

Abstract
Pathophysiological damages and loss of function of dopamine neurons precede their demise and contribute to the early phases of Parkinson’s disease. The presence of aberrant intracellular pathological inclusions of the protein α-synuclein within ventral midbrain dopaminergic neurons is one of the cardinal features of Parkinson’s disease. We employed molecular biology, electrophysiology, and live-cell imaging to investigate how excessive α-synuclein expression alters multiple characteristics of dopaminergic neuronal dynamics and dopamine transmission in cultured dopamine neurons conditionally expressing GCaMP6f. We found that overexpression of α-synuclein in mouse (male and female) dopaminergic neurons altered neuronal firing properties, calcium dynamics, dopamine release, protein expression, and morphology. Moreover, prolonged exposure to the D2 receptor agonist, quinpirole, rescues many of the alterations induced by α-synuclein overexpression. These studies demonstrate that α-synuclein dysregulation of neuronal activity contributes to the vulnerability of dopaminergic neurons and that modulation of D2 receptor activity can ameliorate the pathophysiology. These findings provide mechanistic insights into the insidious changes in dopaminergic neuronal activity and neuronal loss that characterize Parkinson’s disease progression with significant therapeutic implications.
Subject terms: Cellular neuroscience, Neurodegeneration, Neurotransmitters
Introduction
Volitional movement is a fundamental behavior of everyday life that is often taken for granted until control deteriorates. Dopaminergic neurons within the ventral midbrain play a critical role in the initiation and control of volitional movement1,2 and the progressive demise of these neurons is a defining hallmark of Parkinson’s disease (PD)3. Pathophysiological damages and loss of function of these neurons precedes their demise and contribute to the early phases of the movement impairments4. No current therapies reverse or slow the progression of PD or the many related neurodegenerative diseases associated with the demise of dopaminergic neurons due to the incomplete understanding of the etiology of PD.
The presence of aberrant intracellular inclusions comprised of the protein α-synuclein (α-syn) in the form of Lewy bodies and Lewy neurites within ventral midbrain dopaminergic neurons is another cardinal feature of PD5–7. Several missense mutations in the α-syn gene (SNCA), as well as duplication8 or triplication9 of SNCA, are sufficient to cause familial PD and the related disease Lewy body dementia. Thus, only a 50% increase in the expression of wild-type α-syn as in the multiplication of the SNCA gene is sufficient for a detrimental outcome on dopaminergic neurons resulting in disease. Furthermore, some studies indicate that elevated α-syn level also occurs in idiopathic PD, but the pathophysiological mechanisms associated with increased levels of α-syn remain poorly understood.
The location of midbrain dopamine neurons in deep neural structures creates a significant barrier of not only investigation but also control over the experimental milieu. For these reasons, we incorporated a primary culture model system of dopaminergic neurons, which provides unparalleled access and control over the experimental procedure to investigate potential mechanisms of how excessive α-syn level alters dopaminergic neuronal dynamics and dopamine transmission prior to neuronal demise. Importantly, using whole-cell current-clamp recording, we and others have shown that postnatally derived dopaminergic cultures exhibit spontaneous firing properties10–14 similar to those found in ex vivo15–17 and in vivo18–20. Thereby, the experimental model system used in this study provides mechanistic insights not achievable of those found in ex vivo and in vivo models.
The present study utilized complementary approaches in molecular biology, electrophysiology, and live-cell imaging to investigate the hypothesis that elevated α-syn expression in dopaminergic neurons perturbs intracellular calcium signaling, protein homeostasis, and dopamine transmission prior to neuronal demise. We demonstrated that D2 receptor (D2R) autoinhibition contributes to alterations in neuronal homeostatic properties and that modulation thereof can ameliorate the pathophysiology resulting from excessive α-syn levels. These results provide mechanistic insights into the pathobiological impact of α-syn on dopaminergic neuron function and their demise characteristic of PD.
Results and discussion
Tyrosine hydroxylase (TH) promoter-driven adeno-associated virus (AAV) efficiently transduces cultures of midbrain dopamine neurons
In order to investigate the pathophysiological changes associated with α-syn overexpression, we first developed a cell model with high-fidelity AAV-mediated α-syn expression in midbrain dopaminergic neuronal culture. The ventral midbrain neuronal culture contains the dopaminergic nuclei substantia nigra (SNc) and ventral tegmental area (VTA) that have been frequently used to study dopamine transmission10–13,21–25. Notably, SNc dopaminergic neurons are more sensitive than VTA dopaminergic neurons22,26–31. Therefore, midbrain neuronal culture is likely to contain more VTA dopaminergic neurons than SNc dopaminergic neurons22,26–31 (see limitations of this model system in the “Methods” section). We utilized a TH promoter-driven AAV to specifically express wild-type human α-syn in cultured dopaminergic neurons. First, to demonstrate the specificity of the TH promoter vector, cultures were transduced with AAV-TH-GFP for visual confirmation of expression and quantification of dopaminergic specificity. As demonstrated in Fig. 1a, b, 91 ± 3% of neurons expressing green fluorescent protein (GFP) are TH positive, indicating high specificity. The same pAAV1-TH backbone, but with the human α-syn complementary DNA (cDNA) only, was utilized to overexpress human α-syn in dopamine neurons. The transduction of pAAV1-TH-human-αsyn in cultured midbrain dopamine neurons was confirmed via immunocytochemistry (ICC) and western blot analyses, demonstrating elevated expression of α-syn in these neurons (Fig. 1c, e and Supplementary Fig. 1, p = 0.005, two-tailed t test, n = 3 independent experiments). It should be noted that the midbrain neuronal culture contains many cell types, i.e., neuronal and non-neuronal cells. While our model provided 91% transduction specificity of α-syn overexpression in TH-positive neurons, it is likely that α-syn is also expressed in 9% of TH-negative cells—a mix of neuronal and glial cells—which is a limitation of AAV transduction in general. As shown in Fig. 1d, α-syn overexpression decreases neuronal survival as identified by TH-positive neurons expressing GFP (Fig. 1d, p < 0.05). It should be noted that functional analyses are conducted on surviving neurons. Since SNc neurons exhibit increased sensitivity to degeneration, it is possible that the relative proportion of VTA neurons to SNc neurons in cultures is altered by α-syn overexpression. No pre or post hoc techniques were carried out to investigate this ratio or selectively record from a subset of DA cells. Thus, these data may be over-representative of the response of surviving VTA cells; to investigate this question in real-time experiments, future studies with genetic targeting of SNc or VTA subtypes of dopamine neurons will be required (neurod6, pit3x, calbindin, aldh1, and GIRK3).
Fig. 1. Tyrosine hydroxylase (TH) promoter-driven adeno-associated virus (AAV) efficiently transduces human α-synuclein or the control construct (TH-GFP) in midbrain dopamine neurons.

a, b Immunolabeling of TH confirmed 91 ± 3% of TH-positive neurons co-express GFP, suggesting a high fidelity for pAAV1-TH-GFP viral transduction in the TH-positive neurons (n = 3 independent experiments). c, e The transduction specificity was confirmed via immunocytochemistry analysis and western blot (n = 3 independent experiments). Scale bars: 50 μm. d Dopaminergic neuron counts revealed that α-syn overexpression decreases neuronal survival (naive = 223 ± 33.52, α-syn = 113.7 ± 33.52, two-tailed unpaired t test, naive vs. α-syn, p = 0.03), *p < 0.05.
Overexpression of α-synuclein disrupts calcium dynamics and firing activity of dopamine neurons
Increased α-syn burden in dopamine neurons is correlated with neuronal loss in neurodegenerative diseases such as PD32,33. Although extensively studied in cortical neurons, yeast, and heterologous expression systems34–39, α-syn regulation of intracellular calcium and firing activity in dopaminergic neurons prior to cell death remains less clear. The maintenance of calcium homeostasis is a vital process in neurons40–42. Calcium is a ubiquitous second messenger that helps to transmit depolarization status and synaptic activity to the biochemical machinery of a neuron43. Extensive calcium signaling requires high ATP consumption to restore basal (low) intracellular calcium levels. Increased intracellular calcium may also lead to increased generation of mitochondrial reactive oxygen species44,45. Failure to maintain cellular energy levels and to suppress oxygen species may impact calcium signaling during aging and in neurodegeneration5,6. To investigate if α-syn overexpression regulates dopaminergic neuronal activity prior to neuronal demise, we employed live-cell calcium imaging in dopamine neurons conditionally expressing GCaMP6f under the control of the dopamine transporter (DAT) promoter (DAT-GCaMP6f) containing either endogenous levels of α-syn (naive) or overexpressing α-syn. Compared to calcium events in naive dopaminergic neurons, both width and amplitude of calcium peaks were increased in the presence of α-syn overexpression, creating repeated burdens on the neuron (Fig. 2a–c, width—p = 0.0152, unpaired two-tailed t test, n = 33 wild-type neurons, n = 40 α-syn-overexpressing neurons, amplitude—p = 0.0000198, unpaired two-tailed t test, n = 33 wild-type neurons, n = 40 α-syn-overexpressing neurons). These data suggest that increased levels of α-syn in dopamine neurons lead to disturbances in calcium homeostasis that can alter biophysical properties of neurons, neuronal activity, neurotransmission42,43,46–50, and neuronal death, all of which are shared hallmarks in neurodegenerative diseases22,46,51,52.
Fig. 2. Overexpression of α-synuclein disrupts calcium dynamics and firing activity of dopaminergic neurons.
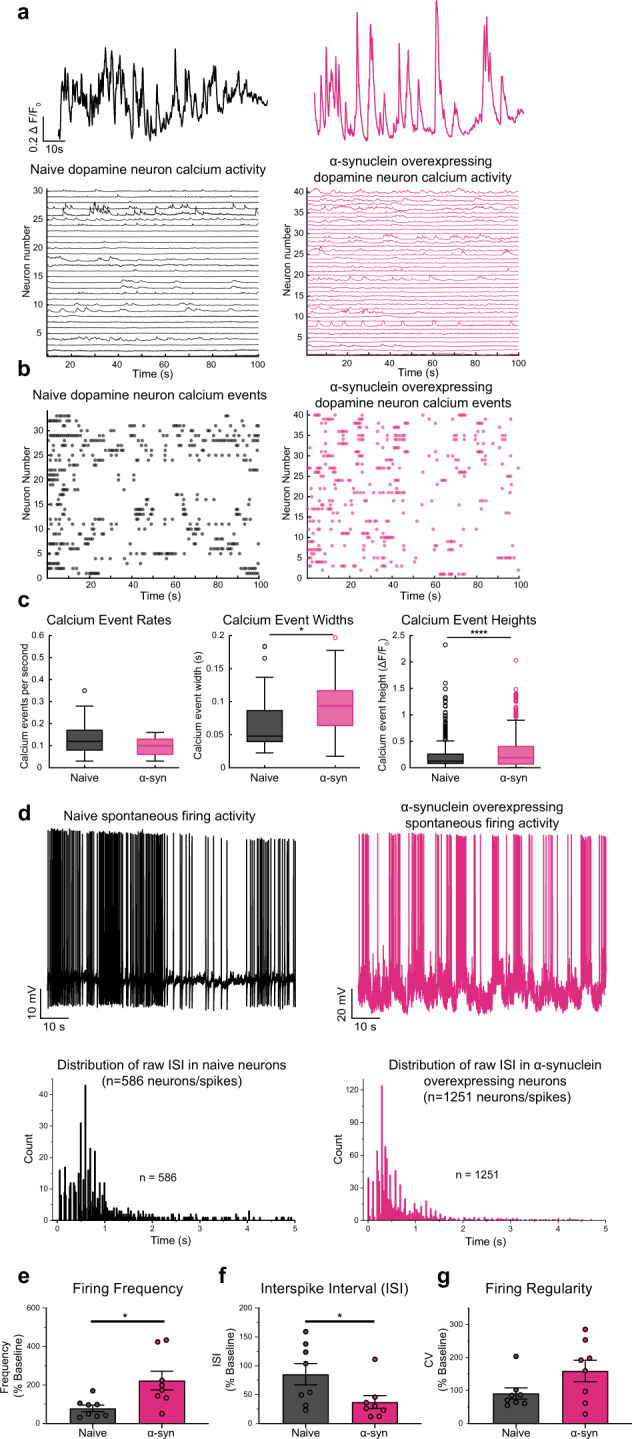
a (Top) Representative spontaneous calcium activity in naive dopaminergic neurons (left, black) and dopaminergic neurons overexpressing α-syn (right, pink) exemplify the alteration in calcium dynamics due to increased levels of α-syn. (Bottom) Spontaneous calcium activity encompassing all neurons recorded in each experimental group (n = 33 wild-type neurons, n = 40 α-syn-overexpressing neurons, form eight biological replicates). b Calcium events in all neurons. c Spontaneous calcium event rate, width, and amplitude. Overexpression of α-syn does not alter calcium event rate (two-tailed unpaired t test, WT vs. α-syn, p = 0.1775, n = 33 wild-type neurons, n = 40 α-syn-overexpressing neurons), α-syn burden broaden calcium events (p = 0.0152, two-tailed unpaired t test, WT vs. α-syn, n = 33 wild-type neurons, n = 40 α-syn-overexpressing neurons) and increases in amplitude (p = 0.0000198, two-tailed unpaired t test, WT vs. α-syn, n = 33 wild-type neurons, n = 40 α-syn-overexpressing neurons). d Representative whole-cell current-clamp recordings of spontaneously active naive (top left, black) compared with overexpressing α-syn dopaminergic neurons (top right, pink). Distribution of raw interspike intervals (ISIs) in naive (bottom left) and α-syn-overexpressing neuron (bottom right) (raw ISI distribution, Kolmogorov–Smirnov test, D = 0.26529, p < 0.001). e Naive compared with α-syn-overexpressing dopaminergic neurons (firing frequency: from eight independent experiments, 100 ± 21.21 for naive neurons vs. 281.7 ± 61.30 for α-syn-overexpressing neurons, two-tailed unpaired t test, p = 0.0142; f interspike interval (ISI): 100 ± 21.63 for naive neurons compared to 43.55 ± 13.28 for α-syn-overexpressing neurons, two-tailed unpaired t test, p = 0.0431) and g firing regularity trend in bursts with intermediated periods of quiescence (CV of ISI—100 ± 18.59 for naive neurons vs. 174.9 ± 35.20 for α-syn-overexpressing neurons, two-tailed unpaired t test, p = 0.0808). Empty circles in panel c represent statistical outliers included in the analyses. Bar graphs ± SEM are overlaid with individually filled data points. *p < 0.05; ****p < 0.0001.
While measurement of calcium activity provides inferential information about the neuronal firing, we did not observe a change in calcium event rates (Fig. 2c, left). The firing rate of dopamine neurons ranges from 0.5 to 20 spike/s10–21. We and others have shown that electrophysiological properties of cultured dopamine neurons are consistent with those of dopamine neurons in vivo or in vitro10,11,14,21. Therefore, next, we investigated whether α-syn overexpression modulates the firing activity of dopaminergic neurons. We utilized whole-cell current-clamp recordings to measure the spontaneous firing activity of cultured DAT-GCaMP6f neurons with endogenous levels or overexpression of α-syn, which were identified by GCAMP6f fluorescence. While dopaminergic neurons containing endogenous levels of α-syn exhibited characteristic pace-making activity53,54, the spontaneous firing activity of α-syn-overexpressing dopamine neurons showed an irregular and clustered firing pattern with increased burst firing activity within the clusters (Fig. 2d, e, data are expressed as the percent of control from eight independent experiments, firing frequency—100 ± 21.21 for naive neurons vs. 281.7 ± 61.30 for α-syn-overexpressing neurons, two-tailed unpaired t test, naive vs. α-syn, p = 0.0142, raw ISI distribution, Kolmogorov–Smirnov test, D = 0.26529, p < 0.001). Thus far, our data suggest that increased α-syn levels in dopaminergic neurons lead to altered calcium dynamics and increased firing activity. Both firing activity and calcium dynamics in dopamine neurons are tightly regulated by the activity of the D2 autoinhibitory receptors53–57. Therefore, we asked whether α-syn-induced dysregulation of dopamine neuronal activity and calcium dynamics is due to reduced D2R autoinhibition.
α-Syn overexpression reduces D2R-mediated autoinhibition
Multiple channels and transporters regulate neuronal activity and intracellular calcium dynamics. Dopamine activation of D2 autoinhibitory receptors on dopamine neurons decreases neuronal activity16,53,54,56,58 and intracellular calcium dynamics52,55,59–62. Therefore, we tested the hypothesis that the observed disturbance in neuronal excitability and calcium dynamics in α-syn-overexpressing neurons is due to dysregulation of canonical D2R autoinhibition in these neurons. In a double-blinded experimental design, we exposed DAT-GCaMP6f cultures to dopamine (1 µM) while monitoring the change in GCaMP6f signal. Consistent with the literature63,64, dopamine induced a decrease in intracellular Ca2+ as measured by a decrease in GCaMP6f fluorescence in non-transduced neurons (Fig. 3a–c (top), n = 14–21, naive neurons, two-way repeated-measures analysis of variance (ANOVA), row factor: time, % of total variation = 7.65, p = 0.0003, column factor: dopamine treated vs. untreated, % of total variation = 6.289, p = 0.0003; interaction: time × treatment, % of total variation = 5.079, p = <0.0001). However, α-syn-overexpressing neurons exhibited an attenuated dopamine-induced reduction of GCaMP6f signal, although still significantly affected (Fig. 3b, n = 17–26, two-way ANOVA where the variables are time and treatment, followed by Tukey’s honestly significant difference (HSD), p = 0.0088, from five independent replicates; α-syn-overexpressing neurons, two-way repeated-measures ANOVA, row factor: time, % of total variation = 3.759, p = 0.0169, column factor: dopamine treated vs. untreated, % of total variation = 3.189, p = 0.0088; interaction: time × treatment, % of total variation = 2.886, p = <0.0001). Further, fold-change responses of naive dopamine neurons were greater than α-syn-overexpressing neurons (Fig. 3c, n = 11–17, two-tailed unpaired t test, naive vs. α-syn, p = 0.0031, from five independent replicates; a comparison was made between average calcium activity 30 s before drug and last 30 s of drug exposure). These data suggest that α-syn overexpression decreases the inhibitory feedback regulation in dopamine neurons.
Fig. 3. α-Synuclein overexpression reduces D2 receptor autoinhibition.

a Representative images of naive dopaminergic neurons (top) and α-syn-overexpressing dopaminergic neurons (bottom); before (left) and during dopamine (1 μM) administration (right). b (Top) In naive neurons, dopamine reduced the ∆F/FGCaMP6f (n = 14–21, two-way ANOVA, p = 0.0003, from five independent replicates). (Bottom) In α-syn-overexpressing neurons, dopamine produced a smaller reduction in ∆F/FGCaMP6f (n = 17–26, two-way ANOVA, p = 0.0088, from five independent replicates). c The fold change in ∆F/FGCaMP6f before and after (n = 11–17, two-tailed t test, p = 0.0031, from five independent replicates). d Representative images of naive dopaminergic neurons (top) and α-syn-overexpressing neurons (bottom), before (left) and after quinpirole (10 μM) (right). e In naive neurons, quinpirole reduced the ∆F/FGCaMP6f (top panel) (n = 11–21, two-way ANOVA, p = 0.0007, from five independent biological replicates). In α-syn-overexpressing neurons, quinpirole produced a smaller reduction in ∆F/FGCaMP6f (bottom panel) (n = 13–26, two-way ANOVA, p = 0.7259, from five independent replicates). f Fold change in ∆F/FGCaMP6f before and after drug (n = 11–13, two-tailed t test, p = 0.0217, from five independent replicates). g (Top left) A representative recording of the spontaneous firing activity of naive dopaminergic neurons before and during quinpirole (10 μM) (n = 6, from three independent biological replicates). (Top right) A representative recording of the spontaneous firing activity of α-syn-overexpressing neurons before and during quinpirole (10 μM) (n = 8, from three independent experiments). (Bottom) Acute quinpirole treatment significantly increases raw interspike interval distributions of naive and α-syn-overexpressing dopaminergic neurons (Kolmogorov–Smirnov test, naive—D = 0.50642, p < 0.001, α-syn overexpressing—D = 0.47776, p < 0.001). h Comparison of the firing frequency of naive (black) and α-syn-overexpressing neurons (pink bar) during quinpirole administration (green) (n = 7 from independent experiments, two-way ANOVA, p = <0.0001). i Interspike interval—n = 7 from independent experiments, two-way ANOVA, p = 0.0136; j firing regularity (CV of ISI)—n = 7 from independent experiments, two-way ANOVA, p = 0.0200). The data are presented as mean ± SEM. h, i are presented as %change from untreated naive. Scale bar = 50 μm. *p < 0.05, **p < 0.01.
While these data support the interpretation that α-syn overexpression decreases the feedback modulation of neuronal activity, they do not unequivocally show a decrease in D2R activity. Dopamine interacts with multiple targets, such as DAT, that also regulate neuronal excitability10,11,25,65 and intracellular calcium activity10,11,21,23,25. Therefore, we utilized quinpirole (10 µM), a D2R-specific agonist, to stimulate D2R in DAT-GCaMP6f cultures containing either endogenous levels of α-syn or its overexpression (Fig. 3d). Consistent with the literature60,62,63,66, quinpirole activation of D2 autoinhibitory receptors induced the canonical suppression of calcium dynamics in naive dopaminergic neurons, whereas the calcium activity in α-syn-overexpressing neurons did not change during quinpirole administration (Fig. 3e, f naive: n = 11–21, two-way repeated-measures ANOVA, row factor: time, % of total variation = 9.923, p = <0.0001, column factor: untreated vs. quinpirole, % of total variation = 4.951, p = 0.007; interaction: time × pharmacology, % of total variation = 7.233, p = <0.0001; α-syn overexpressing: n = 11–21, two-way repeated-measures ANOVA, row factor: time, % of total variation = 1.873, p = 0.4797, column factor: untreated vs. quinpirole, % of total variation = 0.08018, p = 07259; interaction: time × pharmacology, % of total variation = 1.288, p = 0.9999; fold-change comparisons—n = 11–13, two-tailed unpaired t test, naive vs. α-syn, p = 0.0217).
Although live-cell calcium imaging provides a proxy for dopaminergic neuronal activity23, calcium imaging does not reveal changes in firing activity at the resolution of electrophysiological recordings. Therefore, as a complementary approach, we utilized whole-cell current-clamp recordings to compare the firing activity of naive and α-syn-overexpressing dopaminergic neurons following quinpirole administration. Consistent with the literature, quinpirole activation of D2 autoreceptors decreased the firing activity of naive dopamine neurons16,56,67–69 and α-syn-overexpressing dopaminergic neurons (Fig. 3g–j, interspike interval distributions of naive and α-syn-overexpressing dopaminergic neurons (Kolmogorov–Smirnov test, naive—D = 0.50642, p < 0.001, α-syn overexpressing—D = 0.47776, p < 0.001), firing frequency—n = 7 from independent experiments, 100 ± 23.621 baseline and 18.639 ± 5.300 quinpirole for naive neurons vs. 238.638 ± 18.321 baseline and 81.565 ± 56.046 for α-syn treated with quinpirole, two-way repeated-measures ANOVA, row factor: naive vs. α-syn, % of total variation = 28.69, p = 0.0023, column factor: baseline vs. quinpirole, % of total variation = 40.14, p = <0.0001; interaction: phenotype × pharmacology, % of total variation = 4.048, p = 0.0041; interspike interval—n = 7 from independent experiments, 100 ± 13.282 baseline and 782.847 ± 282.0257 quinpirole for naive neurons vs. 51.203 ± 10.741 baseline and 266.5666 ± 124.1509 for α-syn neurons treated with quinpirole, two-way repeated-measures ANOVA, row factor: naive vs. α-syn, % of total variation = 9.071, p = 0.1019, column factor: baseline vs. quinpirole, % of total variation = 20.61, p = 0.0136; interaction: phenotype × pharmacology, % of total variation = 6.025, p = 0.1441; firing regularity (CV of ISI)—n = 7 from independent experiments, 100 ± 14.750 baseline and 149.313 ± 24.054 quinpirole for naive neurons vs. 159.960 ± 28.831 baseline vs. 120.241 ± 28.489 for α-syn neurons treated with quinpirole, two-way repeated-measures ANOVA, row factor: naive vs. α-syn, % of total variation = 0.033, p = 0.9258, column factor: baseline vs. quinpirole, % of total variation = 13.78, p = 0.0200; interaction: phenotype × pharmacology, % of total variation = 4.442, p = 0.1540). While acute quinpirole produced the canonical silencing of naive dopaminergic neurons70,71 (DAT-Cre-GCaMP6f) (Fig. 3g, left), to our surprise, following acute exposure to quinpirole, the firing activity of α-syn-overexpressing dopamine neurons (DAT-Cre-GCaMP6f) began to resemble baseline firing activity observed in naive dopaminergic neurons (Fig. 3g, left black trace: naive dopamine neuron before and after drug application; Fig. 3g—right pink trace: α-syn neurons before and after drug application) (unpaired two-tailed t test, p = 0.6096, naive baseline, 0.972 ± 0.230, vs. α-syn in the presence of quinpirole, 0.793 ± 0.253). Collectively, these data support the hypothesis that α-syn overexpression reduces D2R-mediated autoinhibition in cultured dopamine neurons, and suggests that a more prolonged activation of D2R can potentially restore this deficit.
α-Syn overexpression increases intracellular and extracellular dopamine levels and TH expression
Our findings so far suggest that α-syn may induce a feedforward adaptive mechanism that decreases the ability of inhibitory D2 autoreceptors to act as a brake on neuronal excitability and increase extracellular dopamine levels53,56. To test this hypothesis, we used two complementary approaches of high-performance liquid chromatography (HPLC) analysis and an engineered dopamine sensor to measure intracellular and extracellular dopamine levels, at baseline. First, we used GRABDA2M (G-protein-coupled receptor activation-based DA-expressing HEK293 cells) to measure extracellular dopamine levels. GRABDA2M is a genetically encoded fluorescent dopamine sensor that is engineered by coupling a conformationally sensitive circular-permutated enhanced GFP (cpEGFP) to D2R. In GRABDA2M-expressing HEK293 cells, dopamine binding to the sensor induces a conformational change that results in a robust increase in fluorescence signal in a concentration-dependent manner (Fig. 4a, b).
Fig. 4. Overexpression of α-synuclein increases intracellular and extracellular dopamine levels with concurrent increased tyrosine hydroxylase expression.

a Schematic and representative baseline-subtracted images of GRABDA2M-HEK cells exposed to increasing concentration of dopamine. Scale bar = 20 μm. b A standard curve of GRABDA2M-HEK cells against known extracellular dopamine concentrations (R2 = 0.98). c Constitutive GRABDA2M-HEK cell fluorescence signal in the absence of dopamine neurons (in culture). Scale bar = 10 μm. d Schematic of GRABDA2M-HEK cells seeded into dopaminergic cultures. In the presence of dopamine, GRABDA2M-HEK cells rapidly increase in fluorescence intensity. e Baseline fluorescence levels denote unstimulated and spontaneous dopamine release from the neurons. The average ratio of the fluorescence signal of the cells adjacent to neuron soma and neuronal processes to the average ratio of GRABDA2M-HEK cells (only) were calculated (relative fluorescence = (FGRABDA2M-HEK cells grown with neurons − Fc)/Fc). GRABDA2M-HEK cells cocultured with naive and α-syn-overexpressing neurons. Scale bar = 50 μm. f GRABDA2M-HEK cells cocultured with α-syn-overexpressing neurons show higher basal fluorescence, indicating higher baseline dopamine release (relative fluorescence) compared to naive neurons (n = 10 from three independent replicates; the data are means ± SEM, two-tailed t test, p = 0.0013). g, h HPLC analysis complements the GRABDA2M-HEK results. Extracellular milieu (g) and cell lysate intracellular milieu (h) revealed increased intracellular and extracellular dopamine levels in α-syn-overexpressing neurons compared to naive neurons (n = 8 each, from eight independent replicates, two-tailed t test; intracellular: p = 0.0071; extracellular: p = 0.0139). i Schematic diagram of quantitative ELISA experimental design for TH in dopaminergic neurons. j Standard curve for TH sandwich ELISA shows average absorbance values for each purified TH protein concentration from multiple consecutive experiments (R2 = 0.99). k TH protein levels were detected and quantified in positive control groups, PC12 cells, whereas no protein was detected in the negative control group, HEK293 cells. l α-Syn-overexpressing neurons exhibited increased levels of TH compared to naive (n = 8–10, two-tailed t test, p = 0.0289). These experiments were performed through a double-blinded experimental design. *p < 0.05, **p < 0.01.
Constitutive GRABDA2M fluorescence signal in the absence of dopamine neurons was obtained at the beginning of each experiment, where GRABDA2M-expressing HEK293 cells were plated in similar conditions, but sans neurons (Fc, Fig. 4c). To compare baseline dopamine release amongst the experimental groups, the average ratio of the fluorescence signal of cells adjacent to the soma and neuronal processes to the average ratio of the fluorescence signal of GRABDA2M cells (only) was calculated .
To confirm that the increase in GRABDA2M fluorescence signal is due to dopamine release, we ran a positive control experiment (Supplementary Fig. 2), where GRABDA2M fluorescent signal around the soma and neuronal processes was measured following KCl (90 mM) stimulation of dopamine release72. The average fluorescence signal of cells adjacent to the soma and neuronal processes before and after KCl was calculated (Supplementary Fig. 2A, B). KCl-induced neuronal depolarization73,74 produced a robust fluorescence increase in both experimental groups (Supplementary Fig. 2B, n = 10 from three independent replicates; the data are mean ± SEM, two-tailed t test, p = 0.8991). The KCl-evoked dopamine release was similar in both experimental groups.
After confirming the ability of GRABDA2M cells to detect evoked dopamine release, we measured spontaneous (i.e., baseline, unstimulated) dopamine release in naive and α-syn-overexpressing neurons. The neurons were cocultured with GRABDA2M cells 20–24 h prior to live-cell confocal imaging. This experimental design enables real-time detection of endogenous dopamine released at baseline, i.e., spontaneous dopamine release (Fig. 4d, e). Using a blinded experimental design, we found a significantly higher spontaneous dopamine level, as measured by a higher GRABDA2M fluorescence signal around the soma and neuronal processes of α-syn-overexpressing neurons (Fig. 4f, n = 33 naive, 89 α-syn-overexpressing from three independent replicates; the data are mean ± SEM, two-tailed t test, p = 0.0013). These data support the interpretation that α-syn overexpression increases spontaneous neuronal activity, leading to increased extracellular dopamine levels. We previously reported that α-syn overexpression decreases dopamine uptake via the DAT75 and we have also reported that α-syn overexpression increases the DAT-mediated dopamine efflux24. These data are consistent with the hypothesis that α-syn overexpression can increase extracellular dopamine levels. Collectively, our previous reports72,73, combined with the data shown in Figs. 2–4, provide a reasonable cellular mechanism for the puzzling observation by Lam et al.76 that in mice overexpressing α-syn there is an initial increase in extracellular dopamine levels in the striatum prior to neuronal death.
Because GRABDA readouts report only the relative difference in dopamine release between α-syn and control neurons, we used HPLC to measure absolute dopamine levels in the external milieu of α-syn-transduced and naive DAT-GCaMP6f cultures (i.e., spontaneous dopamine release) via a blinded experimental design. HPLC analysis showed significantly higher extracellular dopamine levels in α-syn-overexpressing neurons compared to naive neurons (Fig. 4g, h; n = 8 each, from eight independent replicates; two-tailed t test, p = 0.0139). Collectively, these data, combined with live-cell detection of extracellular dopamine levels (at baseline), support the notion that α-syn modulation of dopaminergic neuronal activity leads to increased extracellular dopamine levels. An increase in extracellular dopamine could be due to increased neuronal activity, increased dopamine synthesis, or both possibilities. Since we have already examined the former (Figs. 2 and 3) to test the latter possibility, we used HPLC to measure intracellular dopamine levels. The measurement of dopamine in the cell lysate of naive and α-syn-overexpressing neurons revealed significantly higher intracellular dopamine levels (Fig. 4h, n = 8 each, from eight independent replicates, two-tailed t test, p = 0.0071). These data suggest that the decreased autoinhibition of dopamine neurons following α-syn overexpression not only increases neuronal excitability but also dysregulates dopamine synthesis and secretion. Furthermore, in Fig. 2, we showed that increased neuronal α-syn increases the magnitude and duration of intracellular calcium burden, which would promote increased basal dopamine release.
Multiple mechanisms likely contribute to the increased intracellular dopamine following α-syn overexpression. For example, increased dopamine uptake via the DAT, decreased DAT-mediated dopamine efflux, increased expression of TH (a key enzyme involved in dopamine synthesis), or a combination of these mechanisms would possibly contribute to a higher intracellular dopamine level. Previously, we and others have shown that α-syn overexpression reduces dopamine recycling by reducing dopamine uptake75,77,78. In addition, we have shown that α-syn overexpression increases reverse transport of dopamine, i.e., dopamine efflux24, without changing surface DAT levels. Therefore, α-syn regulation of dopamine uptake or dopamine efflux would decrease intracellular dopamine and not increase it.
While α-syn regulation of DAT activity predicts a decrease of intracellular dopamine level24,75,77–79, D2R activity negatively regulates TH protein levels as a compensatory mechanism to downregulate dopamine synthesis56,62,80–83. As shown in Figs. 2 and 3, we found a reduction in the canonical D2R autoinhibition of dopamine neurons, likely modulating downstream signaling cascades that can regulate TH protein levels. Therefore, next, we tested the hypothesis that in α-syn-overexpressing neurons a decrease in the D2 activity (shown in Figs. 2 and 3) leads to increased TH levels that can contribute to increased intracellular dopamine80,82–87. Since the frequently used approaches of western blotting or ICC do not provide purely quantitative data of protein expression to test this hypothesis, we developed an enzyme-linked immunosorbent assay (ELISA)88 to quantify TH levels in α-syn-overexpressing neurons (Fig. 4i). For these experiments, we used HEK293 cells as a negative control group and PC12 cells as a positive control group, and a purified full-length recombinant TH protein was used to generate a standard curve (Fig. 4j, k). α-Syn-overexpressing neurons show significantly higher TH levels compared to naive (Fig. 4l, n = 8–10, two-tailed t test, p = 0.0289). While ELISA provides quantitative data for total TH level across these experimental groups, a limitation of this assay is that it cannot discriminate TH phosphorylation that is associated with TH activity and thus dopamine synthesis80,89,90. Nevertheless, these data support the interpretation that increased intracellular dopamine in α-syn-overexpressing neurons, at least in part, is due to increased TH protein levels.
Altered neural dynamics mediated by α-syn may emerge from altered D2 activity and expression patterns
Our data, thus far, support the interpretation that the canonical D2R-mediated autoinhibition, such as inhibitory modulation of spontaneous firing activity, is reduced in α-syn-overexpressing neurons. While D2R agonist quinpirole silenced naive dopamine neurons, the response to quinpirole in α-syn-overexpressing neurons is significantly reduced, possibly due to desensitization or reduced activity of the D2R (Fig. 3). Therefore, next, we tested the hypothesis that blockade of D2Rs in naive dopaminergic neurons simulates the firing activity observed in α-syn-overexpressing neurons. We performed whole-cell current-clamp recordings to measure spontaneous firing activity of dopaminergic neurons before and during bath application of sulpiride (D2 antagonist, 5 µM). In naive dopaminergic neurons, bath application of sulpiride produced burst firing patterns with intermediated periods of quiescence and firing frequencies similar to α-syn-overexpressing dopaminergic neurons in the presence of sulpiride (Fig. 5a–e, n = 8 from three independent biological replicates, distribution of raw ISIs in naive and α-syn-overexpressing dopaminergic neurons, Kolmogorov–Smirnov test, D = 0.13114, p < 0.001, two-tailed unpaired t test, firing frequency: 100 ± 22.94 naive vs. 158.8 ± 30.37 α-syn-overexpressing neurons, p = 0.148; ISI: 100 ± 14.51 naive vs. 68.15 ± 11.84 α-syn-overexpressing neurons, p = 0.1147; CV of ISI: 100 ± 17.02 naive vs. 85.94 ± 6.599 α-syn-overexpressing neurons, p = 0.456). These data support the hypothesis that α-syn-overexpressing dopamine neurons exhibit reduced functional availability of D2-mediated response, which could be due to receptor desensitization89,90, decreased membrane expression of D2Rs, or a combination of these possibilities.
Fig. 5. D2 receptor antagonism in dopaminergic neurons mimics burst firing pattern with a significantly higher firing frequency observed in α-synuclein-overexpressing dopamine neurons that presents with lower membrane/cytoplasmic D2 ratio.

a, b Representative whole-cell current-clamp recordings of spontaneously active naive (a, top, black) and α-syn-overexpressing (b, top pink) dopaminergic neurons during sulpiride (D2 antagonist, 5 μM) bath application. a, b (Bottom) Distribution of raw ISIs in naive (a, bottom) and α-syn-overexpressing (b, bottom) dopaminergic neurons (Kolmogorov–Smirnov test, D = 0.13114, p < 0.001). c–e The bar graph shows firing frequency (c), interspike interval (ISI) (d), and firing regularity (e) during bath application of sulpiride (5 μM), revealing D2 antagonism in naive dopaminergic neurons promotes firing rates, interspike intervals, and regularity comparable to neurons overexpressing α-syn (n = 8 from three independent biological replicates, two-tailed unpaired t test, firing frequency: 100 ± 22.94 naive vs. 158.8 ± 30.37 α-syn-overexpressing neurons, p = 0.148; ISI: 100 ± 14.51 naive vs. 68.15 ± 11.84 α-syn-overexpressing neurons, p = 0.1147; CV of ISI: 100 ± 17.02 naive vs. 85.94 ± 6.599 α-syn-overexpressing neurons, p = 0.456).
To investigate if α-syn overexpression in dopaminergic neurons alters D2R expression, we performed cell surface biotinylation of D2R via a blinded experimental design with striatal lysate as a positive control group and CHO cells as the negative control group. When total D2R was normalized to HSP60 loading control, there were no significant differences between naive and α-syn-overexpressing neurons (Supplementary Fig. 3, from three independent biological replicates, two-tailed unpaired t test, p = 0.3417). We also found that the ratio of cytoplasmic D2R to total D2R in α-syn-overexpressing neurons was not significantly different from naive neurons (Supplementary Fig. 3, from three independent biological replicates, two-tailed unpaired t test, p = 0.9426). However, when comparing the ratio of membrane-to-cytoplasmic D2R, we found that α-syn-overexpressing neurons have a significantly lower ratio of membrane-to-cytoplasmic D2R than naive neurons (Supplementary Fig. 3B, from three independent biological replicates, two-tailed unpaired t test, p = 0.0039). It should be noted that biotinylation assay detects total (both functional and desensitized receptors). The double ICC of fixed, but not permeabilized, dopamine neurons stained for both D2R and an integral membrane protein such as Na+/K+-ATPase, or GM1-CTxB would have been a suitable complementary approach to examine membrane-localized D2R across the experimental groups in this study. However, the frequently used D2R antibodies in the field91–94 are raised against the intracellular N-terminal domain of the receptor. This limitation decreases the confidence in the identification of membrane vs. intracellular protein levels. A similar technical limitation applies to the single-cell qPCR assay, where total transcript levels do not necessarily reflect functional D2Rs at the membrane. The latter limitation somewhat applies to the biotinylation assay used in this study. Unless an antibody is raised against the active or inactive form of the receptor, a biotinylation assay detects both functional and desensitized receptors. Therefore, although our data suggest that membrane D2Rs are decreased in α-syn-overexpressing neurons, it is possible that the detected membrane D2Rs are desensitized, i.e., a lesser receptor–effector coupling95–98. Therefore, live-cell functional assays, such as electrophysiology and calcium imaging, combined with pharmacological manipulations are more reliable strategies to assess the mechanism of α-syn regulation of neuronal activity.
α-Syn overexpression reduces arborization of dopamine neurons and pretreatment with a D2R agonist partially rescues the detrimental impact of α-syn
Dopaminergic neurons have extensive axonal arborizations and large terminal fields99–101, where one dopamine neuron is estimated to have ~245,000 release sites102,103. Studies in animal models of PD and postmortem data in human PD104 show that decreased axonal complexity and dendritic arborization, reduction of the number of axon terminals, and global neuronal size precede neuronal death77,102,105. Our data suggest that prior to cell death, via a D2R mechanism, α-syn overexpression can induce neuronal disinhibition, leading to increased intracellular and extracellular dopamine levels that are implicated in increased neuronal vulnerability35,106,107. Therefore, we investigated the potential link between α-syn-mediated dopamine neuronal dysfunction and neuronal complexity.
Sholl analysis entails using concentric circles around the soma of a neuron, with neurite fields intersecting these concentric circles counted as a measure of differences in neuronal complexity (Fig. 6a–d). This analytical approach estimates99,106–108 neuronal complexity via assessment of projection area, number of intersections as a measure of neuronal arborization, projection field perimeter, neuronal arborization width, and circularity of neurite arborization108–113. Compared to naive dopaminergic neurons, α-syn-overexpressing neurons exhibit a lower degree of neurite arborization (Fig. 6e, f, h), reduced projection area (Fig. 6j), reduced process arbor circularity (Fig. 6k), smaller projection field perimeter (Fig. 6l), and smaller arborization width (Fig. 6m), but no change in the soma area (Fig. 6i, one-way ANOVA followed by Tukey’s HSD, naive n = 190, α-syn n = 114, intersections: naive vs. α-syn, p = 0.0009, circularity: naive vs. α-syn, p = 0.0021, outer perimeter: naive vs. α-syn, p = 0.0001; width: naive vs. α-syn, p = 0.0001, projection area: naive vs. α-syn, p = 0.0001, soma area: naive vs. α-syn, p = 0.67, from at least three independent biological replicates). The loss of neuronal complexity and decreased dendritic arborization found in this study are consistent with morphological data in postmortem PD samples104, potentially informing the progression of α-syn-induced pathology prior to neuronal loss.
Fig. 6. α-Synuclein overexpression reduces arborization of dopaminergic neurons and treatment with a D2 receptor agonist partially rescues the detrimental impact of α-synuclein.

Experiments conducted in at least three independent biological replicates. a Schematic representation of morphological analysis. b–d Representative binarized images of naive (b), α-syn-overexpressing (c), and quinpirole-pretreated α-syn-overexpressing neurons (d). Sholl intersection profiles of untreated naive (e), untreated α-syn-overexpressing (f), and quinpirole-pretreated α-syn-overexpressing neuron (g) measurement of area under curve (h) (one-way ANOVA, naive n = 190, α-syn, n = 114, naive vs. α-syn, p = 0.0009). Sholl analyses revealed that 48 h quinpirole (0.5 μM) pretreatment partially restores arborization complexity compared to untreated α-syn-overexpressing neurons (one-way ANOVA, naive n = 190, α-syn n = 114, and α-syn + quinpirole n = 32, naive vs. α-syn p = 0.0009, naive vs. α-syn + quinpirole p = 0.5131, and α-syn vs. α-syn + quinpirole p = 0.0041). i Somatic areas were comparable between experimental groups (one-way ANOVA, naive n = 190, α-syn n = 114, naive vs. α-syn p = 0.67). j α-Syn-overexpressing neurons project over smaller area than naive neurons (one-way ANOVA, naive n = 190, α-syn n = 114, naive vs. α-syn p = 0.0001). k–m Detrimental morphological changes in α-syn-overexpressing neurons (one-way ANOVA, naive n = 190, α-syn n = 114, circularity: naive vs. α-syn p = 0.0021, outer perimeter: naive vs. α-syn p = 0.0001; width: naive vs. α-syn p = 0.0001). D2 receptor agonist partially rescues the detrimental impact of α-synuclein. Quinpirole treatment of α-syn-overexpressing neurons rescued changes in (j) projection area (one-way ANOVA, naive n = 190, α-syn n = 114, and α-syn + quinpirole n = 32, naive vs. α-syn p = 0.0001, naive vs. α-syn + quinpirole p = 0.1136, and α-syn vs. α-syn treated with quinpirole p = 0.0486), (k) neuronal circularity, (l) projection field perimeter, and (m) arborization width (one-way ANOVA, naive n = 190, α-syn n = 114, and α-syn + quinpirole n = 32, circularity: naive vs. α-syn p = 0.0021, naive vs. α-syn + quinpirole p = 0.4452, and α-syn vs. α-syn + quinpirole p = 0.0046; outer perimeter: naive vs. α-syn p = 0.0001, naive vs. α-syn + quinpirole p = 0.2250, and α-syn vs. α-syn + quinpirole p = 0.0312; width: naive vs. α-syn p = 0.0001, naive vs. α-syn + quinpirole p = 0.1419, and α-syn vs. α-syn + quinpirole p = 0.0187). n Dopaminergic neuron counts revealed that α-syn overexpression decreases neuronal survival, which is rescued when pretreated with quinpirole (0.5 μM for 48 h) (one-way ANOVA, naive vs. α-syn p = 0.0008, naive vs. α-syn + quinpirole p = 0.1364, and α-syn vs. α-syn + quinpirole p = 0.0021). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The unexpected observation that acute D2 treatment, as shown in Fig. 3g (right), restored firing properties of α-syn-overexpressing neurons to the levels observed in naive dopamine neurons, supports the hypothesis that pharmacological activation of D2R could be a potential target to alleviate α-syn pathology prior to neuronal death. This hypothesis is consistent with reports showing that D2R activation sustains the structural plasticity of dopaminergic neurons by maintaining their dendritic arborization107,114–117. It has been reported that PD is proceeded by retrograde axonal degeneration118,119 and D2Rs’ activity regulates dopaminergic neuronal complexity107,114–117. Therefore, based on the literature and our data in Fig. 3g, next we tested the hypothesis that prolonged D2R activation would ameliorate the reduction of neuronal complexity107,120,121 in the presence of α-syn burdens. We measured neuronal complexity when α-syn-overexpressing neurons were pretreated with quinpirole (0.5 µM) for 48 h. Surprisingly, we found that quinpirole produced a marked improvement in arborization of α-syn-overexpressing neurons (Fig. 6d, g). Detailed morphometric analysis revealed no change in somatic size of neurons across all experimental groups, whereas quinpirole restored arbor circularity, projection area, projection field perimeter, and width of α-syn-overexpressing dopamine neurons, to the level measured in naive untreated neurons shown in Fig. 6b–m (naive n = 190, α-syn n = 114 and α-syn + quinpirole n = 32, from at least three independent replicates; circularity: naive vs. α-syn p = 0.0021, naive vs. α-syn + quinpirole p = 0.4452, and α-syn vs. α-syn + quinpirole p = 0.0046; outer perimeter: naive vs. α-syn p = 0.0001, naive vs. α-syn + quinpirole p = 0.2250, and α-syn vs. α-syn + quinpirole p = 0.0312; shape factor: naive vs. α-syn p = 0.0021, naive vs. α-syn + quinpirole p = 0.5574, and α-syn vs. α-syn + quinpirole p = 0.0086; width: naive vs. α-syn p = 0.0001, naive vs. α-syn + quinpirole p = 0.1419, and α-syn vs. α-syn + quinpirole p = 0.0187, one-way ANOVA followed by Tukey’s HSD).
Next, we examined the impact of α-syn overexpression on neuronal survival by counting TH-positive neurons via ICC. Compared to naive neurons, we found significantly fewer TH-positive neurons following α-syn overexpression; quinpirole-mediated activation of D2Rs (0.5 µM for 48 h) prevented the neuronal loss (Fig. 6n: naive vs. α-syn p = 0.0008, naive vs. α-syn + quinpirole p = 0.1364, and α-syn vs. α-syn + quinpirole p = 0.0021, from six independent biological replicates, one-way ANOVA followed by Tukey’s HSD). The increased neuronal survival following quinpirole pretreatment is consistent with previous reports107,114–117 and supports the interpretation that there is a correlation between α-syn modulation of neuronal complexity, neuronal vulnerability, and neuronal loss122–124. Neuronal survival is not equivalent to neuronal viability. While these data suggest that pretreatment with a D2R agonist increases neuronal survival following α-syn overexpression, they do not demonstrate a restoration of neuronal activity. Therefore, next, we asked whether quinpirole pretreatment prevents the increase in action potential (AP) frequency, increased intrav and extracellular dopamine levels, and elevated intraneuronal calcium dynamics in α-syn-overexpressing neurons.
D2R activation partially restores neuronal activity in α-syn-overexpressing dopamine neurons
The energy homeostasis principle suggests that the balance between energy income, expenditure, and availability is the key parameter in determining neuronal endurance125. APs impose the highest energy demands on neurons102,125,126. In addition, dopamine metabolism is strongly linked to oxidative stress, as its degradation generates reactive oxygen species35,127,128 that have shown to increase the vulnerability of dopamine neurons to oxidative stress101,128–134. So far, we have identified multiple interrelated mechanisms that can potentially lead to the vulnerability of α-syn-overexpressing dopamine neurons. We identified an increase in AP frequency, increased intracellular and extracellular dopamine levels, and elevated intraneuronal calcium dynamics in α-syn-overexpressing neurons that are directly or indirectly related to decreased D2R activity. The unexpected observation that protracted (48 h) application of D2R agonist increased neuronal survival and nearly restored neuronal complexity of α-syn-overexpressing neurons to the levels measured in naive dopaminergic neurons at baseline suggests that the pharmacological activation of D2Rs might be a possible target to alleviate the untoward consequences of α-syn overexpression on neuronal activity prior to neuronal death. To test this hypothesis, we treated α-syn-overexpressing neurons with 0.5 µM quinpirole for 48 h before assessing calcium dynamics, spontaneous firing activity, and dopamine release and synthesis in these neurons (Fig. 7). We compared the results of these experiments to our previous data obtained in α-syn-overexpressing neurons. To reduce the impact of type 1 statistical errors, naive primary midbrain cultures were produced alongside each experimental group.
Fig. 7. Pretreatment with D2 receptor stimulation partially restores neuronal activity in α-synuclein-overexpressing dopamine neurons.

Previous data are overlaid with the dotted line representing averages for untreated naive neurons (black) and untreated α-syn-overexpressing neurons (pink). Shading indicates SEM. a, b Representative ∆F/FGCaMP6f trace (a), calcium activity (b), and event (c) of α-syn-overexpressing neuron pretreated with quinpirole (0.5 μM, 48 h) exhibiting calcium dynamics similar to untreated naive neurons. d–f Event rate, width, and amplitude after quinpirole pretreatment, respectively (n = 28 quinpirole-treated α-syn-overexpressing neurons, two-tailed unpaired t test, α-syn vs. α-syn + quinpirole p = 0.2024 event rate, p = 0.0277 event widths, p = 0.6204 event height, untreated α-syn data presented in Fig. 2). Box plot whiskers represent the 95% confidence interval, the upper and lower bounds of the box represent the 75th and 25th percentiles, respectively; the middle line indicates the median value of the sample. Representative firing activity of an untreated naive (g) and quinpirole-pretreated α-syn-overexpressing neuron (h). i–k Firing frequency (i), interspike interval (j), and firing regularity (k) in quinpirole-pretreated α-syn-overexpressing neuron (n = 7, 1.325 ± 0.2735 Hz for quinpirole-treated α-syn-overexpressing neurons, two-tailed unpaired t test, α-syn vs. α-syn + quinpirole p = 0.0342 for firing frequency, p = 0.1053 for ISI, p = 0.4778 for CV of ISI, untreated α-syn data are presented in Fig. 2). l GRABDA2M-HEKs seeded with untreated naive (left), untreated α-syn-overexpressing (middle), and quinpirole-pretreated α-syn-overexpressing neurons (right). m Quinpirole rescued extracellular dopamine level in α-syn-overexpressing neurons (n = 6, one-way ANOVA, naive vs. α-syn + quinpirole p = 0.9948, α-syn vs. α-syn + quinpirole p = 0.0003, untreated α-syn and naive data presented in Fig. 4). n, o HPLC quantification of dopamine confirm that quinpirole pretreatment of α-syn-overexpressing neurons reduces extracellular (n) and intracellular (o) dopamine levels vs. untreated α-syn-overexpressing neurons (n = 3 each, one-way ANOVA, intracellular: α-syn vs. α-syn + quinpirole p = 0.0325 and naive vs. α-syn + quinpirole p = 0.9959; extracellular: α-syn vs. α-syn + quinpirole p = 0.0449 and naive vs. α-syn + quinpirole p = 0.6197, untreated α-syn and naive data presented in Fig. 4). p Quantitative TH ELISA (n = 3, one-way ANOVA, naive vs. α-syn + quinpirole p = 0.4288 and α-syn vs. α-syn + quinpirole p = 0.6809, untreated α-syn and naive data are presented in Fig. 4). Data are presented as mean ± SEM, from at least three independent biological replicates. n.s. not significant. *p < 0.05, ****p < 0.0001.
While it may be intriguing to study the effects of pretreatment of quinpirole on naive neurons, we found that this was not feasible for spontaneous calcium activity and firing as these neurons were silent in the recording chamber (zero values for firing activity that cannot be included in statistical analyses of the data in Fig. 7). Furthermore, D2 autoreceptor activity has been shown to be a potent regulator of neuronal activity13,58,67,91,135–138, intracellular calcium23,50,91,138–140, neuronal morphology100,141, and protein expression55,90,106,138,140,142–145. Specifically, the purpose of quinpirole pretreatment was to examine whether modulation of D2R activity would restore or augment the properties of neurons with increased α-syn burdens to those of naive neurons, which has previously been shown to be true for MPP + exposure117, and not whether quinpirole pretreatment alters naive neurons. The black dotted line represents the average values measured for untreated naive neurons. The pink dotted line represents the average values for untreated α-syn-overexpressing neurons. The shaded region indicates the respective SEM for each measurement. We found that a prolonged D2R activation partially restores calcium dynamics in these neurons, approximating calcium dynamics measured in untreated naive neurons (Fig. 7a–f, n = 28 quinpirole-treated α-syn-overexpressing neurons, two-tailed unpaired t test, α-syn vs. α-syn pretreated with quinpirole p = 0.2024 event rate, p = 0.0277 event widths, p = 0.6204 event height, untreated α-syn, and naive data are presented in Fig. 2), suggesting a restoration of calcium homeostasis in these neurons that might be causal or a consequence of a shift in neuronal activity. To test this hypothesis, we employed whole-cell current-clamp recordings to measure the spontaneous firing activity of α-syn-overexpressing neurons after treatment with quinpirole (0.5 µM for 48 h). Quinpirole pretreatment on α-syn-overexpressing neurons decreased the burst firing frequency, shortened the intermediate periods of quiescence, and restored firing regularity near to the values measured in untreated naive neurons (Fig. 7g–k, n = 7 from three independent biological replicates, 117.1 ± 21.17 for quinpirole-treated α-syn-overexpressing neurons, two-tailed unpaired t test, α-syn vs. α-syn pretreated with quinpirole p = 0.0342 for firing frequency, p = 0.1053 for ISI, p = 0.4778 for CV of ISI, untreated α-syn, and naive data are presented in Fig. 2). These results suggest that dysregulation of D2R in α-syn-overexpressing dopamine neurons can be partially rescued with prolonged activation of the remaining functional D2Rs on the cell surface.
The observed changes in neuronal responses and calcium activity following extended D2R activation could be predictive of downstream changes in dopamine synthesis in α-syn-overexpressing neurons. To test the hypothesis that D2R activation decreases α-syn modulation of dopamine release, we measured extracellular dopamine levels via two complementary approaches: live-cell imaging utilizing an engineered dopamine sensor and HPLC. To measure D2R-mediated modulation of baseline extracellular dopamine levels (0.5 µM quinpirole 48 h) in α-syn-overexpressing neurons, we cocultured GRABDA2M-expressing cells with the quinpirole-treated, α-syn-overexpressing dopamine neurons for 20–24 h prior to imaging. Quinpirole pretreatment of α-syn-overexpressing neurons decreased basal GRABDA2M fluorescence (used as a proxy to measure basal dopamine release) around the soma and dendritic fields (Fig. 7l, m), comparable to values measured in naive untreated neurons shown in Fig. 4 (represented as a black dotted line in Fig. 7).
As a complementary approach, we used HPLC, as described in the “Methods” section and in Fig. 4, to measure extracellular dopamine level in the external milieu of neurons after quinpirole pretreatment (0.5 µM for 48 h) via a blinded experimental design. HPLC analysis revealed a reduction in basal dopamine release in all quinpirole-treated experimental groups, with the largest fold decrease in α-syn-overexpressing neurons (Fig. 7n, n = 3 from independent biological replicates, one-way ANOVA followed by Tukey’s HSD, α-syn vs. α-syn treated with quinpirole p = 0.0325 and naive vs. α-syn treated with quinpirole p = 0.9959). Therefore, through pharmacological manipulation of D2Rs, the α-syn dysregulation of dopamine transmission is potentially reversible (n = 3 each, from three independent biological replicates, one-way ANOVA followed by Tukey’s HSD, α-syn vs. α-syn treated with quinpirole p = 0.0325 and naive vs. α-syn treated with quinpirole p = 0.9959). The restoration of extracellular dopamine could be due to decreased neuronal activity, decreased dopamine synthesis, or both. Since we have already examined the former (Fig. 7a–k), to test the possibility of decreased dopamine synthesis, we used HPLC to measure intracellular dopamine levels via a blinded experimental design (described in the “Methods” section). Intracellular dopamine levels in quinpirole-treated α-syn-overexpressing neurons were significantly reduced compared to untreated α-syn-overexpressing neurons (Fig. 7o, n = 3 independent biological replicates, one-way ANOVA followed by Tukey’s HSD, α-syn vs. α-syn treated with quinpirole p = 0.0449 and naive vs. α-syn treated with quinpirole p = 0.6197) shown in Fig. 4 (n = 3 each, from 3 independent replicates, one-way ANOVA followed by Tukey’s HSD, α-syn vs. α-syn treated with quinpirole p = 0.0325 and naive vs. α-syn treated with quinpirole p = 0.9959). Since activation of D2R negatively regulates TH55,56,80,89,90,146–148 and neuronal activity16,53–56,58, we then tested the hypothesis that reduced intracellular and extracellular dopamine are, in part, due to decreased TH protein levels. Via a blinded experimental design, we utilized quantitative ELISA, as described in Fig. 4, to measure TH levels. As shown in Fig. 7p, TH protein level is similar in quinpirole-treated, α-syn-overexpressing neurons compared to untreated (0.5 µM for 48 h; n = 3, one-way ANOVA followed by Tukey’s HSD, naive vs. α-syn treated with quinpirole p = 0.4288 and α-syn vs. α-syn treated with quinpirole p = 0.6809). The partial rescue of α-syn-induced neuronal dysregulation after D2R activation is consistent with neuroprotective properties of D2Rs described previously117,149–152. It has been shown that D2 autoreceptors suppress dopamine synthesis through a negative feedback mechanism, and thus reduce oxidative stress caused by a high level of cytoplasmic dopamine149–151. In addition, consistent with our data, activation of D2 autoreceptors mediates neuroprotection by reducing neuronal excitability, cytoplasmic dopamine, and calcium levels117,152 that can restore the balance between energy income, expenditure, and its availability125. The data presented in this study provide a potential druggable target that may revert or prevent the untoward consequences of α-syn burden on dopamine neuronal activity and viability.
To summarize (Fig. 8), we found that α-syn overexpression dysregulates the structural and functional properties of dopaminergic neurons. The untoward consequences of increased α-syn likely cascade across the neuron, protracting the neuronal processes, increasing calcium burdens, and biophysical properties of dopamine neurons as measured by increased burst firing activity. We found that the endogenous self-regulation of dopaminergic neurons fails to restrain the exacerbation of these phenotypes. Thereby, the signaling of these neurons in their networks becomes erratic, potentially creating avalanching neuronal dysfunction. The dysregulation of dopamine signaling within the brain therefore precedes neuronal demise. However, we show that these progressive dysregulations can be reversed through pharmacological manipulation.
Fig. 8. Graphical summary.

α-Syn-mediated pathophysiological damages and loss of function of dopamine neurons precede neuronal demise.
The D2 autoreceptor-mediated feedback mechanism is one of the main autoinhibitory mechanisms regulating dopamine neuronal activity16,56,153. We found that D2 autoreceptor activity is diminished in α-syn-overexpressing dopamine neurons, and prolonged incubation with a D2R agonist, quinpirole (48 h, 0.5 μM), nearly restored the firing activity to its canonical levels, reinstated intracellular and extracellular dopamine levels, and prevented neuronal loss and structural neural complexity. Notably, D2R agonists (full and partial) have attained Food and Drug Administration approval and have made their way into the clinic; however, these are often tested in late-stage PD. Our results suggest that the current treatment timeline may occur too late and that the efficacy of this strategy requires early intervention to reduce the rate of neuronal demise. Most crucially, our results suggest that neuronal loss might be preventable, and future exploration across other mechanistic pathways will reveal intersectional treatments that may have the capacity to ameliorate PD.
Methods
When possible, the experiments are performed via a blinded experimental design. Animals were maintained in the University of Florida animal facilities. Experiments were approved by the Institutional Animal Care and Use Committee at the University of Florida.
Reagents and chemicals
The source, catalog number, and concentration of reagents, antibodies, and chemicals used in this study are outlined in Table 1. All viral vectors utilized in this study are listed in Table 2.
Table 1.
Biochemical assays.
| Dissociation media composition | |||||
|---|---|---|---|---|---|
| Chemical name | Concentration | Vendor | Catalog number | ||
| NaCl | 116 mM | Sigma-Aldrich | S7653 | ||
| NaHCO3 | 26 mM | Sigma-Aldrich | S5761 | ||
| NaH2PO4 | 2 mM | Sigma-Aldrich | S9638 | ||
| d-glucose | 25 mM | Sigma-Aldrich | G8769 | ||
| MgSO4 | 1 mM | Sigma-Aldrich | M7506 | ||
| Cysteine | 1.3 mM | Sigma-Aldrich | C7352 | ||
| Papain | 400 U/ml | Worthington Biochemical Corporation | LS003127 | ||
| Kynurenic acid | 0.5 mM | Sigma-Aldrich | K3375 | ||
| Glia media composition | |||||
|---|---|---|---|---|---|
| Chemical name | Concentration | Vendor | Catalog number | ||
| DMEM | 51.45% | Thermo Fisher Scientific | 11330032 | ||
| Fetal bovine serum | 39.6% | Gemini | 100–106 | ||
| Penicillin/streptomycin | 1% | Thermo Fisher Scientific | 15-140-122 | ||
| Glutamax 100X | 1% | Thermo Fisher Scientific | 35050061 | ||
| Insulin (25 mg/ml stock) | 0.08% | Sigma-Aldrich | I5500 | ||
| DIV0 neuronal media composition | |||||
|---|---|---|---|---|---|
| Chemical name | Concentration | Vendor | Catalog number | ||
| Neurobasal-A | 96.9% | Thermo Fisher Scientific | 10888022 | ||
| B27 Plus | 1.9% | Thermo Fisher Scientific | A3582801 | ||
| GDNF | 0.15% | Sigma-Aldrich | SRP3200 | ||
| Glutamax 100X | 1% | Thermo Fisher Scientific | 35050061 | ||
| Kynurenic acid | 0.08% | Sigma-Aldrich | K3375 | ||
| Maintenance of neuronal media composition | |||||
|---|---|---|---|---|---|
| Chemical name | Concentration | Vendor | Catalog number | ||
| Neurobasal-A | 97.1% | Thermo Fisher Scientific | 10888022 | ||
| B27 Plus | 1.9% | Thermo Fisher Scientific | A3582801 | ||
| Glutamax 100X | 0.97% | Thermo Fisher Scientific | 35050061 | ||
| Immunocytochemistry | |||||
|---|---|---|---|---|---|
| General reagents | |||||
| Chemicals | Concentration | Vendor | Catalog number | ||
| PBS | 1X | Prepared as needed on-site | N/A | ||
| Triton X-100 | 0.50% | Thermo Fisher Scientific | bp151-100 | ||
| NGS | 10% | Lampire Biological Products | 7332500 | ||
| Paraformaldehyde | 4% | Electron Microscopy Sciences | 157-4-100 |
| Primary antibodies | |||||
|---|---|---|---|---|---|
| Primary antibody–antigen | Host species/isotype | Concentration | Vendor | Catalog number | Use |
| Tyrosine hydroxylase | Rabbit/polyclonal | 1:500 | EMD Millipore | AB152 | ICC/WB |
| Alpha-synuclein (syn211) | Mouse/IgG1 | 1:500 | Abcam | AB80627 | ICC |
| DAT | Rat | 1:500 | Millipore Sigma | AB5802 | |
| Calbindin | Rabbit/polyclonal | 1:500 | EMD Millipore | ABN2192 | |
| GFP | Rabbit/IgG | 1:500 | Thermo Fischer Scientific | A-11122 | ICC |
| Human α-syn (C-terminal regions, 130–140) (94-3A10) | Mouse/monoclonal IgG1 | 1:500 | Gifted from Dr. Giasson | N/A | WB |
| Actin C4 | Mouse/monoclonal | 1:500 | EMD Millipore | MAB1501 | WB |
| D2 | Mouse | 1:500 | Neuromab | 75–230 | WB/ICC |
| D2 | Rabbit | 1:500 | Millipore | AB5084P | WB |
| Secondary antibodies | |||||
|---|---|---|---|---|---|
| Conjugate | Host species/target species | Concentration | Vendor | Catalog number | Use |
| Alexa Fluor 568 | Goat/mouse | 1:500 | Life Technologies | A-21124 | ICC |
| Alexa Fluor 647 | Goat/mouse | 1:500 | Life Technologies | A21242 | ICC |
| Alexa Fluor 488 | Goat/rabbit | 1:500 | Life Technologies | A-11006 | ICC |
| HRP | Goat/mouse | 1:2000 | Jackson Immuno Research Labs | AB-10015289 | WB |
| HRP | Goat/rabbit | 1:2000 | Jackson Immuno Research Labs | AB_2307391 | WB |
| IRDye® 800CW | Goat/mouse | 1:15000 | LI-COR | 926-32210 | WB |
| Biotinylation, cell lysis, and ELISA | |||||
|---|---|---|---|---|---|
| ELISA coating buffer, pH 9.6 | |||||
| Chemical name | Concentration | Vendor | Catalog number | ||
| Na2CO3 | 28.3 mM | Sigma-Aldrich | D6546 | ||
| NaHCO3 | 71.42 mM | Sigma-Aldrich | S5761 |
| BufferD lysis buffer, pH 7.6 | |||||
|---|---|---|---|---|---|
| Chemical name | Concentration | Vendor | Catalog number | ||
| Glycerol | 10% (v/v) | Sigma-Aldrich | G5516 | ||
| NaCl | 125 mM | Sigma-Aldrich | S7653 | ||
| EDTA | 1 mM | Sigma-Aldrich | E9884 | ||
| EGTA | 1 mM | Sigma-Aldrich | 3777 | ||
| Biotinylation (buffer pH 7.8) | |||||
|---|---|---|---|---|---|
| Chemical name | Concentration | Vendor | Catalog number | ||
| NaCl | 150 mM | Sigma-Aldrich | S7653 | ||
| CaCl2 | 2 mM | Sigma-Aldrich | 449709 | ||
| Triethanolamine | 10 mM | Sigma-Aldrich | 90279 | ||
| Sulfo-NHS-SS biotin | 1.5 mg/ml | Thermo Fisher Pierce | 22331 | ||
| Monomeric avidin resin | N/A | Thermo Fisher Pierce | 53146 |
| Antibodies for ELISA | |||||
|---|---|---|---|---|---|
| Specificity | Clone/species | Conjugate | Dilution | Vendor | Catalog number |
| TH | Monoclonal/mouse | N/A | 1:1000 | EnCor | MCA-4H2 |
| TH | Polyclonal/rabbit | Biotin | 1:1000 | EnCor | RPCA-TH |
| Biotin | N/A (avidin) | HRP | 1:2500 | Vector Labs | A-2004 |
| Calcium imaging ACSF composition | |||||
|---|---|---|---|---|---|
| pH range: 7.35–7.4; osmolarity range: 305–310 | |||||
| Chemical name | Concentration | Vendor | Catalog number | ||
| NaCl | 126 mM | Sigma-Aldrich | S7653 | ||
| KCl | 2.5 mM | Sigma-Aldrich | P9541 | ||
| CaCl2 | 2 mM | Sigma-Aldrich | 223506 | ||
| NaH2PO4 | 1.25 mM | Sigma-Aldrich | 71505 | ||
| MgSO4 | 2 mM | Sigma-Aldrich | M7506 | ||
| Dextrose | 10 mM | Sigma-Aldrich | D9434 | ||
| NaHCO3 | 24 mM | Sigma-Aldrich | S5761 | ||
| Pharmacological agents | |||||
|---|---|---|---|---|---|
| Resource/reagent | Concentration | Vendor | Catalog number | ||
| Dopamine hydrochloride | 1 μM | Sigma-Aldrich | H8502 | ||
| Quinpirole hydrochloride | 10 μM | Sigma-Aldrich | Q102 |
| Electrophysiology ACSF composition | |||||
|---|---|---|---|---|---|
| pH range: 7.35–7.4; osmolarity range: 305–310 | |||||
| Chemical name | Concentration | Vendor | Catalog number | ||
| NaCl | 126 mM | Sigma-Aldrich | S7653 | ||
| KCl | 2.5 mM | Sigma-Aldrich | P9541 | ||
| CaCl2 | 2 mM | Sigma-Aldrich | 223506 | ||
| NaH2PO4 | 1.25 mM | Sigma-Aldrich | 71505 | ||
| MgSO4 | 2 mM | Sigma-Aldrich | M7506 | ||
| Dextrose | 10 mM | Sigma-Aldrich | D9434 | ||
| NaHCO3 | 26 mM | Sigma-Aldrich | S5761 | ||
| Potassium gluconate | 120 mM | Sigma-Aldrich | P1847 | ||
| MgCl2 | 2 mM | Sigma-Aldrich | M8266 | ||
| HEPES | 10 mM | Sigma-Aldrich | H3375 | ||
| EGTA | 0.1 mM | Sigma-Aldrich | E3889 | ||
| ATPNa2 | 2 mM | Sigma-Aldrich | A2383 | ||
| GTPNa | 0.25 mM | Sigma-Aldrich | A2383 | ||
| Reagents for ELISA | |||||
|---|---|---|---|---|---|
| Reagent | Concentration | Supplier | Catalog number | Purpose | |
| Fat-free milk | 1% or 5%> | Carnation | N/A | WB/ELISA | |
| TMB substrate | Stock | Thermo Fisher | 34028 | ELISA | |
| H2SO4 | 2 N | Sigma | 339741 | ELISA | |
| Tween-20 | 0.002 | Thermo Fisher | MP1Tween201 | TBS-T | |
| Protease inhibitor | 1× | Milipore | 539191 | Cell lysis | |
| DC protein assay | N/A | Biorad | 5000112 | Protein assay | |
| Immulon 4 HBX | N/A | Thermo Fisher | 3855 | ELISA |
| 1× KH buffer (cell lysate) | |||||
|---|---|---|---|---|---|
| pH: 7.4, adjusted with HCl or NaOH as needed | |||||
| Chemical name | Concentration | Vendor | Catalog number | ||
| NaCl | 118 mM | Sigma-Aldrich | S7653 | ||
| KCl | 4.7 mM | Sigma-Aldrich | P9541 | ||
| CaCl2 | 1.25 mM | Sigma-Aldrich | 223506 | ||
| KH2PO4 | 1.2 mM | Sigma-Aldrich | P5655 | ||
| MgSO4 | 1.2 mM | Sigma-Aldrich | M7506 | ||
| d-glucose | 11 mM | Sigma-Aldrich | G8769 | ||
| l-Ascorbic acid | 0.5 mg/mL | Sigma-Aldrich | A5960 | ||
| NaHCO3 | 25 mM | Sigma-Aldrich | S5761 | ||
| HEPES ACSF | |||||
|---|---|---|---|---|---|
| pH: 7.4, adjusted with HCl or NaOH as needed | |||||
| Chemical name | Concentration | Vendor | Catalog number | ||
| NaCl | 92 mM | Sigma-Aldrich | S7653 | ||
| KCl | 2.5 mM | Sigma-Aldrich | P9541 | ||
| CaCl2 | 0.5 mM | Sigma-Aldrich | 223506 | ||
| NaH2PO4 | 1.2 mM | Sigma-Aldrich | P5655 | ||
| NaHCO3 | 30 mM | Sigma-Aldrich | S5761 | ||
| MgSO4 | 10 mM | Sigma-Aldrich | M7506 | ||
| d-glucose | 25 mM | Sigma-Aldrich | G8769 | ||
| HEPES | 20 mM | Sigma-Aldrich | H3375 | ||
| Na-l-ascorbate | 5 mM | Sigma-Aldrich | A4034 | ||
| Na-pyruvate | 3 mM | Sigma-Aldrich | P5280 | ||
| Thiourea | 2 mM | Sigma-Aldrich | T7875 | ||
PBS phosphate-buffered saline, DAT dopamine transporter, GFP green fluorescent protein, NGS normal goat serum, N/A not available.
Table 2.
Virus.
| Viral vectors (AAV) | Concentration | Vendor | Catalog number | Capsid serotype | Other |
|---|---|---|---|---|---|
| TH-α-syn | 3.26.E7 U/μl | Dr. Giasson | N/A | AAV1 | |
| TH-GFP | 3.26.E7 U/μl | Dr. Giasson | N/A | AAV1 | |
| LoxP-Tdtomato | 3.26.E7 U/μl | Addgene | 28306 | AAV1 |
N/A not available.
Animals
All experiments were approved by the Institutional Animal Care and Use Committee at the University of Florida. Mice were housed in the animal care facility at the University of Florida, 2–4 per cage with food and water available ad libitum in the home cage. The room was maintained under 12 h light/dark cycle. Wild-type C57BL/6J mice, or DATIREScre and Ai95(RCL-GCaMP6f)-D (Ai95D) knock-in mice were obtained from The Jackson Laboratory (stock number: 006660 (DATIREScre), 024105 (Ai95D), Bar Harbor, ME, USA). C57BL/6J pups or pups expressing GCaMP6f in dopamine neurons were used for this study. Mice of both sexes were used.
AAV1-TH-α-syn and AAV1-TH-GFP generation
The pAAV2.5-THP-GFP plasmid was purchased from Addgene (#80336)154. Human α-syn cDNA from a pcDNA3.1 plasmid was restriction digested with EcoRI and HindIII (NEB), purified, and ligated in the same sites of the pAAV2.5-THP backbone to generate the pAAV2.5-TH-α-syn plasmid. Purified pAAV2.5-TH-α-syn and pAAV2.5-TH-GFP vectors were utilized to prepare active AAV (capsid 1) using a HEK293T-based transfection method followed by iodixanol gradient purification as previously described155 and these viruses were termed AAV1-TH-α-syn and AAV1-TH-GFP. The genomic titer of each virus was assayed by quantitative PCR as previously described155.
Primary neuronal culture
Primary culture was prepared as previously described, with small distinctions23. Briefly, acutely dissociated mouse midbrains from 0- to 2-day-old male and female pups were isolated and incubated in a dissociation medium at 35–37 °C under continuous oxygenation for 60–90 min. Dissociated cells were triturated with pipettes of decreasing bore size (including a punctured fire-polished pipette), then pelleted by centrifugation at 1500 r.p.m. for 3–5 min, and finally resuspended and plated in glial medium (Table 1). Cells were plated at a density of 100,000 cells/coverslip on a 12 mm coverslip coated with 0.1 mg/ml poly-d-lysine and 5 μg/ml laminin and maintained in neuronal media. After 2 h, cells were supplemented with neuronal media (days in vitro 0 (DIV0) composition). Every 4 days, 1/3 of the media was replaced with fresh media. On DIV5, cultures were transduced with the desired AAV1 (see Table 2). The experiments described in this study were performed on DIV9–11. Reagents and chemicals utilized for midbrain neuronal culture are listed in Table 1.
Electrophysiology
Spontaneous firing activity of midbrain dopamine neurons was examined via whole-cell current-clamp recordings as previously described10,11,21. The neurons were continuously perfused with artificial cerebral spinal fluid (ACSF) (composition is described in Table 1) equilibrated with 95% O2/5% CO2; pH was adjusted to 7.4 at 37 °C. Patch electrodes were fabricated from borosilicate glass (Cat. No. 1B150F-4, 1.5 mm outer diameter; World Precision Instruments, Sarasota, FL) with the P-2000 puller (Sutter Instruments, Novato, CA). The tip resistance was in the range 3–5 MΩ. The electrodes were filled with a pipette solution containing (in mM): 120 potassium gluconate, 20 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA, 2 ATP, and 0.25 GTP, with pH adjusted to 7.25 with KOH. All experiments were performed at 37 °C. To standardize AP recordings, neurons were held at their resting membrane potential (see below) by DC application through the recording electrode. AP was recorded if the following criteria were met: a resting membrane potential more polarized than −35 mV and an AP peak amplitude >60 mV. AP half-width was measured as the spike width at the half-maximal voltage using Clampfit 10 software (Molecular Devices LLC, San Jose, CA). The steady-state basal activity was recorded for 2–3 min before bath application of the drug. Each coverslip was used for only one recording; this is specifically important for experiments involving drug application. The spontaneous spike activity of midbrain dopamine neurons was obtained by averaging 1 min interval activities at baseline and after 3–5 min of drug exposure.
Live-cell calcium imaging
Live-cell calcium imaging and analysis are described previously23. Briefly, naive (non-transduced) and α-syn-overexpressing (transduced with AAV1-TH-α-syn) midbrain neuronal cultures were imaged with a Nikon Eclipse FN1 upright microscope (Nikon Instruments, Melville, NY). A Spectra X (Lumencor, Inc., Beaverton, OR) was used to stimulate GCaMP6f (λex = 470 nm) fluorescence through a custom quad-pass filter (Chroma Technologies, Battleboro, VT), and emission was filtered through visible spectra bandpass filter. Experiments were performed under gravity perfusion of ACSF (Table 1). The average fluorescence of the first 60 s recording is defined as the baseline. After baseline imaging, vehicle (ACSF), 1 μM DA, or 10 μM quinpirole was administered via a perfusion system (flow rate of 2 ml/min) and recorded for an additional 2 min. Background fluorescence was subtracted from each frame. Fold fluorescence change from baseline was calculated and plotted against time. Each coverslip was used for only one recording; this is specifically important for experiments involving drug application. All resources and reagents used for live-cell calcium imaging experiments are listed in Table 1.
Live-cell confocal imaging using GRABDA2M-expressing HEK293 cells to measure extracellular dopamine
These experiments were performed via a blinded experimental design. GRABDA2M is a genetically encoded fluorescent dopamine sensor that is engineered by coupling a conformationally sensitive cpEGFP to D2R. In GRABDA2M-expressing HEK293 cells, dopamine binding to the sensor induces a conformational change that results in a robust increase in fluorescence signal in a concentration-dependent manner156. Flp-In™ 293 T-Rex stable cell lines exhibiting tetracycline-inducible expression of the GRABDA2M dopamine sensor were generously gifted by Dr. Ulrik Gether. The GRABDA2M-expressing HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin. Selection pressure for GRABDA2M-expressing cells was maintained with media containing Hygro-B (1 mg/ml) and blasticidin (0.015 mg/ml). The cells were plated on coverslips in media containing tetracycline (1:1000) to induce the expression for 12–24 h before live-cell imaging under three conditions: (1) only GRABDA2M-expressing HEK293 cells to measure the constitutive fluorescent signal, (2) GRABDA2M cells added to tdTomato-expressing dopaminergic neurons, (3) GRABDA2M cells added to tdTomato-expressing dopaminergic neurons overexpressing α-syn. The tdTomato transduction (λex = 560 nm) is used for better identification of soma and dendritic field and no fluorophore overlap (i.e., excitation and emission of tdTomato does not bleed through GFP channel). Imaging was performed using a Nikon Eclipse FN1 upright microscope (Nikon Instruments, Melville, NY). A Spectra X (Lumencor, Inc., Beaverton, OR) was used to stimulate GFP (λex = 470 nm) and tdTomato (λex = 560 nm) fluorescence through a custom quad-pass filter (Chroma Technologies, Battleboro, VT). Emission was filtered through visible spectra bandpass filter. Regions of interest (ROI) were autodetected via NIS Elements software (Nikon Instruments, Melville, NY). cpEGFP signal was background subtracted using an ROI in an adjacent area of the image devoid of cells or debris.
Generation of a standard curve
To generate a standard curve, the baseline fluorescence signal (Fc), which is the constitutive fluorescent signal in the absence of extracellular dopamine, was recorded. Changes in fluorescence signal after adding various dopamine concentrations (1–20 nM) were plotted against dopamine concentration.
Measurement of basal dopamine release
GRABDA2M cells are cocultured with tdTomato-expressing dopamine neurons (DATIREScre-LoxP-tdTomato) containing endogenous α-syn or its overexpression 20–24 h prior to live-cell confocal imaging.
At the beginning of each experiment, the constitutive GRABDA2M fluorescence signal (Fc) of the cells that are plated in similar conditions sans neurons was obtained. To compare baseline dopamine release amongst the experimental groups, the average fluorescence signal of cells adjacent to the soma and neuronal processes to the average fluorescence signal of GRABDA2M cells (only) were calculated in Eq. (1):
| 1 |
Visualization and quantification of real-time dopamine release following KCl stimulation
Twenty to twenty-four hours prior to live-cell confocal imaging, GRABDA2M cells are cocultured with tdTomato-expressing dopamine neurons containing endogenous α-syn or its overexpression. GRABDA2M fluorescent signal around the soma and neuronal processes were measured before (Fbaseline) and following KCl stimulation (90 mM) of dopamine release69. The average fluorescence signal of cells adjacent to the soma and neuronal processes before and after KCl were calculated in Eq. (2):
| 2 |
Immunocytochemistry
These experiments were performed via a blinded experimental design. On DIV9–11, naive (non-transduced) and α-syn-overexpressing neuronal cultures were fixed with 4% paraformaldehyde (PFA) in PBS for 30 min at room temperature (RT), followed by blocking, permeabilizations, and overnight incubation (at 4 °C) with primary antibodies diluted in blocking buffer, followed by three 20 min phosphate-buffered solution (PBS) washes. Then, a 1 h incubation in blocking buffer with Alexa Fluor-conjugated secondary antibodies at RT, followed by three 20 min washes and an overnight PBS wash at RT. Coverslips were mounted on slides using Fluoromount-G. Images were captured on a Nikon A1 laser-scanning confocal microscope (×20 or ×40 oil-immersion objective). Reagents and chemicals utilized for ICC are listed in Table 1.
Western blot analysis
These experiments were performed via a blinded experimental design. For detection of endogenous and human α-syn, total cell lysates of neurons transduced with AAV1-TH-α-syn (n = 4) or naive (non-transduced) neurons (n = 4) were used, as described previously157. Briefly, the cells were harvested in 200 µl of 2% sodium dodecyl sulfate (SDS) buffer, protein concentrations were determined using the bicinchoninic acid assay (Pierce), and further diluted in sample buffer (10 mM Tris, pH 6.8, 1 mM EDTA, 40 mM dithiothreitol, 0.005% bromophenol blue, 0.0025% pyronin yellow, 1% SDS, 10% sucrose). Following harvest of total cell lysate, samples were heated to 100 °C for 10 min prior to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (13% polyacrylamide gels, 10 µg lysate per well) followed by electrophoretic transfer onto 0.2 µm nitrocellulose membranes as previously described145. Membranes were incubated with block solution (5% milk in TBS) for 1 h, and then with primary antibodies (in block solution) overnight at 4 °C. Membranes were washed with TBS-T and incubated with goat anti-mouse or anti-rabbit secondary antibodies conjugated to horseradish peroxidase (Jackson Immuno Research Labs, Westgrove, PA) diluted in block solution at RT for 1 h. Immunoreactivity was assessed using Western Lightning-Plus ECL reagents (PerkinElmer, Waltham, MA) followed by chemiluminescence imaging (Genegnome XRQ, Syngene, Frederick, MD). For α-syn detection, we used 94-3A10 antibody, which is a mouse monoclonal antibody raised against C-terminal regions (130–140) of α-syn158. For loading control, we used mouse monoclonal anti-actin C4 antibody (EMD Millipore). For TH detection, an affinity-purified rabbit antibody AB152 (EMD Millipore) was used.
Biotinylation assay
These experiments were performed via a blinded experimental design. α-Syn-overexpressing neuronal cultures and naive neuronal cultures were washed three times with cold PBS and incubated with sulfo-NHS-biotin (1.5 mg/ml; Thermo Fisher Pierce, 21331) for 30 min at 4 °C while rocking. The remaining sulfo-NHS-biotin was quenched with cold Quenching Solution (glycine 50 mM in PBS), followed by three washes with cold PBS139,159. Cells were lysed in BufferD lysis buffer (10% glycerol, 125 mM NaCl, 1 mM EDTA, 1 mM EGTA, pH 7.6) containing 1% Triton X-100 and protease inhibitor cocktail (Millipore, 539131) for 1 h at 4 °C while rocking, followed by centrifugation for 15 min at 12,000 × g. The supernatants were divided into three portions—25 μl for protein quantification and 200 μl for incubation with avidin, with the remainder for the whole lysate. After equilibrating monomeric UltraLink Avidin (Thermo Fisher Pierce, 53146) twice with 1 ml BufferD, 40 μl of 50% bead slurry were added to 200 μl lysate and incubated at 4 °C for 1 h while rotating. The supernatant was retained as a cytoplasmic fraction, and beads were washed three times with 1 ml BufferD, eluted with 40 μl Laemmli Sample Buffer 4× (containing 10% beta-mercaptoethanol) at 37 °C for 30 min, and separated by 10% SDS-PAGE, transferred to 0.45 μm nitrocellulose, and probed with antibodies against proteins of interest (see Table 1). Fluorescent images were analyzed using ImageJ (NIH) to measure band optical density. Values were normalized to total protein per lane. Beta-tubulin (Aves, TUJ) was probed to demonstrate membrane fraction isolation during biotinylation.
ELISA quantification of TH
These experiments were performed via a blinded experimental design.
Cell lysis
For total protein quantification via ELISA, neuronal cultures were washed three times with cold PBS, then lysed in BufferD lysis buffer (10% glycerol, 125 mM NaCl, 1 mM EDTA, 1 mM EGTA, pH 7.6) containing 1% Triton X-100 and protease inhibitor cocktail (Millipore, 539131) for 1 h at 4 °C with rocking, followed by centrifugation for 15 min at 12,000 × g. Samples were denatured in Laemmli Sample Buffer 4× (containing 10% beta-mercaptoethanol) at 37 °C for 30 min and separated by 10% SDS-PAGE, transferred to 0.45 μm nitrocellulose, and probed with antibodies against proteins of interest. Values were normalized to total protein per lane.
TH ELISA
Antibodies and concentrations used are given in Table 1. In brief, Immulon 4 HBX High-Binding 96-well plates were coated with 100 μl per well of 1:1000 dilution of mouse anti-TH (EnCor, MCA-4H2) in coating buffer (28.3 mM Na2CO3, 71.42 mM NaHCO3, pH 9.6) for 20 h at 4 °C. Edge lanes 1 and 12 were left empty. Wells were blocked with 5% fat-free milk in 1× TBS (pH 7.4) for 1 h at RT on an orbital shaker set to 90 r.p.m.
Generation of a standard curve
To produce a standard curve, two standard curve lanes were generated, with six serial dilutions, beginning at 10 ng/ml and 1 ng/ml in TBS-T containing 1% fat-free milk (with the last well in each standard curve lane left with incubation buffer only as a blank). The remaining wells were incubated in duplicate with lysates from cells of interest. Incubation was completed for 20 h at 4 °C on an ELISA shaker set to 475 r.p.m. After each well was washed and aspirated six times with TBS-T, anti-TH rabbit (EnCor, RPCA-TH) conjugated to biotin was diluted 1:6000 from a stock concentration of 1.65 mg/ml in TBS-T with 1% fat-free milk and incubated for 1 h at RT by centrifugation at 425 r.p.m. One hundred microliters of Avidin-HRP (Vector labs, A-2004), diluted 1:2500 in TBS-T with 1% fat-free milk, was added to each well following washing, and then incubated for 1 h at RT by centrifugation at 425 r.p.m. Following final washes, 150 μl TMB-ELISA reagent (Thermo Fisher, 34028) was added to each well at RT. The reaction was allowed to continue for 20 min, protected from light, and stopped by the addition of 50 μl 2 N H2SO4. The plate was immediately read at 450 nm. Duplicate standard and sample wells were averaged and background subtracted based on blank wells. TH concentration was calculated using a quadratic curve equation calculated in GraphPad Prism 8, and then normalized to total protein concentration per sample as calculated using Lowry assay. Final TH values shown are presented as pg TH/mg total protein after the multiplication of the nanogram TH value by 1000.
HPLC
These experiments were performed via a blinded experimental design. Midbrain primary cultures were incubated in KH buffer (Table 1), at 37 °C, for 1 h before collecting intracellular and extracellular milieu for HPLC analysis72,160. For extracellular milieu, KH buffer incubated with neurons was collected, treated with 1 M perchloric acid, and snap frozen for analysis. For intracellular milieu, coverslips were washed with KH buffer, scraped, and treated with 1 M perchloric acid, before sonicating. Then, the sample was centrifuged at 12,000 r.p.m. at 4 °C for 10 min, and the supernatant was snap frozen in liquid nitrogen for analysis. The pellet was resuspended using 0.2 NaOH and RIPA buffer161 for protein quantification via Lowry assay. Samples were centrifuged at 16,000 × g for 15 min (4 °C) and the supernatant was filtered through a 0.2 mm pore membrane (Nanosep with 0.2 mm bioinsert, Pall Life Sciences) and 15 μl of the supernatant was injected directly into an HPLC-ECD (HTEC-510; Eicom). Dopamine was separated on a CAX column (EICOMPAK 2.0 i.d. × 200 mm) maintained at 35 °C. The mobile phase consisted of 70% 0.1 M ammonium acetate buffer (pH 6.0) containing sodium sulfate (0.025 M), EDTA-2Na (50 mg/l), and 30% methanol at a flow rate of 250 μl/min. An electrochemical detector that used a glassy working electrode (+450 mV) against a silver-silver chloride reference electrode (WE-3G; Eicom) was used to quantify dopamine in the samples. A dopamine standard was used to identify and quantify the dopamine concentration in the samples.
Morphometric analysis
These experiments were performed via a blinded experimental design. Control (DATIREScre) neurons and α-syn-overexpressing neurons (DATIREScre/α-syn) were transduced with AAV1-LoxP-tdTomato (Addgene) on DIV5. Neurons were fixed with 4% PFA for 30 min at room temperature on DIV10, and coverslips were mounted using Fluoromount-G and allowed to dry. Alternatively, neurons were fixed, co-immunolabeled with TH and GFP antibodies, and mounted for imaging, as described above. Images were captured on a Nikon A1 laser-scanning confocal microscope (visualized through a ×20 oil-immersion objective). Images of neurons with minimal interference from neighboring neurons were analyzed in ImageJ (FIJI) and converted to 8-bit binary images after threshold adjustment. Sholl analysis plugin was used to draw concentric circles starting from 15 μm, followed by 5 μm successive shells in order to identify the number of intersections along the radii108,162,163. Sholl analysis was performed, and the number of intersections was plotted (Fig. 6). Cell area measurements were attained by manually drawing ROI around cell soma using the free polygon selection tool in ImageJ. ROIs were drawn to encompass the complete projection area of the cell and selection was finalized by a convex hull to attain final projection area measurement. Results obtained were plotted to analyze the complexity of morphology.
Statistical analysis
Data analysis was performed using GraphPad Prism version 8.02 and MATLAB version 2020a. Student’s t test, linear regression, one-way, two-way, or repeated-measures ANOVA were used where appropriate and corrected for multiple comparisons. The significance of P < 0.05 was considered statistically significant. Data are presented with a mean and standard error, unless otherwise stated.
Limitations to the methodology and model system used in this study
The primary neuronal culture used in this study is derived from the ventral midbrain, which contains the dopaminergic nuclei SNc and VTA. Notably, it has been shown that SNc dopaminergic neurons are more sensitive than VTA dopaminergic neurons22,23,26–31,164. Therefore, this model is likely to contain more VTA dopaminergic neurons than SNc dopaminergic neurons22,26–31. In addition, α-syn-mediated neuronal loss can also lead to a higher loss of SNc dopaminergic neurons than VTA dopamine neurons. Since functional analyses are conducted on surviving neurons, our data may overrepresent effects in the surviving VTA midbrain dopamine neurons.
In this study, we utilized TH promoter-driven expression of GFP and α-syn to confirm the specificity of TH promoter-dependent viral expression. We also performed calcium imaging, TH expression, and neuronal cell count in dopaminergic neurons expressing control vector (AAV-TH-GFP). We found that, compared to naive neurons, the AAV-TH-GFP transduction did not change neuronal activity and calcium dynamics in response to dopamine and quinpirole exposure, TH levels, and cell count (data not shown); therefore, the control AAV was not used for the rest of the experiments.
In regards to investigating if α-syn overexpression in dopaminergic neurons alters D2R expression, we utilized the biotinylation assay to assess the differences in membrane vs. cytoplasmic D2 levels91–94. Whether or not the detected membrane D2Rs are functional or desensitized remains unclear.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
D.R.M. and A.G. are supported by T32-NS082128. Dr. H.K. is supported by R01-NS071122, R01-DA026947, and 1S10OD020026. Flp-In™ 293 T-Rex stable cell lines exhibiting tetracycline-inducible expression of the GRABDA2M dopamine sensor were generously gifted by Dr. Ulrik Gether. We would like to thank the technical assistance of Madison Francis, Martin Badov, Phillip Mackie, and Dr. Jose A Pino.
Author contributions
A.D. and D.R.M. contributed equally to the study. H.K. and B.I.G. conceived and supervised the study. H.K., B.I.G., A.D., D.R.M., A.G., Z.A.S., J.F.S., and J.J.L. designed and planned the experiments. A.D., D.R.M., F.S., M.L., A.G., S.H., Z.A.S., and J.J.L. collected the data. A.D., D.R.M., S.V., A.R.A., J.A., and C.A.H. analyzed the data. H.K., B.I.G., A.D., and D.R.M. interpreted the data with comments from N.U., H.K., B.I.G., A.D., and D.R.M. prepared the manuscript. H.K., A.G., B.U., and M.B. revised the work and inspected it for rigor and reproducibility. All authors read and approved the final manuscript.
Data availability
The data acquired and analyzed for this study are available from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Abeer Dagra, Douglas R. Miller.
Contributor Information
Benoit I. Giasson, Email: bgiasson@ufl.edu
Habibeh Khoshbouei, Email: habibeh@ufl.edu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41531-021-00210-w.
References
- 1.Cenci MA. Dopamine dysregulation of movement control in l-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Panigrahi B, et al. Dopamine is required for the neural representation and control of movement vigor. Cell. 2015;162:1418–1430. doi: 10.1016/j.cell.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Braak E. Pathoanatomy of Parkinson’s disease. J. Neurol. Suppl. 2000;247:3–10. doi: 10.1007/s004150050002. [DOI] [PubMed] [Google Scholar]
- 4.Burke RE, Dauer WT, Vonsattel JPG. A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann. Neurol. 2008;64:485–491. doi: 10.1002/ana.21541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 6.Uchihara T, Giasson BI. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:49–73. doi: 10.1007/s00401-015-1485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delenclos M, et al. Neonatal AAV delivery of alpha-synuclein induces pathology in the adult mouse brain. Acta Neuropathol. Commun. 2017;5:51. doi: 10.1186/s40478-017-0455-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chartier-Harlin M-C, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 9.Singleton AB, et al. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 10.Lin M, Sambo D, Khoshbouei H. Methamphetamine regulation of firing activity of dopamine neurons. J. Neurosci. 2016;36:10376–10391. doi: 10.1523/JNEUROSCI.1392-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sambo DO, et al. The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat. Commun. 2017;8:2228. doi: 10.1038/s41467-017-02087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasad BM, Amara SG. The dopamine transporter in mesencephalic cultures is refractory to physiological changes in membrane voltage. J. Neurosci. 2001;21:7561–7567. doi: 10.1523/JNEUROSCI.21-19-07561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingram SL, Prasad BM, Amara SG. Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat. Neurosci. 2002;5:971–978. doi: 10.1038/nn920. [DOI] [PubMed] [Google Scholar]
- 14.Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J. Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 17.Howell RD, Dominguez-Lopez S, Ocañas SR, Freeman WM, Beckstead MJ. Female mice are resilient to age-related decline of substantia nigra dopamine neuron firing parameters. Neurobiol. Aging. 2020;95:195–204. doi: 10.1016/j.neurobiolaging.2020.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J. Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat. Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- 20.Kremer Y, Flakowski J, Rohner C, Lüscher C. Context-dependent multiplexing by individual VTA dopamine neurons. J. Neurosci. 2020;40:7489–7509. doi: 10.1523/JNEUROSCI.0502-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin M, et al. Mechanism of manganese dysregulation of dopamine neuronal activity. J. Neurosci. 2020;40:5871–5891. doi: 10.1523/JNEUROSCI.2830-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller DR, et al. Methamphetamine regulation of activity and topology of ventral midbrain networks. PLoS ONE. 2019;14:e0222957. doi: 10.1371/journal.pone.0222957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler B, et al. Dopamine transporter activity is modulated by α-synuclein. J. Biol. Chem. 2015;290:29542–29554. doi: 10.1074/jbc.M115.691592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saha K, et al. Intracellular methamphetamine prevents the dopamine-induced enhancement of neuronal firing. J. Biol. Chem. 2014;289:22246. doi: 10.1074/jbc.M114.563056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lautenschläger J, et al. An easy-to-implement protocol for preparing postnatal ventral mesencephalic cultures. Front. Cell Neurosci. 2018;12:44. doi: 10.3389/fncel.2018.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burke RE, Antonelli M, Sulzer D. Glial cell line-derived neurotrophic growth factor inhibits apoptotic death of postnatal substantia nigra dopamine neurons in primary culture. J. Neurochem. 1998;71:517–525. doi: 10.1046/j.1471-4159.1998.71020517.x. [DOI] [PubMed] [Google Scholar]
- 28.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain. 1999;122(Part 8):1421–1436. doi: 10.1093/brain/122.8.1421. [DOI] [PubMed] [Google Scholar]
- 29.Saper CB, Sorrentino DM, German DC, de Lacalle S. Medullary catecholaminergic neurons in the normal human brain and in Parkinson’s disease. Ann. Neurol. 1991;29:577–584. doi: 10.1002/ana.410290602. [DOI] [PubMed] [Google Scholar]
- 30.Chan CS, et al. “Rejuvenation” protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 31.Nedergaard S, Flatman JA, Engberg I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J. Physiol. 1993;466:727–747. [PMC free article] [PubMed] [Google Scholar]
- 32.Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005;2:14. doi: 10.1186/1742-2094-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.St Martin JL, et al. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 2007;100:1449–1457. doi: 10.1111/j.1471-4159.2006.04310.x. [DOI] [PubMed] [Google Scholar]
- 34.Emanuele M, Chieregatti E. Mechanisms of alpha-synuclein action on neurotransmission: cell-autonomous and non-cell autonomous role. Biomolecules. 2015;5:865–892. doi: 10.3390/biom5020865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dias V, Junn E, Mouradian MM. The role of oxidative stress in parkinson’s disease. J. Parkinson’s Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira C, Bessa C, Soares J, Leão M, Saraiva L. Contribution of yeast models to neurodegeneration research. J. Biomed. Biotechnol. 2012;2012:941232. doi: 10.1155/2012/941232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen KE, et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volles MJ, Lansbury PT., Jr. Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 2007;366:1510–1522. doi: 10.1016/j.jmb.2006.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McBurney RN, Neering IR. Neuronal calcium homeostasis. Trends Neurosci. 1987;10:164–169. doi: 10.1016/0166-2236(87)90042-7. [DOI] [Google Scholar]
- 41.Gleichmann M, Mattson MP. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 2011;14:1261–1273. doi: 10.1089/ars.2010.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grienberger C, Konnerth A. Imaging calcium in neurons. Neuron. 2012;73:862–885. doi: 10.1016/j.neuron.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 44.Ekstrand MI, Galter D. The MitoPark Mouse - an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat. Disord. 2009;15(Suppl. 3):S185–S188. doi: 10.1016/S1353-8020(09)70811-9. [DOI] [PubMed] [Google Scholar]
- 45.Ricke KM, et al. Mitochondrial dysfunction combined with high calcium load leads to impaired antioxidant defense underlying the selective loss of nigral dopaminergic neurons. J. Neurosci. 2020;40:1975–1986. doi: 10.1523/JNEUROSCI.1345-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angelova PR, et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 2016;129:1792–1801. doi: 10.1242/jcs.180737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/S0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- 48.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 49.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 1999;9:305–313. doi: 10.1016/S0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]
- 50.Duda J, Pötschke C, Liss B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochemistry. 2016;139(Suppl.):156–178. doi: 10.1111/jnc.13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barzilai A, Melamed E. Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol. Med. 2003;9:126–132. doi: 10.1016/S1471-4914(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 52.Catoni C, Calì T, Brini M. Calcium, dopamine and neuronal calcium sensor 1: their contribution to Parkinson’s disease. Front. Mol. Neurosci. 2019;12:55. doi: 10.3389/fnmol.2019.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jackson, D. M. & Westlind-Danielsson, A. Dopamine receptors: molecular biology, biochemistry and behavioural aspects. Pharmacol. Ther. 10.1016/0163-7258(94)90041-8 (1994). [DOI] [PubMed]
- 54.Paladini CA, Robinson S, Morikawa H, Williams JT, Palmiter RD. Dopamine controls the firing pattern of dopamine neurons via a network feedback mechanism. Proc. Natl Acad. Sci. USA. 2003;100:2866–2871. doi: 10.1073/pnas.0138018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 56.Ford CP. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience. 2014;282:13–22. doi: 10.1016/j.neuroscience.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: What do D2 receptors have to do with it? Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 58.Pucak ML, Grace AA. Evidence that systemically administered dopamine antagonists activate dopamine neuron firing primarily by blockade of somatodendritic autoreceptors. J. Pharmacol. Exp. Ther. 1994;271:1181–1192. [PubMed] [Google Scholar]
- 59.Kramer PF, Williams JT. Calcium release from stores inhibits GIRK. Cell Rep. 2016;17:3246–3255. doi: 10.1016/j.celrep.2016.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seabrook GR, et al. Depression of high-threshold calcium currents by activation of human D2 (short) dopamine receptors expressed in differentiated NG108-15 cells. Br. J. Pharmacol. 1994;111:1061–1066. doi: 10.1111/j.1476-5381.1994.tb14852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montmayeur JP, Borrelli E. Transcription mediated by a cAMP-responsive promoter element is reduced upon activation of dopamine D2 receptors. Proc. Natl Acad. Sci. USA. 1991;88:3135–3139. doi: 10.1073/pnas.88.8.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Missale C, Russel Nash S, Robinson SW, Jaber M, Caron MG. Dopamine receptors:from structure to function. Physiol. Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 63.Lee AK. Dopamine (D2) receptor regulation of intracellular calcium and membrane capacitance changes in rat melanotrophs. J. Physiol. 1996;495(Part 3):627–640. doi: 10.1113/jphysiol.1996.sp021621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei C, et al. Response dynamics of midbrain dopamine neurons and serotonin neurons to heroin, nicotine, cocaine, and MDMA. Cell Discov. 2018;4:60. doi: 10.1038/s41421-018-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richardson BD, et al. Membrane potential shapes regulation of dopamine transporter trafficking at the plasma membrane. Nat. Commun. 2016;7:10423. doi: 10.1038/ncomms10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Innis RB, Aghajanian GK. Pertussis toxin blocks autoreceptor-mediated inhibition of dopaminergic neurons in rat substantia nigra. Brain Res. 1987;411:139–143. doi: 10.1016/0006-8993(87)90690-1. [DOI] [PubMed] [Google Scholar]
- 67.Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J. Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kennedy RT, Jones SR, Wightman RM. Simultaneous measurement of oxygen and dopamine: coupling of oxygen consumption and neurotransmission. Neuroscience. 1992;47:603–612. doi: 10.1016/0306-4522(92)90169-3. [DOI] [PubMed] [Google Scholar]
- 69.Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-Z. [DOI] [PubMed] [Google Scholar]
- 70.Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL. Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J. Neurosci. 2008;28:8908–8913. doi: 10.1523/JNEUROSCI.1526-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vallar L, Meldolesi J. Mechanisms of signal transduction at the dopamine D2 receptor. Trends Pharmacol. Sci. 1989;10:74–77. doi: 10.1016/0165-6147(89)90082-5. [DOI] [PubMed] [Google Scholar]
- 72.Jensen, K. L. et al. PICK1-deficient mice exhibit impaired response to cocaine and dysregulated dopamine homeostasis. eNeuro5, ENEURO.0422-17.2018 (2018). [DOI] [PMC free article] [PubMed]
- 73.Nieoullon A, Cheramy A, Glowinski J. Release of dopamine in vivo from cat substantia nigra [28] Nature. 1977;266:375–377. doi: 10.1038/266375a0. [DOI] [PubMed] [Google Scholar]
- 74.Hoffman AF, Gerhardt GA. Differences in pharmacological properties of dopamine release between the substantia nigra and striatum: an in vivo electrochemical study. J. Pharmacol. Exp. Ther. 1999;289:455–463. [PubMed] [Google Scholar]
- 75.Swant J, et al. α-Synuclein stimulates a dopamine transporter-dependent chloride current and modulates the activity of the transporter. J. Biol. Chem. 2011;286:43933–43943. doi: 10.1074/jbc.M111.241232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lam HA, et al. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein. J. Neurosci. Res. 2011;89:1091–1102. doi: 10.1002/jnr.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lundblad M, Decressac M, Mattsson B, Björklund A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl Acad. Sci. USA. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yavich L, et al. Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-synuclein. Neurobiol. Dis. 2005;20:303–313. doi: 10.1016/j.nbd.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 79.Butler, B., Sambo, D. & Khoshbouei, H. Alpha-synuclein modulates dopamine neurotransmission. J. Chem. Neuroanat.10.1016/j.jchemneu.2016.06.001 (2016). [DOI] [PMC free article] [PubMed]
- 80.Lindgren N, et al. Dopamine D(2) receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. Eur. J. Neurosci. 2001;13:773–780. doi: 10.1046/j.0953-816x.2000.01443.x. [DOI] [PubMed] [Google Scholar]
- 81.Alerte TNM, et al. Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci. Lett. 2008;435:24–29. doi: 10.1016/j.neulet.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salvatore MF, Garcia-Espana A, Goldstein M, Deutch AY, Haycock JW. Stoichiometry of tyrosine hydroxylase phosphorylation in the nigrostriatal and mesolimbic systems in vivo: effects of acute haloperidol and related compounds. J. Neurochem. 2000;75:225–232. doi: 10.1046/j.1471-4159.2000.0750225.x. [DOI] [PubMed] [Google Scholar]
- 83.Salvatore MF, Calipari ES, Jones SR. Regulation of tyrosine hydroxylase expression and phosphorylation in dopamine transporter-deficient mice. ACS Chem. Neurosci. 2016;7:941–951. doi: 10.1021/acschemneuro.6b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dadalko OI, et al. mTORC2/rictor signaling disrupts dopamine-dependent behaviors via defects in striatal dopamine neurotransmission. J. Neurosci. 2015;35:8843–8854. doi: 10.1523/JNEUROSCI.0887-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Håkansson K, et al. Regulation of striatal tyrosine hydroxylase phosphorylation by acute and chronic haloperidol. Eur. J. Neurosci. 2004;20:1108–1112. doi: 10.1111/j.1460-9568.2004.03547.x. [DOI] [PubMed] [Google Scholar]
- 86.Onali P, Mosca E, Olianas MC. Presynaptic dopamine autoreceptors and second messengers controlling tyrosine hydroxylase activity in rat brain. Neurochem. Int. 1992;20(Suppl.):89S–93S. doi: 10.1016/0197-0186(92)90217-F. [DOI] [PubMed] [Google Scholar]
- 87.el Mestikawy S, Hamon M. Is dopamine-induced inhibition of adenylate cyclase involved in the autoreceptor-mediated negative control of tyrosine hydroxylase in striatal dopaminergic terminals? J. Neurochem. 1986;47:1425–1433. doi: 10.1111/j.1471-4159.1986.tb00775.x. [DOI] [PubMed] [Google Scholar]
- 88.Gopinath A, et al. TNFα increases tyrosine hydroxylase expression in human monocytes. npj Parkinsons Dis. 2021;7:62. doi: 10.1038/s41531-021-00201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kehr W, Carlsson A, Lindqvist M, Magnusson T, Atack C. Evidence for a receptor-mediated feedback control of striatal tyrosine hydroxylase activity. J. Pharm. Pharmacol. 1972;24:744–747. doi: 10.1111/j.2042-7158.1972.tb09104.x. [DOI] [PubMed] [Google Scholar]
- 90.Wolf ME, Roth RH. Autoreceptor regulation of dopamine synthesis. Ann. NY Acad. Sci. 1990;604:323–343. doi: 10.1111/j.1749-6632.1990.tb32003.x. [DOI] [PubMed] [Google Scholar]
- 91.Beckstead MJ, Williams JT. Long-term depression of a dopamine IPSC. J. Neurosci. 2007;27:2074–2080. doi: 10.1523/JNEUROSCI.3251-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barton AC, Black LE, Sibley DR. Agonist-induced desensitization of D2 dopamine receptors in human Y-79 retinoblastoma cells. Mol. Pharmacol. 1991;39:650–658. [PubMed] [Google Scholar]
- 93.Salinas AG, Davis MI, Lovinger DM, Mateo Y. Dopamine dynamics and cocaine sensitivity differ between striosome and matrix compartments of the striatum. Neuropharmacology. 2016;108:275–283. doi: 10.1016/j.neuropharm.2016.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Davis MI, et al. The cannabinoid-1 receptor is abundantly expressed in striatal striosomes and striosome-dendron bouquets of the substantia nigra. PLoS ONE. 2018;13:e0191436. doi: 10.1371/journal.pone.0191436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 96.Robinson BG, et al. Desensitized D2 autoreceptors are resistant to trafficking. Sci. Rep. 2017;7:4379. doi: 10.1038/s41598-017-04728-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 98.Bartlett SE, et al. Dopamine responsiveness is regulated by targeted sorting of D2 receptors. Proc. Natl Acad. Sci. USA. 2005;102:11521–11526. doi: 10.1073/pnas.0502418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matsuda W, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Giguère N, et al. Increased vulnerability of nigral dopamine neurons after expansion of their axonal arborization size through D2 dopamine receptor conditional knockout. PLoS Genet. 2019;15:e1008352. doi: 10.1371/journal.pgen.1008352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pacelli C, et al. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 2015;25:2349–2360. doi: 10.1016/j.cub.2015.07.050. [DOI] [PubMed] [Google Scholar]
- 102.Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov. Disord. 2012;27:1478–1483. doi: 10.1002/mds.25135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ducrot C, et al. Dopaminergic neurons establish a distinctive axonal arbor with a majority of non-synaptic terminals. FASEB J. 2021;35:e21791. doi: 10.1096/fj.202100201RR. [DOI] [PubMed] [Google Scholar]
- 104.Kordower JH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Decressac M, Mattsson B, Lundblad M, Weikop P, Björklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of α-synuclein in midbrain dopamine neurons. Neurobiol. Dis. 2012;45:939–953. doi: 10.1016/j.nbd.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 106.Chen S, et al. D2/D3 receptor agonist ropinirole protects dopaminergic cell line against rotenone-induced apoptosis through inhibition of caspase- and JNK-dependent pathways. FEBS Lett. 2008;582:603–610. doi: 10.1016/j.febslet.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 107.Bono F, Fiorentini C. Exploring pre-degenerative alterations in humans using induced pluripotent stem cell-derived dopaminergic neurons. Neural Regen. Res. 2017;12:1068–1070. doi: 10.4103/1673-5374.211184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shaerzadeh F, et al. Microglia senescence occurs in both substantia nigra and ventral tegmental area. Glia. 2020;68:2228–2245. doi: 10.1002/glia.23834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- 110.Benavides-Piccione R, Hamzei-Sichani F, Ballesteros-Yáñez I, DeFelipe J, Yuste R. Dendritic size of pyramidal neurons differs among mouse cortical regions. Cereb. Cortex. 2006;16:990–1001. doi: 10.1093/cercor/bhj041. [DOI] [PubMed] [Google Scholar]
- 111.O’Neill KM, et al. Assessing effects on dendritic arborization using novel Sholl analyses. Front. Cell Neurosci. 2015;9:285. doi: 10.3389/fncel.2015.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Levy M, Lu Z, Dion G, Kara P. The shape of dendritic arbors in different functional domains of the cortical orientation map. J. Neurosci. 2014;34:3231–3236. doi: 10.1523/JNEUROSCI.4985-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brown KM, Gillette TA, Ascoli GA. Quantifying neuronal size: summing up trees and splitting the branch difference. Semin. Cell Dev. Biol. 2008;19:485–493. doi: 10.1016/j.semcdb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ballion B, et al. D2 receptor stimulation, but not D1, restores striatal equilibrium in a rat model of Parkinsonism. Neurobiol. Dis. 2009;35:376–384. doi: 10.1016/j.nbd.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 115.Kihara T, et al. Protective effect of dopamine D2 agonists in cortical neurons via the phosphatidylinositol 3 kinase cascade. J. Neurosci. Res. 2002;70:274–282. doi: 10.1002/jnr.10426. [DOI] [PubMed] [Google Scholar]
- 116.Hill MP, et al. Antiparkinsonian effects of the novel D3/D2 dopamine receptor agonist, S32504, in MPTP-lesioned marmosets: Mediation by D2, not D3, dopamine receptors. Mov. Disord. 2006;21:2090–2095. doi: 10.1002/mds.21106. [DOI] [PubMed] [Google Scholar]
- 117.Wiemerslage L, Schultz BJ, Ganguly A, Lee D. Selective degeneration of dopaminergic neurons by MPP(+) and its rescue by D2 autoreceptors in Drosophila primary culture. J. Neurochem. 2013;126:529–540. doi: 10.1111/jnc.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.O’Malley KL. The role of axonopathy in Parkinson’s disease. Exp. Neurobiol. 2010;19:115–119. doi: 10.5607/en.2010.19.3.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tagliaferro P, Burke RE. Retrograde axonal degeneration in Parkinson disease. J. Parkinson’s Dis. 2016;6:1–15. doi: 10.3233/JPD-150769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Du F, Li R, Huang Y, Li X, Le W. Dopamine D3 receptor-preferring agonists induce neurotrophic effects on mesencephalic dopamine neurons. Eur. J. Neurosci. 2005;22:2422–2430. doi: 10.1111/j.1460-9568.2005.04438.x. [DOI] [PubMed] [Google Scholar]
- 121.Bono F, et al. Role of dopamine D2/D3 receptors in development, plasticity, and neuroprotection in human iPSC-derived midbrain dopaminergic neurons. Mol. Neurobiol. 2018;55:1054–1067. doi: 10.1007/s12035-016-0376-3. [DOI] [PubMed] [Google Scholar]
- 122.Koller W, Herbster G, Anderson D, Wack R, Gordon J. Quinpirole hydrochloride, a potential anti-parkinsonism drug. Neuropharmacology. 1987;26:1031–1036. doi: 10.1016/0028-3908(87)90245-0. [DOI] [PubMed] [Google Scholar]
- 123.Swarzenski BC, Tangt L, Oht YJ, O’malleyt KL, Todd D. Morphogenic potentials of D2, D3, and D4 dopamine receptors revealed in transfected neuronal cell lines (neuronal development/mesencephalic cells) Proc. Natl Acad. Si. USA. 1994;91:649–653. doi: 10.1073/pnas.91.2.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim SY, et al. The dopamine D2 receptor regulates the development of dopaminergic neurons via extracellular signal-regulated kinase and Nurr1 activation. J. Neurosci. 2006;26:4567–4576. doi: 10.1523/JNEUROSCI.5236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vergara RC, et al. The energy homeostasis principle: neuronal energy regulation drives local network dynamics generating behavior. Front. Comput. Neurosci. 2019;13:49. doi: 10.3389/fncom.2019.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013;7:13. doi: 10.3389/fncom.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013;11:34. doi: 10.1186/1478-811X-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Delcambre, S., Nonnenmacher, Y. & Hiller, K. in Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease (ed. Buhlman, L. M.) 25–47 (Springer International Publishing, 2016).
- 129.Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res. 2003;989:205–213. doi: 10.1016/S0006-8993(03)03354-7. [DOI] [PubMed] [Google Scholar]
- 130.Hastings TG. Enzymatic oxidation of dopamine: the role of prostaglandin H synthase. J. Neurochem. 1995;64:919–924. doi: 10.1046/j.1471-4159.1995.64020919.x. [DOI] [PubMed] [Google Scholar]
- 131.Spencer JP, et al. Conjugates of catecholamines with cysteine and GSH in Parkinson’s disease: possible mechanisms of formation involving reactive oxygen species. J. Neurochem. 1998;71:2112–2122. doi: 10.1046/j.1471-4159.1998.71052112.x. [DOI] [PubMed] [Google Scholar]
- 132.Mattammal MB, Haring JH, Chung HD, Raghu G, Strong R. An endogenous dopaminergic neurotoxin: implication for Parkinson’s disease. Neurodegeneration. 1995;4:271–281. doi: 10.1016/1055-8330(95)90016-0. [DOI] [PubMed] [Google Scholar]
- 133.Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl Acad. Sci. USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roy S. Synuclein and dopamine: the Bonnie and Clyde of Parkinson’s disease. Nat. Neurosci. 2017;20:1514–1515. doi: 10.1038/nn.4660. [DOI] [PubMed] [Google Scholar]
- 135.Rice ME, Patel JC. Somatodendritic dopamine release: recent mechanistic insights. Philos. Trans. R. Soc. Lond. Ser. B. 2015;370:20140185. doi: 10.1098/rstb.2014.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Avelar AJ, Cao J, Newman AH, Beckstead MJ. Atypical dopamine transporter inhibitors R-modafinil and JHW 007 differentially affect D2 autoreceptor neurotransmission and the firing rate of midbrain dopamine neurons. Neuropharmacology. 2017;123:410–419. doi: 10.1016/j.neuropharm.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhou Y, Bunney BS, Shi W-X. Differential effects of cocaine on firing rate and pattern of dopamine neurons: role of alpha1 receptors and comparison with L-dopa and apomorphine. J. Pharmacol. Exp. Ther. 2006;317:196–201. doi: 10.1124/jpet.105.094045. [DOI] [PubMed] [Google Scholar]
- 138.Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 2004;24:165–205. doi: 10.1081/RRS-200029981. [DOI] [PubMed] [Google Scholar]
- 139.Kabbani N, Negyessy L, Lin R, Goldman-Rakic P, Levenson R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J. Neurosci. 2002;22:8476–8486. doi: 10.1523/JNEUROSCI.22-19-08476.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J. Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zeng X, et al. Long-term changes in morphology, D2R expression and targets of regenerated dopaminergic terminals in the striatum after a partial lesion in the substantia nigra in the rat. Brain Res. 2012;1450:166–173. doi: 10.1016/j.brainres.2012.02.047. [DOI] [PubMed] [Google Scholar]
- 142.Urs NM, et al. Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl Acad. Sci. USA. 2015;112:E2517–26. doi: 10.1073/pnas.1502740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lüscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010;11:301–315. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen JF, Aloyo VJ, Weiss B. Continuous treatment with the D2 dopamine receptor agonist quinpirole decreases D2 dopamine receptors, D2 dopamine receptor messenger RNA and proenkephalin messenger RNA, and increases mu opioid receptors in mouse striatum. Neuroscience. 1993;54:669–680. doi: 10.1016/0306-4522(93)90238-B. [DOI] [PubMed] [Google Scholar]
- 145.Yan Z, Feng J, Fienberg AA, Greengard P. D(2) dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc. Natl Acad. Sci. USA. 1999;96:11607–11612. doi: 10.1073/pnas.96.20.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hadjiconstantinou M, Neff NH, Zhou LW, Weiss B. D2 dopamine receptor antisense increases the activity and mRNA of tyrosine hydroxylase and aromatic L-amino acid decarboxylase in mouse brain. Neurosci. Lett. 1996;217:105–108. [PubMed] [Google Scholar]
- 147.Pothos EN, Przedborski S, Davila V, Schmitz Y, Sulzer D. D2-Like dopamine autoreceptor activation reduces quantal size in PC12 cells. J. Neurosci. 1998;18:5575–5585. doi: 10.1523/JNEUROSCI.18-15-05575.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sibley DR. New insights into dopaminergic receptor function using antisense and genetically altered animals. Annu. Rev. Pharmacol. Toxicol. 1999;39:313–341. doi: 10.1146/annurev.pharmtox.39.1.313. [DOI] [PubMed] [Google Scholar]
- 149.Carter AJ, Müller RE. Pramipexole, a dopamine D2 autoreceptor agonist, decreases the extracellular concentration of dopamine in vivo. Eur. J. Pharmacol. 1991;200:65–72. doi: 10.1016/0014-2999(91)90666-E. [DOI] [PubMed] [Google Scholar]
- 150.Schapira AHV, Olanow CW. Rationale for the use of dopamine agonists as neuroprotective agents in Parkinson’s disease. Ann. Neurol. 2003;53(Suppl. 3):S149–57. doi: 10.1002/ana.10514. [DOI] [PubMed] [Google Scholar]
- 151.Park SK, et al. Par-4 links dopamine signaling and depression. Cell. 2005;122:275–287. doi: 10.1016/j.cell.2005.05.031. [DOI] [PubMed] [Google Scholar]
- 152.Parvez S, Winkler-Stuck K, Hertel S, Schönfeld P, Siemen D. The dopamine-D2-receptor agonist ropinirole dose-dependently blocks the Ca2+-triggered permeability transition of mitochondria. Biochim. Biophys. Acta. 2010;1797:1245–1250. doi: 10.1016/j.bbabio.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 153.Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J. Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Oh MS, Hong SJ, Huh Y, Kim K-S. Expression of transgenes in midbrain dopamine neurons using the tyrosine hydroxylase promoter. Gene Ther. 2009;16:437–440. doi: 10.1038/gt.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chakrabarty P, et al. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS ONE. 2013;8:e67680. doi: 10.1371/journal.pone.0067680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sun F, et al. A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell. 2018;174:481–496.e19. doi: 10.1016/j.cell.2018.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sorrentino ZA, et al. Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J. Biol. Chem. 2018;293:18914–18932. doi: 10.1074/jbc.RA118.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dhillon J-KS, et al. A novel panel of α-synuclein antibodies reveal distinctive staining profiles in synucleinopathies. PLoS ONE. 2017;12:e0184731. doi: 10.1371/journal.pone.0184731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vickery RG, von Zastrow M. Distinct dynamin-dependent and -independent mechanisms target structurally homologous dopamine receptors to different endocytic membranes. J. Cell Biol. 1999;144:31–43. doi: 10.1083/jcb.144.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mauna JC, et al. G protein βγ subunits play a critical role in the actions of amphetamine. Transl. Psychiatry. 2019;9:81. doi: 10.1038/s41398-019-0387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Koutzoumis DN, et al. Alterations of the gut microbiota with antibiotics protects dopamine neuron loss and improve motor deficits in a pharmacological rodent model of Parkinson’s disease. Exp. Neurol. 2020;325:113159. doi: 10.1016/j.expneurol.2019.113159. [DOI] [PubMed] [Google Scholar]
- 162.Heindl S, et al. Automated morphological analysis of microglia after stroke. Front. Cell. Neurosci. 2018;12:106. doi: 10.3389/fncel.2018.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu H, et al. Environmental enrichment potently prevents microglia-mediated neuroinflammation by human amyloid β-protein oligomers. J. Neurosci. 2016;36:9041–9056. doi: 10.1523/JNEUROSCI.1023-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Miller, D. et al. Dopamine transporter is a master regulator of dopaminergic neural network connectivity. J. Neurosci. 10.1523/JNEUROSCI.0223-21.2021 (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data acquired and analyzed for this study are available from the corresponding authors upon reasonable request.