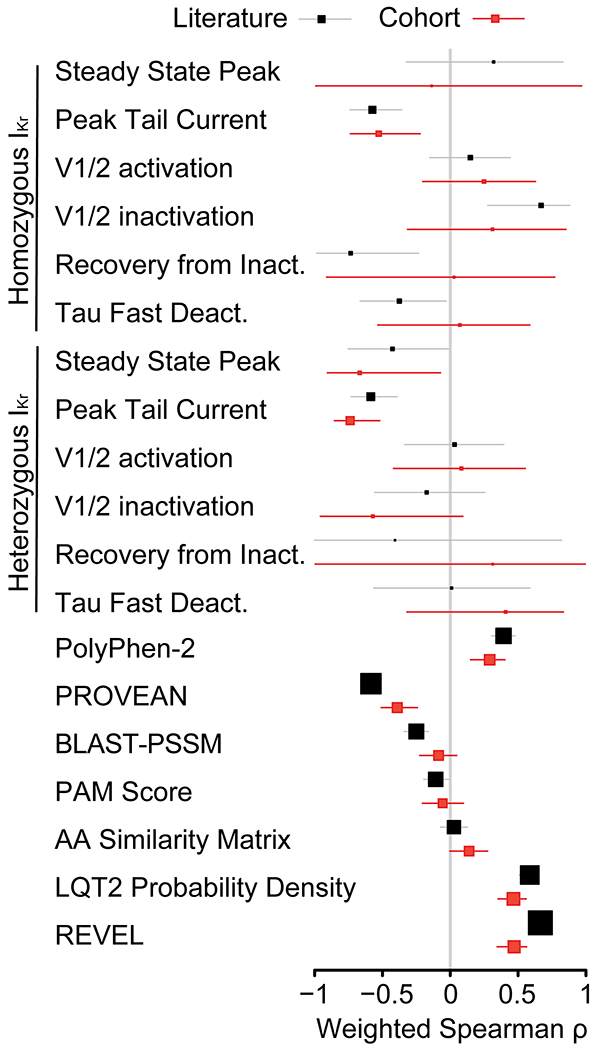
Weighted Spearman correlations between the fraction of heterozygotes diagnosed with LQT2 in the literature or cohort and the listed features for each variant. Weighted Spearman correlations between the fraction of heterozygotes diagnosed with LQT2 in the literature and listed features for each variant. Black and red squares indicate the estimate for the weighted Spearman correlation, weighted by , for the literature and cohort dataset, respectively. The grey and red lines indicate 95% confidence intervals (obtained by bootstrap), respectively. Larger box sizes indicate greater number of variants included in calculation.
Two broad in silico variant classifiers, PROVEAN and PolyPhen-2, had lower Spearman ρ point estimates than structure and peak tail currents (weighted Spearman ρ of 0.39 and −0.59 in the literature dataset and 0.33 and −0.46 in the cohort set for PolyPhen-2 and PROVEAN, respectively), though still statistically significant. Repeating this analysis in the cohort dataset produced mostly lower coefficients, though many retained statistical significance (Figure 1). Several biophysical properties were not statistically significant in either dataset.
Magnitude of LQT2 probability varies by residue location in three-dimensional space.
Given the relatively high correlation between LQT2 probability density and observed LQT2 probability, we mapped LQT2 probability density on to the structure of KV11.1 (Figures 2, S2, and S3). Figures 2 and S2 demonstrate LQT2 penetrance is not uniformly distributed over the major domains in KV11.1 (see Figure S2 for specific examples). This is in contrast to averaging over variants in sequence space as shown in Figure S4 and done previously.14, 15 For instance, the transmembrane segment in KV11.1 includes the voltage-sensing domains and pore domains, each of which have their own subdomains with high or low LQT2 penetrance. Some of these subregions are very small, localized to only a few contacting residues. For example, the voltage-sensing domain has a relatively low penetrance density in the intracellular half of helices S2 and S3 (Figure S2), while the most highly penetrant residues in this domain are in helices S1 and S4, which contact the pore domain near the midpoint of the membrane bilayer. Similarly, the pore domain shows the highest penetrance density near the selectivity filter and decreases towards the intracellular face of the pore. Additionally, the N-terminal Per-Arnt-Sim (PAS) domain and C-terminal cyclic nucleotide binding homology domain (CNBhD), both having relatively high observed LQT2 penetrance overall (Figure S4), are also heterogeneous. The most highly penetrant residues in these domains exist near the contacting surfaces between and among these domains. These trends are more muted in the in the observed LQT2 probability from cohort and literature data viewed linearly (Figure S4).
Figure 2.
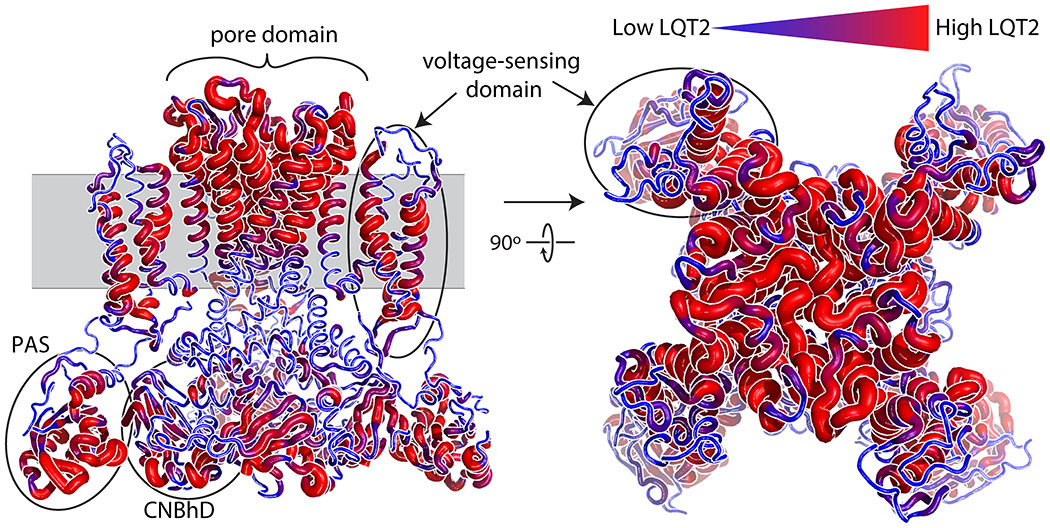
LQT2 probability density mapped on to KV11.1 structure. LQT2 probability density mapped on to the three-dimensional structure of the KV11.1 channel. Larger and redder segments indicate higher LQT2 probability density; smaller and bluer segments indicate lower LQT2 probability density. The model illustrates structural information regarding amino acids predicted to increase disease probability. Unlike linear graphical displays identifying pathogenic loci, LQT2 probability density provides novel, three-dimensional, insights into the specific structural components of the PAS, CNBhD, voltage-sensing, and pore domains that are associated with increased prevalence of LQT2.
Estimated post-test probability of LQTS based on KCNH2 variants found in only one heterozygote is predictive.
Variants found in only a single known heterozygous individual are the largest class of variants in the literature and our cohort data. Accordingly, we split the data into two groups: 1) variants with two or more heterozygous individuals and 2) variants with only one heterozygous individual (Figure S5). We then estimated the post-test probabilities of LQTS based on KCNH2 variants from group 1 (those with at least two heterozygous individuals). A Bayesian model was fit using an expectation maximization algorithm (see Materials and Methods for details and ref. 11). The predictive ability of our post-test LQT2 probability estimates were evaluated using the area under a Receiver Operating Characteristic curve (AUC) from group 2, those found in only one heterozygous individual (Figures 3 and S5). Additionally, we evaluated models fit on the full literature dataset on variants found in the cohort dataset (Figure 3, bottom, and Figures S5–S6). In all cases, the estimated AUC from our method outperformed other existing algorithms (LQT2 probability density, REVEL, PROVEAN, PolyPhen-2, BLAST-PSSM, and PAM score). For single heterozygotes in the literature, we observed AUCs of 0.87, 0.84, and 0.83 for our post-test probability model, REVEL, and LQT2 probability density, respectively. PROVEAN and PolyPhen-2, with AUCs of 0.74 and 0.73, respectively, were lower than our method and REVEL, as expected, since REVEL included PROVEAN and PolyPhen-2 as predictive covariates during construction.16 For variants with single heterozygotes in the cohort dataset, (Figure 3, bottom), AUC point estimates were lower overall, 0.78 and 0.77 for our method and LQT2 probability density, respectively. Surprisingly, REVEL scores were much less predictive in this group of variants, producing an AUC of 0.65, compared to 0.84 in the literature dataset. These differences in AUCs were also present when all variants were included and evaluated at various observed probability cutoffs (Figure S6). Previously published in silico predictors PROVEAN and PolyPhen-2 each had an AUC of 0.70, similar for the literature and cohort.
Figure 3.
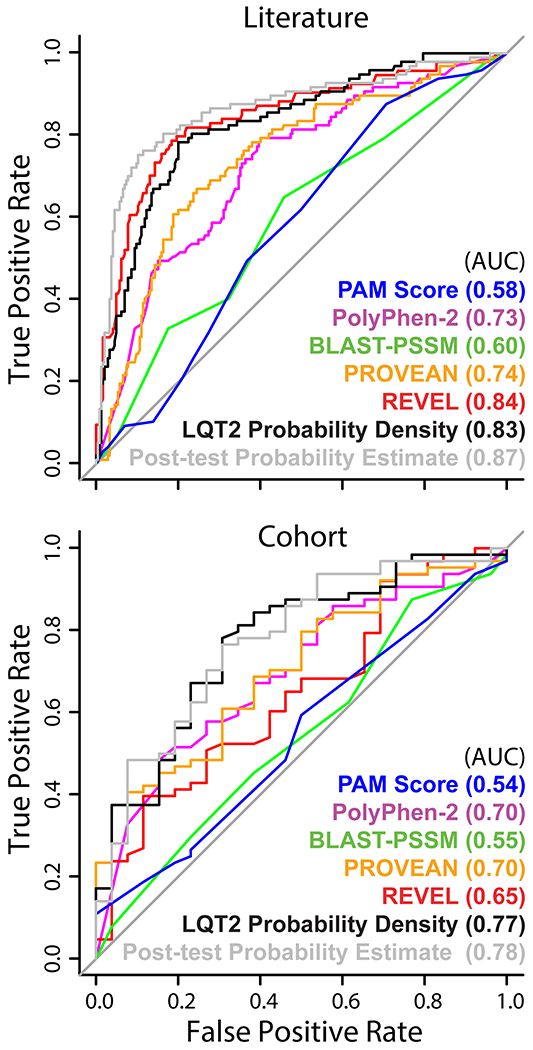
Receiver operating characteristic curves of features sorting variants with only one heterozygote observed. Receiver operating characteristic curves from predictors against variants with only one observation, an individual affected with LQT2 or not, in the cohort (Figures S5). The carriers come from either the literature (above) or the cohort (below). The estimated post-test probability and LQT2 probability density were not exposed to the evaluation variants, those whose heterozygote count is equal to one, during training. All cohort data were withheld from the EM and LQT2 probability density during construction (Figure S5).
Post-test LQT2 probability estimates are improved by including KCNH2 variant features.
The R2 between our LQT2 probability estimates and the cohort observed LQT2 probability is 0.30 when all variants are included, higher than in silico classifiers or LQT2 probability density; R2 estimates are higher overall when restricting to the set of variants where heterozygously collected peak tail current is known (Tables 1 and S1).
Table 1.
Weighted R2 between the fraction of heterozygotes diagnosed with LQT2 in the literature and cohort with estimates.
| LQT2 Probability Estimates | Literature (n = 706)† | >Cohort (n = 246)† |
|---|---|---|
|
| ||
| LQT2 probability density | 0.49 [0.39-0.60] | 0.23 [0.12-0.34] |
| REVEL | 0.38 [0.31-0.44] | 0.21 [0.11-0.33] |
| Post-test LQT2 Probability | 0.82 [0.77-0.86] | 0.30 [0.19-0.43] |
Weighted R2 [95% Confidence Interval] for the same subset of variants, weighted by , n is the number of unique KCNH2 variants
Since probability estimates are generally more reliable as the number of phenotyped heterozygous individuals increases, we calculated R2 at varying cutoffs of heterozygote count (Figure 4). As we restrict the analysis to variants with higher numbers of heterozygotes, we see R2 between our LQT2 post-test probability predictions and observed LQT2 probability substantially increase in both datasets (Figure 4). This shows that LQT2 probability predictions are statistically significant across sources.
Figure 4.
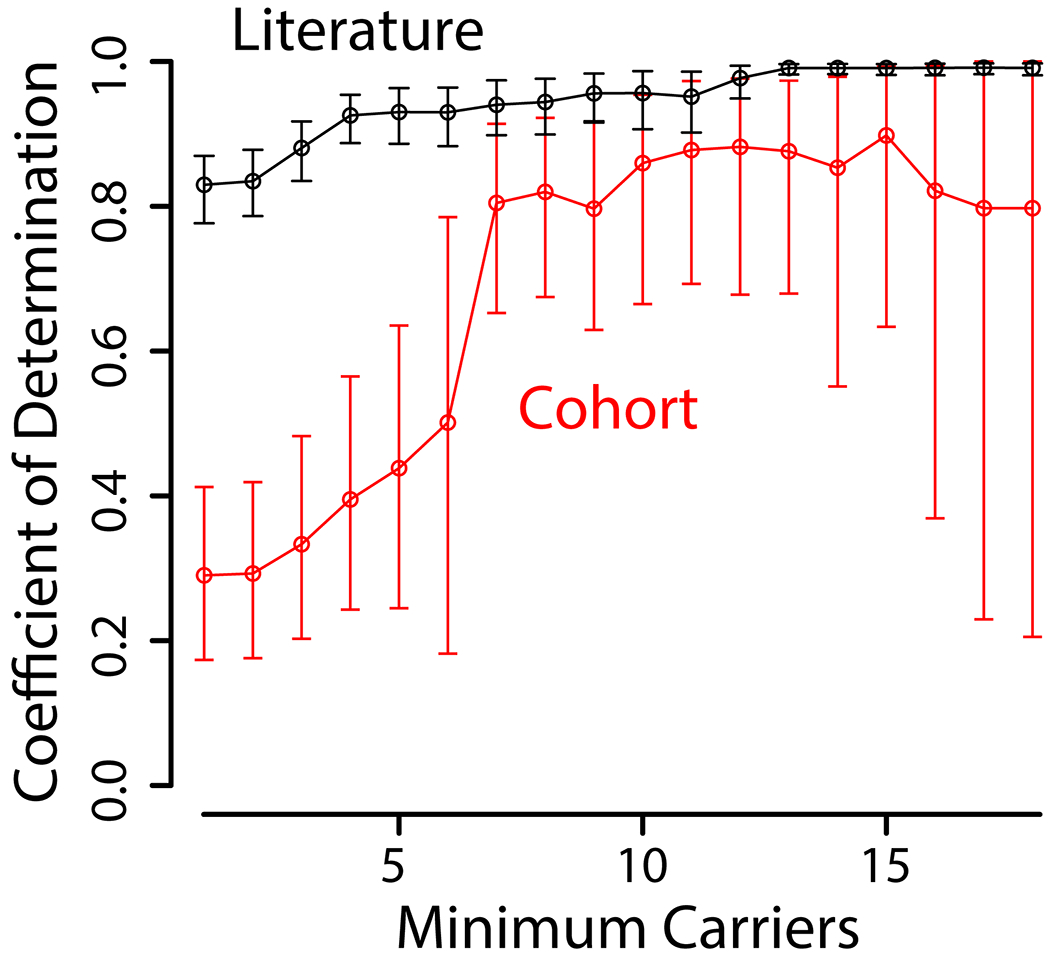
Coefficient of determination determined for LQT2 post-test probability predictions and fraction of homozygotes diagnosed with LQT2 from the cohort or literature. Coefficient of determination determined between EM LQT2 probability predictions and observed LQT2 probability from the cohort or literature. There are fewer variants to analyze as we restrict to variants with higher heterozygote counts.
GnomAD data are critical to build the most robust LQT2 probability estimates.
When gnomAD heterozygotes are removed from the literature dataset, the mean weighted probability observed in the cohort and the literature sets are closer to each other; this is also reflected in empirical probability distributions (Figure S7). However, rank order correlation between the literature and the cohort was reduced: without gnomAD, Spearman’s ρ between literature and cohort was 0.26 [95% confidence interval of 0.01-0.50]; when gnomAD was added to the literature ρ = 0.35 [95% CI of 0.11-0.58]; and when gnomAD was added to the cohort ρ = 0.33 [95% CI of 0.09-0.55]. In addition, predictive models trained from the literature without gnomAD resulted in lower AUCs and R2s, due in part to the reduced information in the LQT2 probability density feature (Table S2 and Figures S8–S9). These results demonstrate the importance of including control variants, such as those from gnomAD, in the LQT2 post-test probability estimates.
Example LQT2 probability estimates for a segment of KV11.1.
The outcome of our analysis is a range of data-driven post-test probabilities for each variant, initial probabilities conditioned on variant-specific properties, and posterior probabilities after heterozygous individuals are added. Each estimate is a probability distribution with a 95% credible interval. We illustrate the outcome of this method for variants in a segment within KV11.1 from p.Leu622 (c.1866) to p.Arg752 (c.2255) in Figure 5. Residues towards the extracellular face of KV11.1 have a higher prior and posterior estimated probability. The LQT2 probability prior probabilities conditioned on heterozygously-collected peak tail current, LQT2 probability density, and REVEL score, are near the observed probability (LQT2/total heterozygotes) for most variants (Figure 5).
Figure 5.
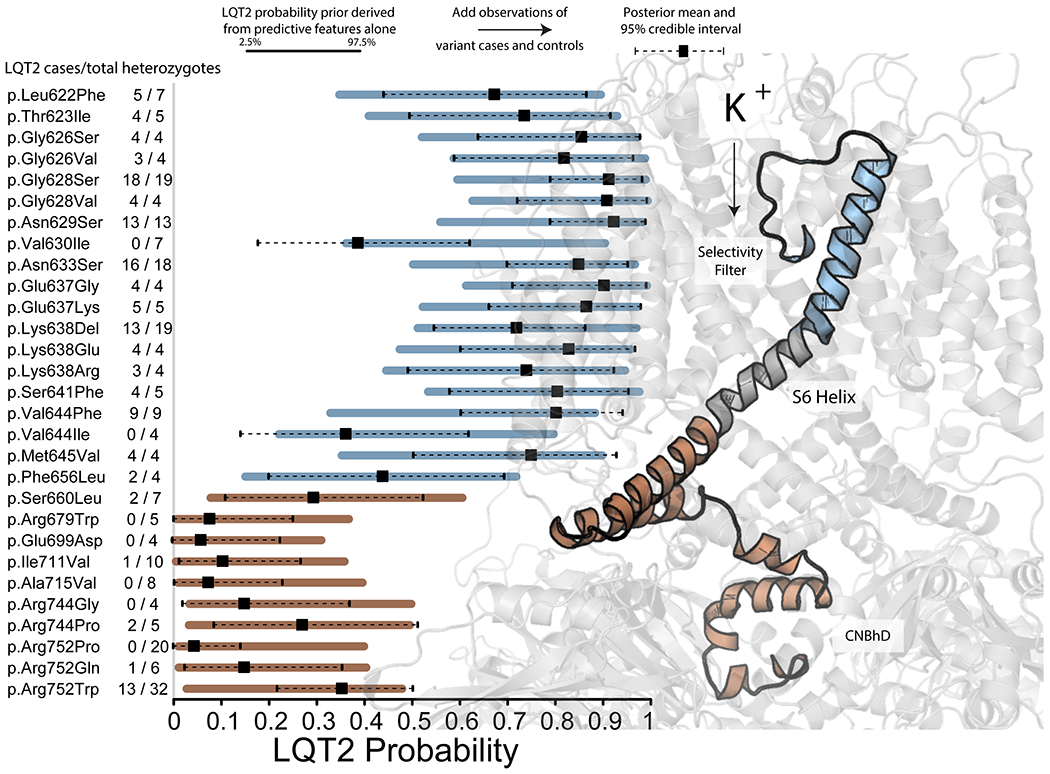
Example probability predictions for segment of KCNH2 including the selectivity filter and S6 helix. Example probability predictions for segment of KCNH2 including the selectivity filter and S6 helix. Priors were generated from in vitro (when available) and in silico covariates. Bars indicate the 95% interval of the prior. The dot and lines reflect the point estimate and 95% credible interval of the posterior, after the observations of affected individuals and those not meeting the threshold for affected status are included. Number of heterozygotes follows variant name on the left side of the figure. To the right, is a translucent structure of the KV11.1 channel. KV11.1 from p.Ser621 (c.1863) to p.Arg752 (c.2255) is represented as a solid cartoon, with two segments highlighted in different colors. The blue region highlights a segment with higher overall LQT2 probability while the red region highlights a segment with lower overall LQT2 probability.
Equivalence between KCNH2 variant features and clinically phenotyped heterozygotes.
The width of initial prior probability intervals conditioned on variant features (Figure 5, solid-colored lines) are determined by choice of ν in Equation 1 as previously described 11:
| Equation 1 |
where ν represents the number of observations in the beta binomial model, in this case, clinically phenotyped individuals heterozygous for variants in KCNH2, σi is the variance in the beta-binomial model and μi is the mean penetrance estimate for the ith variant. As ν grows, the prior 95% interval narrows; as ν decreases, prior 95% intervals expand. For very large ν, e.g. ν = 100, the posterior estimates of LQT2 post-test probability are heavily influenced by the prior such that very many observations of heterozygotes (1,000-10,000) are required to significantly change the posterior. At the other extreme of very small ν, e.g. ν = 1, the posterior estimates of LQT2 probability are largely independent of the priors. Acceptable values ofν would be those where 95% of variants have true LQT2 probabilities within the posterior 95% credible interval. To find values ofν where this was the case, we calculated posterior coverage rates by adding hypothetical heterozygotes sampled at the observed LQT2 probability to the prior generated with multiple values of ν, as described in the supplemental text and shown in Figures S10–S12. This procedure resulted in a range of acceptable ν values near ν = 10. Heuristically, for each variant, the post-test estimate of LQT2 probability carries the information equivalent to clinically phenotyping approximately 10 heterozygotes.
Discussion
Spectrum and example of LQT2 diagnosis probability attributable to KCNH2 variants.
Few KCNH2 coding variants have been discovered in a sufficiently large population to reliably estimate their post-test probability of developing LQT2 as defined in the Materials and Methods. However, variants such as p.Lys897Thr (c.2690A>C), p.Arg176Trp (c.526C>T), p.Val822Met (c.2464G>A), and p.Ala561Val (c.1682C>T) have been observed in many clinically phenotyped individuals both in the literature and in our assembled cohort. These variants span both the spectrum of channel defect (measured as heterozygously-collected peak tail current compared to WT) and spectrum of LQT2 disease probability. The most common KCNH2 coding variant, p.Lys897Thr, induces a very modest channel phenotype (peak tail current 78% of WT)17 and is common enough (5-24% of alleles)18 to preclude a large influence in LQT2 diagnosis, though its presence may modify risk.17 p.Arg176Trp induces peak tail current between 50-75% of WT 19, 20 and has a well-established LQT2 probability, estimated at 20%.21 We observe a similar LQT2 probability of 35% in the literature and 43% in the cohort, higher values likely reflecting a bias in ascertainment (also discussed below). p.Val822Met induces a significant channel defect, peak tail current of 44% of WT,22 and we correspondingly observe a higher LQT2 probability from the literature (65%) 23, 24 and cohort (60%). p.Ala561Val induces a severe channel defect, peak tail current between 0 and 46% of WT with a mean near 20% 25–28; we observed a LQT2 probability of 91% from the literature and 88% from the cohort. These variants illustrate that molecular defects induced by genetic variants in KCNH2 place heterozygotes at higher risk for LQT2.
Our framework allows us to exploit this relationship in part by conditioning estimates of disease probability on these defects, directly (in vitro data) or indirectly (in silico data). Our resulting model is informed largely by variants with many classified heterozygotes, like the variants just mentioned, but is most useful for variants with few or no known heterozygous carriers. Here, we validated our method with a diverse, international cohort of clinically phenotyped KCNH2 variant heterozygotes curated from among five centers; the cohort was withheld during all training stages and all potential overlapping individuals were removed. We tested how well our prior LQT2 probability estimates discriminate variants observed once in individuals who do or do not meet the criteria for LQT2 diagnosis (Figure 3) and correlate with the observed LQTS probability/penetrance (Figure 4). Lastly, all performance statistics reported for the probability density covariate were generated using leave-one-out cross validation, i.e. the probability density derived for each variant was never exposed to the observed LQT2 probability/penetrance for that variant.
Structure combined with previously described variants produced the most predictive feature of observed cohort LQT2 probability.
Variant position in KV11.1 domains such as transmembrane, pore, or intracellular is associated with differential risk of events.14, 29, 30 Expanding on this observation, and leveraging the recently determined KV11.1 channel structure (PDBID: 5VA1),31 we developed a metric to quantitate average LQT2 probability in the three-dimensional space surrounding each residue (Figures 2 and S2). The resulting metric, LQT2 probability density, was comparable to the in silico predictor REVEL in terms of AUC and R2 (Figure 3 and Table 1). Alone, LQT2 probability density could explain 50% of the variance in LQT2 probability as observed in the literature (Table 1); this reduced to 23% in the cohort but was still more predictive than even in vitro heterozygously-collected peak tail current data (Table S1). We attribute at least part of this decrease to greater ascertainment bias and lower heterozygous carrier counts in the cohort dataset. In addition, the R2 of 0.23 (up to 0.3 when including all covariates) is the most pessimistic coefficient of determination. When restricting to variants with greater heterozygote counts, the R2 improves to around 0.8 and so we estimate the generalized R2 is likely closer to a clinically meaningful value.
Bias in data collected from the literature, gnomAD, and cohort.
The clinical environment taxonomizes KCNH2 variants disproportionately from patients who present with disorders.32, 33 For example, individuals heterozygous for KCNH2 p.Arg176Trp (annotated in ClinVar variously as a risk factor, Likely Benign, and Variant of Uncertain Significance) have a mean QTc of 459 +/− 40 ms in clinical LQT2 families, those with at least one proband, but a mean QTc of 433 +/− 27 ms in a cross-sectional cohort of unselected heterozygotes.34 Similarly, we found most variants in the cohort have higher LQT2 probabilities than what we observed in the literature (Figures 6 and S13). Some of these variants have statistically-significant differences in observed LQT2 probability between the datasets (Figure 6B). Though all datasets have biases, adding heterozygous individuals from gnomAD to the available literature yields LQT2 probability estimates more consistent with the cohort (Tables S2 and Figures S8–S9) and we therefore suggest the combined datasets produce the most accurate, though flawed, estimate of variant-specific LQT2 probability.
Figure 6.
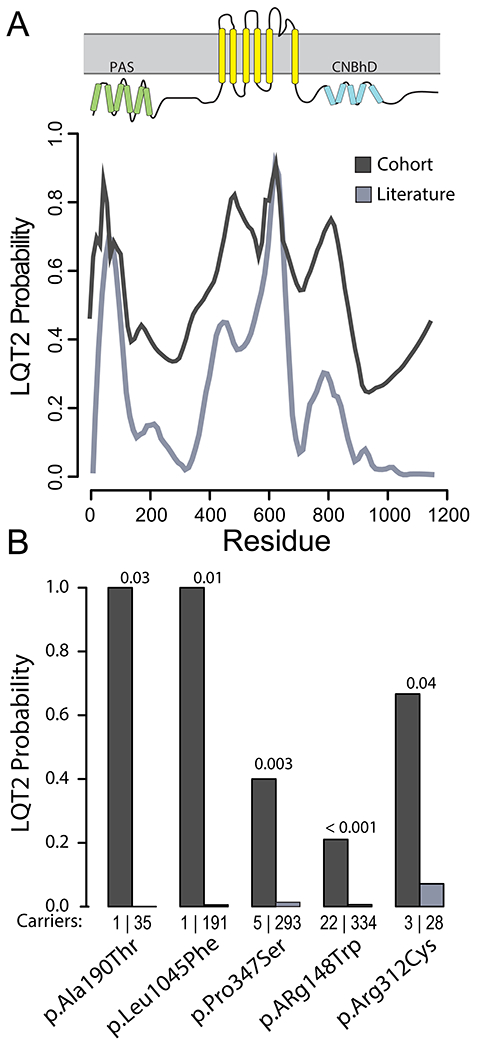
Ascertainment bias in LQT2 probability for KCNH2 variants from the arrhythmia center cohort compared to the literature. (A) The 30-residue moving average of LQT2 probability for each amino acid over the entire KV11.1 channel for data acquired from the cohort (dark gray) and collected from the literature (light gray) (see Figure S4 for the distribution of variants used in the calculation). Ascertainment bias is evident by the higher overall LQT2 probability in the cohort dataset compared to the literature (which includes gnomAD). (B) Observed probability (literature and the cohort) for a selected set of KCNH2 variants, p-values from Fisher’s exact test between the observations of heterozygotes in the literature and in the cohort. The observed probability for many variants from the cohort is significantly higher than that calculated from the literature. The number of heterozygous carriers discovered in each group is shown directly above the variant names.
Evidence that some variants classified as Benign increase the probability of LQT2 to higher than the general population rate.
Similar to KCNH2 p.Arg176Trp, variants such as p.Pro347Ser (c.1039C>T), p.Arg148Trp (c.442C>T), p.Ala913Val (c.2738C>T), and p.Arg328Cys (c.982C>T) were previously associated with LQT2,35–39 but are also more common in the general population than is expected for highly penetrant variants.18 This trend, observed in several variants which also produce a functional perturbation consistent with LQTS, has prompted some to use the label “LQT-lite”.40, 41 We also observed these variants in affected individuals in the cohort (2 out of 5, 4 out of 19, 1 out of 4, and 1 out of 2 heterozygotes, respectively). The most recent classifications for these variants in ClinVar are Benign or Likely benign. However, we estimate the probability of LQTS for heterozygous carriers of these variants at around 2% or higher, much greater than the ~0.04% in the general population. These data suggest some variants classified as Benign or Likely Benign are truly disease-causing for a small fraction of patients.
Application of Bayesian Probability to Arrhythmia Genetics Clinics.
Bayesian reasoning has long been at the core of clinical diagnosis. Given that the majority of heterozygous carriers of rare KCNH2 variants found in arrhythmia genetics clinics (or elsewhere) carry ultra-rare or novel variants, we anticipate that this prior, trained on variant-specific features, will have direct clinical utility to the clinician. The addition of 10 equivalent observations for a previously unreported or seldomly reported variant can directly guide clinical management and establish a threshold of intervention with drug treatment or simple clinical observation. Additionally, this method helps overcome ascertainment bias prevalent in clinically-obtained data since the prior is trained on variant-specific features agnostic to clinical information (Figures 5 and 6). While there is no intention to replace clinical phenotyping, we do anticipate the ability to augment clinical reasoning through a more accurate prior when combining clinical and population features with variant-specific features.
As an example, p.Pro347Ser, a variant with an observed 40% penetrance in the clinical cohort, would likely result in treatment intervention if clinically encountered by a clinician familiar with the variant through families seen in their clinic (Figure 6). However, from variant-specific features, our analysis generated a prior for p.Pro347Ser equivalent to observing only 1 in 10 heterozygous individuals diagnosed with LQT2. This new information could permit a more flexible approach to workup if no other information were known. A relatively high number of observations of p.Pro347Ser in the literature and gnomAD, which also suggest a LQT2 probability/penetrance of less than 5% for this variant (Figure 6), helps illustrate the calibration of our variant-informed priors. In this way, joint clinical phenotyping and tool utilization can be used in a mutually beneficial way for patients heterozygous for rare variants. We have developed an online searchable tool, the KCNH2 Variant Browser (https://variantbrowser.org/KCNH2/), to allow rapid access to the estimated penetrance based on variant-specific features.
Limitations
One limitation is the bias inherent to each of the data sources used. We may be able to observe more carriers of these KCNH2 variants as greater numbers of individuals are exome sequenced; however, for many variants, we may never observe more carriers and will be underpowered to estimate LQT2 probability by observation of heterozygotes alone. This fact is further motivation to establish a framework where experimental data is included quantitatively in the estimate of disease probability. The availability of functional data is also biased in that most variants which have these data available are from variants discovered in individuals presenting with a phenotype (Figure S14), however, high-throughput variant functional characterization has the potential to overcome this bias.42 Additionally, many factors influence the ultimate presentation of LQT2 in an individual, including genetic and environmental factors,4, 43, 44 though we did not observe significant differences in predictive performance across nationality (Table S3). Although the largest effect sizes for LQTS-associated variants come from rare variants, some of the variability in LQTS presentation can be explained by variability in common variants. Recently, two publications by Lahrouchi et al.45 and Turkowski et al.46 concluded polygenic risk scores accounted for ~15% and < 2% of the variability in LQTS susceptibility. Though in either case the contribution is relatively small, it is possible polygenic risk is potentiated in the rare variant context and could therefore explain a greater portion of the variability in disease presentation. Lastly, though beyond the scope of the present study, we envision this method will enable improved prognostication of more severe presentations of LQT2 including arrhythmic events. Future work will address these exciting possibilities.
Conclusions
We have shown how variant-specific features can inform a prior probability of disease for rare variants even in the absence of clinically phenotyped heterozygotes. We have demonstrated this framework on the classical Mendelian disease-gene pair, LQT2 and KCNH2. We exploit in vitro functional studies, LQT2 probability density, and broad in silico predictors to calculate these priors. We then combine these estimates with patient data to form the post-test probability of disease for each variant. We have demonstrated that these in vitro and in silico variant features are equivalent to approximately 10 clinically characterized heterozygotes when used to understand KCNH2 variant-specific LQT2 disease probability. Presenting these data in this way allows us to encode both the probability of disease and the uncertainty in our estimates: we do not claim to have as much certainty as you would have if you phenotyped 100 heterozygotes; however, we do claim greater certainty than a single observation of a phenotyped heterozygote.
Supplementary Material
Sources of Funding:
Funding: This work was supported by the National Institutes of Health grant number R00HL135442 (BMK), the American Heart Association training grant 16FTF30130005 (JDM) and the Leducq Foundation for Cardiovascular Research grant 18CVD05 “Towards Precision Medicine with Human iPSCs for Cardiac Channelopathies” (CLE, LC, PJS, BCK, and BMK), National Institutes of Health grant number R35HL144980 (BCK), R01 HL149826 (DMR), K99 HG010904 (AMG), and Marie Skłodowska-Curie Individual Fellowship (H2020-MSCA-IF-2017; Grant Agreement No. 795209, LS).
Nonstandard Abbreviations and Acronyms:
- LQT2
long QT syndrome type 2
- ACMG
The American College of Medical Genetics and Genomics
- PPV
positive predictive value
- ECG
electrocardiogram
- indel
in-frame insertion/deletion
- MAF
minor allele frequency
- PAS
Per-Arnt-Sim
- CNBhD
domain and C-terminal cyclic nucleotide binding homology domain
- AUC
area under a Receiver Operating Characteristic curve
- WT
wildtype
Footnotes
Disclosures: None
References:
- 1.Tennessen JA, Bigham AW, O’Connor TD, Fu WQ, Kenny EE, Gravel S, McGee S, Do R, Liu XM, Jun G, et al. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science. 2012;337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O’Dushlaine C, Van Hout CV, Staples J, Gonzaga-Jauregui C, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR Study. Science. 2016;354. [DOI] [PubMed] [Google Scholar]
- 3.Lacaze P, Sebra R, Riaz M, Tiller J, Revote J, Phung J, Parker EJ, Orchard SG, Lockery JE, Wolfe R, et al. Medically actionable pathogenic variants in a population of 13,131 healthy elderly individuals. Genet Med. 2020;22:1883–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Crotti L, George AL, Jr. Modifier genes for sudden cardiac death. Eur Heart J. 2018;39:3925–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, Biesecker LG; ClinGen Sequence Variant Interpretation Working G. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–7. [DOI] [PubMed] [Google Scholar]
- 8.Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002;3:779–89. [DOI] [PubMed] [Google Scholar]
- 9.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, Jamal SM, Szybowska M, Morel CF, Bowdin S, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation. 2018;138:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroncke BM, Smith DK, Zuo Y, Glazer AM, Roden DM, Blume JD. A Bayesian method to estimate variant-induced disease penetrance. PLoS Genet. 2020;16:e1008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrin MJ, Subbiah RN, Vandenberg JI, Hill AP. Human ether-a-go-go related gene (hERG) K+ channels: function and dysfunction. Prog Biophys Mol Biol. 2008;98:137–48. [DOI] [PubMed] [Google Scholar]
- 13.Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol Rev. 2017;97:89–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh R, Lahrouchi N, Tadros R, Kyndt F, Glinge C, Postema PG, Amin AS, Nannenberg EA, Ware JS, Whiffin N, et al. Enhancing rare variant interpretation in inherited arrhythmias through quantitative analysis of consortium disease cohorts and population controls. Genet Med. 2021;23:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL Jr., et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–8. [DOI] [PubMed] [Google Scholar]
- 18.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fodstad H, Bendahhou S, Rougier JS, Laitinen-Forsblom PJ, Barhanin J, Abriel H, Schild L, Kontula K, Swan H. Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann Med. 2006;38:294–304. [DOI] [PubMed] [Google Scholar]
- 20.Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen-Mattila M, Kerkela E, Hyttinen J, Kontula K, Swan H, Conklin BR, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5:220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fodstad H, Swan H, Laitinen P, Piippo K, Paavonen K, Viitasalo M, Toivonen L, Kontula K. Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann Med. 2004;36Suppl 1:53–63. [DOI] [PubMed] [Google Scholar]
- 22.Cui J, Kagan A, Qin D, Mathew J, Melman YF, McDonald TV. Analysis of the cyclic nucleotide binding domain of the HERG potassium channel and interactions with KCNE2. J Biol Chem. 2001;276:17244–51. [DOI] [PubMed] [Google Scholar]
- 23.Benhorin J, Moss AJ, Bak M, Zareba W, Kaufman ES, Kerem B, Towbin JA, Priori S, Kass RS, Attali B, et al. Variable expression of long QT syndrome among gene carriers from families with five different HERG mutations. Ann Noninvasive Electrocardiol. 2002;7:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satler CA, Walsh EP, Vesely MR, Plummer MH, Ginsburg GS, Jacob HJ. Novel missense mutation in the cyclic nucleotide-binding domain of HERG causes long QT syndrome. Am J Med Genet. 1996;65:27–35. [DOI] [PubMed] [Google Scholar]
- 25.Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci U S A. 1996;93:2208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–73. [DOI] [PubMed] [Google Scholar]
- 27.Mehta A, Sequiera GL, Ramachandra CJ, Sudibyo Y, Chung Y, Sheng J, Wong KY, Tan TH, Wong P, Liew R, et al. Re-trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovasc Res. 2014;102:497–506. [DOI] [PubMed] [Google Scholar]
- 28.Li G, Shi R, Wu J, Han W, Zhang A, Cheng G, Xue X, Sun C. Association of the hERG mutation with long-QT syndrome type 2, syncope and epilepsy. Mol Med Rep. 2016;13:2467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimizu W, Moss AJ, Wilde AA, Towbin JA, Ackerman MJ, January CT, Tester DJ, Zareba W, Robinson JL, Qi M, et al. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol. 2009;54:2052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W, MacKinnon R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell. 2017;169:422–430 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright CF, West B, Tuke M, Jones SE, Patel K, Laver TW, Beaumont RN, Tyrrell J, Wood AR, Frayling TM, et al. Assessing the Pathogenicity, Penetrance, and Expressivity of Putative Disease-Causing Variants in a Population Setting. Am J Hum Genet. 2019;104:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minikel EV, Zerr I, Collins SJ, Ponto C, Boyd A, Klug G, Karch A, Kenny J, Collinge J, Takada LT, et al. Ascertainment bias causes false signal of anticipation in genetic prion disease. Am J Hum Genet. 2014;95:371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marjamaa A, Salomaa V, Newton-Cheh C, Porthan K, Reunanen A, Karanko H, Jula A, Lahermo P, Vaananen H, Toivonen L, et al. High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann Med. 2009;41:234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christiansen M, Hedley PL, Theilade J, Stoevring B, Leren TP, Eschen O, Sorensen KM, Tybjaerg-Hansen A, Ousager LB, Pedersen LN, et al. Mutations in Danish patients with long QT syndrome and the identification of a large founder family with p.F29L in KCNH2. BMC Med Genet. 2014;15:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–17. [DOI] [PubMed] [Google Scholar]
- 37.Mechakra A, Vincent Y, Chevalier P, Millat G, Ficker E, Jastrzebski M, Poulin H, Pouliot V, Chahine M, Christe G. The variant hERG/R148W associated with LQTS is a mutation that reduces current density on co-expression with the WT. Gene. 2014;536:348–56. [DOI] [PubMed] [Google Scholar]
- 38.Saenen JB, Paulussen AD, Jongbloed RJ, Marcelis CL, Gilissen RA, Aerssens J, Snyders DJ, Raes AL. A single hERG mutation underlying a spectrum of acquired and congenital long QT syndrome phenotypes. J Mol Cell Cardiol. 2007;43:63–72. [DOI] [PubMed] [Google Scholar]
- 39.Millat G, Chevalier P, Restier-Miron L, Da Costa A, Bouvagnet P, Kugener B, Fayol L, Gonzalez Armengod C, Oddou B, Chanavat V, et al. Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome. Clin Genet. 2006;70:214–27. [DOI] [PubMed] [Google Scholar]
- 40.Lane CM, Giudicessi JR, Ye D, Tester DJ, Rohatgi RK, Bos JM, Ackerman MJ. Long QT syndrome type 5-Lite: Defining the clinical phenotype associated with the potentially proarrhythmic p.Asp85Asn-KCNE1 common genetic variant. Heart Rhythm. 2018;15:1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giudicessi JR, Roden DM, Wilde AAM, Ackerman MJ. Classification and Reporting of Potentially Proarrhythmic Common Genetic Variation in Long QT Syndrome Genetic Testing. Circulation. 2018;137:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozek KA, Glazer AM, Ng CA, Blackwell D, Egly CL, Vanags LR, Blair M, Mitchell D, Matreyek KA, Fowler DM, et al. High-throughput discovery of trafficking-deficient variants in the cardiac potassium channel KV11.1. Heart Rhythm. 2020;17:2180–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castel SE, Cervera A, Mohammadi P, Aguet F, Reverter F, Wolman A, Guigo R, Iossifov I, Vasileva A, Lappalainen T. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat Genet. 2018;50:1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lahrouchi N, Tadros R, Crotti L, Mizusawa Y, Postema PG, Beekman L, Walsh R, Hasegawa K, Barc J, Ernsting M, et al. Transethnic Genome-Wide Association Study Provides Insights in the Genetic Architecture and Heritability of Long QT Syndrome. Circulation. 2020;142:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turkowski KL, Dotzler SM, Tester DJ, Giudicessi JR, Bos JM, Speziale AD, Vollenweider JM, Ackerman MJ. The QTc-Polygenic Risk Score (QTc-PRS) and its Contribution to Type 1, Type 2, and Type 3 Long QT Syndrome in Probands and Genotype-Positive Family Members. Circ Genom Precis Med. 2020;13:e002922. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–4. [DOI] [PubMed] [Google Scholar]
- 50.Kroncke BM, Mendenhall J, Smith DK, Sanders CR, Capra JA, George AL, Blume JD, Meiler J, Roden DM. Protein structure aids predicting functional perturbation of missense variants in SCN5A and KCNQ1. Comput Struct Biotechnol J. 2019;17:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.