

These results identify CIP2A as a nongenetic driver and therapeutic target in basal-like breast cancer that regulates DNA damage–induced G2–M checkpoint and proliferative signaling.
Abstract
Basal-like breast cancers (BLBC) are characterized by defects in homologous recombination (HR), deficient mitotic checkpoint, and high-proliferation activity. Here, we discover CIP2A as a candidate driver of BLBC. CIP2A was essential for DNA damage–induced initiation of mouse BLBC-like mammary tumors and for survival of HR–defective BLBC cells. CIP2A was dispensable for normal mammary gland development and for unperturbed mitosis, but selectively essential for mitotic progression of DNA damaged cells. A direct interaction between CIP2A and a DNA repair scaffold protein TopBP1 was identified, and CIP2A inhibition resulted in enhanced DNA damage–induced TopBP1 and RAD51 recruitment to chromatin in mammary epithelial cells. In addition to its role in tumor initiation, and survival of BRCA-deficient cells, CIP2A also drove proliferative MYC and E2F1 signaling in basal-like triple-negative breast cancer (BL-TNBC) cells. Clinically, high CIP2A expression was associated with poor patient prognosis in BL-TNBCs but not in other breast cancer subtypes. Small-molecule reactivators of PP2A (SMAP) inhibited CIP2A transcription, phenocopied the CIP2A-deficient DNA damage response (DDR), and inhibited growth of patient-derived BLBC xenograft. In summary, these results demonstrate that CIP2A directly interacts with TopBP1 and coordinates DNA damage–induced mitotic checkpoint and proliferation, thereby driving BLBC initiation and progression. SMAPs could serve as a surrogate therapeutic strategy to inhibit the oncogenic activity of CIP2A in BLBCs.
Significance:
These results identify CIP2A as a nongenetic driver and therapeutic target in basal-like breast cancer that regulates DNA damage–induced G2–M checkpoint and proliferative signaling.
Introduction
One of the most aggressive and clinically challenging breast cancer subtypes is the basal-like breast cancer (BLBC; refs. 1–3). On the basis of transcriptional signatures of the breast cancer subtypes (4), the hallmarks of BLBCs are high proliferation activity, G2–M checkpoint dysregulation, ATR/BRCA pathway activity, and high DNA replication. In addition, molecular characteristics of BLBCs include high genetic instability, BRCA mutations, TP53 inactivation, and dysregulation of EGFR (1–4). About 75% of BLBCs belong to the triple-negative breast cancer subtype (BL-TNBC), devoid of ER, PR, and HER2 (1). In addition to their frequently aggressive clinical appearance, the lack of these targetable receptors makes BLBCs therapeutically very challenging. Despite the near saturated genetic knowledge of breast cancer, like some other subtypes as well, no clear genetic oncogenic drivers have been identified for the BLBCs (1, 3).
Among the breast cancer subtypes, BLBCs have the highest mutational burden as a result of acquisition of BRCA mutations, or other defects in the homologous recombination (HR) pathways (1–4). Normal HR-proficient cells respond to double-stranded DNA breaks (DSB) by activating the G2–M cell-cycle checkpoint, resulting in mitotic arrest (5). To allow mitotic progression under DNA-damaging conditions, transformed cells instead have developed strategy to dampen G2–M checkpoint signaling (5, 6). On the basis of the high mutational burden observed in BLBC, it could be hypothesized that a potential BLBC driver mechanism has the ability to coordinately dampen the DNA damage–induced G2–M checkpoint, and to support high-proliferation activity in premalignant mammary epithelial cells. One of the DDR proteins involved in G2–M checkpoint signaling is DNA topoisomerase II binding protein 1 (TopBP1; ref. 7), which is a scaffold protein that interacts with the checkpoint kinase ATR (8). In the presence of DSBs, TopBP1 promotes RAD51 chromatin loading, resulting in G2–M arrest (9–12). RAD51 foci formation has been associated in BLBCs as a marker of BRCA deficiency and HR impairment (4, 13). These features make TopBP1-mediated RAD51 regulation a candidate G2–M checkpoint mechanism in BLBC (5, 7, 10).
Recently, the serine/threonine phosphatase PP2A has gained attention as a druggable tumor suppressor (14–16). Of specific relevance to this work, is the role of serine/threonine phosphatases in the DNA-damage response (DDR) at chromatin (6), which could link them to cancer types with HR defects, and high mutational burden, such as BLBCs. PP2A is inhibited in most cancers by nongenetic mechanisms, including high expression of endogenous inhibitor proteins such as CIP2A, PME-1 or SET, or changes in carboxymethylation to the C-terminal tail of the catalytic domain (15, 17). CIP2A is expressed at low levels in normal mammary gland tissue (18). However, CIP2A transcription is induced by TP53 mutation via increased E2F1 activity (18, 19), and by EGFR pathway activation (20, 21), features closely linked to BLBC (1). However, it is currently unclear what role CIP2A plays in BLBC initiation, or progression. Notably, understanding of CIP2A-related cancer initiation mechanisms is also therapeutically relevant due to the recent development of small-molecule activators of the CIP2A-inhibited PP2A-B56 heterotrimer that demonstrate potent antitumor activities (14, 16).
In this study, we provide the first evidence for an essential role for CIP2A in tumor initiation in cancer. Specifically, we demonstrate by using both chemical and transgenic tumor models that CIP2A is essential for the initiation of mouse BLBC tumors, but not for the initiation of skin, ovarian, lung or stomach tumors. Furthermore, among transformed breast cancer cell types, CIP2A is essential for survival of BRCA/TP53-mutant BLBC cells. Mechanistically, the role for CIP2A in driving BLBC initiation and progression can be explained by its capacity to coordinately regulate key BLBC hallmarks; specifically the G2–M checkpoint and cellular proliferation. Molecularly, we discover direct CIP2A interaction with TopBP1 and provide evidence for CIP2A-mediated inhibition of both TopBP1 and RAD51 recruitment to chromatin upon DNA damage in premalignant mammary epithelial cells. CIP2A also promotes pro-proliferative MYC and E2F1 activities in BLBC cells. Finally, we discover that small molecules shown previously to activate PP2A (14, 16), transcriptionally inhibit CIP2A expression, and serve as candidate therapeutics for CIP2A-positive BLBCs.
Materials and Methods
Mouse experiments
All animal work protocols were approved either by the Project Authorization Board of the Regional State Administrative Agency for Southern Finland, the Animal Ethics Committee of the Netherlands Cancer Institute or the Institutional Animal Care and Use Committee at the Case Western Reserve University, which is certified by the American Association of Accreditation for Laboratory Animal Care under protocol # 2013–0132.
For DMBA (7,12-dimethylbenz[a]anthracene)-induced tumors in wild-type (WT) and Cip2a−/− female mice, several independent cohorts of the mice were administered with 1 mg of DMBA dissolved in 200 μL of corn oil by oral gavage once a week for 6 weeks starting at 12–14 weeks of age as previously described (22). The mice were monitored twice a week for tumor formation until morbidity. Mice were sacrificed upon tumor burden and/or when they showed general signs of illness. For DMBA-induced mutation load and Cip2a mRNA expression in WT and Cip2a−/− premalignant mammary gland tissues, the mice were sacrificed 2 weeks after the last DMBA treatment. DMBA/TPA protocol for skin tumorigenesis and experiments with Cip2a−/− mice crossed with an ovarian cancer mouse model TgMISIIR-Tag are described in Supplementary Materials and Methods. Tissue samples collected for extraction of RNA and genomic DNA were snap-frozen into liquid nitrogen. Tissue samples for histochemical and for immunohistochemical analyses were fixed in formalin.
Mouse tumor cell lines were generated from spontaneous mammary tumors of following breast cancer mouse models: K14Cre; Brca1F/F; Trp53F/F(KB1P; ref. 23), K14Cre; Cdh1F/F; Trp53F/F(KEP; ref. 24), and Wap-cre; Cdh1F/F; Akt1E17K(WEA; ref. 25). Tumor cell lines were generated by collecting tumors in cold PBS and minced by chopping with scalpels. Aggregates were plated out. KEP and WEA tumor cell line cultures were incubated at 37°C with 5% CO2 and 20% O2. KB1P cell lines were incubated at 37°C with 5% CO2 and 3% O2. Homogenous epithelial cell morphology was obtained after cultures were passaged 2–3 times. Used cell culture media are described previously in Supplementary Table S1.
Analysis of human breast cancer patient sample cohorts
The FinHer study (HUCH 426/E6/00) was approved by an ethics committee of the Helsinki University Central Hospital (Helsinki, Finland). Study participants provided written informed consent before study entry. The study was conducted in accordance of the Declaration of Helsinki. The role of CIP2A in disease-free survival of patients with breast cancer in the GSE21653 cohort was analyzed by using an online platform “R2: Genomics Analysis and Visualization Platform” (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). More detailed description of analysis of both cohorts can be found from Supplementary Data.
Cell culture and transfections
All the commercial cell lines used in this article were purchased from the ATCC or Leibniz Institute's German Collection of Microorganisms and Cell Cultures (DSMZ). All the cells in culture were negative on periodically testing for Mycoplasma using the Mycoplasma Detection Kit (Roche). All the human and mouse cells, their culture conditions and supplements used for cell culture are listed in Supplementary Table S1. Breast cancer stem-like cells were isolated from patients with TNBC who received standard chemotherapy and cultured as described previously (26).
Antibodies, RNAs, primers, DNA constructs and drugs
Antibodies (along with dilutions for each application), plasmids and sequences of siRNAs, gRNAs, and primers used are listed in Supplementary Table S2. All chemical inhibitors and drugs used are listed in the Supplementary Table S3.
Mitotic index experiments
Mitotic index experiments were conducted by modifying previously published protocol described in (7). Briefly, MCF10A cells were transfected with indicated siRNAs for 24 hours, following which, they were seeded into ibidi 8-well μ slides (ibiTreat #80826) for 24 hours. Cells were irradiated with 10Gy radiation followed by nocodazole block (100 ng/mL), one hour after IR for 18 hours. After the indicated time points, cells were stained for phosphohistone H3 (Ser10) using similar immunofluorescence protocols as mentioned above. Images were taken on Zeiss Axiovert or EVOS fl Microscope with ×10 objective and quantified using ImageJ software (RRID:SCR_003070). Experiment was repeated 3 times.
Results
Cip2a is selectively required for initiation of DMBA-induced mammary tumors in mice
To address whether CIP2A is truly essential for the initiation of cancer in vivo, we challenged several independent cohorts of the previously described Cip2a−/− mice (19, 27) with a chemical carcinogenesis protocol consisting of six consecutive doses of the genotoxic agent DMBA (Fig. 1A). Similar to other polyaromatic hydrocarbons, DMBA forms covalent DNA adducts, and induces a DDR, including activation of γH2AX, ATR, and RAD51 (28). Oral exposure of mice to DMBA induces mouse BLBCs (22), but also several other cancers, allowing us to assess the relative importance of Cip2a to tumor development across different cancer types. As compared with a model combining DMBA and medroxyprogesterone acetate, the DMBA-only mammary tumors are initiated with much longer latency (22). Importantly, the tumor-initiating cell type of mammary tumors induced by DMBA-only is basaloid cells, and molecularly these tumors faithfully resemble human BLBCs (22).
Figure 1.

Cip2a knockout mice are selectively resistant to DMBA-induced mammary tumorigenesis. A, DMBA was orally administered to WT and Cip2a−/− mice once a week for 6 consecutive weeks, after which, mice were monitored for signs of spontaneous tumor formation. B, Number of genetic variants in exons of the expressed genes in nontreated (control) and DMBA-treated WT (n = 3) and Cip2a−/− (n = 3) mouse mammary glands. P value by the Wilcoxon test. C, Incidences of tumor formation in different tissues in sacrificed DMBA-administered WT (n = 18) and Cip2a−/− (n = 14) mice. P values between WT and Cip2a−/− groups calculated by the Fisher exact test. D, Incidence of mammary tumors in WT (n = 18) and Cip2a−/− (n = 14) mice after starting administration of DMBA. P value by the log-rank test. E, Mitotic index analysis of MCF10A cells transfected with the indicated siRNAs. Cells were treated with 10 Gy radiation dose and nocodazole (100 ng/mL) block 1 hour after IR for 18 hours. Mitotic cells were stained using phosphohistone H3 at Ser10. Scale bar, 100 μm. Bar graph shows the percentage of H3pS10-positive nuclei from three replicates, expressed as mean ± SD. Statistical analyses were conducted using unpaired t test for unequal variances. *, P < 0.05; ***, P < 0.001. F, Heat map of fraction of gene-trap insertions in the sense orientation compared with the total (sense and antisense) insertions in untreated HAP1 cells and HAP1 cells treated with successive low doses of IR (5 × 1 Gy). Color coding indicates essentiality of the indicated PP2A inhibitor protein for cell survival. n.s., nonsignificant.
As expected (28), DMBA treatment induced a significant increase in the mutational load in nontumorigenic mammary gland tissue as soon as only 2 weeks after the last DMBA dose; however, the mutational load (Fig. 1B; Supplementary Table S4), or overall survival (Supplementary Fig. S1A) was not associated with the Cip2a genotype. When assessed by palpation, external observation, and by tissue pathology analysis upon autopsy of the mice with any symptoms of reduced well-being, tumors in five different tissue types were observed in the DMBA-treated mice (Fig. 1C). Notably, although incidences of tumors in ovary, lung, skin or stomach were not altered in Cip2a−/− mice, mammary tumors showed almost absolute dependence on Cip2a for tumor initiation (Fig. 1C and D; Supplementary Fig. S1B).
To control for the possibility that lack of genotype dependence of other cancer types on Cip2a was a result of leakage of the genetrap cassette used for Cip2a gene silencing (27), we confirmed the absence of CIP2A protein expression in ovarian cancer tissues from Cip2a−/−mice (Supplementary Fig. S1C). We further confirmed that Cip2a was dispensable for skin and ovarian tumorigenesis by independent in vivo models. To this end, we crossed Cip2a−/− mice with the MISIIR-Tag mouse model producing tumors resembling high-grade ovarian cancer (29), and did not observe any notable difference in ovarian tumorigenesis between Cip2a WT or Cip2a−/− mice by PET/CT-imaging or by visual inspection after autopsy (Supplementary Fig. S1D and S1E). For the skin tumorigenesis model, we used a classical DMBA/TPA two-stage skin tumorigenesis protocol and again did not observe any differences in skin tumor initiation between the Cip2a genotypes (Supplementary Fig. S1F).
Combined, these results in multiple independent in vivo models strongly suggest that CIP2A is required for the propagation of DNA-damaged mammary basaloid epithelial cells. To validate that this is a cell intrinsic property of CIP2A, we tested the impact of CIP2A silencing on mitotic progression of MCF10A basal-like immortalized mammary epithelial cells treated with ionizing radiation (IR). Notably, whereas inhibition of checkpoint kinase CHK1 abrogated the G2–M checkpoint and CIP2A silencing did not impact mitotic progression of untreated MCF10A cells, CIP2A was found to be indispensable for G2–M progression in IR-treated MCF10A cells (Fig. 1E; Supplementary Fig. S1G). To provide independent validation to these results, we surveyed results from a genetic screen in HAP1 cells (see Supplementary Fig. S1H for technical description; ref. 30). Directly supportive of the role for CIP2A in allowing cell propagation under DNA damage, CIP2A was the only tested PP2A inhibitor protein that was essential under repeated low-dose irradiation (Fig. 1F).
These results establish essentiality for Cip2a for the initiation of DNA damage–induced mammary tumors previously confirmed to faithfully represent mouse BLBCs (22). As such the results represent the first evidence for a critical role for CIP2A in tumor initiation.
Cip2a is induced by DMBA in premalignant mammary gland tissue and drives initiation of mouse BLBC-like tumors
A key criterion for a cancer driver candidate involved in tumor initiation is expression in premalignant tissue prior tumorigenesis. Consistent with negligible CIP2A protein expression in normal human mammary glands (18), Cip2a mRNA was expressed at a very low level in WT mouse mammary glands (Fig. 2A). Importantly, nontumorous WT mammary glands sampled 2 weeks after the last dose of DMBA (Fig. 1A) displayed significantly increased Cip2a mRNA expression (Fig. 2A). In line with a suggested role as a disease driver, Cip2a mRNA expression was induced significantly more in mammary tumors from DMBA-treated WT mice (Fig. 2A). However, as mammary tumors were induced in only some of the mammary glands in each of the DMBA-treated WT mice, we conclude that Cip2a expression is essential, but not sufficient alone to induce tumorigenesis.
Figure 2.

Cip2a drives initiation of mouse BLBC-like tumors. A, qRT-PCR analysis of Cip2a mRNA expression normalized to Actb and Gapdh from WT and Cip2a−/− nontreated (Ctrl) and DMBA-administered mouse nontumorigenic mammary glands (MG), and from WT DMBA-induced mammary tumors. Shown is mean ± SD of 10 WT and 9 Cip2a−/− nontreated mammary glands (Ctrl MG), 3 WT, and 3 Cip2a−/− mammary glands from DMBA-administered mice, and 16 mammary tumors. P values calculated by the Mann–Whitney test. B, Representative images of immunohistochemical staining of keratin-14 (K14), keratin-8 (K8), CIP2A, Ki67 and MYC proteins and hematoxylin and eosin (H&E) from DMBA-induced mammary tumors from WT mice. Scale bar, 50 μm. C, Semiquantitative analysis of receptor status from 10 WT tumors. D, Top five enriched hallmark gene sets based on differentially regulated genes in nontumorigenic Cip2a KO mouse mammary glands treated with six doses of DMBA.
Based upon the molecular characterization of the mammary tumors from DMBA-treated WT mice, the majority of the characterized tumors had BLBC or BL-TNBC phenotypes (Fig. 2A and C; Supplementary Fig. S2A). This is consistent with a previous report demonstrating that the tumor-initiating cells from this DMBA model are of basaloid origin (22). The small number of estrogen receptor (ER)–positive tumors observed is consistent with The Cancer Genome Atlas (TCGA) data that about 10% of human BLBCs are ER positive. Furthermore, although use of different DMBA in vivo protocol was recently shown to result in ER-positive tumors (31), it was demonstrated by using the DMBA protocol used in our study that serial transplantation of ER-positive tumors yielded ER-negative secondary tumors, suggesting that ER expression was not a driver mechanism in these tumors (22). To study the impact of Cip2a on DMBA-induced mammary gland gene expression profiles, we performed RNA-sequencing analysis of non-tumorigenic mammary glands from mice housed for more than 4 months after the last dose of DMBA. Fully consistent with basaloid and homologous recombination–defective (HRD) phenotype, Cip2a was found to control G2–M checkpoint, and proliferative signaling via the MYC and E2F1 pathways (Fig. 2D). The tumors in WT mice were also highly proliferative based on Ki67 staining, and displayed both CIP2A and MYC protein overexpression potentially indicative of their known feed-forward regulatory loop (Fig. 2B; ref. 32). Evaluating the CIP2A positivity in DMBA-treated pre-tumorigenic mammary gland duct epithelial cells and from mammary tumors in WT mice, revealed predominantly cytoplasmic, but also nuclear CIP2A protein expression (Supplementary Fig. S2B). This pattern is consistent with previous analysis, indicating that there is a small pool of nuclear CIP2A in propagating cells (33), and indicate a potential function for nuclear CIP2A in early mammary tumorigenesis. Notably, the lack of predominantly BLBC tumors in Cip2a−/− mice was not related to any genotype-associated alterations in the basal and luminal epithelial cell ratio in the mammary gland (Supplementary Fig. S2C–S2F). Furthermore, we did not observe any notable differences in the mammary gland development and branching morphogenesis between WT and Cip2a−/− mice (Supplementary Fig. S2G).
Collectively, these results demonstrate that although Cip2a is dispensable for normal mouse mammary development, DMBA-elicited induction of Cip2a mRNA expression is required for initiation of mouse BLBC-like tumors.
Codependence analysis reveals a functional association between CIP2A, TopBP1, and G2–M checkpoint regulation
Although our results strongly indicate that the previously reported CIP2A-MYC feed-forward loop (32, 34, 35) is highly relevant for proliferation in DMBA-induced BLBCs (Fig. 2B; Supplementary Fig. S2A), MYC regulation is unlikely to explain either the newly discovered role for CIP2A in G2–M checkpoint regulation (Fig. 1E and F; Supplementary Fig. S2A), or its essentiality for in vivo propagation of DNA damaged mammary epithelial cells (Fig. 1C and D; Supplementary Fig. S2G). Furthermore, as TPA/DMBA-induced skin tumorigenesis is dependent on MYC (36), but totally independent of CIP2A (Supplementary Fig. S1F), we hypothesized that CIP2A is involved in the regulation of a yet uncharacterized MYC-independent, but HRD-related mechanism. To identify such mechanism in an unbiased manner, we surveyed a CRISPR/Cas9-based dropout screen repository from DepMap (Avana 2020 Q1; https://depmap.org), to identify genes that are most significantly similar in their essentiality with CIP2A.
Remarkably, across 739 human cancer cell lines all the top 10 co-dependent genes with CIP2A (i.e., functionally most similar to CIP2A) were DDR genes (Fig. 3A). Notably, out of these top 10 CIP2A-associated DDR factors, CIP2A was at the genome-wide level the most significantly similar gene for RHNO1, TOPBP1, POLQ, NBN, and PARP1 (Fig. 3A). In the case of TOPBP1, the codependency with CIP2A was greater than with ATR (Fig. 3B), which is the bona fide TopBP1 DDR effector (7, 8). On the other hand, strongly supporting the selectivity of CIP2A's association with DDR mechanisms, and arguing against MYC being the sole effector of CIP2A in tumorigenesis, there was no overlapping genes found among the top ten co-dependent genes between CIP2A and MYC (Fig. 3A; Supplementary Fig. S3A).
Figure 3.

Codependence analysis reveals functional association of CIP2A with critical DNA damage response proteins. A, Top 10 codependencies with CIP2A across 739 cell lines genome-wide from CRISPR Avana screen. CIP2A's own codependency rank for the top 10 genes is also listed. Data extracted from DepMap portal (Avana 2020Q1). B, Genome-widely, CIP2A is the closest functional homolog to RHNO1 and TOPBP1. C, STRING functional protein association network analysis of CIP2A codependent proteins from A. By using the highest data confidence score (0.9), except for APEX2, DSCC1, and CIP2A, the other proteins form a highly connected protein network. NBN phosphorylation indicated by red dot was found to be regulated by CIP2A based on ref. 37. D, Top 10 Reactome pathways associated with genes from A. E, Correlation between CIP2A and TOPBP1 mRNA expression across 1,156 cell lines from Cancer Cell Line Encyclopedia. F, Pair-wise correlation of dependence of either BLBC or HGSOC cell lines of the indicated genes from DepMap portal (Avana 2020Q1). The values for BLBC and HGSOC indicate correlation (max. 1) in dependence of the cells for the genes in the gene pair; the higher number indicates higher similarity in the dependence. The color-coded numbers indicate the difference in the codependence between BLBC and HGSOC cells for the indicated gene pair.
When analyzed for functional protein association networks by STRING database (https://string-db.org), the top CIP2A-associated proteins (Fig. 3A) formed a tight protein network (Fig. 3C), that was functionally linked with HRD-associated processes such as “G2–M DNA damage checkpoint,” “homology-directed repair,” and “processing of DNA DSB ends” (Fig. 3D). Interestingly, in a recent PP2A-related phosphoproteome survey (37), CIP2A was found to prevent the dephosphorylation of Nibrin (NBN), which was one of the TopBP1 protein network members (Fig. 3C). NBN is known to cooperate with TopBP1 in ATR activation, and dephosphorylation of serine 432 of NBN, as observed in CIP2A-depleted cells (Supplementary Fig. S3B; ref. 37), impairing cell survival under IR (38). CIP2A was also recently found to be a NBN codependent gene in PARP inhibitor–treated cells (39).
Additional evidence for the intertwining of CIP2A with the TopBP1 complex was obtained by mRNA coexpression analysis across 1156 cell lines from the Broad institute Cancer Cell line Encyclopedia. Of the CIP2A codependent genes (Fig. 3A), TOPBP1 and POLQ were also among the 25 most significantly coexpressed genes with CIP2A (Fig. 3E; Supplementary Fig. S4A). Both TOPBP1 and POLQ showed very significant coexpression with CIP2A also when only the BLBC cell lines were surveyed (Supplementary Fig. S4B). The DepMap codependence data were also used to understand the interesting difference in CIP2A dependence in the initiation of mammary and ovarian cancers (Fig. 1). To this end, we analyzed in a pair-wise fashion the correlation between dependence on either CIP2A, or one of the functionally most similar genes RHNO1, TOPBP1, POLQ, NBN, and PARP1 (rank 1 genes from Fig. 3A) across either BLBC or HGSOC cell lines. TOPBP1 and NBN had higher codependence with CIP2A in BLBC than in HGSOC cells, whereas in HGSOC, RHNO1 was more co-dependent with CIP2A (Fig. 3F). These differences may provide one plausible explanation for the differential requirement of Cip2a for DMBA-induced BLBC-like, but not ovarian cancer initiation (Fig. 1C; Supplementary Fig. S1B). Notably, TOPBP1 did not show CIP2A codependence in HGSOC cells, but was codependent in BLBC cells (Fig. 3F).
CIP2A dampens TopBP1–RAD51 function under DNA damage
Although the results above identify a potential novel role for CIP2A in TopBP1-complex–mediated G2–M arrest, there is currently no evidence for a direct mechanistic link between CIP2A and TopBP1. Here, by using a genome-wide Y2H assay with a human breast cancer cDNA library, TopBP1 was identified with very high confidence as an interaction partner for CIP2A (Fig. 4A; Supplementary Table S5). As Y2H assay only detects direct interactions between two proteins, we conclude that the CIP2A-TopBP1 association does not involve PP2A. However, as indicated by NBN phosphorylation data (Supplementary Fig. S3B; ref. 37), CIP2A may still protect proteins in the TopBP1 complex from PP2A-mediated dephosphorylation.
Figure 4.

CIP2A is an interacting partner of TopBP1 and promotes mitotic progression of DNA damaged cells. A, Schematic presentation of breast cancer cell line cDNA fragments coding for TopBP1 domains that interact with full length CIP2A in a yeast two-hybrid assay. Numbers in the TopBP1 drawing refer to BRCT domains 1–8. AAD, ATR activation domain. Analysis of the minimal common overlapping region between the TopBP1 fragments interacting with CIP2A reveal the TopBP1 aa. 829–853 as a candidate CIP2A interaction domain. B, Coimmunoprecipitation of endogenous CIP2A and γH2AX in HEK293 cells transiently overexpressing GFP or full-length TopBP1-GFP as indicated. Input 5% of total IP. C, Coimmunoprecipitation of CIP2A in HEK293 cells transiently overexpressing V5-tagged CIP2A and GFP-tagged empty vector (EV) or TopBP1 truncated mutants T0, T1, T2, T3 as indicated in D. Input 5% of total IP. D, Schematic representation of TopBP1 mutants used in B and C. Relative interaction efficiencies are estimated from the experiment, where all indicated mutants were included. E, Immortalized MCF10A cells transfected with nontargeting (SCR) or CIP2A siRNAs for 48 hours. Immunoblot of whole-cell extracts probed for pATR, total ATR, and CIP2A. Vinculin was used as a loading control. Relative quantifications of pATR/ATR and CIP2A plotted as mean± SD from five replicates. F, MDA-MB-231 cells transfected with nontargeting (SCR) and CIP2A targeting siRNAs for 72 hours and overexpressing TopBP1 mutants T0 and T1 as indicated for 48 hours. Immunoblot of whole-cell extracts probed for pATR, γH2AX, and CIP2A. Actin was used as a loading control. Relative quantifications of γH2AX plotted as mean± SD from two replicates. G, IR-induced TopBP1 foci formation in MCF10A cells transfected with SCR or CIP2A siRNA as indicated for 48 hours. Cells were treated with 5 Gy radiation for 1 hour and stained for CIP2A or TopBP1. H, Quantifications of the nuclear foci from G expressed as mean ± SD from representative experiment of three experiments with similar results I, IR-induced RAD51 foci formation in mouse mammary epithelial cells (MMEC) isolated from WT and Cip2a−/− mice cultured in vitro for 48 hours, treated with 5 Gy radiation for 2 hours. J, Quantifications of the foci expressed as mean ± SD of representative experiment. G–J, Images were taken at ×63 on 3i spinning disk confocal and at least 150 cells quantified per each condition using speckle counter pipeline on CellProfiler; scale bar, 10 μm. E–J, All statistical analyses were conducted with the Welch Student t test for unequal variances; *, P < 0.05; **, P < 0.01. K, Schematic presentation of the role of CIP2A in directly inhibiting TopBP1/RAD51-elicited G2–M checkpoint activation in nontransformed mammary epithelial cells.
CIP2A is predominantly a cytoplasmic protein, but based on our current (Supplementary Fig. S2B) and previous data (33), there is also a nuclear CIP2A pool in propagating cells in vitro and in vivo. Interaction between TopBP1 and endogenous nuclear CIP2A was confirmed from propagating cells by coimmunoprecipitation and proximity ligation analysis (Fig. 4B; Supplementary Fig. S5A). γH2AX also coimmunoprecipitated with TopBP1 and CIP2A from DNAse-treated cellular lysates, indicating that direct TopBP1–CIP2A interaction occurs at chromatin (Fig. 4B). By narrowing down the minimal shared region between TopBP1 fragments interacting with CIP2A in the Y2H assay, the interaction with CIP2A was delineated to be mediated by the stretch of amino acids 829–853 located between 5th and 6th BRCT domain of TopBP1 (Fig. 4A; Supplementary Table S5). The region between the 5th and 6th BRCT domain was also essential for interaction by coimmunoprecipitation analysis (Fig. 4C and D). Notably, the interaction was greatly strengthened by the presence of the ATR-activation domain (AAD) of TopBP1 adjacent to 6th BRCT repeat (Fig. 4C and D). As, no other DNA repair–related proteins were identified in CIP2A Y2H screen (Supplementary Table S5), the results support a model where direct binding to the scaffold protein TopBP1 mediates CIP2A interaction with the DDR network.
Directly linking CIP2A to TopBP1-regulated DDR, CIP2A inhibition significantly increased ATR phosphorylation in the propagating premalignant mammary cell line MCF10A (Fig. 4E), and the highest phosphorylation of the ATR target H2AX (γH2AX; ref. 40) was observed in CIP2A-depleted cells overexpressing AAD variant of TopBP1 (Fig. 4F). The role of CIP2A in dampening IR-induced γH2AX was validated in primary mammary epithelial cells isolated from WT and Cip2a−/− mice (Supplementary Fig. S5B). Although PP2A has been validated as a γH2AX phosphatase in replication stressed cells (41), we interpret that increased γH2AX phosphorylation in CIP2A-depleted replicating cells is rather due to increased TopBP1-associated ATR activity (7, 8, 40). Further supporting the role of CIP2A in dampening TopBP1 function, CIP2A depletion in IR-treated proliferating cells resulted in significantly enhanced chromatin recruitment of TopBP1 (Fig. 4G and H). This was specific to TopBP1, as CIP2A did not impact IR-induced p53BP1 chromatin recruitment (Supplementary Fig. S5C). As CIP2A depletion did not induce ATM phosphorylation (Supplementary Fig. S5D) that is a known mechanism increasing TopBP1 chromatin recruitment (8), we postulate that CIP2A prevents TopBP1 chromatin binding by direct interaction with its BRCT-domains (Fig. 4A). In BRCA WT cells, TopBP1 mediates G2–M arrest in response to IR by promoting RAD51 chromatin loading (10–12, 42). Consistent with the G2–M arrest phenotype (Figs. 1E and 2D), and increased TopBP1 foci formation (Fig. 4G and H), the Cip2a−/− mammary epithelial cells exposed to IR displayed significantly enhanced RAD51 chromatin recruitment 2 hours after irradiation (Fig. 4I and J). Importantly, increased RAD51 retention at chromatin was observed still 6 hours after irradiation in Cip2a−/− mammary epithelial cells, indicating for a long-term defect in DNA repair foci clearance (Supplementary Fig. S5E).
Together with the established role for TopBP1/RAD51 complex in DNA damage–induced G2–M checkpoint activity (7, 9–11, 42), the discovered CIP2A-mediated inhibition of TopBP1 and RAD51 chromatin recruitment provides a mechanism for dampening of the G2–M checkpoint in CIP2A-positive premalignant mammary epithelial cells (Fig. 4K).
CIP2A is essential for survival of TP53/BRCA-mutant BLBC cells
Next, we assessed whether the role of CIP2A as a mouse BLBC driver candidate translates to human BLBC. First, both CIP2A and TOPBP1 mRNA were found to be highest expressed in BLBC across the human breast cancer subtypes (Fig. 5A; Supplementary Fig. S6A and S6B). TP53 mutations in BLBC may result in activation of CIP2A gene promoter activity through the p21-E2F1 pathway (19) and accordingly, a significant correlation between TP53 mutation, and high CIP2A expression was confirmed in the GSE21653 cohort (Supplementary Fig. S6C). The clinical relevance and selectivity of CIP2A for human BLBC was evident from patient survival analysis. Both high mRNA and protein expression of CIP2A predicted poor disease-free and overall survival only in BL-TNBC, but not in non-BL-TNBC breast cancers (Fig. 5B–E; Supplementary Fig. S6D–S6H). CIP2A neither had a predictive role among patients with ER-positive tumors, or in unselected breast cancers (Supplementary Fig. S6D and S6G). Notably, the 5-year survival of patients with highly CIP2A-positive BL-TNBC tumor was only about 50% in both patient cohorts (Fig. 5B and D), indicating that these high CIP2A tumors are particularly aggressive. Furthermore, high CIP2A expression significantly associated with mutation load in TNBC tumors based on TCGA data (Supplementary Fig. S6I). This finding indicates that although CIP2A expression is not affecting mutation frequency per se (Fig. 1B), it allows survival of DNA damaged cells (Fig. 1F), and thereby accumulation of tumor mutational load in fully developed TNBCs.
Figure 5.

CIP2A is essential for survival of TP53/BRCA-mutant BLBC cells and drives proliferative signaling. A, Expression of CIP2A mRNA in indicated molecular breast cancer subtypes. Data derived from TCGA. P values by the unpaired t test. B, Disease-free survival of CIP2A high (n = 15) and CIP2A low (n = 45) expressing basal-like patients with TNBC in GSE21653 cohort. C, Disease-free survival of CIP2A high (n = 45) and CIP2A low (n = 132) expressing non-basal-like (HER2+, luminal A, luminal B, and normal-like) patients with breast cancer in GSE21653 cohort. D, Overall survival of CIP2A high (n = 12) and CIP2A low (n = 51) basal-like patients with TNBC in FinHer cohort. E, Overall survival of CIP2A high (n = 17) and CIP2A low (n = 47) non-basal like TNBC patients based IHC analysis from FinHer cohort. B–E,P values calculated by the log-rank test. F,CIP2A dependence of breast cancer cell lines with CERES score < −0.4 from DepMap portal (Avana 2020Q1). Lower CERES scores indicate that the cell line is more dependent on CIP2A. Color coding indicates the breast cancer subtype of the cell line based on PAM50 classification. G, Colony growth assays conducted on mammary tumor cell lines isolated from basal-type (KB1P#1 and KB1P#2: Brca1 and Trp53 mutant) and invasive lobular carcinoma (ILC)-type (KEP#1 and KEP#2: E-cadherin and Trp53 mutant) mouse models; Cip2a was knocked out using CRISPR/Cas9 using two unique gRNAs. Western blots from the same samples probed for CIP2A below. Shown are representative images of at least two independent biological repeats for each cell line. H, Summary of CIP2A dependence on colony growth of indicated TP53-mutant TNBC cell lines transfected with Mock, nontargeting siRNA (siSCR), or three unique CIP2A-targeting siRNAs (siCIP2A #1, #2, #3). Colony areas were quantified and normalized to siSCR. I, Gene set enrichment analysis (GSEA) conducted on differentially expressed genes obtained from RNA-seq of HCC38 cells depleted with three unique CIP2A siRNAs. J, HCC38 cells transfected with SCR or CIP2A siRNAs for 72 hours and immunoblotted for indicated protein.
To assess the BLBC cell dependence on CIP2A, we surveyed the Dep-Map essentiality database across 33 breast cancer cell lines. Among the 12 cell lines with CERES gene dependency score less than −0.4 for CIP2A loss, the great majority of cell lines were found to be BLBC cells (Fig. 5F; Supplementary Table S6). Notably, all except one of these most CIP2A-dependent BLBC cells carried either a BRCA1 or BRCA2 mutation that is a hallmark of BLBCs (Fig. 5F). Furthermore, in a genetically defined CRISPR/Cas9 model, Cip2a was found to be essential for colony growth of mouse mammary tumor cells depleted for Trp53 and Brca1 (KB1P; basal-type; Fig. 5G; ref. 23). However, Cip2a was dispensable for growth of either Trp53/E-cadherin mutant mammary tumor cells (KEP; invasive lobular carcinoma-type; Fig. 5G; ref. 24), or cells from the mice with activated AKT and loss of E-cadherin in mammary tumor cells (WEA; invasive lobular carcinoma-type; ref. 25; Supplementary Fig. S7A). Furthermore, RNA-sequencing analysis from the most CIP2A-dependent TP53-mutant BLBC cell line (Fig. 5H; Supplementary Fig. S7B) and HCC38 (TP53 mutant/BRCA1 promoter methylation/BRCA2 mutant), revealed that CIP2A drove similar BLBC and HRD-associated gene expression programs as was observed from DMBA-treated mouse mammary tissue (Figs. 2D and 5I). The role of CIP2A in inhibiting the dephosphorylation of the activating phosphorylation sites in both MYC and E2F1 was confirmed by Western blot analyses (Fig. 5J). Finally, consistent with recent identification of CIP2A as an essential gene in PARP inhibitor talazoparib-treated cells (39), CIP2A depletion hypersensitized BRCA-proficient MDA-MB-231 cells to two different PARP inhibitors (Supplementary Fig. S7C).
These data reveal essentiality of CIP2A for the survival BRCA-deficient BLBC cells, and thus fully support the mouse data, indicating the driver role of CIP2A in BLBC. Clinically, the data introduce CIP2A as a potential biomarker to identify patients with BL-TNBC with particularly aggressive disease.
Transcriptional CIP2A targeting by SMAPs as potential BLBC therapy
Effective treatment of BLBCs represents a significant unmet medical need. As our data indicate that CIP2A regulates PP2A activity toward both the TopBP1 complex (Fig. 3C; Supplementary Fig. S3B), as well as toward MYC and E2F1 (Fig. 5J), we tested whether a recently developed series of small-molecule activators of PP2A (SMAP; refs. 14, 16), could be used to target CIP2A-expressing BLBC. SMAPs activate the CIP2A-regulated PP2A-B56alpha heterotrimer (14, 43), and the cellular effects of SMAPs in cancer cells are both correlated with PP2A reactivation capacity, and can be rescued by concomitant PP2A inhibition (22, 44).
Treatment with two independent SMAPs (DBK-1154 and DT-061) resulted in a robust inhibition of cell viability in eight established BL-TNBC cell lines (Fig. 6A; Supplementary Fig. S8A), and in five BLBC patient-derived cancer stem cell-like lines (Fig. 6B; ref. 26). Notably, consistent with notion that these cell lines were derived from tumors of patients that had undergone neoadjuvant chemotherapy, these CIP2A-positive (Supplementary Fig. S8B) patient-derived BLBC cell lines showed resistance to classical chemotherapies (Fig. 6B). Directly supportive of the therapeutic relevance of these observations, oral DT-061 therapy resulted in significant inhibition of tumor growth of an orthotopic patient-derived xenograft (PDX) model from a patient with TP53 mutant, EGFR+ BLBC over a 40-day treatment period (Fig. 6C). Similar to other in vivo studies with SMAPs (14, 45, 46), we did not observe any treatment-related adverse effects in mice. Importantly, the control tumors were CIP2A positive, whereas tumors from DT-061–treated mice showed a clear trend for reduced CIP2A protein levels (Supplementary Fig. S8C and S8D).
Figure 6.

CIP2A targeting by SMAPs as potential BLBC therapy. A, SMAP (DBK-1154) sensitivity profiles of eight BL-TNBC cell lines. Cell viabilities were measured using CellTiterGlo Luminescence Assay after 24 hours of drug treatment. EC50 values are listed in parentheses. B, Screening of patient-derived BLBC stem cell like cells for chemotherapy and SMAP responses. Heatmap indicates the drug sensitivity scores (DSS) of these cells across standard chemotherapeutics and three SMAPs (DBK-1154, DT-061, NZ-1160). Higher DSS value indicates higher sensitivity. C, Tumor growth of an orthotopic PDX model of basal triple-negative breast cancer treated with DMA or 5mpk BID SMAP DT-061 for 43 days. Respective quantifications are represented as mean ± SD. D and E, CIP2A Western blots from MDA-MB-468 (D) and patient-derived stem cell-like cells BCSC1 (E) on treatment with indicated SMAPs for 24 hours. DT-061 and DBK-1154 concentration, 20 μmol/L. F, Kinetics of CIP2A mRNA expression from MDA-MB-468 cells after treatment with 20 μmol/L of DT-061. n = 3 expressed as mean ± SD. G, Kinetics of pT202/Y204-ERK, CIP2A, and pS62-MYC from the MDA-MB-468 cell line treated with SMAP DT-061 (20 μmol/L) for indicated time points. Representative Western blot data shown in Supplementary Fig. S8H. n = 3 expressed as mean ± SD. H, Quantification of Western blots of the MDA-MB-468 cell line treated with 20 μmol/L SMAP DT-061 for 24 hours and probed for γH2AX, represented as mean ± SD from n = 3 replicates normalized to the untreated controls. I, Quantification of Western blots from the MDA-MB-468 cell line treated with 20 μmol/L of DT-061 for 24 hours, displayed in Supplementary Fig. S9A. Data expressed as mean ± SD from n = 3 replicates normalized to the untreated controls. J, Time course of CIP2A and γH2AX protein expression in MDA-MB-468 treated with DT-061 (20 μmol/L) for indicated time periods. Western blot data are shown in Supplementary Figs. S8F and S9B. K, CIP2A overexpression in the MCF10A cell line rescues the SMAP-elicited γH2AX activation. Western blots of parental and CIP2A OE MCF10A cells treated with SMAP DBK-1154 for 24 hours and probed for γH2AX. GAPDH is used as loading control; γΗ2AX quantifications from n = 4 replicates displayed below. L, Dose–response curve of control and CIP2A OE stable cell line (CIP2A OE) MCF10A cells on treatment with concentration series of DBK-1154 for 24 hours. IC50 values are indicated in parenthesis. A–L,P values calculated using the unpaired t test, *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Related to a potential link between SMAP response and CIP2A, Western blot analyses revealed a surprising inhibition of CIP2A protein expression by SMAPs at 24 hours (Fig. 6D and E; Supplementary Fig. S8E–S8G). Indicative of transcriptional level regulation, CIP2A protein inhibition was accompanied with inhibition of CIP2A mRNA expression (Fig. 6F; Supplementary Fig. S8G). The candidate mechanism for SMAP-elicited CIP2A inhibition was evaluated by studying the time course of CIP2A inhibition in relation to its two known upstream activators ERK and MYC (20, 21, 32), which both are inhibited by SMAPs (44). Whereas inhibition of ERK phosphorylation by SMAP preceded inhibition of CIP2A expression, MYC was inhibited in SMAP-treated cells after CIP2A inhibition (Fig. 6G; Supplementary Fig. S8H).
SMAPs, representing surrogate CIP2A inhibitors, were next tested for possible effects on biomarkers of CIP2A activity. Consistent with results in CIP2A-depleted cells (Fig. 4), SMAPs induced potent checkpoint signaling exemplified by phosphorylation of H2AX, ATR, and CHK2 (Fig. 6H and I; Supplementary Fig. S9A and S9B). SMAP treatment for 24 hours also resulted in potent inhibition of MYC protein expression (Supplementary Fig. S9C). Regarding causality between SMAP-induced checkpoint activation and CIP2A inhibition, SMAP-elicited CHK2 phosphorylation preceded CIP2A inhibition (Supplementary Fig. S9D and S9F), whereas the ToPBP1-related p-ATR and γH2AX induction occurred only after CIP2A protein inhibition (Fig. 6J; Supplementary Fig. S9D, S9E, S9G–S9H). Importantly, exogenous overexpression of CIP2A in MCF10A cells, that were used to link the results to G2–M arrest (Fig. 1E), and TopBP1 effects (Fig. 4G and H), abrogated SMAP-elicited γH2AX induction (Fig. 6K; Supplementary Fig. S9I). CIP2A overexpression also shifted the SMAP IC50 response in these cells (Fig. 6L). These results reveal that SMAPs have bi-phasic therapeutic activity consisting of direct PP2A activation (14, 16), followed by transcriptional inhibition of CIP2A expression discovered here. SMAP-elicited CIP2A inhibition may thus halt the growth of BLBC cells by both prolonging the PP2A reactivation effects, but also via removing direct CIP2A-mediated direct effects on TopBP1 (Fig. 7).
Figure 7.
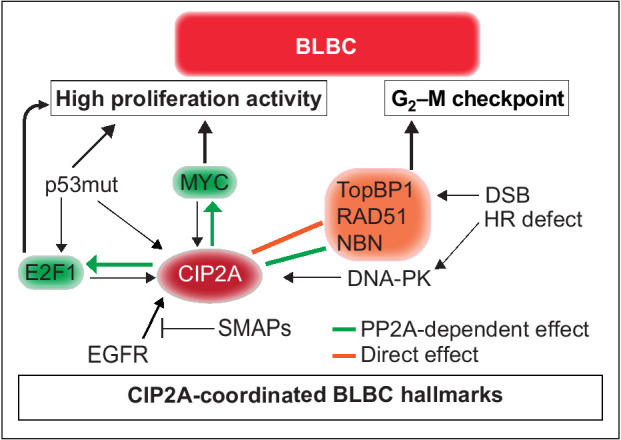
Schematic presentation of the role for CIP2A in coordinating BLBC molecular hallmarks. The capacity of CIP2A to coordinately regulate G2–M checkpoint, and proliferative signaling by MYC and E2F1, provides a molecular basis for its role as BLBC driver. Black arrows indicate known mechanisms implicated in BLBC that either regulate CIP2A expression and/or promote BLBC progression. Green arrows indicate PP2A inhibition-dependent mechanisms by which CIP2A increase proliferation capacity of BLBC cells. The PP2A dependence of these mechanisms has been demonstrated previously (19, 35). The orange block arrow indicates direct binding of CIP2A to TopBP1 and inhibition of TopBP1-mediated checkpoint activity in response to DSB in premaligant mammary epithelial cells. Green block arrow indicates potential PP2A-dependent regulation of NBN phosphorylation. Collectively, CIP2A both respond to hallmarks of BLBCs (p53 inhibition, EGFR activity, and HR defects) and coordinately control the essential functional hallmarks, G2–M checkpoint and high proliferation activity. The relationship between CIP2A and BLBC hallmarks is also a plausible explanation for the selective role for CIP2A in BLBC as compared with other cancer types.
Discussion
In this study, we provide comprehensive evidence that CIP2A overexpression can be a driver mechanism for BLBC initiation and malignant progression (Fig. 7). Consistent with the notion that even saturated genetic analysis of human breast cancers has failed to identify genetic BLBC driver, CIP2A gene sequence is not altered in BLBCs (https://cancer.sanger.ac.uk/cosmic). Instead, CIP2A expression is enhanced due to constitutive DNA-PK/CHK1 activity (47), TP53 inactivation (19), and EGFR pathway activation (20), which are all molecular hallmarks of BLBC (Fig. 7; refs. 1, 3). Transcriptional CIP2A induction early in DMBA-induced tumorigenesis is also fully supportive of its role in BLBC tumor initiation. Together these findings provide an explanation for high CIP2A expression in BLBC, whereas its newly discovered interaction with TopBP1 forms a molecular basis for its essential function in allowing malignant progression of DNA-damaged mammary epithelial cells toward BLBCs.
As opposed to previous assumptions that CIP2A is involved in the development of multiple human solid cancers (17), our results demonstrate selective involvement of CIP2A in initiation and progression of BLBCs both in human and mice. In addition to in vivo tumorigenesis results, in genetically defined cell culture models, only the Brca1/Trp53 mutant basal-like cells were dependent on Cip2a for their colony growth. Also in human breast cancer samples, high CIP2A expression predicted for poor patient survival exclusively in BLBCs. The selectivity of CIP2A for BLBCs among the tumor types studied here can be molecularly explained by the notion that CIP2A is able to coordinate BLBC hallmarks via promoting proliferative MYC and E2F1 activities, and at the same time blunting the G2–M checkpoint via its effects on the TopBP1/RAD51 complex (Fig. 7). In addition to TopBP1, other highly CIP2A codependent DDR genes (Fig. 3A), not studied in this work, might confer its BLBC selectivity. One such candidate gene is POLQ (Fig. 3A; Supplementary Fig. S4A and S4B), which is upregulated, and promotes genetic instability in BL-TNBC cells (48).
The functional homology of CIP2A with a number of critical DNA-damage proteins (Fig. 3A), is likely one of the most important contributions of this work for the future studies. This is supported by the recent results demonstrating essentiality of CIP2A in different DDR-related genomic screens (39, 49). Here, we focused on validation of the novel interaction between nuclear CIP2A and TopBP1. On the basis of our results, TopBP1 can induce effective DDR in CIP2A deficient cells whereas in CIP2A-positive cells the TopBP1/RAD51 complex chromatin recruitment is dampened allowing for continued mitotic activity (Fig. 4K). This mechanism is fully in line with previous data related to TopBP1-mediated G2–M checkpoint regulation (5, 7, 9, 11, 42). As a notion, we validated the CIP2A function in TopBP1/RAD51 complex and in G2–M checkpoint activity in BRCA-proficient cells (Figs. 1E, 4E–J; Supplementary Fig. S5B–S5E). Whether the same mechanism is behind essentiality of CIP2A for HRD cells, including BRCA-deficient BLBC cells, remains to be studied. Importantly, in our model the enhanced TopBP1/RAD51 chromatin recruitment is due to loss of a direct and PP2A-independent CIP2A–TopBP1 interaction (Fig. 7). On the other hand, CIP2A can protect proteins in the TopBP1-associated complex from PP2A-mediated dephosphorylation as indicated with NBN S432 phosphorylation data (37). In addition, similar to other models (19, 32, 34), CIP2A inhibited dephosphorylation of both MYC and E2F1 in BLBC cells. CIP2A expression is in turn driven by MYC and E2F1 (19, 32), and we postulate that these positive feedback loops have critical role in maintaining high proliferative activity in BLBC. Collectively, we conclude that CIP2A-mediated BLBC initiation and progression results from a mixture of its PP2A-dependent and -independent effects (Fig. 7).
In addition to identifying CIP2A as a driver candidate for BLBC, we demonstrate that SMAPs (14) function as transcriptional inhibitors of CIP2A expression. Our results reveal a model where SMAPs initially directly activate PP2A-B56 (14, 16), and this followed by a prolonged PP2A activation due to transcriptional downregulation of CIP2A. Importantly, we were also able to demonstrate that CIP2A overexpression rescued the effects of SMAPs as assessed by both decreases in cell viability and γH2AX regulation. However, it is important to note that we consider SMAPs as surrogate CIP2A inhibitors that also have acute effects not mediated by CIP2A inhibition, thereby explaining the anticancer effects of SMAPs noted in other cancer types (45, 46). Importantly, we validated the therapeutic effects of three SMAPs across 15 different cell lines, including 6 individual patient-derived lines and a PDX model, together minimizing concerns related to compound specific effects, and known intratumoral heterogeneity of BLBC tumors.
Together these results credential CIP2A as a driver protein for one of the most aggressive human cancer types, BLBCs. We also discover a novel link between CIP2A and DDR via direct interaction with TopBP1. Generally, these results emphasize the importance in characterizing proteome level signaling dysregulation in the cancer subtypes for which genetic drivers are lacking.
Authors' Disclosures
No disclosures were reported.
Supplementary Material
Acknowledgments
The authors thank Taina Kalevo-Mattila for excellent technical assistance. Erica Nyman is thanked for help in IHC analysis. Professor Wojciech Niedzwiedz and Dr. Andrew Blackford are thanked for sharing research tools and protocols. The authors are very grateful to Ruth Keri Laboratory from Case Western Reserve University for sharing the PDX model. Dr. Michael Ohlmeyer (Atux Iskay LLC) is acknowledged for DBK-1154 and DBK-1160. Johanna Ivaska and Dipanjan Chowdhury are acknowledged for their valuable comments to the data. We acknowledge Finnish Functional Genomics Center, Cell Imaging and Cytometry core facility, and Genome Editing Core at Turku Bioscience Center, and Turku Center for Disease Modeling (TCDM) all supported by Biocenter Finland, and/or ELIXIR Finland. The Central Laboratory Animal Facilities of University of Turku are acknowledged for help with the mouse models. The project was funded by Academy of Finland (323096 to E. Peuhu; 296801, 314443, and 310561 to L.L. Elo), W81XWH-19-BCRP-BTA12 DOD (to G. Narla and J. Westermarck), Finnish Cancer Foundations (to J. Westermarck), Finnish Cultural Foundation (to L.L. Elo), Sigrid Juselius Foundation (to J. Westermarck and E. Peuhu), Breast Cancer Now (2017NovPCC1067 to J. Westermarck and K. Wennerberg), and Governmental Research Funding for Turku University Hospital (to T.J. Grönroos). D.C. Connolly is supported by the Fox Chase Cancer Center FCCC Core grant NCI P30 CA006927. G. Narla is supported by R01 CA181654, HL144741, CA240993, and Rogel Cancer Gift Funds. J. Maurer is supported by Deutsche Forschungsgemeinschaft [DFG, German Research Foundation; PN (407869199)]. A. Laine was supported by Oncode Institute, Svenska Kulturfonden, Orion Research Foundation, Relander Foundation, Inkeri and Mauri Vänskä's Foundation, Finnish Cultural Foundation's Varsinais-Suomi Regional Fund, and by K. Albin Johanssons Stiftelse. S.G. Nagelli was supported by the University of Turku Graduate School (UTUGS), Instrumentarium Science Foundation, and Ida Montin Foundation.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
A. Laine: Conceptualization, formal analysis, investigation, writing–original draft. S.G. Nagelli: Conceptualization, formal analysis, investigation, writing–review and editing. C. Farrington: Formal analysis, investigation, writing–review and editing. U. Butt: Investigation. A.N. Cvrljevic: Investigation. J.P. Vainonen: Investigation. F.M. Feringa: Investigation. T.J. Grönroos: Investigation. P. Gautam: Investigation. S. Khan: Investigation. H. Sihto: Investigation. X. Qiao: Investigation. K. Pavic: Resources. D.C. Connolly: Formal analyses, resources, writing–review and editing. P. Kronqvist: Investigation. L.L. Elo: Supervision. J. Maurer: Resources. K. Wennerberg: Resources, methodology. R.H. Medema: Resources. H. Joensuu: Resources. E. Peuhu: Investigation, writing–review and editing. K. de Visser: Formal analysis, supervision, writing–review and editing. G. Narla: Conceptualization, supervision, funding acquisition, writing–review and editing. J. Westermarck: Conceptualization, resources, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017;389:2430–42. [DOI] [PubMed] [Google Scholar]
- 2. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol 2008;26:2568–81. [DOI] [PubMed] [Google Scholar]
- 3. Duijf PHG, Nanayakkara D, Nones K, Srihari S, Kalimutho M, Khanna KK. Mechanisms of genomic instability in breast cancer. Trends Mol Med 2019;25:595–611. [DOI] [PubMed] [Google Scholar]
- 4. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121:2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giunta S, Jackson SP. Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle 2011;10:1215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng XF, Kalev P, Chowdhury D. Emerging role of protein phosphatases changes the landscape of phospho-signaling in DNA damage response. DNA Repair 2015;32:58–65. [DOI] [PubMed] [Google Scholar]
- 7. Cotta-Ramusino C, McDonald ER III, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 2011;332:1313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sokka M, Parkkinen S, Pospiech H, Syvaoja JE. Function of TopBP1 in genome stability. Subcell Biochem 2010;50:119–41. [DOI] [PubMed] [Google Scholar]
- 9. Kim WJ, Lee H, Park EJ, Park JK, Park SD. Gain- and loss-of-function of Rhp51, a Rad51 homolog in fission yeast, reveals dissimilarities in chromosome integrity. Nucleic Acids Res 2001;29:1724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y, Smolka MB. TOPBP1 takes RADical command in recombinational DNA repair. J Cell Biol 2016;212:263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moudry P, Watanabe K, Wolanin KM, Bartkova J, Wassing IE, Watanabe S, et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J Cell Biol 2016;212:281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ogiwara H, Ui A, Onoda F, Tada S, Enomoto T, Seki M. Dpb11, the budding yeast homolog of TopBP1, functions with the checkpoint clamp in recombination repair. Nucleic Acids Res 2006;34:3389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple-negative breast cancer. Nat Commun 2020;11:2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leonard D, Huang W, Izadmehr S, O'Connor CM, Wiredja DD, Wang Z, et al. Selective PP2A enhancement through biased heterotrimer stabilization. Cell 2020;181:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kauko O, Westermarck J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int J Biochem Cell Biol 2018;96:157–64. [DOI] [PubMed] [Google Scholar]
- 16. Vainonen JP, Momeny M, Westermarck J. Druggable cancer phosphatases. Sci Transl Med 2021;13:eabe2967. [DOI] [PubMed] [Google Scholar]
- 17. Khanna A, Pimanda JE. Clinical significance of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancers. Int J Cancer 2016;138:525–32. [DOI] [PubMed] [Google Scholar]
- 18. Côme C, Laine A, Chanrion M, Edgren H, Mattila E, Liu X, et al. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res 2009;15:5092–100. [DOI] [PubMed] [Google Scholar]
- 19. Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemela M, et al. Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov 2013;3:182–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khanna A, Okkeri J, Bilgen T, Tiirikka T, Vihinen M, Visakorpi T, et al. ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PLoS ONE 2011;6:e17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chao TT, Wang CY, Chen YL, Lai CC, Chang FY, Tsai YT, et al. Afatinib induces apoptosis in NSCLC without EGFR mutation through Elk-1-mediated suppression of CIP2A. Oncotarget 2015;6:2164–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim S, Roopra A, Alexander CM. A phenotypic mouse model of basaloid breast tumors. PLoS ONE 2012;7:e30979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A 2007;104:12111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006;10:437–49. [DOI] [PubMed] [Google Scholar]
- 25. Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019;572:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Metzger E, Stepputtis SS, Strietz J, Preca BT, Urban S, Willmann D, et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res 2017;77:5900–12. [DOI] [PubMed] [Google Scholar]
- 27. Ventelä S, Côme C, Mäkelä JA, Hobbs RM, Mannermaa L, Kallajoki M, et al. CIP2A promotes proliferation of spermatogonial progenitor cells and spermatogenesis in mice. PLoS ONE 2012;7:e33209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A compendium of mutational signatures of environmental agents. Cell 2019;177:821–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res 2003;63:1389–97. [PubMed] [Google Scholar]
- 30. Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015;350:1092–6. [DOI] [PubMed] [Google Scholar]
- 31. Abba MC, Zhong Y, Lee J, Kil H, Lu Y, Takata Y, et al. DMBA induced mouse mammary tumors display high incidence of activating Pik3caH1047 and loss of function Pten mutations. Oncotarget 2016;7:64289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khanna A, Böckelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst 2009;101:793–805. [DOI] [PubMed] [Google Scholar]
- 33. Myant K, Qiao X, Halonen T, Come C, Laine A, Janghorban M, et al. Serine 62-phosphorylated MYC associates with nuclear lamins and its regulation by CIP2A is essential for regenerative proliferation. Cell Rep 2015;12:1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc Natl Acad Sci U S A 2014;111:9157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Niemela M, Kauko O, Sihto H, Mpindi JP, Nicorici D, Pernila P, et al. CIP2A signature reveals the MYC dependency of CIP2A-regulated phenotypes and its clinical association with breast cancer subtypes. Oncogene 2012;31:4266–78. [DOI] [PubMed] [Google Scholar]
- 36. Oskarsson T, Essers MA, Dubois N, Offner S, Dubey C, Roger C, et al. Skin epidermis lacking the c-Myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21Cip1 gene. Genes Dev 2006;20:2024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kauko O, Imanishi SY, Kulesskiy E, Yetukuri L, Laajala TD, Sharma M, et al. Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J Biol Chem 2020;295:4194–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wohlbold L, Merrick KA, De S, Amat R, Kim JH, Larochelle S, et al. Chemical genetics reveals a specific requirement for Cdk2 activity in the DNA damage response and identifies Nbs1 as a Cdk2 substrate in human cells. PLos Genet 2012;8:e1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeWeirdt PC, Sangree AK, Hanna RE, Sanson KR, Hegde M, Strand C, et al. Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat Commun 2020;11:752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol 2006;26:6056–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell 2005;20:801–9. [DOI] [PubMed] [Google Scholar]
- 42. Yamane K, Chen J, Kinsella TJ. Both DNA topoisomerase II-binding protein 1 and BRCA1 regulate the G2–M cell-cycle checkpoint. Cancer Res 2003;63:3049–53. [PubMed] [Google Scholar]
- 43. Wang J, Okkeri J, Pavic K, Wang Z, Kauko O, Halonen T, et al. Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. EMBO Rep 2017;18:437–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 2017;127:2081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kauko O, O'Connor CM, Kulesskiy E, Sangodkar J, Aakula A, Izadmehr S, et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci Transl Med 2018;10:eaaq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Farrington CC, Yuan E, Mazhar S, Izadmehr S, Hurst L, Allen-Petersen BL, et al. Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J Biol Chem 2020;295:757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khanna A, Kauko O, Böckelman C, Laine A, Schreck I, Partanen JI, et al. Chk1 targeting reactivates PP2A tumor suppressor activity in cancer cells. Cancer Res 2013;73:6757–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prodhomme MK, Pommier RM, Franchet C, Fauvet F, Bergoglio V, Brousset P, et al. EMT transcription factor ZEB1 represses the mutagenic POLtheta-mediated end-joining pathway in breast cancers. Cancer Res 2020;81:1595–606. [DOI] [PubMed] [Google Scholar]
- 49. Hustedt N, Alvarez-Quilon A, McEwan A, Yuan JY, Cho T, Koob L, et al. A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol 2019;9:190156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.