

Abstract
Triple-negative breast cancer (TNBC) is the most aggressive breast cancer subtype with low survival rate and a lack of biomarkers and targeted treatments. Here we target pyruvate kinase M2 (PKM2), a key metabolic component of oncogenesis. In TNBC patients, PKM2pS37 was identified as a prominent phosphoprotein corresponding to the aggressive breast cancer phenotype that showed a characteristic nuclear staining pattern and prognostic value. Phosphorylation of PKM2 at S37 was connected with a cyclin-dependent kinase (CDK) pathway in TNBC cells. In parallel, pyruvate kinase activator TEPP-46 bound PKM2pS37 and reduced its nuclear localization. In a TNBC mouse xenograft model, treatment with either TEPP-46 or the potent CDK inhibitor Dinaciclib reduced tumor growth and diminished PKM2pS37. Combinations of Dinaciclib with TEPP-46 reduced cell invasion, impaired redox balance, and triggered cancer cell death. Collectively, these data support an approach to identify PKM2pS37-positive TNBC and target the PKM2 regulatory axis as a potential treatment.
Keywords: Pyruvate Kinase M2, phosphorylation, cyclin-depended kinases, cancer metabolism, triple-negative breast cancer, cancer biomarkers, cancer therapy
Introduction
Pyruvate kinase (PK) enzymes catalyze a key, high energy step in central carbon metabolism that is an essential control point for the generation of ATP in all cells. PKM2, a splice isoform of the normal muscle type PKM, has long been recognized as a unique biomarker in cancer and has been proposed to be the catalytic driver of the Warburg effect (1–3). According to this model, a switch to PKM2 isoform in cancer cells elevates aerobic metabolism which increases the consumption of glucose and the production of glycolytic intermediates and lactate (3,4). However, little is known on how oxidative phosphorylation (OXPHOS) is reprogrammed in this new metabolic state as higher pyruvate kinase amounts are anticipated to provide more pyruvate and thus increase the flux through TCA cycle. Accumulated evidence suggests that PKM2 isoform can potentially adopt altered catalytic mechanisms to support the demands of a new metabolic steady state that retains the flux through OXPHOS and at the same time builds up anabolic intermediates for rapid cell proliferation (5–7).
PKM2 phosphorylation has been linked with different regulatory pathways that coordinate proliferative and survival signaling during tumorigenesis (4,7–9). PKM2pS37 is the best studied phosphorylated form to date yet is missing critical mechanistic insight into its role in cancer. PKM2pS37 is proposed to increase dimer/tetramer ratio, lower PKM2 enzymatic activity, and dictate nuclear localization. However, each of these properties and their role in cancer needs further investigation (7,10–12). In addition, the role of specific kinases in these processes are only beginning to be defined (8). For instance, in glioblastomas activated ERK2 directly phosphorylates PKM2, and not PKM1, in position S37 (13). Interestingly, more recent observations directly link the cyclin-dependent kinase complex D3/CDK6 with PKM2pS37 in lymphomas, where it was found to be effectively reversed by a CDK4/6 antagonist, Palbociclib (10). This suggests that further studies on PKM2pS37 in more cancer types may better define prominent PKM2 signaling pathways that could improve cancer prognosis and dictate new therapies.
In addition to post-translational modifications, numerous metabolites can modulate PKM2 enzymatic properties in order to rewire cellular metabolic regimes (5,7,14). Fructose 1,6-bisphosphate (FBP), a glycolytic metabolite, is the most well-known physiologic PKM2 activator which stabilizes the active tetrameric enzyme (15–17). Recent studies, initially focused on PKM2 activity in cancer, discovered a class of small molecules that mimic FBP activation. The pyruvate kinase activator TEPP-46 was found to selectively bind PKM2 isoform and change its enzymatic activity (12,18–21). Preclinical studies have shown positive antitumor effects of TEPP-46 (12,22–24), therefore this small molecule could be ideal for further development of targeted therapies against PKM2 expressing cancers. However, the role of PKM2 phosphorylation and the actions of TEPP-46 remain to be explored in most cancer types.
Herein, we characterize PKM2pS37 in TNBC and connect it to cancer cell metabolism, tumor growth, and invasion. TNBC is the most aggressive breast cancer subtype. Lacking hormone receptors (HR) and human epidermal growth factor receptor 2 (HER2), TNBC cases are often not responsive to conventional breast cancer therapies. While cyclin-dependent kinase 4/6 (CDK4/6) inhibitors, such us Palbociclib, have been recently approved as first-line therapy in conjunction with estrogen receptor modulators or aromatase inhibitors for metastatic HR+/HER2− breast cancer, TNBC patients do not benefit from such combinations (25–27). Nevertheless, the novel CDK1/2/5/9 inhibitor, Dinaciclib (SCH727965) (28,29), is currently being investigated in combination with other specific agents as an alternative approach for metastatic TNBC (30,31). This strongly suggested that alternative CDK pathways could dictate PKM2 phosphorylation and activity in TNBC. We investigated this possibility and showed that PKM2pS37 could act as a prognostic biomarker for TNBC patients. We found that PKM2pS37 in TNBC cells was sensitive to Dinaciclib and that PKM2pS37 was directly targeted by TEPP-46 both in vitro and in vivo. Our work identifies PKM2pS37 as a prognostic indicator of TNBC outcomes with the potential to use CDK inhibitors and pyruvate kinase activators alone or in combination for immediate impact on TNBC patients.
Materials and Methods
Cell lines and tissues
All human breast and ovarian cancer cell lines were obtained from ATCC and cultured in the recommended medium. MCF-7 (Cat# HTB-22, RRID: CVCL_0031) were cultured in EMEM (ATCC) supplemented with 10% (v/v) FBS and 0.01 mg/mL bovine insulin (Sigma). MDA-MB-231 (Cat# HTB-26, RRID: CVCL_0062) were cultured in DMEM-high glucose (Gibco) supplemented with 10% (v/v) FBS. MDA-MB-468 (Cat# HTB-132, RRID: CVCL_0419), HCC1937 (Cat# CRL-2336, RRID: CVCL_0290), DU4475 (Cat# HTB-123, RRID: CVCL_1183) were cultured in RPM1–1640 (Gibco) supplemented with 10% (v/v) FBS and OVCAR-3 (Cat# HTB-161, RRID: CVCL_0465) ovarian cell line used the same medium supplemented with 0.01 mg/ml bovine insulin. INS-1 pancreatic insulinoma (Millipore, Cat# SCC208, RRID: CVCL_ZL55) and Jurkat T-cell leukemia (Invitrogen, Cat# R72207, RRID: CVCL_D588) cell lines were cultured in RPM1–1640 (Gibco) supplemented with 10% (v/v) FBS. All experiments performed in cells tested negative for mycoplasma and passage 5 to 15 (post-thaw).
MCF-7 PKM2-KD cell line was generated by lentiviral transduction. 3rd generation lentiviral vectors were obtained from Addgene (#12251, #12253, #12259). shRNA against 3’-UTR of PKM2 transcript and control scramble shRNA were designed and cloned into pLKO.1 plasmid (Addgene #84530). Plasmid construct information are provided in the supplement. Lentiviral and expression plasmids were co-transfected (1:1 ratio) into HEK293T/17 cells (ATCC Cat# CRL-11268, RRID: CVCL_1926) using Lipofectamine 3000 transfection reagent (Invitrogen). The viral containing supernatant were collected 24- and 48-hours post transfection, filtered and used to infect MCF-7 cells in the presence of 10 ug/ml polybrene (Millipore). Stable clones were selected for 9 days with 2 ug/ml puromycin (Gibco).
To generate wild-type PKM2 (WT), mutant PKM2 (S37A or S37E), and nuclear directed PKM2 (NLS) TNBC cells, MDA-MB-468 cells were transfected with pFUGW (Addgene #63592) plasmids encoding FLAG/HA-tagged (N-term) PKM2-WT, -S37A, -S37E, or -NLS (PAAKRVKLD, C-term) using the FuGENE® HD Transfection Reagent (Promega) according to manufacturer’s guidelines. Plasmid construct information are provided in the supplement.
Tumor and normal tissues were collected immediately after surgical excision and directly formalin-fixed and embedded in paraffin by the Yale Pathology Tissue Services. Written informed consent or waiver of consent was provided by all the patients. The study was approved by the Yale Human Investigation Committee protocol #9505008219 and conducted in accordance with the Declaration of Helsinki. The YTMA128 and YTMA341 tissue arrays contained 101 and 94 tumors respectively resected between 2001 and 2012 from breast cancer patients. Patient characteristics are presented in the supplement. Protein expression on tissues was determined by quantitative immunofluorescence using the AQUA Method (32). Detailed AQUA methods and analyses results are included in the supplement. Sample randomization and analyses were performed by researchers blinded to the patient’s intervention and medical background.
Immunoprecipitation and Immunoblotting
MCF7, MDA-MB-231, MDA-MB-468, DU4475 cells were grown in 15 cm plates until reach 90% confluency. Then lysed in 1 ml ice cold 1x RIPA lysis buffer (Boston BioProducts) supplemented with protease and phosphatase inhibitors (cOmplete, Roche; Phosphatase Inhibitor Cocktail 1, Sigma) for 30 min, sonicated (15s ON / 10s OFF for 2 cycles at 40% amplitude) and centrifuged (4 °C, 10 min at 13,000 rpm) to remove cell debris. PKM2 protein complexes were bound to anti-PKM2 antibody (RRID: AB_1904096) at 4 °C O/N under rotation. Immunocomplexes were then pulled down by coupling to SureBeads™ Protein A Magnetic Beads (Bio-Rad) for 3 hours at 4 °C. Normal rabbit IgG antibody (Cell Signaling Technology, Cat# 2729, RRID: AB_1031062) was used as a negative control.
All cells used in immunoblot experiments were lysed in 1x RIPA buffer as described above. Preparation of nuclear and cytoplasmic extracts was performed by using a Nuclear Extraction Kit (abcam) according to manufacturer’s guidelines. Excised xenograft tumors were lysed in 0.5 ml 1x Cytobuster reagent (Millipore) supplemented with protease and phosphatase inhibitors, sonicated and normalized using a Bradford Assay (Bio-Rad).
All samples were resolved by SDS-PAGE electrophoresis on 4–15% TGX precast gels (Bio-Rad). Proteins were transferred to PVDF membranes using semi-dry transfer cells (Bio-Rad), and blocked in either 5% BSA or milk (Sigma). Antibodies used in the analysis include PKM2 (Cell Signaling Technology, Cat# 4053, RRID: AB_1904096), PKM1 (Cell Signaling Technology, Cat# 7067, RRID: AB_2715534), PKM (Santa Cruz Biotechnology, Cat# sc-365684, RRID: AB_10844484), CDC2 p34 (Santa Cruz Biotechnology, Cat# sc-54, RRID: AB_627224), Cyclin B1 (Cell Signaling Technology, Cat# 4138, RRID: AB_2072132), FLAG (Sigma-Aldrich, Cat# F3165, RRID: AB_259529), beta-actin (Abcam, Cat# ab6276, RRID: AB_2223210), PKM2pS37 (rabbit polyclonal, custom-made); more information for custom antibody production and validation are included in the supplement. The antibody is available to other researchers upon request to the authors. Protein band quantification was performed using the ImageQuant TL (version 8.1, RRID:SCR_018374) image analysis software.
PKM2 Oligomerization Assay
Glutaraldehyde (GA) crosslinking assay was performed as described previously with modifications (11). In brief, MDA-MB-231 cells (1 × 106) untreated or treated with TEPP-46 (5 uM) for 24 hours were lysed in 120 ul 1x NP-40 buffer (50 mM Tris-HCl pH=7.4, 150 mM NaCl, 1% NP-40). Samples (25 ul from each lysate) were cross-linked with 0.05% GA (Sigma) for 3 minutes at 37 °C. Reactions were next terminated with the addition of 60 mM Tris-HCl pH=8.0 and analyzed by immunoblotting for total PKM2 and PKM2pS37.
Immunofluorescence analysis
7 to 8 × 104 cells were seeded in ibiTreat chamber slides (Ibidi). Fixed with 4% PFA for 15 min, after treatment with Dinaciclib (15 nM) or TEPP-46 (10 uM) or their combination for 6 or 20 hours, washed three times with 1x DPBS, permeabilized with 0.1% Triton X-100 for 15 minutes and blocked with 10% goat serum (constituted in 0.1% Triton X-100) at room temperature. Then stained O/N at 4 °C with primary antibodies against PKM2pS37 and total PKM2 using a 1:200 and 1:100 dilution in blocking solution respectively, or FLAG antibody (1:200) followed by Alexa Fluor 488 (Thermo Fisher Scientific Cat# A-11034, RRID: AB_2576217) or Alexa Fluor 594 (Thermo Fisher Scientific Cat# A-11032, RRID: AB_2534091) conjugated secondary antibody and Hoechst 33342 (Invitrogen) labeling. All samples were imaged by confocal microscopy on a Leica Laser Scanning SP8 Microscope (20x or 63x oil objective) at room temperature and same settings. Taken images were analyzed using ImageJ software (Fiji, RRID: SCR_002285). Fluorescence intensity determined with identically adjusting brightness and contrast, using manual masking.
Cell apoptosis and ROS detection assay
Cell apoptosis was quantified using Caspase-Glo3/7 and RealTime-Glo Annexin V Apoptosis detection assays (Promega). For both assays, 0.5 or 1 × 104 cells were seeded in 50 ul complete medium per well (96-well solid white plates, Corning). Cells were incubated with the treatment for 6 or 20 hours and mixed with equal volume of the detection mixture. Luminescence was measured after 1.5 hours of incubation. ROS-Glo assay (Promega) was used to measure intracellular levels of Hydrogen Peroxide (H2O2). 104 cells were seeded in 70 ul complete medium per well. Treatment was added after 4 hours and cells incubated for 24 hours. H2O2 Substrate solution was added 18 hours after treatment initiation. Reactions mixed with equal amount of detection solution at treatment end point and luminescence was measured after 20–40 minutes. All measurements were taken on a VICTOR Multilabel Plate Reader (PerkinElmer) using an exposure time of 1s.
Matrigel Invasion Assays
Invasion assays were carried out using Biocoat Matrigel invasion chambers (BD Biosciences). Cells were serum starved for 6 hours and seeded on the chamber well inserts (5 × 104 cells per insert) in presence of Dinaciclib (5 or 2.5 nM), TEPP-46 (5 μM) or their combination. Cells let to invade for 20–24 hours. Uninvaded cells were removed from the inner surface of the Matrigel chamber using a moistened cotton swab. The remaining invaded/migrated cells were fixed directly on chamber by soaking in 4% PFA. After washed with 1x DPBS, membranes were stained with Hoechst 33342 (Invitrogen) and mounted on slides using ProLong™ Gold Antifade Mountant (Invitrogen). Imaging was performed using a Fluorescence Microscope (10x objective) and taken images (6–8 fields per membrane) were analyzed with ImageJ software.
TNBC xenograft model
Five million MDA-MB-231 cells were implanted subcutaneously into the right flank of immune deficient Rag2 / IL2RG double knockout mice (Envigo) in the presence of Matrigel (Corning). Fifteen mice with palpable, similar sized tumors were randomized to three experimental groups: vehicle, Dinaciclib (50 mg/kg) and TEPP-46 (40 mg/kg). Dinaciclib was obtained from Cayman chemical (Cat# 14707) and TEPP-46 was a gift from Dr. Craig J. Thomas, NCATS, NIH. Both drugs were formulated in 20% (2-Hydroxypropyl)-β-cyclodextrin (Sigma) water solution and administrated daily by intraperitoneal injection in a 100 ul volume for two weeks. Tumor volumes were recorded by caliper measurements at three-day intervals. The mice were euthanized at the end of treatment and the tumors analyzed for total PKM2 and PKM2pS37 levels. Animal sample sizes and maximum treatment doses was determined according to previous animal studies (18,28). Mice randomization to treatments, data collection, and analysis were performed by investigators blinded to the groups. All procedures were performed at the Yale Center for Precision Cancer Modeling under the protocols approved by the Yale Institutional Animal Care and Use Committee. IHC staining was performed on Leica Bond Rx using Leica Refine Polymer Detection Kit as per manufacturer’s instructions. Detailed experimental protocol is provided in the supplement. H-score determination was performed by a pathologist who was blinded to the experimental procedure.
TCGA Data Analysis
RNA-seq data from clinical human specimens were retrieved from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) and analyzed via Xena Functional Genomics Explorer from the UCSC Genomics Institute Data Analysis Center (https://xenabrowser.net; RRID: SCR_018938). mRNA expression levels of PKM2, CDK1, CCNB1, CDK6 and CCND3 genes are presented as log2(x+1) transformed RSEM normalized count. Sample IDs are available in the supplement.
Proteomics analysis
Phosphoproteomics workflow was carried out as described previously (33). CDK Kinase reactions and LC-MS/MS analysis were performed as described previously (34). Detailed experimental information is provided in the supplement.
Statistical analysis
All analyses were performed using the GraphPad Prism software (version 8.0.0; RRID: SCR_002798). All plotted values represent mean values with SD or SEM as indicated in the figure legends. Significance across mean values of each replicate group was determined by Turkey’s multiple comparison test or unpaired t test. A Log-rank (Mantel-Cox) test was used to compare survival curves. Relative risk was assessed using the multivariate Cox-proportional hazards model. Significance was considered at P < 0.05 (95% confidence interval).
Results
PKM2pS37 phosphorylation signature in cancer cells
Previous studies support that PKM2 isoform expression drives both tumor cell growth and invasion in breast and ovarian cancer (35–37). To evaluate the biological relevance of PKM2 expression in tumors of different type and aggressiveness we performed a pan-cancer analysis of RNA-seq data obtained from patient-derived tumor samples in The Cancer Genome Atlas (TCGA) database (Table S1). We observed elevated levels of the PKM2 transcript in all primary tumors of selected tissue types in comparison with the adjacent normal tissue. Significantly higher PKM2 expression was found in more aggressive tumor phenotypes, such as glioblastoma multiform (GMB) and metastatic melanoma (MM) (Figure 1A). Phosphoproteomic data sets matching TCGA samples are not available, so we carried out phosphoproteomic analysis of representative PKM2 isoform-expressing cancer cell lines, MCF-7, Jurkat, and OVCAR-3, to examine PKM2 phosphorylation. We confirmed PKM2 expression across cell types and PKM2pS37 as the most prominent phosphorylation site in common. Notably, PKM2pS37 was absent from a pancreatic insulinoma cell line (INS-1) used as a control where all PKM1/PKM2 and PKL isoforms are present yet expected to have a contrasting phenotype (Figure 1B, Table S2) (38,39).
Figure 1:
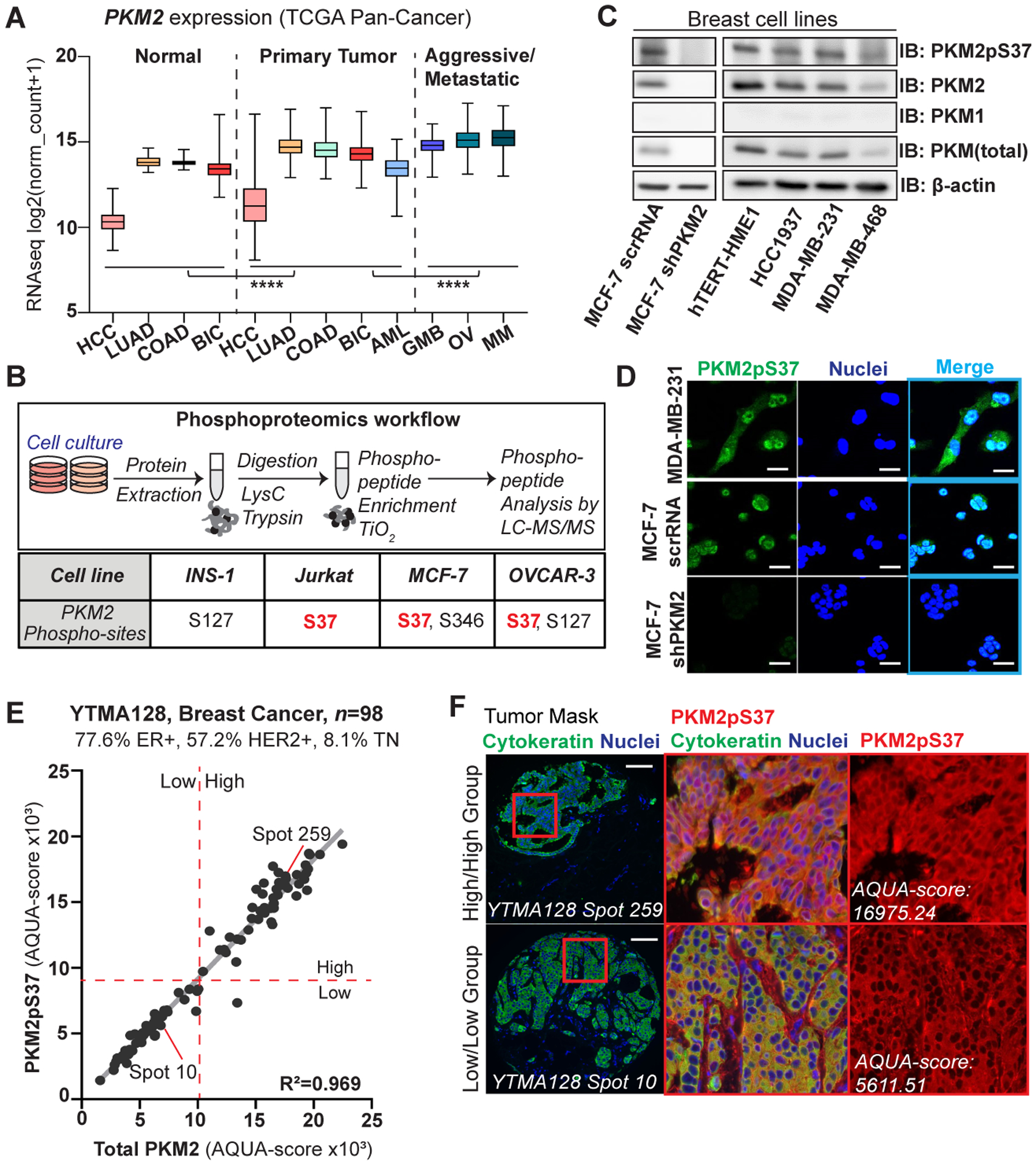
PKM2pS37 is present across different PKM2 expressing tumor types and highly expressed in breast cancer. A) PKM2 gene expression in different tumor types (source: TCGA). Tumor types: hepatocellular carcinoma (HCC; normal, n = 50, tumor, n = 371), lung adenocarcinoma (LUAD; normal n = 59, tumor, n = 515), colon adenocarcinoma (COAD; normal n = 41, tumor, n = 452), breast invasive carcinoma (BIC; normal n = 114, tumor, n = 1097), acute myeloid leukemia (AML; n = 173), glioblastoma multiforme (GBM; n = 154), ovarian serous cystadenocarcinoma (OV; n = 303), metastatic melanoma (MM; n = 369). One-way ANOVA (Tukey’s HSD), ****P < 0.0001. B) (Upper panel) Overview of phospho-proteomics experimental workflow. (Lower panel) Mass spectrometry analysis of site-specific phosphorylation on PKM2. C) Immunoblot analysis showing PKM expression and PKM2pS37 status in breast cell lines. D) Immunofluorescence analysis of MDA-MB-231 or MCF-7 cell lines show enhanced nuclear PKM2pS37, absent from a PKM2-KD cell line (MCF-7 sh-PKM2); Nuclear staining, Hoechst 33342 (blue); Scale bar, 20 μm. E) Correlation between AQUA (QIF) scores for PKM2pS37 and total PKM2 quantified in tumor mask on each YTMA128 spots. Dashed red lines correspond to median cut-points used for patient Low/High grouping. F) Representative YTMA128 spots stained for PKM2pS37. PKM2/PKM2pS37 high- (upper panel) and low- (lower panel) expressing patient; Nuclear staining, DAPI (blue); Scale bar, 100 μm.
Previous studies have linked PKM2 phosphorylation with tumor promotion in breast cells (40). Moreover, PKM2 phosphorylation seems to play a critical role in regulatory pathways that support breast cancer cell metabolic reprogramming (11). However, the exact regulatory mechanisms involved remain largely elusive. These observations prompted us to utilize breast cancer as tumor model to better characterize PKM2 phosphorylation in human cancer. Immunoblot experiments showed PKM2 as the dominant isoform in both non-tumorigenic (hTERT-HME1) and cancer cells of different subtypes and grades (Figure 1C). We next generated a PKM2pS37 phospho-specific antibody (Figure S1A–B), to detect PKM2pS37 in different breast cell phenotypes and explore differential expression patterns (Figure 1C–D). Immunofluorescence studies using the same antibody showed a strong, specific nuclear localization of PKM2pS37 in MDA-MB-231 and MCF-7 cells that was confirmed with experiments in MCF-7 cells with PKM2 knocked down via sh-RNA (Figure 1C–D). To further validate this striking staining pattern in vivo we performed immunohistochemistry on freshly dissected, paraffin-embedded tissue from TNBC patients and found prominent PKM2pS37 staining in the nucleus and cytoplasm (Figure S1C). These observations demonstrate that our antibody can assess PKM2pS37 expression and localization in breast cancer in vitro and in vivo.
PKM2pS37 as a biomarker for triple negative breast cancer
PKM2pS37 phosphorylation has been characterized in other tumor types but a quantitative assessment with a highly specific antibody has not been performed (10,13). Furthermore, other studies suggest that PKM2 phosphorylation in breast cancer cells is potentially linked with stem-like cell properties and mediation of cancer metabolic reprogramming by supporting the formation of tumorigenic PKM2 oligomers (11,40). To investigate a possible correlation of PKM2pS37 with breast cancer subtypes and aggressiveness we utilized two different tissue microarrays (TMAs), one containing 101 tumor cores with high proportions of ER+/HER2+ subtypes (YTMA128, 77.6% ER+, 57.2% HER2+, 8.1% TN) and one containing 94 cores of the TNBC subtype (YTMA341) (Table S3, Table S4). Quantitative Immunofluorescence (QIF) analysis of the ER+/HER2+ TMA showed high correlation between PKM2 and PKM2pS37 expression in tumor (R2 = 0.969) (Figure 1E). Notably, elevated PKM2pS37 nuclear staining was observed in the PKM2/PKM2pS37-high patient group (Figure 1F). Similar QIF analysis indicated a substantial increase in PKM2 expression in the TNBC subtype (Figure 2A). In contrast to ER+/HER2+ patients, correlation between tumor PKM2 and PKM2pS37 expression was weak for TNBC patients (R2 = 0.20) (Figure 2B). This suggests PKM2pS37 amplification is unrelated to total PKM2 levels. Moreover, QIF scores of total PKM2 and PKM2pS37 revealed a continuous distribution in the tumor and stromal compartments (Table S4). Unlike total PKM2 expression, PKM2pS37 showed distinct compartmental variations within the tumor area of TNBC patients (Figure 2C–D), while similar patterns weren’t observed for the ER+/HER2+ patients (Figure S2A). Interestingly, for all subtypes, nuclear PKM2pS37 was elevated in PKM2pS37-high patient groups (Figure 1F, Figure 2D). This suggests that PKM2pS37 is possibly a signal for nuclear accumulation which is in agreement with previous studies (11). Here we validated this observation in human tumors and investigate a potential connection with poor prognosis.
Figure 2:
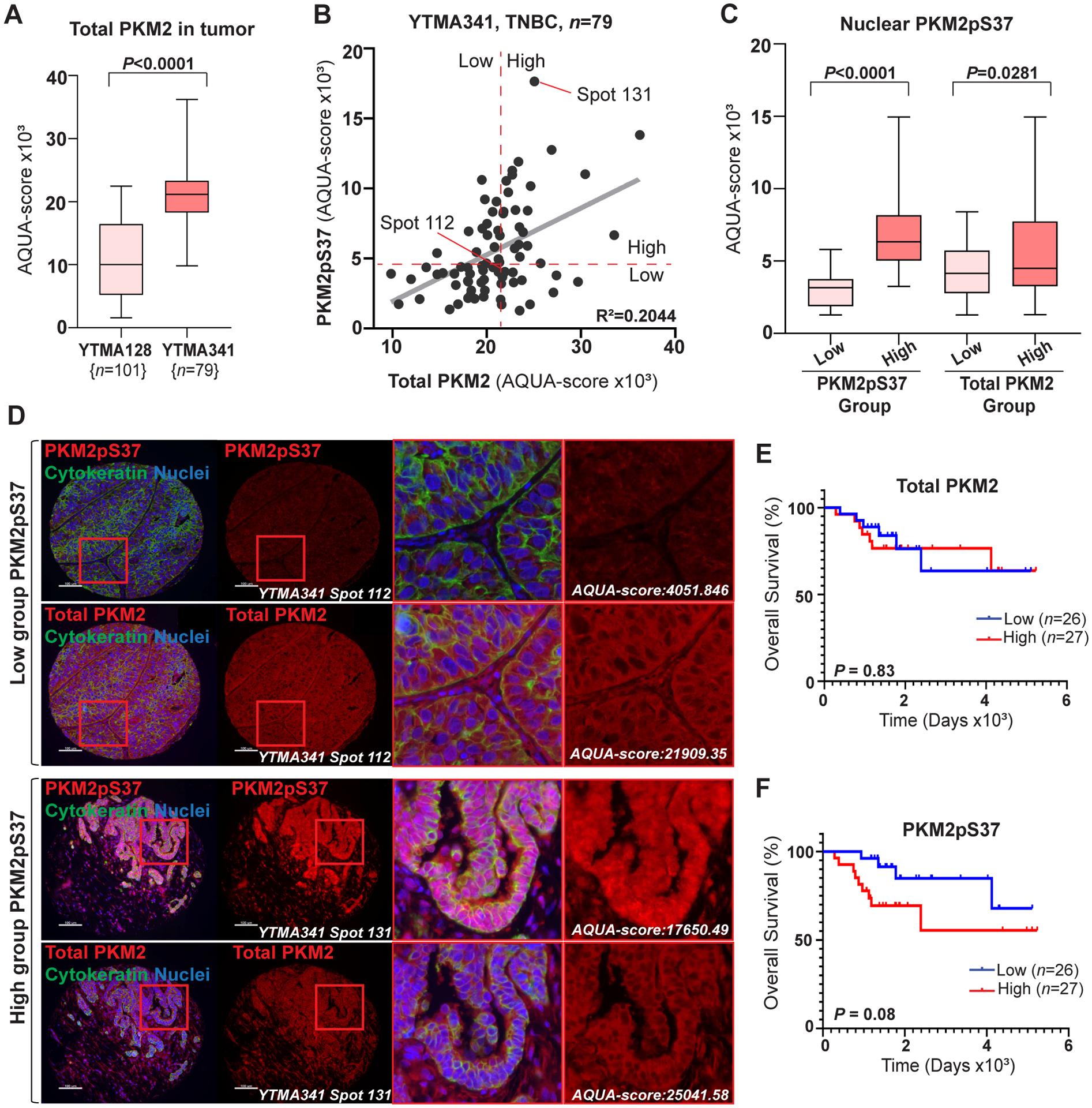
PKM2pS37 presence in the nucleus correlates with decreased overall survival in TNBC patients. A) Comparison of PKM2 expression in tumor using AQUA scores obtained from YTMA128 (n = 101) and YTMA341 (n = 79) tumor array analysis (t test, ****P < 0.0001). B) Correlation between AQUA (QIF) scores for PKM2pS37 and total PKM2 quantified in tumor mask on each YTMA341 spots. Dashed red lines correspond to median cut-points used for patient Low/High grouping. C) Comparison of nuclear PKM2pS37 expression in tumor between Low- (n = 40) and High- (n = 39) expressing PKM2pS37 or total PKM2 patient groups using AQUA scores obtained from YTMA341 analysis. Significance, one-way ANOVA (Tukey’s HSD). D) Representative YTMA341 spots stained for PKM2pS37 or total PKM2. PKM2pS37 low- (upper panel) and high- (lower panel) expressing patient; Nuclear staining, DAPI (blue); Scale bar, 100 μm. E), F) Kaplan-Meier curves for overall survival with respect to total PKM2 and PKM2pS37 expression. Significance, Log-rank (Mantel-Cox) test.
In order to examine the prognostic potential of PKM2 phosphorylation in breast cancer subtypes we next categorized the patients into high and low groups by applying a visual cut-point based on total PKM2 and PKM2pS37 QIF scores in tumor areas of the tissue sections (Table S4). For the TNBC subtype, no trend towards patient overall survival was observed for total PKM2 expression (Hazard Ratio [HR], 0.88; 95% CI, 0.3–2.6; P = 0.83) (Figure 2E). Importantly, we observed a trend for decreased overall survival (Hazard Ratio [HR], 0.38; 95% CI, 0.13–1.12; P = 0.08) for PKM2pS37-high as compared to PKM2pS37-low expressing patients (Figure 2F). Similar analysis of the ER+/HER2+ cohort showed no significant correlation of either total PKM2 or PKM2pS37 with overall survival (Figure S2B–C). Notably, we observed weak correlation between nuclear expression of total PKM2 and PKM2pS37 in TNBC patients (R2 = 0.2427) (Figure S2D). Patients in the PKM2pS37-high expressing group showed a prominent nuclear and cytoplasmic staining pattern, while patients in the PKM2pS37-low group showed reduced cytoplasmic signals and no visible nuclear staining (Figure 2D). On the contrary, a predominantly cytoplasmic staining for total PKM2 was observed for all patients regardless their grouping (Figure 2D). Finally, for the TNBC subtype, we additionally observed a trend towards decreased disease-free survival for PKM2pS37-high expressing patients (Hazard Ratio [HR], 0.37; 95% CI, 0.13–1.06; P = 0.06), while no significant association with stage and grade was found for either total PKM2 or PKM2pS37 (Figure S2E–H). Collectively, our analyses suggest that PKM2pS37 expression and nuclear accumulation have a significant association with the TNBC subtype and could act as a more information rich prognostic marker for aggressive cancer phenotypes compared to total PKM2.
CDK inhibitors selectively target and dephosphorylate PKM2pS37
Studies focusing on lymphomas have shown that CDK6-cyclinD3 complex can directly phosphorylate PKM2 at S37 (10). However, it is unknow whether similar CDK-PKM2 interactions occur in breast cancer or across its different subtypes. To investigate this further we categorized breast cancer clinical specimens with available RNA-seq data found in TCGA Breast Invasive Carcinoma (BRCA) database according to their ER status (Table S5). For each group (ER+ or ER−) we assessed the expression of CDK6 or CDK1 genes and their cyclin partners, CCND3 or CCNB1 respectively, in comparison to annotated RNA-seq data from normal breast specimens found in the same database. We confirmed that CDK6-cyclinD3 complex components showed either no significant changes in comparison with the normal tissue or were reduced. On the contrary CDK1-cyclinB1 gene expression was elevated in all tumor data sets and higher expression was observed for the ER− group (Figure 3A). Moreover, a series of immunoprecipitation experiments using a panel of breast cell lines of different subtypes, either ER/HER2 status or aggressiveness, verified that PKM2 can associate with CDK1-cyclin B1 complexes in whole cell lysates (Figure 3B, Figure S3A).
Figure 3:
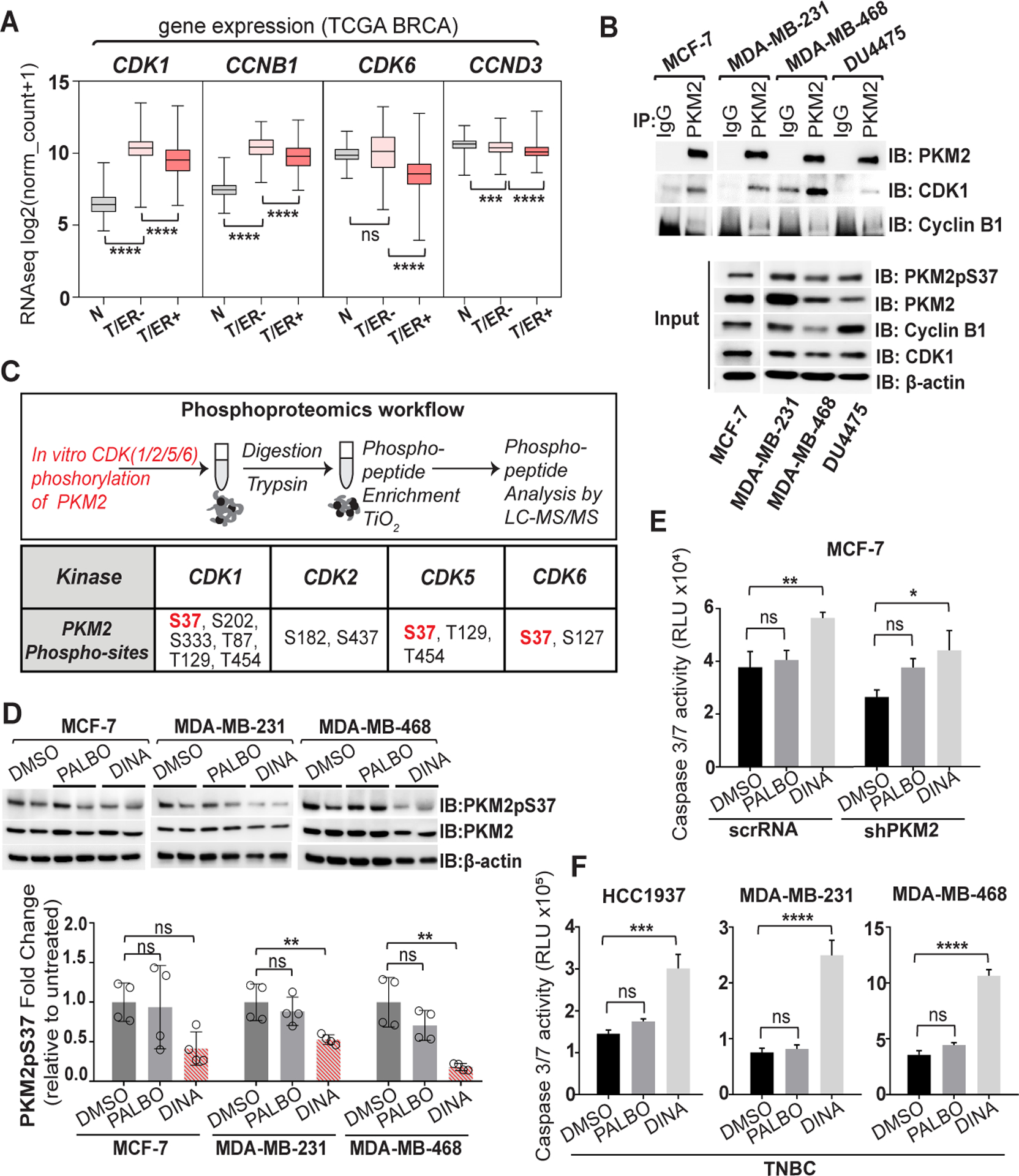
Dinaciclib suppresses PKM2pS37 phosphorylation by selective inhibition of CDK-cyclin complexes and induces apoptosis in TNBC cells. A) Comparison of CDK1, CCNB1, CDK6 and CCND3 gene expression in breast cancer subtypes (Source: TCGA-BRCA); N: normal, T/ER−: tumor, estrogen receptor negative, T/ER+: tumor, estrogen receptor positive. One-way ANOVA (Tukey’s HSD), ****P < 0.0001; ns, nonsignificant. B) Immunoprecipitation of PKM2 and CDK1/cyclinB1 complexes in whole cell lysates with anti–PKM2 or normal IgG antibodies. C) (Upper panel) Overview of phospho-proteomics experimental workflow. (Lower panel) Mass spectrometry analysis of the enriched phosphorylated peptides of the recombinant human PKM2 protein upon CDK treatment. D) Immunoblot analysis of PKM2pS37 in whole cell lysate after treatment with Dinaciclib (1 uM) or Palbociclib (1 uM) for 20 h. Bars, mean values, n = 4; error bars, SD; one-way ANOVA (Tukey’s HSD), *P < 0.05, **P = 0.002; ns, nonsignificant. E), F) Apoptosis in breast cancer cells treated with Dinaciclib (10 nM) or Palbociclib (0.5 uM) for 20 hours. Bars, mean values, n = 3; error bars, SD; one-way ANOVA (Tukey’s HSD), *P < 0.05, **P = 0.002, ***P = 0.0002, ****P < 0.0001; ns, nonsignificant.
To address whether different CDKs could directly phosphorylate PKM2 at S37 we performed in vitro phosphorylation of the recombinant human PKM2 protein with CDK1/2/5/6. Mass spectrometry analysis showed that CDKs 1, 5 and 6 phosphorylated PKM2 at S37 (Figure 3C, Table S6). We hypothesized that breast cancer types that were non-responsive to CDK4/6 inhibitor, Palbociclib (10,41) yet contained high levels of PKM2pS37, would be uniquely sensitive to CDK1/2/5/9 inhibitors. Indeed, treatment with a potent CDK1/2/5/9 inhibitor Dinaciclib, significantly diminished PKM2pS37 phosphorylation in TNBC cell lines (MDA-MB-231, MDA-MB-468) in comparison to Palbociclib. We observed a similar trend for MCF-7, ER+/HER2− breast cancer cell line (Figure 3D, Figure S3B). Our analysis of PKM2 phosphorylation and the contrasting response of PKM2pS37 levels to CDK inhibitors suggested that PKM2pS37 expressing breast cancer cells would be resistant to Palbociclib, and sensitive to Dinaciclib. Consistent with this hypothesis, Dinaciclib effectively triggered apoptosis in the MCF-7 cell line while Palbociclib had no effect. PKM2 knockdown (MCF-7 sh-PKM2) reduced the magnitude of the Dinaciclib effect although did not affect the sensitivity to Palbociclib (Figure 3E). Similarly, Dinaciclib significantly induced apoptosis across different TNBC cell lines albeit MDA-MB-231 and MDA-MB-468 cells exhibit the highest sensitivity. Moreover, all tested TNBC cell lines were unresponsive to Palbociclib (Figure 3F, Figure S3C).
Our findings suggest that PKM2pS37 accounts for a strong regulatory signal in TNBC cells which assists vital central metabolic pathways both in the cytoplasm and the nucleus (Figure 4A). Based on this model the CDK1-Cyclin B1 complex phosphorylates PKM2 at S37 and increases PKM2/PKM2pS37 oligomers in the cytoplasm which support tumor metabolic reprogramming and suppress cell death. In parallel, elevation of PKM2pS37 assists conformational changes that direct PKM2 to the nucleus where it mediates gene regulation that further supports tumor metabolism and survival (13,42–44). Therefore, Dinaciclib treatment is expected to reverse the effects of PKM2pS37 in both the cytoplasm and the nucleus. Immunofluorescence analysis of MDA-MB-231 cells treated with Dinaciclib showed a robust decrease of PKM2pS37 signal in the nucleus and the cytoplasm (Figure 4B). Dinaciclib treatment also decreased the proportion of total PKM2 translocated to the nucleus (Figure 4C).
Figure 4:
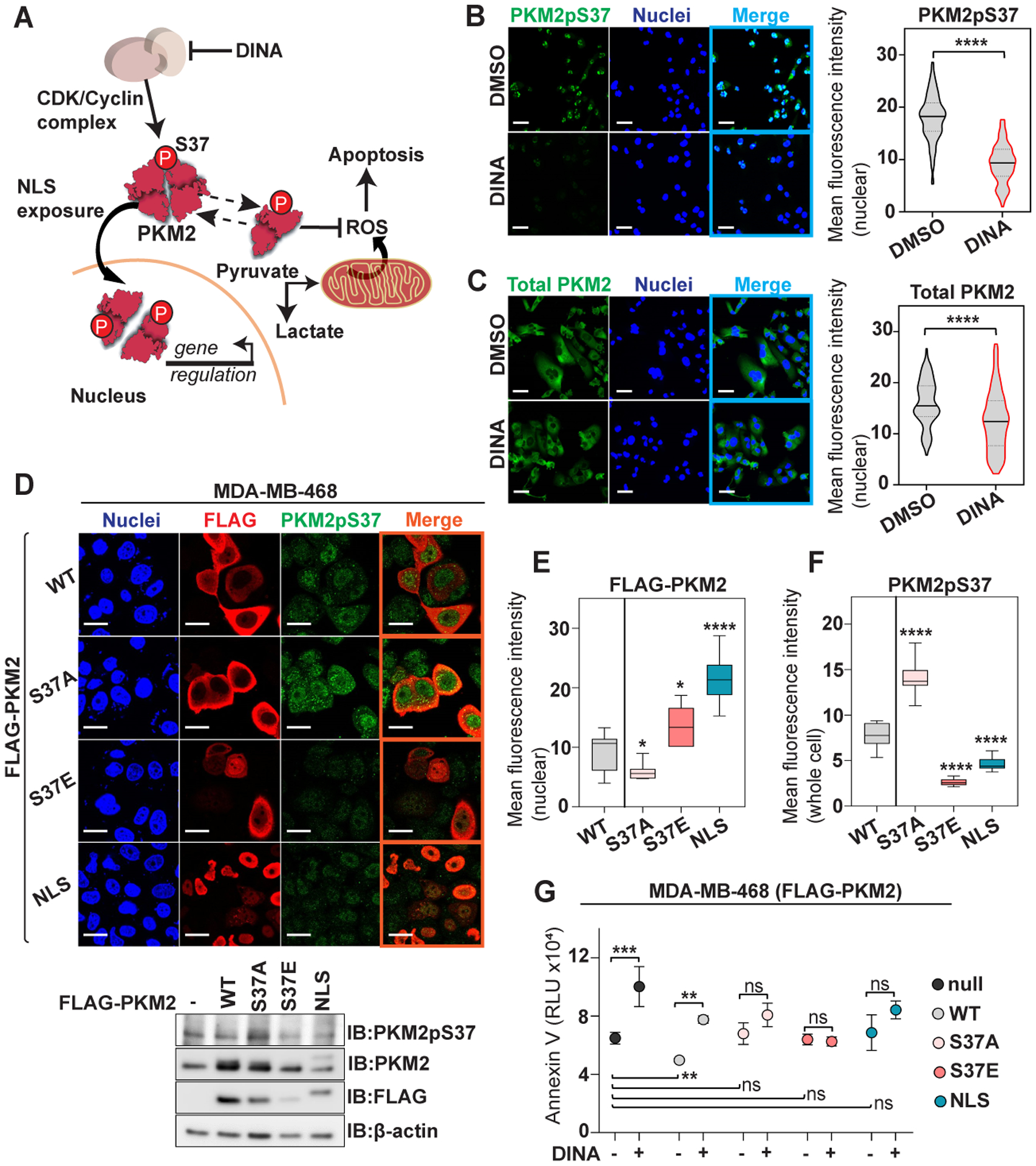
PKM2pS37 is a strong signal responsible for PKM2 nuclear trafficking and cancer metabolic reprograming in TNBC cells. A) Schematic representation of a model proposing the involvement of PKM2pS37 in tumor cell metabolism. B), C) Immunofluorescence analysis of nuclear PKM2pS37 and total PKM2 in MDA-MB-231 cells treated with Dinaciclib (15 nM) for 20 hours. Nuclear staining, Hoechst 33342 (blue); Scale bar, 20 μm. D) Immunofluorescence and immunoblot analysis of MDA-MB-468 cells expressing wild-type PKM2 (WT), PKM2 S37 mutants (S37A, S37E) and the PKM2-NLS fusion; Nuclear staining, Hoechst 33342 (blue); Scale bar, 20 μm. E) Comparative nuclear localization analysis of ectopically expressed PKM2 proteins in MDA-MB-468 cells and F) PKM2pS37 quantification in the whole cell (t test, *P < 0.05, ****P < 0.0001). G) Apoptosis in MDA-MB-468 PKM2-WT, -S37A, -S37E, -NLS cells upon Dinaciclib (5 nM) treatment (6 hours). Error bars, SD, n = 3; one-way ANOVA (Tukey’s HSD), **P = 0.002, ***P = 0.0002; ns, nonsignificant.
To further investigate whether PKM2 nuclear localization in TNBC cells relies on phosphorylation in position S37 we ectopically expressed FLAG-tagged PKM2 mutants in MDA-MB-468 cells. These cells were selected because they express less endogenous PKM2 (Figure 1C) and are the most sensitive to Dinaciclib treatment (Figure 3F). An alanine mutant (S37A) was used as phospho-deficient PKM2 and a glutamate mutant (S37E) as phospho-mimetic PKM2. Moreover, we employed a strategy to maximize nuclear localization independent of S37 phosphorylation by fusing a nuclear localization signal (NLS) sequence to the C-terminus of PKM2 (PKM2-NLS) (Figure 4D). Immunofluorescence analysis showed that the S37A mutant has reduced ability to enter the nucleus in contrast to WT, while S37E has enhanced nuclear localization. Likewise, PKM2-NLS displayed a profound nuclear localization as expected (Figure 4D, Figure 4E). These data are consistent with a mechanism described in other cancer cell types where PKM2pS37 phosphorylation triggers nuclear localization, albeit by forming a complex with a second nuclear import protein (13). The unnaturally strong localization of the PKM2-NLS fusion protein supports the model that PKM2pS37 phosphorylation is the endogenous mechanism controlling the balance of nuclear/cytoplasmic distribution in TNBC.
Surprisingly, ectopic expression of the PKM2 S37A mutant increased endogenous PKM2 phosphorylation (Figure 4F). This upregulation of PKM2pS37 phosphorylation presumably compensates for increasing non-phosphorylated PKM2 oligomers in the cytoplasm. On the other hand, ectopic expression of S37E or NLS PKM2 variants decreased the endogenous PKM2pS37 and, in turn, its translocation to the nucleus (Figure 4F). These results suggest that both nuclear and cytoplasmic localization of PKM2 is controlled by S37 phosphorylation. Moreover, the presence of non-phosphorylatable (S37A, S37E) or nuclear restricted (NLS) PKM2 oligomers reduced sensitivity to Dinaciclib (Figure 4G). This suggests that the nuclear/cytoplasmic balance of PKM2, controlled by phosphorylation at S37, is critical to TNBC cell metabolism and survival. Collectively, our observations indicate an alternative CDK pathway regulating TNBC cell survival via PKM2pS37 and are consistent with a previously described model (10) in which CDKs can regulate PKM2 via direct phosphorylation and, in turn, control cancer cell viability and confer sensitivity to CDK inhibitor therapy (Figure 4A&G).
TEPP-46 reduces PKM2 nuclear localization in TNBC cells.
Our QIF analysis on tissue cores of breast cancer clinical specimens showed that PKM2pS37 is accumulated in the nucleus (Figure 2C–D, Figure S1C). Uniquely, we observed that PKM2pS37 is distributed in both the cytoplasm and the nucleus in PKM2pS37-high-expressing patient group (Figure 1F, Figure 2D). These observations fit the model that PKM2 enzyme transfers incrementally from a compact tetramer to a disassembled dimeric/monomeric state, which finally enters the nucleus (7,11,19). It has been argued that these two states partition a low activity PKM2 state in the cytoplasm and a novel, potentially non-metabolic, PKM2 state in the nucleus (19).
Recently, a small molecule TEPP-46 was shown to target and activate PKM2 in lung cancer cell lines by stabilizing the tetramer formation (12). Along with our TNBC data, previous studies have also shown that PKM2 nuclear translocation relies on PKM2pS37 phosphorylation (11,13,19). These observations prompted us to examine whether TEPP-46 could target PKM2pS37 in TNBC cells to alter PKM2 oligomeric structure and nuclear localization. Image analysis showed that TEPP-46 treatment reduced nuclear levels of both PKM2pS37 and total PKM2 in TNBC cells (Figure 5A–B). Moreover, immunoblot analysis of nuclear and cytoplasmic fractions showed that while TEPP-46 treatment reduced nuclear PKM2pS37, its cytoplasmic levels appear to be unaffected (Figure 5C). Further analysis of the cytoplasmic fractions after treatment with TEPP-46 indicated that PKM2 equilibrium shifts from monomer/dimer to dimer/tetramer, with PKM2pS37 present in the different oligomeric states (Figure 5D). These results show, for the first time, that the PKM2pS37 pool is targeted by TEPP-46 and that this small molecule could alter PKM2 localization in TNBC cells making it an attractive therapeutic approach for further validation. Therefore, we next examined whether TEPP-46 has a direct impact on cell growth and survival. We observed no significant effect of TEPP-46 on cell proliferation and clonogenicity in all breast cancer cell lines tested (Figure S3D–E). Moreover, TEPP-46 treatment does not seem to significantly contribute to changes in redox balance or enhancing apoptosis in TNBC cells (Figure 5E–F). Previous studies showed that the effectiveness of TEPP-46 in enhancing apoptosis in lung cancer cells relies on chemotherapeutic manipulation of p53 activity and the oxidation state of PKM2 (22). However, it is unknown whether the phosphorylation state of PKM2 could impact the efficacy of TEPP-46 in triggering cell death in different cancers. While we have observed that TEPP-46 can efficiently target an elevated pool of PKM2pS37 in TNBC, our work suggests that a combination with another therapeutic agent could be exploited to elevate cancer cell death and, in turn, tumor regression.
Figure 5:
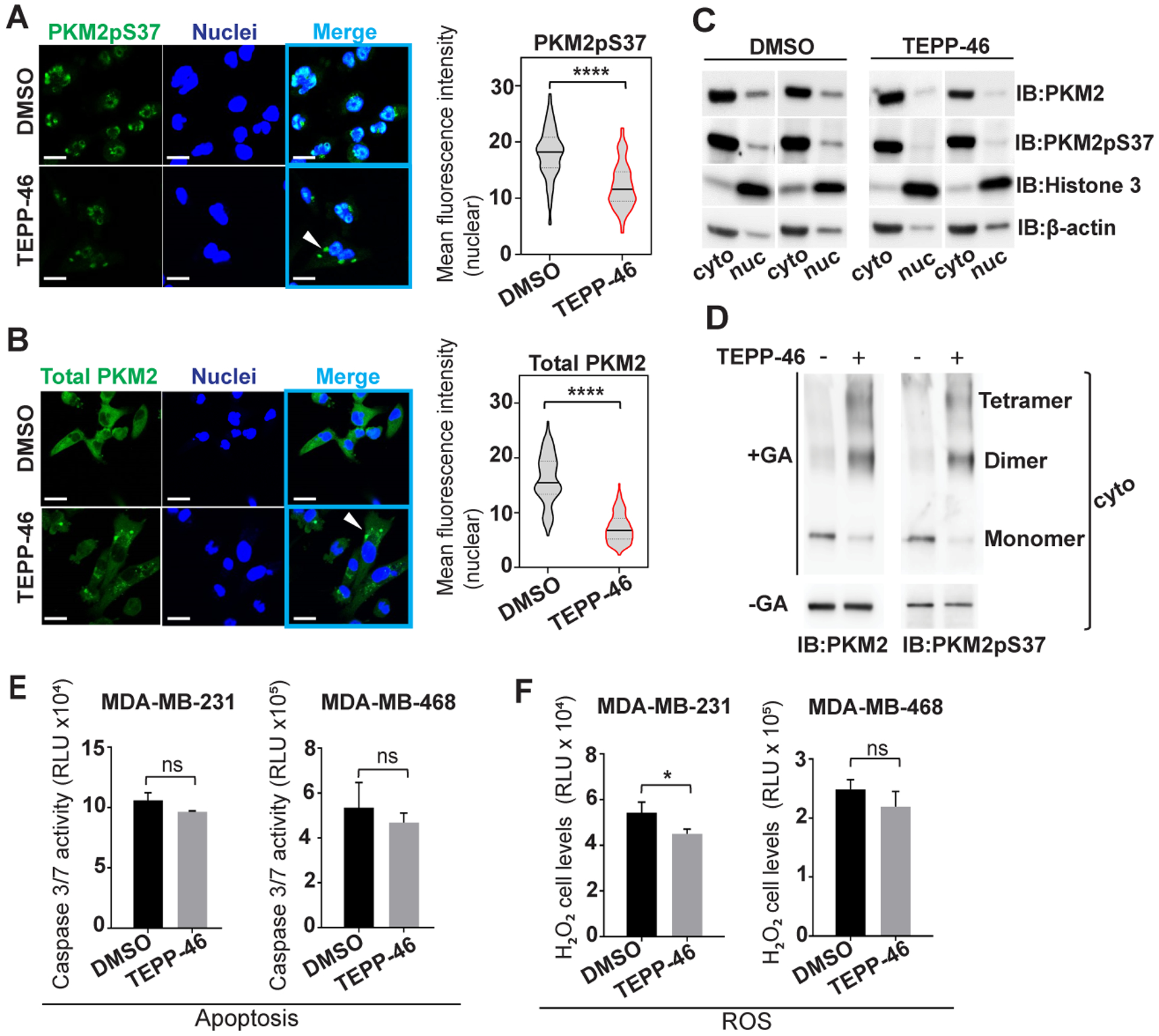
TEPP-46 activator stabilizes a PKM2 state unable to enter the nucleus. A), B) MDA-MB-231 cells, treated or untreated with TEPP-46 (10 uM, 20 hours), analyzed for PKM2pS37 or total PKM2 localization; Nuclear staining, Hoechst 33342 (blue); Scale bar, 20 μm. White arrows show indeterminate PKM2/PKM2pS37 formations in the cytoplasm. C) Immunoblot analysis of nuclear and cytoplasmic fractions of MDA-MB-231 cells upon TEPP-46 treatment. D) PKM2 oligomer state analysis in the cytoplasm of TEPP-46 treated cells. GA, glutaraldehyde crosslinking. E) Apoptosis and F) detection of ROS levels in TNBC cells treated with TEPP-46 (10 uM) for 20 hours. Bars, mean values, n = 3; error bars, SD; t test, *P < 0.05; ns, nonsignificant.
Dinaciclib and TEPP-46 target and reduce TNBC PKM2pS37 and tumorigenesis in vivo
To further establish a potential therapeutic effect of Dinaciclib or TEPP-46 on targeting PKM2pS37 in TNBC in vivo we utilized a Rag2 / IL2RG Double Knockout (R2G2) mouse xenograft model. We first examined the tolerability of each drug treatments in a 5-day dosing regimen. No sign of weight loss was observed at any tested dose of Dinaciclib or TEPP-46 ruling out overt toxicity due to treatment with the drug (Figure S4A–B). For tumor efficacy studies, we implanted MDA-MB-231 cells into R2G2 mice and when the tumors reached a size of approximately 125 mm3, randomized them into three treatment arms. One group was treated with 50 mg/kg of Dinaciclib daily for two weeks, another with 40 mg/kg TEPP-46, and the third group with the vehicle (Figure 6A). We then assessed the efficacy of both treatments on tumor growth. The Dinaciclib and TEPP-46 treatment groups each showed decreased tumor growth compared to the vehicle control (Figure 6B). Our observations showed that treatment with Dinaciclib and TEPP-46 reduced PKM2pS37 localization and abundance in vitro. To determine if the drugs caused a similar effect in vivo in the TNBC xenografts, we examined PKM2 localization and phosphorylation in 14 day treated tumors. We observed a robust decrease in total PKM2pS37 expression in tumors after either Dinaciclib or TEPP-46 treatment demonstrating that targeting CDKs upstream of PKM2pS37 or the phosphorylated PKM2 directly is effective at reducing PKM2pS37 in vivo (Figure 6C, Figure S4C). We also examined PKM2pS37 localization and showed that either Dinaciclib or TEPP-46 could significantly reduce nuclear PKM2pS37 with a pronounced reduction in cytoplasmic expression of PKM2pS37 (Figure 6D). Moreover, no significant changes in total PKM2 expression were observed after treatment with either of the drugs (Figure 6C–D). These findings are consistent with our in vitro results and suggest that Dinaciclib and TEPP-46 could be explored as treatment options for TNBC.
Figure 6:
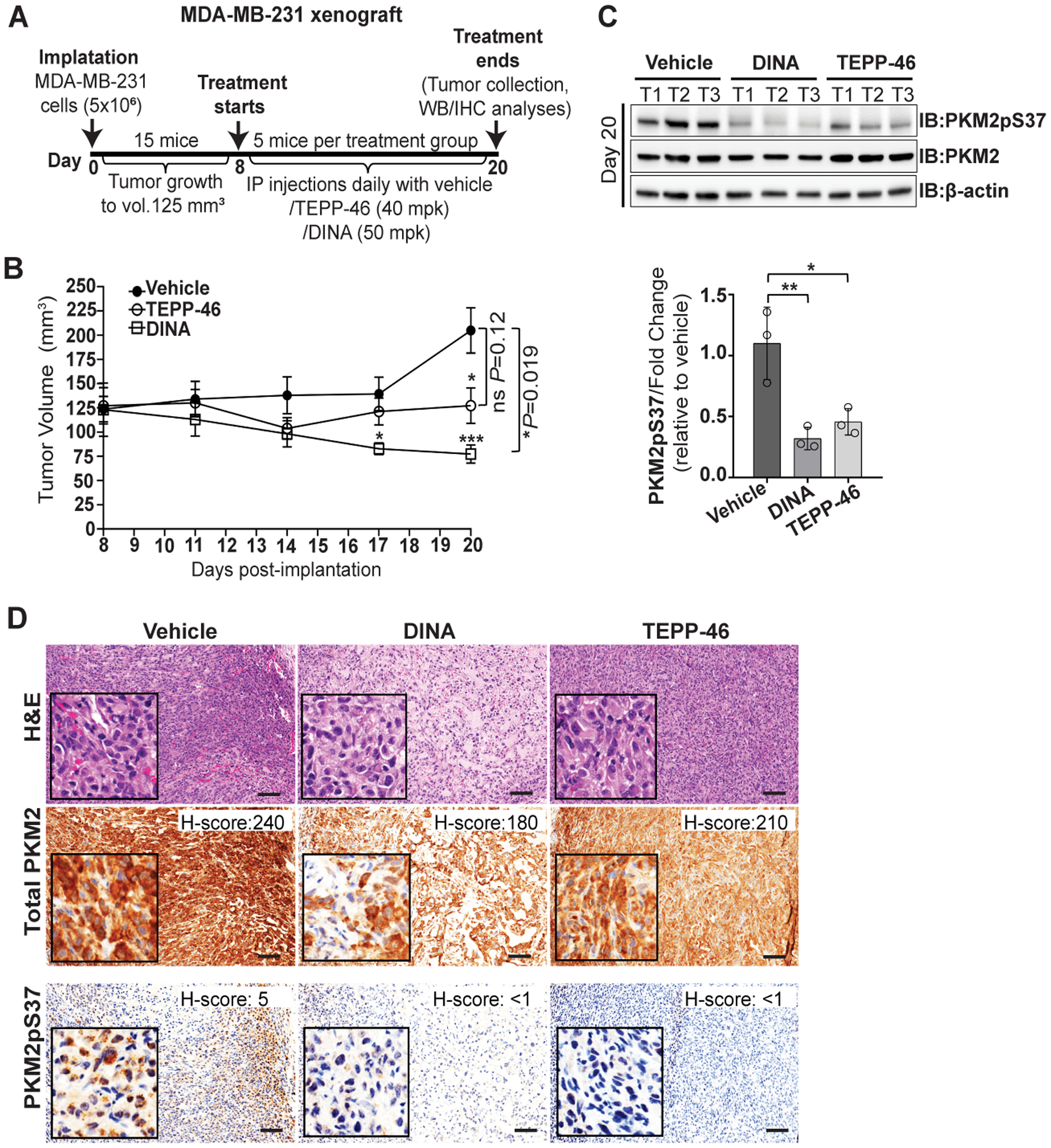
Sustenance of PKM2pS37 supports tumor growth in vivo. A) Overview of MDA-MB-231 xenograft model generation and experimental workflow. B) Tumor volume measurement for two weeks of treatment with Dinaciclib (50 mg/kg) or TEPP-46 (40 mg/kg). Error bars, SEM; one-way ANOVA (Tukey’s HSD), *P = 0.03, ***P = 0.0002. t test, comparison of vehicle group to treatment groups; ns, nonsignificant. C) Immunoblot analysis and D) immunohistochemical analysis of dissected tumors at treatment end point (day 20 post-implantation). Bars, mean values, n = 3; error bars, SD; one-way ANOVA (Tukey’s HSD), *P = 0.03, **P = 0.002; T, tumor. H&E, Hematoxylin and Eosin staining. Scale bar, 100 μm.
Dinaciclib and TEPP-46 combination reduces nuclear translocation of PKM2pS37 and TNBC cell invasion
PKM2pS37 has been described previously as an important integrator of tumorigenic signals in EGFR driven cancer models (13). Given that tumorigenic signals are complex, and frequently intertwined, we aimed to systematically address the potential roles of an EGFR/MEK/ERK/CDK signaling axis with PKM2pS37 in TNBC cells. Treatment of MDA-MB-231 cells with Trametinib (a strong MEK1/2 inhibitor)(45) had little effect on PKM2pS37 levels and suggested that MEK/ERK activation was not a major pathway. In contrast, CDK inactivation with Dinaciclib can lower PKM2pS37 levels while ERK is active (Figure S5A).
We next examined the role of EGFR, and the associate PI3K/Akt pathway in the context of the CDK/PKM2 axis (Figure S5B–C). We performed a series of tests with compounds that target this axis and assessed their apoptotic effects in TNBC cells. These tests revealed that only Dinaciclib could significantly trigger apoptosis in MDA-MB-231 cells (Figure S5D). We also examined combinations of TEPP-46 and inhibitors targeting components of the PI3K/Akt axis. No significant effect in apoptosis was observed when TEPP-46 was combined with these agents (Figure S5E).
Collectively, our findings suggested that TNBC cells could be uniquely targeted by both Dinaciclib and TEPP-46 to lower PKM2pS37 levels and reduce tumorigenesis. Furthermore, this suggests a potent synergistic approach to target PKM2pS37 rich TNBC. Localization studies in TNBC cells showed that Dinaciclib and TEPP-46 can independently deplete nuclear PKM2pS37 (Figure 7A). A significant synergistic effect observed only for MDA-MB-468 cells but not for the highly aggressive MDA-MB-231 cells (Figure 7B). On the contrary we observed a substantial effect on triggering apoptosis when TEPP-46 was used in combination with Dinaciclib in both MDA-MB-231 and MDA-MB-468 cells (Figure 7C). To further evaluate the effects of this combination we tested it in different breast cancer cell lines. The combination of TEPP-46 and Dinaciclib dramatically decreased the clonogenicity of all tested breast cancer cell lines after 2 weeks of treatment (Figure S3D). Importantly, highly aggressive TNBC cells (MDA-231-MB) seemed to be the most responsive to Dinaciclib/TEPP-46 as they showed a continuous apoptotic response, unrelated to necrosis, after 28 hours of treatment (Figure S6A–B). We also examined intracellular levels of ROS and found a significant synergistic effect of Dinaciclib and TEPP-46 on ROS production (Figure 7D). This suggests that, while TEPP-46 binds and alters PKM2pS37 directly in TNBC cells, Dinaciclib treatment further increases the non-phosphorylated PKM2 pool for an enhanced effect of TEPP-46 treatment. The increased intracellular levels of ROS are consistent with the increased apoptosis observed with the combination treatment (Figure 7C). These observations suggest that the major synergy could be nuclear depletion combined with effects on the cytoplasmic pool of PKM2.
Figure 7:
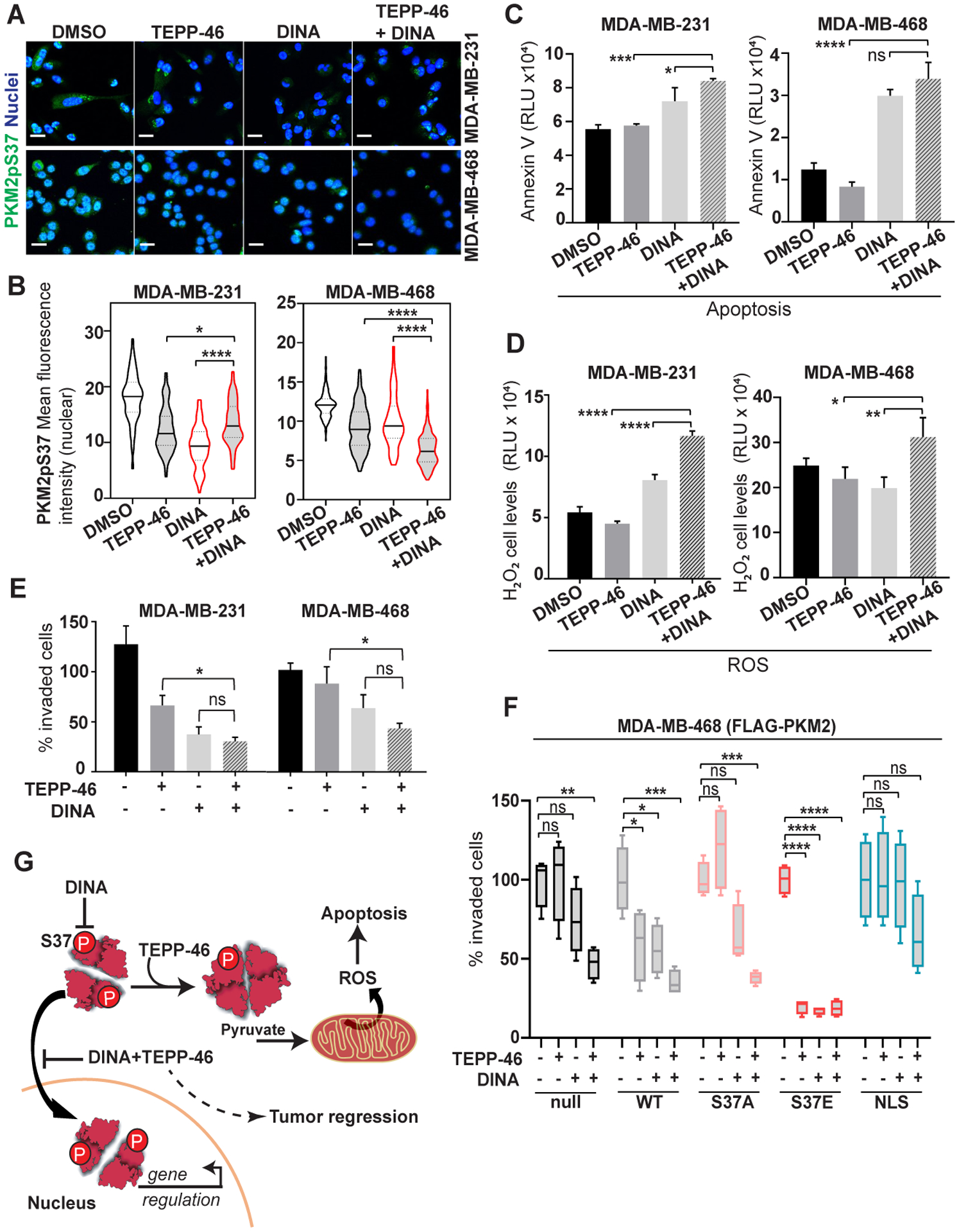
Dinaciclib and TEPP-46 small molecule combination treatment synergistically diminishes PKM2 phosphorylation and its regulatory effects in TNBC cells. A), B) Immunofluorescence analysis of nuclear PKM2pS37 and total PKM2 in TNBC cells treated with Dinaciclib (15 nM) or TEPP-46 (10 uM) or their combination; Nuclear staining, Hoechst 33342 (blue); Scale bar, 20 μm. C) Apoptosis and D) detection of ROS levels in TNBC cells treated with Dinaciclib (100 nM) or TEPP-46 (5 uM) or their combination for 20 hours. Bars, mean values, n = 3; error bars, SD; one-way ANOVA (Tukey’s HSD), *P < 0.05, **P = 0.002, ***P = 0.0002, ****P < 0.0001; ns, nonsignificant. Matrigel invasion assay of E) TNBC cells or F) MDA-MB-468 cells expressing wild-type PKM2 (WT), PKM2 S37 mutants (S37A, S37E) and the PKM2-NLS fusion, treated with Dinaciclib (2.5 nM) or TEPP-46 (5 uM) or their combination. One-way ANOVA (Tukey’s HSD), *P < 0.05, **P = 0.002, ***P = 0.0002, ****P < 0.0001; ns, nonsignificant. G) Schematic representation of a model proposing a therapeutic strategy to reduce the invasive properties of TNBC cells by targeting PKM2pS37.
A prominent feature of the TNBC cell type is enhanced, aggressive migratory phenotypes that characterize the metastatic tumors (46). We assessed the migratory capacity of MDA-MB-231 and MDA-MB-468 cells through Matrigel surfaces under different treatment regimens. Dinaciclib reduced both MDA-MB-231 and MDA-MB-468 cell invasion and showed a synergistic effect when used in combination with TEPP-46 (Figure 7E). To further investigate the effect of the drugs and their combination on the invasive properties of TNBC cells we used MDA-MB-468 with ectopically expressed variants of PKM2. Additional PKM2 expression in MDA-MB-468 cells increased the antimigratory effects of TEPP-46, Dinaciclib, or the combination (Figure 7F). This increased sensitivity for Dinaciclib was suppressed by an S37A mutation, but the effect persisted for TEPP-46 and the combination. On the contrary, cells expressing a phospho-mimetic S37E mutation, which mimics endogenous phosphorylation but shifts PKM2 localization more nuclear (Figure 4D), were profoundly sensitive in all treatments. This mutant lead to reduction of endogenous PKM2pS37 levels (Figure 4D&F) and the striking anti-migratory effects of the drug treatments suggests the low levels of endogenous PKM2pS37, together with the “fixed” phospho-mimetic mutation, produce a strong synergistic effect (Figure 7F). These effects were not observed with ectopic expression of the PKM2-NLS variant which pushes the PKM2 expression more nuclear but maintains endogenous levels of PKM2 S37 phosphorylation and localization. These results suggest that nuclear/cytoplasmic location is dictated by S37 phosphorylation and is required for TNBC cell migration. Furthermore, the phenotypic changes observed by genetic manipulations of the nuclear/cytoplasmic PKM2pS37 pool supports our therapeutic strategy to reduce the invasive properties TNBC cells (Figure 7G).
Discussion
PKM2 has long been identified as a promising drug target in numerous cancer types while PKM2pS37 has only recently been suggested as a therapeutic target in lymphomas and gliomas (10,13). While PKM2 is a well-established cancer biomarker, insight into the role of PKM2pS37 more broadly in cancer is just beginning to emerge. To provide insight into the role of PKM2pS37 in breast cancer we generated a phospho-specific antibody against PKM2pS37 and validated it in TNBC cell lines and patient-derived tumor tissue. This new antibody was used with quantitative immunofluorescence to characterize PKM2pS37 in TNBC patients. This work establishes PKM2pS37 as a prominent feature in TNBC with the potential to stratify breast cancer patients by overall survival. Further studies including a greater number of patients and the development of a monoclonal antibody will give a more definitive assessment of the prognostic value of PKM2pS37 for TNBC. Future work will focus on these aspects as well as examine a broader range of PKM2 positive tumor types for PKM2pS37 expression and localization.
While details of the signaling pathways that control PKM2 phosphorylation are beginning to emerge (10,13), pathways and mechanistic insights were lacking in breast cancer. To better understand the relevant signaling pathways likely to drive PKM2 phenotypes in TNBC we screened candidate small molecules gleaned from the literature and our own knowledgebase. We showed that the potent CDK1/2/5/9 kinase inhibitor Dinaciclib significantly reduces PKM2pS37 in aggressive TNBC cells and notably PKM2pS37 was intact in the absence of detectable MEK/ERK activity. This suggests that PKM2pS37 phosphorylation in breast cancers may be unrelated to the EGFR/MEK/ERK axis which operates in other cancers (13). Moreover, another broadly used CDK4/6 inhibitor against metastatic breast cancer (25–27), Palbociclib, failed to impair PKM2pS37 phosphorylation in TNBC cell lines. This suggests that cancers, non-responsive to Palbociclib and with high levels of PKM2pS37, would benefit from alternative therapies that target CDK1/2/5/9.
Numerous studies have shown that PKM2 regulation is intimately tied to its oligomeric state and intracellular localization. We undertook detailed analysis of these important aspects of PKM2 biology in various breast cancer models armed with our new knowledge of the prominent role of CDK1/2/5/9 kinase regulation. We confirmed that PKM2pS37 dictates nuclear localization in breast cancer akin to other cancers and CDK1/2/5/9 kinases play a unique role. Future efforts with more selective CDK inhibitors could further narrow the list, however, our co-immunoprecipitation studies showed direct interactions with CDK1/cyclin B1 complexes in multiple breast cancer cell lines. We then examined how the PKM2 activator TEPP-46 would target PKM2pS37 in TNBC and potentially synergize with CDK targeting therapies. We showed that TEPP-46 associated with PKM2pS37 in TNBC cells and prevented PKM2 nuclear translocation and altered its oligomeric status. These results suggested that TNBC cells have a prominent pool of PKM2pS37 that is accessible to small molecules that target PKM2 directly and could synergize with CDK inhibitors such as Dinaciclib. We then showed that Dinaciclib and TEPP-46 had synergistic effects to target PKM2pS37 in highly aggressive TNBC cells. The combination treatment showed significant effects on redox balance, invasiveness, and promoted cell death. The two small molecules appear to work together to reduce nuclear PKM2 accumulation and thereby shift a more active pool of PKM2 to the cytoplasm. In support of this mechanism, we found that ectopic expression of PKM2 mutants in MDA-MB-468 TNBC cells altered the endogenous pool of phosphorylated PKM2 with direct effects on the migratory phenotype and sensitivity to both TEPP-46 and Dinaciclib. We also showed that reducing PKM2 phosphorylation with Dinaciclib increased ROS production and cell death in combination with TEPP-46. This mechanism is in clear contrast to steady state PKM2pS37 which is largely nuclear in TNBC and consistent with studies in other cancers that show less active cytoplasmic PKM2 pool sustains cancer cell viability (6,7,12,13,47,48).
Our results consistently mark a nuclear fraction of PKM2pS37 in TNBC cell lines, xenografts, and patient derived tumors that point to a new therapeutic opportunity. We have identified small molecules that manipulate this pool of phosphorylated PKM2 in vitro and in vivo and provide mechanistic insight into the role of PKM2pS37 in breast cancer. Importantly, we have suggested an effective way to identify TNBC tumors which may be susceptible to combination treatments with Dinaciclib and TEPP-46. Given that these agents have been used separately in other unrelated pre-clinical and clinical applications with positive outcomes (22–24,31,49–51), our approach against TNBC could be rapidly translated into the clinic.
Supplementary Material
Significance.
PKM2 phosphorylation marks aggressive breast cancer cell phenotypes and targeting PKM2pS37 could be an effective therapeutic approach for treating triple-negative breast cancer.
Acknowledgments
We thank Svetlana Rogulina for technical support and overall help. We thank Yalai Bai, Lisa Gras, Yan Song, Melissa Leggio of Yale Pathology Tissue Services and Specialized Translational Services Laboratory and Yuping Qian and Man Li of Yale Center for Precision Cancer Modeling for their services. We thank Terence Wu of the West Campus Analytical Core and Joerg Nikolaus of the West Campus Imaging Core for training on the use and maintenance of equipment. We thank Dr. Craig Thomas, leader of chemistry technologies at the National Institutes of Health (NIH), National Center for Advancing Translational Sciences (NCATS), for providing us with the TEPP-46 compound and sharing critical guidance for formulation. This study was also supported by the National Institutes of Health (NIH) / National Cancer Institute (NCI) U54 Grant #CA209992 to J.R. and a Lion Heart Foundation grant to J.R.
Footnotes
Declaration of Interests
D.L.R. has served as an advisor for Astra Zeneca, Agendia, Amgen, BMS, Cell Signaling Technology, Cepheid, Danaher, Daiichi Sankyo, Genoptix/Novartis, GSK, Konica Minolta, Merck, NanoString, PAIGE.AI, Perkin Elmer, Roche, Sanofi, Ventana and Ultivue. The other authors declare no competing interests.
References
- 1.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324(5930):1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008;452(7184):230–3. [DOI] [PubMed] [Google Scholar]
- 3.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013;155(2):397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahra K, Dey T, Ashish, Mishra SP, Pandey U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol 2020;10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep 2016;17(12):1721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris RA, Fenton AW. A critical review of the role of M(2)PYK in the Warburg effect. Biochim Biophys Acta Rev Cancer 2019;1871(2):225–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, function and expression and regulation. Cell Biosci 2019;9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prakasam G, Iqbal MA, Bamezai RNK, Mazurek S. Posttranslational Modifications of Pyruvate Kinase M2: Tweaks that Benefit Cancer. Front Oncol 2018;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2009;2(97):ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017;546(7658):426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Liu J, Jin X, Zhang D, Li D, Hao F, et al. O-GlcNAcylation destabilizes the active tetrameric PKM2 to promote the Warburg effect. Proc Natl Acad Sci U S A 2017;114(52):13732–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 2012;8(10):839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 2012;14(12):1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol 2015;43:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashizawa K, Willingham MC, Liang CM, Cheng SY. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J Biol Chem 1991;266(25):16842–6. [PubMed] [Google Scholar]
- 16.Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005;44(27):9417–29. [DOI] [PubMed] [Google Scholar]
- 17.Srivastava D, Razzaghi M, Henzl MT, Dey M. Structural Investigation of a Dimeric Variant of Pyruvate Kinase Muscle Isoform 2. Biochemistry 2017;56(50):6517–20. [DOI] [PubMed] [Google Scholar]
- 18.Jiang J, Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, et al. ML265: A potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. Probe Reports from the NIH Molecular Libraries Program. Bethesda MD: 2010. [PubMed] [Google Scholar]
- 19.Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, et al. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4(+) T Cell Pathogenicity and Suppresses Autoimmunity . Cell Metab 2020;31(2):391–405 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med 2017;23(6):753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le S, Zhang H, Huang X, Chen S, Wu J, Chen S, et al. PKM2 Activator TEPP-46 Attenuates Thoracic Aortic Aneurysm and Dissection by Inhibiting NLRP3 Inflammasome-Mediated IL-1beta Secretion. J Cardiovasc Pharmacol Ther 2020:1074248420919966. [DOI] [PubMed] [Google Scholar]
- 22.Saleme B, Gurtu V, Zhang Y, Kinnaird A, Boukouris AE, Gopal K, et al. Tissue-specific regulation of p53 by PKM2 is redox dependent and provides a therapeutic target for anthracycline-induced cardiotoxicity. Sci Transl Med 2019;11(478). [DOI] [PubMed] [Google Scholar]
- 23.Tee SS, Park JM, Hurd RE, Brimacombe KR, Boxer MB, Massoud TF, et al. PKM2 activation sensitizes cancer cells to growth inhibition by 2-deoxy-D-glucose. Oncotarget 2017;8(53):90959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Bailleul J, Yazal T, Dong K, Sung D, Dao A, et al. PK-M2-mediated metabolic changes in breast cancer cells induced by ionizing radiation. Breast Cancer Res Treat 2019;178(1):75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roskoski R Jr. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol Res 2016;107:249–75. [DOI] [PubMed] [Google Scholar]
- 26.Roskoski R Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res 2019;139:471–88. [DOI] [PubMed] [Google Scholar]
- 27.Rugo HS, Finn RS, Diéras V, Ettl J, Lipatov O, Joy AA, et al. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res Treat 2019;174(3):719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 2010;9(8):2344–53. [DOI] [PubMed] [Google Scholar]
- 29.Criscitiello C, Viale G, Esposito A, Curigliano G. Dinaciclib for the treatment of breast cancer. Expert Opin Investig Drugs 2014;23(9):1305–12. [DOI] [PubMed] [Google Scholar]
- 30.Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer 2014;14(3):169–76. [DOI] [PubMed] [Google Scholar]
- 31.Mitri Z, Karakas C, Wei C, Briones B, Simmons H, Ibrahim N, et al. A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Invest New Drugs 2015;33(4):890–4. [DOI] [PubMed] [Google Scholar]
- 32.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med 2002;8(11):1323–7. [DOI] [PubMed] [Google Scholar]
- 33.Gassaway BM, Petersen MC, Surovtseva YV, Barber KW, Sheetz JB, Aerni HR, et al. PKCε contributes to lipid-induced insulin resistance through cross talk with p70S6K and through previously unknown regulators of insulin signaling. Proc Natl Acad Sci U S A 2018;115(38):E8996–E9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gassaway BM, Cardone RL, Padyana AK, Petersen MC, Judd ET, Hayes S, et al. Distinct Hepatic PKA and CDK Signaling Pathways Control Activity-Independent Pyruvate Kinase Phosphorylation and Hepatic Glucose Production. Cell Rep 2019;29(11):3394–404 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci U S A 2014;111(43):15526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan Y, Wang W, Huang S, Ni W, Wei Z, Cao Y, et al. Beta-elemene inhibits breast cancer metastasis through blocking pyruvate kinase M2 dimerization and nuclear translocation. J Cell Mol Med 2019;23(10):6846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng B, Geng L, Zeng L, Liu F, Huang Q. AKT2 contributes to increase ovarian cancer cell migration and invasion through the AKT2-PKM2-STAT3/NF-κB axis. Cell Signal 2018;45:122–31. [DOI] [PubMed] [Google Scholar]
- 38.Wong CC, Au SL, Tse AP, Xu IM, Lai RK, Chiu DK, et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS One 2014;9(12):e115036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou HL, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019;565(7737):96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Z, Li M, Zhang L, Zhao H, Şahin Ö, Chen J, et al. Oncogenic Kinase-Induced PKM2 Tyrosine 105 Phosphorylation Converts Nononcogenic PKM2 to a Tumor Promoter and Induces Cancer Stem-like Cells. Cancer Res 2018;78(9):2248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pernas S, Tolaney SM, Winer EP, Goel S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther Adv Med Oncol 2018;10:1758835918786451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011;145(5):732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011;480(7375):118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012;150(4):685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schafer JM, Lehmann BD, Gonzalez-Ericsson PI, Marshall CB, Beeler JS, Redman LN, et al. Targeting MYCN-expressing triple-negative breast cancer with BET and MEK inhibitors. Sci Transl Med 2020;12(534). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koedoot E, Fokkelman M, Rogkoti VM, Smid M, van de Sandt I, de Bont H, et al. Uncovering the signaling landscape controlling breast cancer cell migration identifies novel metastasis driver genes. Nat Commun 2019;10(1):2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011;334(6060):1278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan HP, O’Reilly FJ, Wear MA, O’Neill JR, Fothergill-Gilmore LA, Hupp T, et al. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci U S A 2013;110(15):5881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep 2016;17(9):2367–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y, et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res 2018;78(3):742–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajput S, Khera N, Guo Z, Hoog J, Li S, Ma CX. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget 2016;7(35):56864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.