

Abstract
We disclose a nickel-catalyzed reaction, which enabled us to difunctionalize unactivated γ,δ-alkenes in ketones with alkenyl triflates and arylboronic esters. The reaction was made feasible by the use of 5-chloro-8-hydroxyquinoline as a ligand along with NiBr2•DME as a catalyst and LiOtBu as base. The reaction proceeded with a wide range of cyclic, acyclic, endocyclic and exocyclic alkenyl ketones, and electron-rich and electron-deficient arylboronate esters. The reaction also worked with both cyclic and acyclic alkenyl triflates. Control experiments indicate that carbonyl coordination is required for the reaction to proceed.
Keywords: Alkenyl ketone, alkenylarylation, dicarbofunctionalization, nickel-catalyzed, 8-hydroxyquinoline
Graphical Abstract
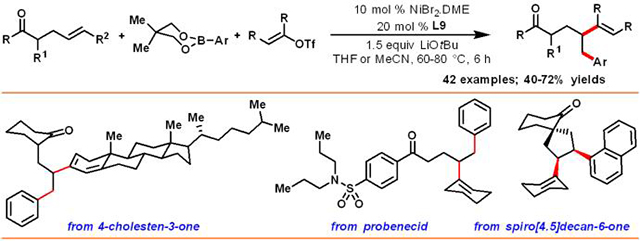
A nickel-catalyzed alkenylarylation reaction of γ,δ-alkenylketones with alkenyl triflates and arylboronic esters is described. The reaction was made feasible by the use of 5-chloro-8-hydroxyquinoline as a ligand along with NiBr2•DME as a catalyst and LiOtBu as base, and works with cyclic, acyclic, endocyclic and exocyclic alkenyl ketones. Control experiments indicate that carbonyl coordination is required for the reaction to proceed.
Metal-catalyzed alkene dicarbofunctionalization[1] is an emerging synthetic method for complex molecule synthesis[2] with a promise to reduce a multistep process to a few-step process.[3] This reaction integrates two carbon sources into alkenes with high regiofidelity, and provides branched molecules in one step.[4] However, its development has been met with formidable challenges largely due to the formation of Heck products by β-H elimination (Scheme 1).[5] The reaction selectivity toward alkene difunctionalization and the Heck product depend on the difference in kinetic barriers between a single-step β-H elimination and a two-step transmetallation/reductive elimination process. In the majority of catalytic processes, β-H elimination has been shown to occur with a much lower kinetic barrier than both the transmetallation and reductive elimination steps.[6]
Scheme 1.

Alkene 1,2-dicarbofunctionalization and complications by β-H elimination
Early work focused on difunctionalizing 1,1-disubstituted terminal[7] and bicyclic[8] alkenes to avoid β-H elimination. Recently, a broad class of substrates were functionalized using a coordination approach[1a–f] in which alkylmetal intermediates were stabilized as transient metallacycles.[9] However, this strategy requires installation and removal of strong coordinating groups like imines,[10] pyridines,[11] 2-aminopyrimidines,[12] and 8-aminoquinolines[13] for stabilizing metallacycles.[14] Additionally, this strategy generally works with substrates that generate rigid and planar five-membered metallacycles. Limited examples of reactions involving six-membered metallacycles are known but they either require strongly coordinating bidentate groups[15] or are derived from vinylarenes containing activated alkenes[10] to planarize and rigidify metallacycles. Alkene difunctionalization reactions that involve nonplanar and fluxional six-membered metallacycles form rearranged products through contraction of metallacycles by β-H elimination.[5a]
We recently introduced a cationic catalysis[16] to suppress the contraction of metallacycles. While the reaction was successful, the cationization of organometallic intermediates is also known to promote β-H elimination.[6a, 6b, 17] As such, when weakly coordinating groups are used, the cationization of metallacycles promotes β-H elimination faster than the transmetallation/reductive elimination steps.[5b] For that reason, regioselective alkene dicarbofunctionalization in weakly coordinating ketone substrates are not known.[18] Herein, we disclose a new Ni-catalyzed reaction in the presence of 5-chloro-8-hydroxyquinoline that addresses the issue of β-H elimination, and materializes alkenylarylation of unactivated γ,δ-alkenes in simple ketones. To the best of our knowledge, the current reaction represents the first example of keto carbonyl-assisted dicarbofunctionalization of unactivated alkenes.[19]
In our initial studies, we examined various ancillary ligands (L1-L7) for alkenylarylation of γ,δ-alkene in 2-allylcyclohexanone (Scheme 2). We were pleased to discover that a combination of 20 mol % 8-hydroxyquinoline (L7) and 10 mol % NiBr2•DME, along with LiOtBu as a base, generated the alkenylarylation product 3 with cyclohexenyl triflate and phenylboronic ester B3 in 48% yield. Control experiments conducted in the absence of L7 and in the presence of L8, a neutral variant of L7, produced no product, confirming that the anionic ancillary ligand played a critical role in the reaction. Upon further evaluation (L9-L12), we found that 5-chloro-8-hydroxyquinoline (L9) performed optimally and furnished the product 3 in 75% yield. Examination of different arylboron reagents showed that phenylboronic ester B3 afforded the product 3 in best yield. Replacing 2-allylcyclohexanone with 2-vinyl- and 2-butenylketones also did not furnish expected products (Scheme 2).
Scheme 2.

Examination of reaction parametersa
aReactions run at 0.10 mmol scale in 0.5 mL THF. 1H NMR yields with pyrene as a standard. bThe yield of isolated product from a 0.50 mmol scale reaction in 2.5 mL THF in parenthesis. dr is 1.1:1.
Examination of different alkoxide bases indicated that the reaction was best performed with LiOtBu or LiOAd (Table 1, entries 1–6). NiBr2•DME could be replaced with NiCl2•DME or NiI2 (entry 7). Other Ni-catalysts furnished the product in lower yields (entries 8–10) and other metal-catalysts such as FeCl2, Co(OAc)2, CuI and Pd(OAc)2 generated no product at all (entry 11). The reaction was optimally performed at 80 °C in THF since other solvents and lower temperatures either decreased the yield or formed no product (entries 12–17).
Table 1.
Further reaction optimizationa
| ||
|---|---|---|
|
| ||
| entry | deviation in reaction condition | yields of 3 (%)b |
|
| ||
| 1 | none | 75 (72)c |
| 2 | Lithium-1-adamantanolate (LiOAd) instead of LiOtBu | 75 |
| 3 | LiOMe instead of LiOtBu | 23 |
| 4 | LiOiPr instead of LiOtBu | 15 |
| 5 | 1 equiv LiOtBu instead of 1.5 equiv | 42 |
| 6 | 2 equiv LiOtBu instead of 1.5 equiv | 65 |
| 7 | NiI2 or NiCl2·DME instead of NiBr2.DME | 70–72 |
| 8 | Ni(TMHD)2 instead of NiBr2·DME | 53 |
| 9 | Ni(PPh3)4 instead of NiBr2·DME | 12 |
| 10 | Ni(cod)2 instead of NiBr2·DME | 52 |
| 11 | other metals instead of NiBr2·DMEd | 0 |
| 12 | room temperature instead of 80 °C | 10 |
| 13 | 60 °C instead of 80 °C | 62 |
| 14 | 2-Me-THF instead of THF | 70 |
| 15 | 1,4-dioxane instead of THF | 60 |
| 16 | MeCN or toluene instead of THF | 35–38 |
| 17 | NMP, DMF, DMSO or dioxane instead of THF | 0–25 |
Reactions run at 0.10 mmol scale in 0.5 mL solvent.
1H NMR yields with pyrene as a standard.
The yield of isolated product from a 0.50 mmol scale reaction in 2.5 mL THF in parenthesis. dr is 1.1:1.
Pd(OAc)2, CuI, Co(OAc)2, FeCl2.
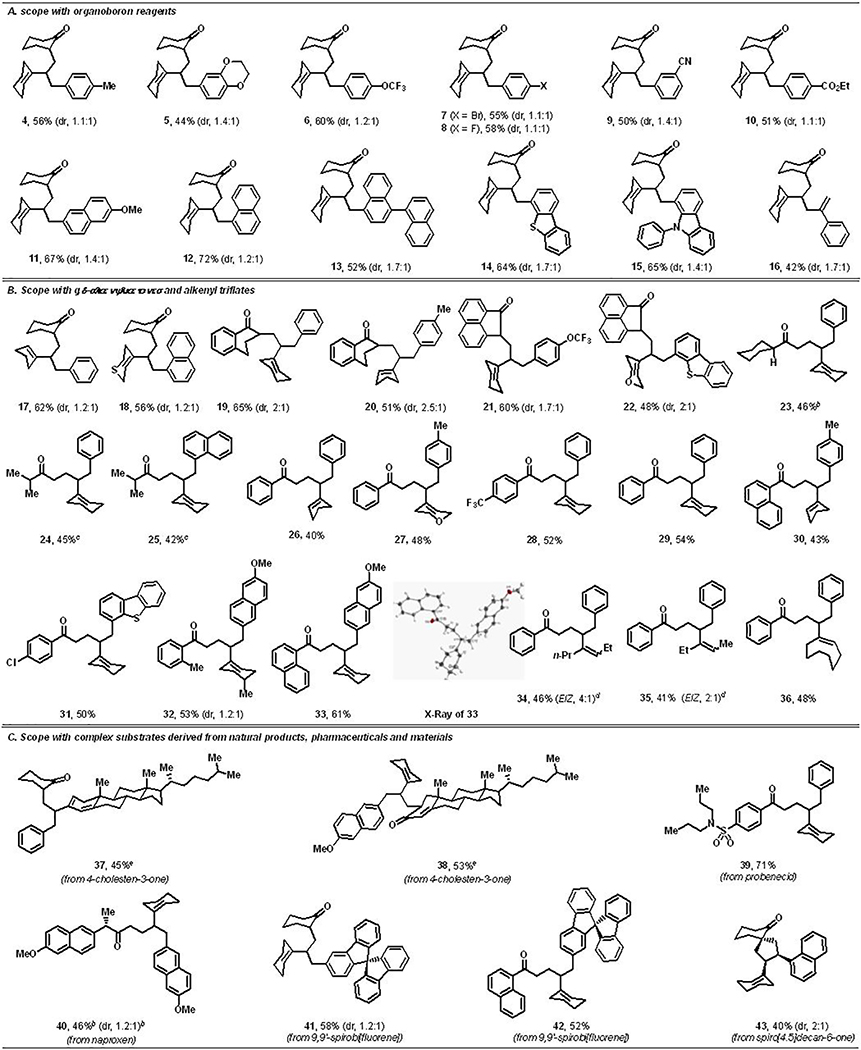
After optimization of the reaction conditions, we explored the scope of the alkenylarylation reaction. Examination of different arylboronic esters indicated that the reaction tolerated both electron-rich and electron-deficient arenes, and the products were formed in good yields with moderate diastereoselectivity (Table 2, A). The reaction was compatible with various substituents like Me, dioxanyl, OMe, OCF3 and F, and transition metal-sensitive functional groups such as Br, nitrile and ester (4-11). This method can be applied to couple arylboronic esters having polyaromatic hydrocarbons including naphthyl, methoxynaphthyl and binaphthyl (11-13). The reaction also tolerates heterocyclic arylboronic esters, such as thiazolyl and carbozolylboronic esters (14-15), and alkenylboronic esters (16).
Table 2.
Reaction scopea
|
Reactions run in 0.50 mmol scale in 2.5 mL solvent. Reactions for alkylketones (products 4–22, 34–36) run in THF at 80 °C while those for arylketones (products 26–33, 39 and 42) run in MeCN 60 °C for 6 h.
Reaction run under MeCN conditions.
Reactions run under THF conditions at 60 °C for 6 h.
20 mol % NiBr2·DME. Alkenyl triflate E/Z: 2:1.
A mixture of 4 diastereomers based on 13C NMR (see SI for details).
Moreover, we evaluated reaction scope with regard to γ,δ-alkenyl ketones and alkenyl triflates (Table 2, B). Various γ,δ-alkenyl endocyclic ketones containing both monocyclic and bicyclic scaffolds, such as cyclohexenonyl, tetralonyl and acenaphthylenonyl, could be implemented as substrates along with different alkenyl triflates and arylboronic esters (17-22). The reaction could also be performed with γ,δ-alkenyl exocyclic ketones bearing both cyclic and acyclic aliphatic backbones such as cyclohexyl and isopropyl (23-25). Surprisingly, γ,δ-alkenyl arylketones furnished products in low yields when the reactions were conducted in THF (20–30%). Unlike γ,δ-alkenyl alkylketones for which THF was the solvent of choice at 80 °C, γ,δ-alkenyl arylketones furnished products in best yields in MeCN at 60 °C. γ,δ-Alkenyl arylketones with p-CF3, o-Me and p-Cl substituents and a naphthyl group afforded dicarbofunctionalized products in good yields with different alkenyl triflates and arylboronic esters (26-33). Reactions of these diverse γ,δ-alkenyl ketones could be conducted with carbocyclic alkenyl triflates containing cyclopentenyl, cyclohexenyl, cyclooctenyl and 4-methylcyclohexenyl rings, and heterocyclic alkenyl triflates, such as 3,6-dihydro-2H-thiopyran-4-yl (18) and 3,6-dihydro-2H-pyran-4-yl triflates (22 and 27). The reaction condition was also amenable for difunctionalizing acyclic alkenyl triflates (34-35). These types of product derivatives form the cores of pharmaceutical molecules and natural products.[20]
We have further demonstrated synthetic applications by difunctionalizing unactivated alkenes in natural products, pharmaceuticals and complex spirocyclic molecules, and by using alkenyl triflates and arylboronic esters derived from complex molecules (Table 2, C). For example, an alkenyl triflate derived from a steroid, cholestenone, was incorporated into γ,δ-alkene in cyclohexanone (37). In addition, γ,δ-alkenes contained in complex molecules such as cholestenone, probenecid (a uricosuric drug) and naproxen (a NSAID drug) could also be difunctionalized with cyclohexenyl triflate and various arylboronic esters (38-40). Arylboronic ester derived from 9,9’-spirobifluorene also afforded products (41-42) with alkenylcyclohexanone and alkenylarylketone. Furthermore, we examined an internal γ,δ-alkene contained in a spirobicyclic ketone, which reacted to furnish a cis-difunctionalized product (43).
Control experiments with an alkene lacking carbonyl group support carbonyl coordination during reaction (Scheme 3). Moreover, alkenyl ketones bearing less sterically bulky substituents on the carbonyl group predominantly produced alkenylarylated products 24 and 29 along with trace amounts of Heck products 44 and 45 (Scheme 4). In contrast, a sterically demanding tert-butyl group on the carbonyl group generated the Heck product 47 in significant amounts (22%) in addition to the alkenylarylation product 46 in 35% yield. These experiments indicate that carbonyl coordination to nickel is required to generate alkenylarylated products and that the Heck products are generated due to equilibrium between the cyclic and acyclic alkylnickel species 48 and 49 (Scheme 4).[21] Intimate analyses of regioselectivity in products indicated that the reaction should be catalyzed by a Ni(I) catalyst and transmetallation should precede the reaction of alkenyl triflates (see SI for details).[22]
Scheme 3.

Carbonyl coordination effect
Scheme 4.

Carbonyl steric effect
In summary, we report a nickel-catalyzed reaction, which addresses the issue of β-H elimination in alkene difunctionalization in carbonyl-assisted alkene dicarbofunctionalization reaction. The success of the reaction relied upon the use of a combination of 5-chloro-8-hydroxyquinoline and Ni(cod)2 in the presence of LiOtBu. This catalysis enabled us to difunctionalize unactivated γ,δ-alkenes in ketones with alkenyl triflates and arylboronic esters. The reaction proceeded with a wide range of cyclic, acyclic, endocyclic and exocyclic alkenyl ketones along with electron-rich and electron-deficient arylboronate esters, and cyclic and acyclic alkenyl triflates. Control experiments with a substrate lacking a carbonyl group and with a sterically hindered carbonyl group indicated that the carbonyl coordination was required for the reaction to proceed.
Supplementary Material
Acknowledgements
We gratefully acknowledge the NIH NIGMS (R35GM133438) and The Pennsylvania State University for support of this work, and the PSU NMR facility for NMR support. We thank Dr. Christy George for help with stereochemistry determination of compound 43 by NMR.
Footnotes
Supporting information for this article and crystallographic data (CCDC number: 2059802) for compound 33 are given via a link at the end of the document.
References
- [1].For reviews, see:Dhungana RK, KC S, Basnet P, Giri R, Chem. Rec. 2018, 18, 1314–1340;Giri R, Kc S, J. Org. Chem. 2018, 83, 3013–3022;Derosa J, Apolinar O, Kang T, Tran VT, Engle KM, Chem. Sci. 2020, 11, 4287–4296;Badir SO, Molander GA, Chem 2020, 6, 1327–1339;Tu H-Y, Zhu S, Qing F-L, Chu L, Synthesis 2020, 52, 1346–1356;Qi X, Diao T, ACS Catal. 2020, 10, 8542–8556;Herath A, Li W, Montgomery J, J. Am. Chem. Soc. 2008, 130, 469–471;Montgomery J, Acc. Chem. Res. 2000, 33, 467–473.
- [2].Nicolaou KC, Rigol S, Nat. Prod. Rep. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3] a).Chemler SR, Karyakarte SD, Khoder ZM, J. Org. Chem. 2017, 82, 11311–11325; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wolfe JP, Synlett 2008, 2008, 2913–2937; [Google Scholar]; c) Ahmed W, Zhang S, Yu X, Feng X, Yamamoto Y, Bao M, Angew. Chem. Int. Ed. 2019, 58, 2495–2499; [DOI] [PubMed] [Google Scholar]; d) Peng J-B, Wu F-P, Li D, Geng H-Q, Qi X, Ying J, Wu X-F, ACS Catal. 2019, 9, 2977–2983. [Google Scholar]
- [4] a).Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS, Science 2016, 352, 801–805; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang L, Lovinger GJ, Edelstein EK, Szymaniak AA, Chierchia MP, Morken JP, Science 2016, 351, 70–74; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nakamura M, Hirai A, Nakamura E, J. Am. Chem. Soc. 2000, 122, 978–979; [Google Scholar]; d) Neufeldt SR, Sanford MS, Org. Lett. 2013, 15, 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For selected examples on three-component alkene dicarbofunctionalization, see:Basnet P, Dhungana RK, Thapa S, Shrestha B, Kc S, Sears JM, Giri R, J. Am. Chem. Soc. 2018, 140, 7782–7786;Dhungana RK, Kc S, Basnet P, Aryal V, Chesley LJ, Giri R, ACS Catal. 2019, 9, 10887–10893;Jeon J, Ryu H, Lee C, Cho D, Baik M-H, Hong S, J. Am. Chem. Soc. 2019, 141, 10048–10059;Liao L, Jana R, Urkalan KB, Sigman MS, J. Am. Chem. Soc. 2011, 133, 5784–5787;Saini V, Sigman MS, J. Am. Chem. Soc. 2012, 134, 11372–11375;Orlandi M, Hilton MJ, Yamamoto E, Toste FD, Sigman MS, J. Am. Chem. Soc. 2017, 139, 12688–12695;Huang H-M, Bellotti P, Pflüger PM, Schwarz JL, Heidrich B, Glorius F, J. Am. Chem. Soc. 2020, 142, 10173–10183;Li Y, Wei H, Wu D, Li Z, Wang W, Yin G, ACS Catal. 2020, 10, 4888–4894.
- [6] a).Menezes da Silva VH, Braga AAC, Cundari TR, Organometallics 2016, 35, 3170–3181; [Google Scholar]; b) Campbell AN, Gagné MR, Organometallics 2007, 26, 2788–2790; [Google Scholar]; c) Veerakumar P, Thanasekaran P, Lu K-L, Lin K-C, Rajagopal S, ACS Sustain. Chem. Eng. 2017, 5, 8475–8490. [Google Scholar]
- [7].For alkene dicarbofunctionalization by cyclization/coupling, see:Burns B, Grigg R, Ratananukul P, Sridharan V, Stevenson P, Sukirthalingam S, Worakun T, Tetrahedron Lett. 1988, 29, 5565–5568;Fretwell P, Grigg R, Sansano JM, Sridharan V, Sukirthalingam S, Wilson D, Redpath J, Tetrahedron 2000, 56, 7525–7539;Grigg R, Sansano J, Santhakumar V, Sridharan V, Thangavelanthum R, Thornton-Pett M, Wilson D, Tetrahedron 1997, 53, 11803–11826;Grigg R, Mariani E, Sridharan V, Tetrahedron Lett. 2001, 42, 8677–8680;Wilson JE, Tetrahedron Lett. 2012, 53, 2308–2311;Fan P, Lan Y, Zhang C, Wang C, J. Am. Chem. Soc. 2020, 142, 2180–2186;Jin Y, Wang C, Angew. Chem. Int. Ed. 2019, 58, 6722–6726;Wang K, Ding Z, Zhou Z, Kong W, J. Am. Chem. Soc. 2018, 140, 12364–12368;Zhou L, Li S, Xu B, Ji D, Wu L, Liu Y, Zhang Z-M, Zhang J, Angew. Chem. Int. Ed. 2020, 59, 2769–2775;j) Marchese AD, Larin EM, Mirabi B, Lautens M, Acc. Chem. Res. 2020, 53, 1605–1619.
- [8] a).Kosugi M, Tamura H, Sano H, Migita T, Tetrahedron 1989, 45, 961–967; [Google Scholar]; b) Masanori K, Tomoyuki K, Hiroshi O, Toshihiko M, Bull. Chem. Soc. Jpn. 1993, 66, 3522–3524; [Google Scholar]; c) Kosugi M, Tamura H, Sano H, Migita T, Chem. Lett. 1987, 16, 193–194; [Google Scholar]; d) Catellani M, Chiusoli GP, Concari S, Tetrahedron 1989, 45, 5263–5268; [Google Scholar]; e) Kang S-K, Kim J-S, Choi S-C, Lim K-H, Synthesis 1998, 1998, 1249–1251; [Google Scholar]; f) Shaulis KM, Hoskin BL, Townsend JR, Goodson FE, Incarvito CD, Rheingold AL, J. Org. Chem. 2002, 67, 5860–5863; [DOI] [PubMed] [Google Scholar]; g) Kadam AA, Metz TL, Qian Y, Stanley LM, ACS Catal. 2019, 9, 5651–5656. [Google Scholar]
- [9].For examples of alkene dicarbofunctionalization without a coordinating group, see:García-Domínguez A, Li Z, Nevado C, J. Am. Chem. Soc. 2017, 139, 6835–6838;Gao P, Chen L-A, Brown MK, J. Am. Chem. Soc. 2018, 140, 10653–10657;Kc S, Dhungana RK, Shrestha B, Thapa S, Khanal N, Basnet P, Lebrun RW, Giri R, J. Am. Chem. Soc. 2018, 140, 9801–9805;Liu L, Lee W, Youshaw CR, Yuan M, Geherty MB, Zavalij PY, Gutierrez O, Chem. Sci. 2020, 11, 8301–8305;Anthony D, Lin Q, Baudet J, Diao T, Angew. Chem. Int. Ed. 2019, 58, 3198–3202;Campbell MW, Compton JS, Kelly CB, Molander GA, J. Am. Chem. Soc. 2019, 141, 20069–20078;Zhang W, Lin S, J. Am. Chem. Soc. 2020, 142, 20661–20670;Jeon J, He Y-T, Shin S, Hong S, Angew. Chem. Int. Ed. 2020, 59, 281–285;Lee K, Lee S, Kim N, Kim S, Hong S, Angew. Chem. Int. Ed. 2020, 59, 13379–13384;j) Sakurai S, Matsumoto A, Kano T, Maruoka K, J. Am. Chem. Soc. 2020, 142, 19017–19022;Huang D, Olivieri D, Sun Y, Zhang P, Newhouse TR, J. Am. Chem. Soc. 2019, 141, 16249–16254;Yong X, Han Y-F, Li Y, Song R-J, Li J-H, Chem. Commun. 2018, 54, 12816–12819;Ouyang X-H, Hu M, Song R-J, Li J-H, Chem. Commun. 2018, 54, 12345–12348;Chierchia M, Xu P, Lovinger GJ, Morken JP, Angew. Chem. Int. Ed. 2019, 58, 14245–14249;Myhill JA, Wilhelmsen CA, Zhang L, Morken JP, J. Am. Chem. Soc. 2018, 140, 15181–15185;Ishii T, Ota K, Nagao K, Ohmiya H, J. Am. Chem. Soc. 2019, 141, 14073–14077;Wang X-G, Li Y, Liu H-C, Zhang B-S, Gou X-Y, Wang Q, Ma J-W, Liang Y-M, J. Am. Chem. Soc. 2019, 141, 13914–13922.
- [10].Shrestha B, Basnet P, Dhungana RK, Kc S, Thapa S, Sears JM, Giri R, J. Am. Chem. Soc. 2017, 139, 10653–10656. [DOI] [PubMed] [Google Scholar]
- [11].Thapa S, Dhungana RK, Magar RT, Shrestha B, Kc S, Giri R, Chem. Sci. 2018, 9, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li W, Boon JK, Zhao Y, Chem. Sci. 2018, 9, 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13] a).Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM, J. Am. Chem. Soc. 2017, 139, 10657–10660; [DOI] [PubMed] [Google Scholar]; b) Yang T, Jiang Y, Luo Y, Lim JJH, Lan Y, Koh MJ, J. Am. Chem. Soc. 2020, 142, 21410–21419; [DOI] [PubMed] [Google Scholar]; c) Bai Z, Bai Z, Song F, Wang H, Chen G, He G, ACS Catal. 2020, 10, 933–940. [Google Scholar]
- [14].For radical-related alkene dicarbofunctionalization and related reactions, see:Xu C, Cheng R, Luo Y-C, Wang M-K, Zhang X, Angew. Chem. Int. Ed. 2020, 59, 18741–18747;Gu J-W, Min Q-Q, Yu L-C, Zhang X, Angew. Chem. Int. Ed. 2016, 55, 12270–12274;Wang L, Wang C, Org. Lett. 2020, 22, 8829–8835;Apolinar O, Tran VT, Kim N, Schmidt MA, Derosa J, Engle KM, ACS Catal. 2020, 10, 14234–14239;Guo L, Yuan M, Zhang Y, Wang F, Zhu S, Gutierrez O, Chu L, J. Am. Chem. Soc. 2020, 142, 20390–20399;Semba K, Ohta N, Nakao Y, Org. Lett. 2019, 21, 4407–4410;Zheng S, Chen Z, Hu Y, Xi X, Liao Z, Li W, Yuan W, Angew. Chem. Int. Ed. 2020, 59, 17910–17916;Koy M, Bellotti P, Katzenburg F, Daniliuc CG, Glorius F, Angew. Chem. Int. Ed. 2020, 59, 2375–2379;Weires NA, Slutskyy Y, Overman LE, Angew. Chem. Int. Ed. 2019, 58, 8561–8565;j) Liu J, Wu S, Yu J, Lu C, Wu Z, Wu X, Xue X-S, Zhu C, Angew. Chem. Int. Ed. 2020, 59, 8195–8202;White DR, Hinds EM, Bornowski EC, Wolfe JP, Org. Lett. 2019, 21, 3813–3816;Bornowski EC, Hinds EM, White DR, Nakamura Y, Wolfe JP, Org. Process Res. Dev. 2019, 23, 1610–1630.
- [15] a).Derosa J, van der Puyl VA, Tran VT, Liu M, Engle Keary M., Chem. Sci. 2018, 9, 5278–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Basnet P, Kc S, Dhungana RK, Shrestha B, Boyle TJ, Giri R, J. Am. Chem. Soc. 2018, 140, 15586–15590. [DOI] [PubMed] [Google Scholar]
- [17].Chen L, Sanchez DR, Zhang B, Carrow BP, J. Am. Chem. Soc. 2017, 139, 12418–12421. [DOI] [PubMed] [Google Scholar]
- [18].For dicarbofunctionlization of carboxylates and carboxamides, see:Zhao X, Tu H-Y, Guo L, Zhu S, Qing F-L, Chu L, Nat. Commun. 2018, 9, 3488;Tu H-Y, Wang F, Huo L, Li Y, Zhu S, Zhao X, Li H, Qing F-L, Chu L, J. Am. Chem. Soc. 2020, 142, 9604–9611;Derosa J, Kang T, Tran VT, Wisniewski SR, Karunananda MK, Jankins TC, Xu KL, Engle KM, Angew. Chem. Int. Ed. 2020, 59, 1201–1205;Derosa J, Kleinmans R, Tran VT, Karunananda MK, Wisniewski SR, Eastgate MD, Engle KM, J. Am. Chem. Soc. 2018, 140, 17878–17883.
- [19].While our manuscript was under review, one example of ketone-directed alkene diarylation reaction appeared in a preprint. See: DOI: 10.26434/chemrxiv.14150174. [DOI] [Google Scholar]
- [20] a).Cini E, Banfi L, Barreca G, Carcone L, Malpezzi L, Manetti F, Marras G, Rasparini M, Riva R, Roseblade S, Russo A, Taddei M, Vitale R, Zanotti-Gerosa A, Org. Process Res. Dev. 2016, 20, 270–283; [Google Scholar]; b) Li Y-P, Yang Y-C, Li Y-K, Jiang Z-Y, Huang X-Z, Wang W-G, Gao X-M, Hu Q-F, Fitoterapia 2014, 95, 214–219. [DOI] [PubMed] [Google Scholar]
- [21].Quaternarization of α-C proximal to alkenes suppressed the reaction regardless of presence or absence of α-H in alkenylketones. Therefore, the role of α-H in the reaction could not be established unambiguously.
- [22].For mechanisms of Ni-catalyzed alkene difunctionalization reactions, see:Lin Q, Diao T, J. Am. Chem. Soc. 2019, 141, 17937–17948;Diccianni J, Lin Q, Diao T, Acc. Chem. Res. 2020, 53, 906–919. For general mechanisms of Ni-catalyzed C-C bond-forming reactions, see:Yuan M, Song Z, Badir SO, Molander GA, Gutierrez O, J. Am. Chem. Soc. 2020, 142, 7225–7234;Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P, Vicic DA, J. Am. Chem. Soc. 2006, 128, 13175–13183;Weix DJ, Acc. Chem. Res. 2015, 48, 1767–1775;Li Y, Luo Y, Peng L, Li Y, Zhao B, Wang W, Pang H, Deng Y, Bai R, Lan Y, Yin G, Nat. Commun. 2020, 11, 417.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.