

Abstract
Lymphadenopathies can be part of the clinical spectrum of several primary immunodeficiencies, including diseases with immune dysregulation and autoinflammatory disorders, as the clinical expression of benign polyclonal lymphoproliferation, granulomatous disease or lymphoid malignancy. Lymphadenopathy poses a significant diagnostic dilemma when it represents the first sign of a disorder of the immune system, leading to a consequently delayed diagnosis. Additionally, the finding of lymphadenopathy in a patient with diagnosed immunodeficiency raises the question of the differential diagnosis between benign lymphoproliferation and malignancies. Lymphadenopathies are evidenced in 15–20% of the patients with common variable immunodeficiency, while in other antibody deficiencies the prevalence is lower. They are also evidenced in different combined immunodeficiency disorders, including Omenn syndrome, which presents in the first months of life. Interestingly, in the activated phosphoinositide 3‐kinase delta syndrome, autoimmune lymphoproliferative syndrome, Epstein–Barr virus (EBV)‐related lymphoproliferative disorders and regulatory T cell disorders, lymphadenopathy is one of the leading signs of the entire clinical picture. Among autoinflammatory diseases, the highest prevalence of lymphadenopathies is observed in patients with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) and hyper‐immunoglobulin (Ig)D syndrome. The mechanisms underlying lymphoproliferation in the different disorders of the immune system are multiple and not completely elucidated. The advances in genetic techniques provide the opportunity of identifying new monogenic disorders, allowing genotype–phenotype correlations to be made and to provide adequate follow‐up and treatment in the single diseases. In this work, we provide an overview of the most relevant immune disorders associated with lymphadenopathy, focusing on their diagnostic and prognostic implications.
Keywords: activated phosphoinositide 3‐kinase δ syndrome, autoimmune lymphoproliferative syndrome, common variable immunodeficiency, CTLA‐4, LRBA
Lymphadenopathies can be part of the clinical spectrum of several primary immunodeficiencies, diseases with immune dysregulation, and autoinflammatory disorders, as the clinical expression of benign polyclonal lymphoproliferation, granulomatous disease, or lymphoid malignancy. In this work, we provide an overview of the most relevant immune disorders associated with lymphadenopathy, focusing on their diagnostic and prognostic implications.
INTRODUCTION
Lymphadenopathy can be part of the clinical spectrum of different primary immunodeficiency disorders (PIDs), including diseases with immune dysregulation, and autoinflammatory disorders. In these conditions, the altered immune or inflammatory function can be responsible for a polyclonal lymphoproliferation presenting with lymphadenopathy, hepatosplenomegaly or peripheral lymphocytosis. Lymphadenopathy and other clinical features of lymphoproliferation can represent the first clinical presentation or the leading sign of immune disorders, causing frequent misdiagnosis with hematological malignancies and delayed diagnosis [1, 2]. It is relevant to note that immune diseases themselves, particularly if associated with benign lymphoproliferation, also show an increased risk of lymphoid malignancies, and [3] therefore the presence of lymphadenopathy in patients with a diagnosis of PID can be difficult to interpret.
Knowledge of the multiple molecular mechanisms underlying lymphoproliferation in immune disease has been rapidly progressing, together with the identification of new monogenic forms of PIDs, through the use of next‐generation and whole‐exome sequencing techniques [4, 5].
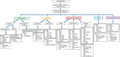
In this paper, we review the genetic background, the clinical pictures and laboratory features of the most relevant disorders of the immune and inflammatory response associated with lymphadenopathy, including those characterized by immune dysregulation and defects of innate immunity (Figure 1). Moreover, we describe the main warning signs to guide the diagnostic approach and prognostic implications, including the risk either of extranodal disease or malignancies in each condition.
FIGURE 1.

Immunodeficiency, immune dysregulation and autoinflammatory disorders associated with lymphadenopathy. The figure shows the classification of primary immunodeficiency disorders associated with lymphadenopathy, including immune dysregulation and autoinflammatory disorders. In particular, the significant overlap between T and B cell immunodeficiency and immune dysregulation disorders is highlighted
Lymphadenopathies in T and B cell immunodeficiencies
Among primary antibody deficiencies, the most significant prevalence of lymphadenopathy is reported in patients with common variable immunodeficiency (CVID), while it is described with lower frequency in patients with other disorders, including X‐liked agammaglobulinemia (XLA), hyper‐immunoglobulin (Ig)M syndrome (HIGM) and selective IgA deficiency (Figure 2).
FIGURE 2.

Frequency of lymphadenopathy in immunodeficiency, immune dysregulation and autoinflammatory disorders. *Fewer than 10 described patients. **Highly variable clinical phenotype
Primary antibody deficiencies
CVID is defined by the presence of clinical signs (increased risk of infection, immune dysregulation with autoimmunity, lymphoproliferation, granulomatous disease) accompanied by a reduction of two immunoglobulin isotypes and poor response to vaccinations or reduced switched‐memory B cells, in the absence of a T cellular deficiency, where other causes of hypogammaglobulinemia have been excluded [6]. Patients with CVID can develop lymphadenopathies at disease onset or during the clinical course as the clinical expression of benign polyclonal lymphoproliferation, granulomatous disease phenotype or lymphoma [7]. In a significant percentage of patients, the involvement of lymph nodes precedes the other clinical features of CVID [8], complicating and delaying the diagnostic process. Overall, lymphoproliferation has been reported to occur in 15–20% of patients [9, 10, 11], with splenomegaly and lymphadenopathy being frequently associated [12]. The pathogenesis of lymphoproliferation in CVID is not completely elucidated, and the role of genetic background and immunological features is currently being investigated [9, 13]. Although a clear genotype–phenotype association is not demonstrated, nuclear factor kappa B1 (NF‐κB1) and transmembrane activator and calcium‐modulator and cyclophilin ligand (CAML) interactor (TACI) mutations seem to be related to the development of lymphoproliferation and autoimmunity, with NF‐κB1 mutation being the most common molecular defect observed in a large cohort of CVID patients with lymphoproliferation [14, 15]. Concerning the immunological parameters, it emerges that in patients with benign lymphoproliferation a significant expansion of transitional B cells is common [12]. Additionally, a relationship between lymphoproliferation and preserved IgM levels has been suggested [9, 10].
Granulomatous disease, featured by non‐caseating and sterile granulomas in different systems, is described in 8–22% of the patients with CVID and typically affects the lungs, spleen, lymph nodes and skin [16]. Moreover, granulomas can be detected in the liver, bone marrow, kidney, brain, gastrointestinal tract, eye and parotid glands [17]. Granulomatous CVID is more frequently diagnosed in patients with lymphoproliferation, being strongly associated with the presence of splenomegaly [12], suggesting a common pathogenic mechanism between the two disease phenotypes. Accordingly, recent studies have revealed that patients with the granulomatous disease have a marked reduction of switched‐memory B cells and IgM‐only memory B cells, which can also be observed in patients with lymphoproliferation (particularly those with splenomegaly) [18].
The finding of lymphadenopathy in patients with CVID carries significant clinical and prognostic implications. They have an increased susceptibility to the development of both nodal and extranodal lymphomas, particularly non‐Hodgkin’s lymphomas (NHL), which is reported in 4% of the patients [19] (Table 1). The risk of lymphoid neoplasms is higher in CVID patients with other features of lymphoproliferation [9, 19, 20], thus highlighting the need of an accurate diagnostic approach to lymph node enlargement in this disease. Although most lymphadenopathies in CVID are benign [11], in the absence of adequate non‐invasive predictors of malignancy the finding of lymphadenopathy often requires performing a lymph‐nodal biopsy, the interpretation of which could be complicated by the variable degree of histological alterations observed in benign lymph nodes of CVID patients [21]. Indeed, although the most common histological finding is lymphoid hyperplasia with reduced plasma cells, some patients can show disrupted germinal centers and a clonal expansion in non‐malignant lymph nodes, potentially mimicking lymphomas [20]. Moreover, patients with CVID and lymphoproliferation or granulomatous disease have a higher risk of developing pulmonary involvement, described as ‘granulomatous lymphocytic interstitial lung disease’ (GLILD), which is responsible for significant mortality in these patients [22]. GLILD is frequently misdiagnosed as sarcoidosis, but a correct diagnosis is mandatory to provide adequate immunosuppressive treatment. Currently, azathioprine, mycophenolate mofetil and rituximab are the most widely used medications for GLILD [23].
TABLE 1.
Risk of lymphoid and non‐lymphoid malignancies in primary immunodeficiency disorder
| Disease | Risk of malignancies |
|---|---|
| CVID [3, 19] |
|
|
|
| XLA [25, 26] |
|
|
|
| RAG deficiency [34] |
|
| APDS‐1, APDS‐2 [42] |
|
|
|
| APDS‐L [46] |
|
| ALPS [55, 56] |
|
| CTLA‐4 deficiency [70] |
|
|
|
| LATAI [70] |
|
| STAT3 GOF [77] |
|
|
|
| STAT1 GOF [79] |
|
| SOCS1 deficiency [80] |
|
| TET2 deficiency [60] |
|
| XLP‐1 [99, 100] |
|
| XMEN [94] |
|
|
|
| STK4 deficiency [88] |
|
| ITK deficiency [3, 84] |
|
|
|
| CD70 deficiency [93] |
|
| CD27 deficiency [93] |
|
| CTPS1 deficiency [84] |
|
| CD137 deficiency [87] |
|
| RASGRP1 deficiency [3, 84] |
|
| RALD [53, 54] |
|
| WAS [48] |
|
| AT [51] |
|
|
|
| NBS [49] |
|
|
|
|
|
| Bloom syndrome [50] |
|
|
|
|
|
| CHH [52] |
|
|
Abbreviations: ALPS = autoimmune lymphoproliferative syndrome; APDS = activated phosphoinositide 3‐kinase δ syndrome; APDS‐L = activated phosphoinositide 3‐kinase δ syndrome‐like; AT = ataxia‐teleangectasia ; CHH = cartilage‐hair hypoplasia; CTLA‐4 = cytotoxic T lymphocyte antigen‐4; CTPS1 = CTP synthase 1; CVID = common variable immunodeficiency; EBV = Epstein–Barr virus; GOF = gain‐of‐function; HEM‐1 = hematopoietic protein 1; HL = Hodgkin’s lymphomas; ITK = interleukin‐2‐inducible T cell kinase; LATAI = LRBA deficiency with autoantibodies, regulatory T cell (Treg) cell defects, autoimmune infiltration and enteropathy; NBS = Nijmegen breakage syndrome ; NHL = non‐Hodgkin’s lymphomas; RAG = recombinase activating genes; RALD = RAS‐associated autoimmune leukoproliferative disease; RASGRP1 = RAS guanyl‐releasing protein 1; SOCS1 = suppressor of cytokine signaling1; STAT = signal transduced and activator of transcription; STK4 = serine/threonine kinase 4; TET‐2 = 10–11 translocation methylcytosine dioxygenase 2; WAS = Wiskott–Aldrich syndrome; XLA = X‐linked agammaglobulinemia; XLP = X‐linked proliferative syndrome; XMEN = X‐linked immunodeficiency with magnesium defect, EBV infection and neoplasia.
Differently from CVID, benign lymphoproliferation is not part of the classical clinical picture of XLA, although it has been described in a reduced number of patients [24]. Consequently, in patients with XLA the finding of lymphadenopathy should alert the clinician regarding the high risk of neoplastic disease, including NHL and gastrointestinal neoplasia, which are reported with increased frequency in this disease [3, 25, 26]. Concerning HIGM, the association between infections and polyclonal lymphoproliferation is more commonly reported in patients with activation‐induced deaminase (AID) or uracil‐DNA glycosylase (UNG) mutations, in which the molecular defect impairs the class‐switch recombination and somatic hypermutation [27].
Combined immunodeficiencies
Lymphadenopathy can be part of the clinical picture of several diseases classified among the group of combined immunodeficiency disorders (CID), including conditions that share common features with immune dysregulation disorders (Table 2) [4, 28].
TABLE 2.
Genetic background of combined immunodeficiencies associated with lymphadenopathies and clinical implications
| Disease | Molecular defect | Other clinical features and implications | Main laboratory findings |
|---|---|---|---|
| Omenn syndrome [29, 30, 31] | Mutation of the RAG genes cause altered VDJ recombination | Life‐threatening infections in the first months of life Splenomegaly, hepatomegaly, generalized erythroderma | ↑ Peripheral eosinophils, serum IgE |
| Other molecular defects reported: mutations of AK2, JAK3, IL‐7R. | ↓ Circulating B cells, TRECs in neonatal screening | ||
| RAG‐associated disorders [29, 33, 34] | Mutation of the RAG genes cause altered VDJ recombination | Splenomegaly, hepatomegaly, susceptibility to viral and bacterial infections, autoimmune cytopenia, organ‐specific autoimmunity | ↓ Peripheral lymphocytes, impaired T cell function |
| Granulomas in skin, mucosae, solid organs. GLILD | Hypogammaglobulinemia/HIGM | ||
| BENTA [37, 38] | Gain of function mutation of CARD11, leading to over activation of the NF‐kB pathway | Splenomegaly. Recurrent sinopulmonary infections. Increased risk of viral infections (herpesviruses) | ↑ Peripheral lymphocytes, B CD19 cells; |
| ↓ Memory B cells, class‐switched B cells | |||
| Hypogammaglobulinemia | |||
| IL‐2 Rβ deficiency [39] | IL‐R2B mutation causes reduced STAT‐5 activity and a reduced expansion of Tregs | Combined immunodeficiency; bowel inflammation, dermatological abnormalities, endocrinopathy. Susceptibility to herpesvirus infections (cytomegalovirus disease) | ↓ T cell proliferative response |
| ↑ NK cells, serum Ig, autoantibodies | |||
| HEM‐1 deficiency [40] | Mutation of HEM‐1 causes impaired regulation of actin cytoskeleton, with abnormal lymphocyte development and apoptosis | Recurrent otitis and sinopulmonary infections, splenomegaly, hepatomegaly. Possible development of HLH‐like clinical picture | Inverted CD4/CD8 ratio |
| ↑ Senescent T cells, memory T cells | |||
| Hypergammaglobulinemia | |||
| APDS‐1 [42, 43] | PIK3CD gain of function mutation causes enhanced activation of the mTOR pathway, promoting the proliferation of effector T cells | Splenomegaly, hepatomegaly, recurrent sinopulmonary infections, susceptibility to herpesvirus infections, enteropathy, autoimmunity | ↑ Senescent T cells, transitional B cells |
| ↓ Naive T cells | |||
| Hypogammaglobulinemia/HIGM | |||
| APDS‐2 [42, 43] | PIK3R1 loss of function mutation causes enhanced activation of the mTOR pathway, promoting the proliferation of effector T cells | Splenomegaly, hepatomegaly, recurrent sinopulmonary infections, susceptibility to herpesvirus infections, enteropathy, autoimmunity, short stature | ↑ Senescent T cells, transitional B cells |
| ↓ Naive T cells | |||
| Hypogammaglobulinemia/HIGM | |||
| APDS‐L [42, 46] | Loss of function of PTEN causes enhanced activation of the PI3K pathway | Enteropathy, facial dysmorphisms, macrocephaly, neurodevelopmental delay, recurrent respiratory infections. Increased risk of several malignancies | ↑ Transitional B cells |
| ↓ Class‐switched memory B cells | |||
| Hypogammaglobulinemia |
Abbreviations: AK2 = adenylate kinase 2; APDS = activated phosphoinositide 3‐kinase δ syndrome; APDS‐L = activated phosphoinositide 3‐kinase δ syndrome‐like; BENTA = B cell expansion with nuclear factor kappa B (NF‐κB) and T cell anergy; CARD11 = caspase recruitment domain family member 11; GLILD = granulomatous lymphocytic interstitial lung disease; HEM‐1 = hematopoietic protein 1; HIGM = hyper‐IgM syndrome; HL = Hodgkin’s lymphoma; IL‐7R = interleukin‐7 receptor; IL‐R2B = interleukin‐2 receptor β chain; JAK3 = Janus kinase 3; NHL = non‐Hodgkin’s lymphoma; NK = natural killer; mTOR = mammalian target of rapamycin; PIK3CD = phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit δ; PIK3R1 = phosphoinositide‐3‐kinase regulatory subunit 1; PTEN = phosphatase and tensin homolog; RAG = recombinase activating genes; STAT = signal transduced and activator of transcription; TRECs = T cell receptor excision circles.
In young infants, the finding of features of lymphoproliferation (commonly hepatosplenomegaly, but lymphadenopathies are also reported) in the context of a suspected PID strongly suggests the diagnosis of Omenn syndrome (OS), a subtype of severe combined immunodeficiency disorder (SCID) featured by the peripheral expansion of oligoclonal T cells, which is most frequently caused by mutations of the recombinase activating genes 1 and 2 (RAG 1 and RAG 2) [29]. Other molecular defects [mutations of adenylate kinase 2 (AK2), Janus kinase 3 (JAK3), interleukin 7 receptor (IL7R) genes and others] have been associated with the clinical picture of OS, although in rare cases [30]. In OS, the susceptibility to infections is accompanied by severe eczema (caused by skin infiltration of eosinophils, lymphocytes and histocytes), lymphadenopathy and hepatosplenomegaly [31]. Laboratory findings include peripheral eosinophilia, usually accompanied by absent circulating B cells and elevation of serum IgE [31]. Similarly to the other SCIDs, it is known that patients with OS, unless promptly initiated for hematopoietic stem‐cell transplantation (HSCT), die in early life from severe infections [32].
Benign lymphoproliferation is described with considerable frequency in other rare monogenic CIDs (Table 2). Partial mutations of RAG genes have marked genetic and clinical heterogeneity, which is far from being fully understood and can be responsible for a clinical picture of combined immunodeficiency associated with immune dysregulation, lymphadenopathy, hepatosplenomegaly and granulomatous disease [33]. In patients with RAG‐dependent immunodeficiency, the risk of lymphoid malignancies, and particularly NHL, is higher compared to the general population [29, 34]. The histological finding of granulomas is of relevance in the diagnostic process. Notably, in some patients with partial RAG deficiency, the rubella virus vaccine strain has been detected in granulomas [35]; therefore, testing for rubella in the histological specimen could provide further help in the diagnostic approach. Another useful diagnostic tool is represented by the study of T cell receptor (TCR) variability, diversity and joining [V(D)J] recombination through Vα7.2 analysis with fluorescence‐activated cell sorting (FACS), which significantly helps to differentiate RAG deficiencies from other CIDs [36].
The prevalence of lymphadenopathy, both as a presentation sign and during the clinical course, is highly relevant among the diseases caused by mutations in the caspase recruitment domain family member 11–B cell lymphoma/leukemia 10–mucosa‐associated lymphoid tissue lymphoma translocation protein 1 (CARD11–BCL10–MALT1) molecular complex [37], including B cell expansion with NF‐κB and T cell anergy (BENTA), deriving from gain‐of‐function (GOF) mutations of CARD11 [38].
Although extremely rare, mutations of the IL‐2 receptor β chain (IL‐2RB) gene and hematopoietic protein 1 (HEM‐1) deficiency are also associated with complex clinical phenotypes, including lymphadenopathy, autoimmune manifestations and enhanced inflammation (Table 2) [39, 40].
Activated phosphoinositide 3‐kinase δ syndrome and related disorders
The activated phosphoinositide 3‐kinase δ syndrome (APDS), a recently described autosomal‐dominant primary immunodeficiency associated with lymphoproliferation and autoimmunity, represents a paradigmatic condition. The disease is caused by mutations in the molecular complex of the phosphoinositide 3‐kinase (PI3K), particularly by phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit δ (PIK3CD) GOF mutations (APDS1) and phosphoinositide‐3‐kinase regulatory subunit 1 (PIK3R1) loss of function (LOF) mutation (APDS2) [41]. The hyperactivation of the PI3K complex, mainly through the enhanced function of the mechanistic target of rapamycin (mTOR) pathway, leads to a combined immune defect, often associated with reduced naive T cells, elevation of senescent lymphocytes and hypogammaglobulinemia, although the finding of a HIGM is common [42]. Patients with APDS experience multiple sinopulmonary infections and increased susceptibility to viral infections, in particular caused by herpesviruses. The molecular defect is also responsible for a marked immune dysregulation and lymphoproliferation: lymphadenopathies are evidenced in almost all patients, frequently in association with hepatomegaly and/or splenomegaly, and patients have a higher risk of developing lymphomas (mainly NHL) than the healthy population [43, 44]. Moreover, patients show high susceptibility for the development of autoimmunity, mainly autoimmune cytopenias and enteropathy [42, 45]. Lymphadenopathy is one of the leading signs of APDS, frequently being the presentation sign of the disease, and the differential diagnosis with autoimmune lymphoproliferative syndrome (ALPS) or lymphomas is extremely difficult. Pathological examination of lymph nodes often shows disrupted germinal centers, follicular hyperplasia and reduced mantle zones [42]. A similar clinical picture (APDS‐like) is observed in patients with loss of function of the phosphatase and tensin homolog (PTEN) gene, implicated in the regulation of the PI3K molecular pathway. In APDS‐L lymphoproliferation is less common, and patients are often affected by neurodevelopmental delay and increased risk of different non‐lymphoid neoplasms (Table 1) [42, 46].
Recognizing APDS is fundamental for the outcome of the disease, as the treatment with immunosuppressive agents, such as sirolimus, specific PI3K inhibitors (leniolisib, nemiralisib) or, in selected patients, hematopoietic stem cell transplantation (HSCT) [42], can improve the clinical course.
Rare PIDs presenting with lymphadenopathy
The finding of a clinically relevant non‐infectious lymphadenopathy in patients with defects of the innate immune response such as phagocytic function is rare, being described only in isolated case reports and series of patients with chronic granulomatous disease [47]. Patients with Wiskott–Aldrich syndrome (WAS) rarely develop benign polyclonal lymphoproliferation, as the classical clinical phenotype is featured by immunodeficiency, eczema and thrombocytopenia, but have a higher risk of lymphoid malignancies compared to the general population [48]. Other immunodeficiencies associated with syndromes, including those caused by molecular defects impairing DNA repair [ataxia‐teleangectasia (AT), Nijmegen breakage syndrome (NBS), Bloom syndrome and others] and short telomere syndromes [cartilage‐hair hypoplasia (CHH)], are associated with a remarkable risk for the development of lymphoid neoplasms (Table 1). Because, in these syndromes, lymphadenopathy is not part of the classic clinical picture [3, 49, 50, 51, 52], its finding in patients diagnosed with one of these disorders is highly suggestive of an underlying neoplastic disease.
Among the disorders classified as ‘phenocopies’ of PIDs [4], RAS‐associated autoimmune leukoproliferative disease (RALD) is of particular interest, and could present a clinical picture resembling ALPS. RALD, caused by somatic mutations in the RAS signaling pathway (often NRAS or KRAS) is clinically featured by an early onset of a systemic lupus erythematosus (SLE)‐like picture, accompanied by hematological involvement (cytopenias), hepatosplenomegaly and lymphadenopathy. In patients with RALD, the development of NHL has been described [53, 54].
Immune dysregulation syndromes
In syndromes associated with immune dysregulation, lymphadenopathies can be one of the leading signs of the clinical spectrum (Table 3). Immune dysregulation syndromes are caused by defective lymphocyte apoptosis, accumulation of self‐reactive lymphocytes or altered function of regulatory T cells (Tregs) (Figure 3). A distinct subclass of immune dysregulation disorders includes diseases associated with increased susceptibility to Epstein–Barr virus (EBV)‐induced lymphoproliferation.
TABLE 3.
Genetic background of immune dysregulation syndromes associated with lymphadenopathies and clinical implications
| Disease | Molecular defect | Other clinical features and implications | Main laboratory findings |
|---|---|---|---|
| ALPS‐FAS | Mutation of FAS, sFAS, FASL or CAPS‐10 finally cause defective lymphocyte apoptosis | Splenomegaly, hepatomegaly, autoimmune cytopenia, other autoimmune diseases (thyroiditis, hepatitis, uveitis) | ↑ abDNT cells, serum Ig levels, serum vitamin B12, serum IL‐10, IL‐18; serum FAS ligand |
| ALPS‐sFAS | |||
| ALPS‐FASL | |||
| ALPS‐CASP10 | |||
| ALPS‐U [55, 56, 57, 62] | |||
| TET‐2 deficiency [60] | TET‐2 mutation causes reduced DNA methylation with consequently altered lymphocyte homeostasis, differentiation and apoptosis | Increased susceptibility to infection, splenomegaly, hepatomegaly, autoimmune cytopenia, ALPS‐like clinical phenotype | ↑ abDNT, serum FAS ligand, serum IL‐10 |
| ↓ Th17, Th1, class‐switched B cells | |||
| Hypogammaglobulinemia | |||
| IPEX syndrome [64, 65] | Mutations of FoxP3 lead to defective maturation of Tregs | Autoimmune enteropathy, endocrinopathy, eczematous dermatitis, lymphoproliferation, food allergies, autoimmune disorders (type 1 diabetes, cytopenias, thyroiditis), arthritis | ↑ Peripheral eosinophils, serum IgE, CD4 cells |
| CD25 deficiency [63, 71, 72] | Mutations of the IL‐R2A gene causes reduced activity of the IL‐2 receptor, with a consequent deficit in the T cell adaptive response and proliferation of Tregs | IPEX‐like phenotype. Enteropathy, eczema, splenomegaly, hepatomegaly | ↓ T cell proliferative response, CD4/CD8 ratio |
| Susceptibility to viral and bacterial infections | |||
| CTLA‐4 deficiency [63, 66, 67, 70] | Mutation of CTLA‐4 causes reduced Tregs suppressive activity, with ineffective inhibition of co‐stimulatory molecules CD80/CD86 on APCs | IPEX‐like phenotype. Enteropathy, splenomegaly, respiratory infections, autoimmunity (cytopenia, thyroiditis, arthritis, uveitis), psoriasis, GLILD | ↓ Peripheral lymphocytes, naive T cells, Tregs CD19 cells, switched‐memory B cells, NK cells |
| Hypogammaglobulinemia | |||
| Potentially normal laboratory findings | |||
| LATAI [63, 68, 69, 70] | LRBA mutation causes reduced CTLA‐4 expression on Tregs surface, with consequent altered Tregs suppressive activity | IPEX‐like or ALPS‐like phenotype Splenomegaly, hepatomegaly. Enteropathy, autoimmunity (cytopenia, hepatitis, uveitis, diabetes), respiratory infections, bronchiectasis. GLILD | ↓ Peripheral lymphocytes, Tregs, memory B cells |
| Hypogammaglobulinemia | |||
| Potentially normal laboratory findings | |||
| BRIDA [63, 73] | BACH‐2 mutation causes impaired germ center reactions (B cell maturation, class‐switching) and T cell differentiation, with Tregs deficiency | CVID‐like phenotype, enteropathy, sinopulmonary infections, splenomegaly | ↑ Th1 cells, transitional B cells |
| ↓ Tregs, T cell proliferation, memory B cells, class‐switched B cells | |||
| Hypogammaglobulinemia | |||
| STAT1 GOF [63, 74, 78, 79] | The mutation causes defective production of Th17 cells | IPEX‐like phenotype, predisposition to mucocutaneous candidiasis and autoimmunity (thyroiditis) | ↓ Th17 cells |
| STAT3 GOF [63, 74, 77] | The mutation causes impaired Treg proliferation (via indirect inhibition of STAT5), reduced CD25 expression. Probably altered Th17 proliferation and function | IPEX‐like phenotype. Enteropathy, eczema, severe infections, growth retardation, and multiorgan autoimmunity (i.e. diabetes, thyroiditis, arthritis), GLILD | ↓ Memory B cells, Tregs, double‐negative T cells |
| Hypogammaglobulinemia | |||
| Potentially normal laboratory findings | |||
| SOCS‐1 deficiency [80] | SOCS1 mutations cause an uncontrolled activation of STAT‐dependent pathways, enhanced T cell proliferation and reduced Treg integrity and function | Early‐onset autoimmunity (cytopenia, SLE‐like phenotype, arthritis, thyroiditis), hepatomegaly, splenomegaly | ↓ Tregs, switched‐memory B cells |
| Anti‐nucleous antibodies, other autoantibodies (anti‐dsDNA, anti‐phospholipid) | |||
| PKCD [61] | PKCD mutations cause defective lymphocyte apoptosis, enhanced IL‐2 production and T cell proliferation, enhanced BCR signaling. Altered immune tolerance | Hepatomegaly, splenomegaly, SLE‐like phenotype (articular, hematological, cutaneous, and renal involvement), antiphospholipid syndrome | Hypogammaglobulinemia |
| Anti‐nucleous antibodies, other autoantibodies (anti‐dsDNA, anti‐phospholipid) |
Abbreviations: abDNT = αβ double‐negative T cells; ALPS = autoimmune lymphoproliferative syndrome; APCs = antigen‐presenting cells; BACH‐2 = BTB domain and CNC homolog 2; BCR = B cell receptor; BRIDA = BACH2‐related immunodeficiency and autoimmunity; CASP10 = caspase 10; CTLA‐4 = cytotoxic T lymphocyte antigen‐4; FAS = first apoptosis signal, FASL = FAS ligand; FoxP3 = forkhead box protein 3; GLILD = granulomatous lymphocytic interstitial lung disease; GOF = gain‐of‐function; HL = Hodgkin’s lymphoma; IGF‐1 = insulin growth factor 1; IL‐R2A = ‐nterleukin‐2 receptor α chain; IPEX = immune dysregulation, polyendocrinopathy, enteropathy, X‐linked; LATAI = LRBA deficiency with autoantibodies, Treg cell defects, autoimmune infiltration and enteropathy; LRBA = lipopolysaccharide (LPS)‐responsive and beige‐like anchor; NHL = non‐Hodgkin’s lymphoma; PKCD = protein kinase C δ deficiency; sFAS = somatic mutation of FAS; SLE = systemic lupus erythematosus; SOCS1 = suppressor of cytokine signaling1 ;STAT = signal transduced and activator of transcription; Tregs = regulatory T cells, TET‐2 = 10–11 translocation methylcytosine dioxygenase 2.
FIGURE 3.

Summary of the molecular mechanisms leading to immune dysregulation syndromes. (a) The molecular mechanisms associated with the development of immune dysregulation in disorders with ineffective apoptosis. When the first apoptosis signal (FAS) binds FAS ligand (FASL), the apoptotic pathway is initiated, as FAS‐associated protein with death domain (FADD) promotes the activation of pro‐caspase 8 and pro‐caspase 10, responsible for the initiation of apoptosis. In autoimmune lymphoproliferative syndrome‐FAS (ALPS‐FAS), ALPSFASL) and ALPS‐caspase 10 (CASP10) the molecular defect is responsible for an impairment of the first molecular steps of the apoptotic cascade. Protein kinase C δ (PKC‐δ) promotes the apoptosis through a caspase‐3 dependent mechanism, and acts by down‐regulating the B cell receptor (BCR) and T cell receptor (TCR) signaling, thus reducing the lymphocyte proliferation. Therefore, in PKC‐δ deficiency (PKCD) the apoptotic defect is accompanied by enhanced lymphocyte proliferation. (b) Mechanisms responsible for immune dysregulation and lymphadenopathy in diseases affecting regulatory T cells (Tregs) functioning. Forkhead box protein 3 (FoxP3), which is deficient in immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX), is central for Tregs proliferation and activity. CD25 is important in activating interleukin (IL)‐2‐dependent signaling, and therefore in promoting the transcription of FoxP3, which is impaired in CD25 deficiency. Signal transducer and activator of transcription (STAT)‐5b is an activator of FoxP3, which is inhibited by STAT‐3. In STAT‐3 gain‐of‐function (GOF), the enhanced inhibition of STAT‐5 finally leads to reduced FoxP3 activity and Tregs proliferation. Also, the BTB domain and CNC homolog 2 (BACH‐2) has a role in promoting Treg proliferation, which is altered in patients with BACH2‐related immunodeficiency and autoimmunity (BRIDA). Cytotoxic T lymphocyte antigen 4 (CTLA‐4), through interaction with the co‐stimulatory molecules CD80/CD86 on the antigen‐presenting cells (APC) surface, reducing their availability for the interaction with CD28 expressed by T cells. CTLA‐4 deficiency results in ineffective inhibition of co‐stimulatory signaling, leading to enhanced activation of T cells. Finally, lipopolysaccharide‐responsive and beige‐like anchor (LRBA) inhibits the lysosomal degradation of CTLA‐4, and in LRBA deficiency with autoantibodies, Treg cell defects, autoimmune infiltration and enteropathy (LATAI) a secondary CTLA‐4 deficiency is observed
Disorders with ineffective apoptosis
ALPS is an inherited disease determined by mutations in genes involved in the first apoptosis signal (FAS) molecular pathway [55], which includes the FAS, FAS ligand (FASL) and caspase‐10 (CASP‐10) genes, although in approximately 20–30% of patients the molecular diagnosis remains unknown [56]. The defective apoptosis causes the accumulation of circulating and nodal (paracortical) αβ double‐negative T CD8+ cells (abDNT), and self‐reactive T cells can also be observed. Patients with ALPS commonly present to clinical attention in the first year of life, with lymphoproliferation being the leading sign of the entire picture. In particular, patients show multiple chronic lymphadenopathies associated with splenomegaly and, frequently, hepatomegaly [56]. Patients can also present with autoimmune cytopenia, more frequently hemolytic anemia and immune thrombocytopenia, while organ‐specific autoimmunity is described in a lower percentage of patients [55, 56]. Other relevant laboratory findings include altered levels of serum immunoglobulin, more frequently showing polyclonal elevation of IgG; however, hypogammaglobulinemia is also described in a condition partially overlapping with CVID [55]. Reduced soluble FAS ligand levels, impaired apoptotic function, elevated abDNT and high vitamin B12 levels are highly suggestive for the diagnosis [57]. Although histological features are non‐specific, in most cases lymph nodes show follicular hyperplasia accompanied by the expansion of small lymphocytes (abDNT) with high proliferative rate and plasma cells in the paracortical area [55, 58]. A correct diagnosis of ALPS is fundamental to provide a strict follow‐up for the patients, as the disease is associated with a high risk of developing lymphomas, predominantly Hodgkin’s lymphoma (HL) [59]. Interestingly, an ALPS‐like clinical picture with susceptibility to HL and NHL and impairment of the FAS‐dependent apoptotic pathway can be observed in other monogenic disorders, such as 10–11 translocation methylcytosine dioxygenase 2 (TET2) deficiency [60] (Table 3).
Another disease caused by impaired apoptosis is protein kinase C δ deficiency (PKCD). PKCD is featured by defective apoptosis of B and T cells, increased lymphocyte proliferation and impaired immune tolerance. The disease presents with a clinical feature at a crossroad between PIDS and immune dysregulation syndromes, with lymphoproliferation triggered by viral infections being the leading signs of the disease. Moreover, patients can produce different specificities of autoantibodies, resulting in a systemic lupus erythematosus (SLE)‐like phenotype [61]. Both ALPS and PKCD are commonly treated with immunosuppressive agents, with HSCT being reserved for refractory cases of ALPS [62].
Disorders of regulatory T cells
The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome, caused by mutations leading to a loss of function of forkhead box protein 3 (FoxP3), is the main condition in the group of Tregs‐associated disorders (Tregopathies) [63]. IPEX is featured by the classical clinical triad of autoimmune enteropathy, endocrinopathy and early‐onset severe eczematous dermatitis [64]. Given the central role of FoxP3 in both the development and function of Tregs, the consequence carried by the molecular defect is an immune dysregulation [63], which can be accompanied by lymphoproliferation. Other classical signs and symptoms include the presence of peripheral eosinophilia, food allergies and autoimmune disorders, particularly type I diabetes and autoimmune cytopenias [64]. Patients with IPEX show lymphadenopathies in approximately 15% of cases [65], while lymphomas have been described only in isolated cases [64].
In other conditions featuring impaired function of Tregs, but showing wild‐type FoxP3, patients can present with an IPEX‐like clinical phenotype, whose differences in the prevalence of the single disease manifestations depend upon the specific molecular defect [65]. In particular, the group of Tregopathies includes cytotoxic T lymphocyte antigen‐4 (CTLA‐4) deficiency, lipopolysaccharide (LPS)‐responsive and beige‐like anchor (LRBA) deficiency with autoantibodies, Treg cell defects, autoimmune infiltration and enteropathy (LATAI), BTB domain and CNC homolog 2 (BACH‐2) haploinsufficiency, CD25 deficiency and signal transducer and activator of transcription (STAT)‐related disorders (Table 3) [63].
The molecular pathogenesis of the Treg alterations in CTLA‐4 deficiency and LATAI is strictly connected. In particular, CTLA‐4, expressed by Tregs, is pivotal in causing the reduction of the expression of the co‐stimulatory molecules CD80 and CD86 on the antigen‐presenting cell (APC) surface [66], and LRBA inhibits the lysosomal degradation of CTLA‐4 (Figure 3). Therefore, the clinical phenotypes of the diseases share many common features. They are both associated with hypogammaglobulinemia, increased risk of infections (with a higher frequency of life‐threatening infections in LATAI) and autoimmune manifestations, including autoimmune cytopenia and enteropathy. Additionally, up to 50% of the patients with CTLA‐4 mutations show lymphadenopathy [67], which is observed in a similar percentage of patients with LATAI [65, 68]. Therefore, both diseases could present a clinical picture that can resemble both IPEX and ALPS. In CTLA‐4 and LATAI, patients show an increased risk of HL and NHL [67, 68, 69]. Interestingly, a recent systematic review evidenced that malignant lymphoproliferation is more common in patients with CTLA‐4 deficiency [70] (Table 1).
Immune dysregulation and lymphoproliferation are part of the clinical spectrum observed in patients with CD25 deficiency, which have ineffective IL‐2 signaling with consequently reduced activation of T cells and reduced proliferation of Tregs [71, 72], and in those affected by the BACH2‐related immunodeficiency and autoimmunity (BRIDA), a rare condition featured by impaired reactions of the gem center and enhanced proliferation of Th1 cells with a reduced number of circulating Tregs [73].
A clinical picture of immune dysregulation is also observed in patients with molecular defects involving the STAT genes, which are implicated in the signaling pathways activated by different cytokines through JAK‐mediated phosphorylation. In particular, GOF mutations of STAT‐1 and STAT‐3 can show features of immunodeficiency, autoimmunity and lymphoproliferation [74]. Both STAT‐1 and STAT‐3 are activated by IL‐2, while IL‐6 participates only in the induction of STAT‐3 phosphorylation [74].
In STAT‐3 GOF, reduced proliferation of Tregs, decreased expression of CD25 and altered T helper type 17 (Th17) cell proliferation are observed [74]. Clinically, patients present lymphadenopathy and experience severe infections, growth retardation and multi‐organ autoimmunity (i.e. diabetes, thyroiditis arthritis) [75, 76, 77]. Moreover, an increased risk of HL and NHL has been described [63]. Patients with STAT‐1 GOF show reduced Th17 proliferation, resulting in increased susceptibility to different infections, mainly mucocutaneous candidiasis [74]. The disease is also associated with immune dysregulation and autoimmunity [78]. The molecular mechanisms leading to immune dysregulation and Treg impairment have not been completely elucidated. Although lymphadenopathy is not part of the classical clinical picture of STAT‐1 GOF, it has been recently described in two patients in whom HL was also documented [79]. Notably, as suppressor of cytokine signaling1 (SOCS1) is implicated in the down‐regulation of the JAK/STAT pathway, its haploinsufficiency is associated with lymphocyte hyperactivity, autoimmunity and lymphoproliferation [80].
Although there is a significant clinical overlap between the different Tregopathies and their definitive treatment is represented by HSCT, reaching a molecular diagnosis can help in providing specific targeted treatments, such as the CTLA‐4 analogue abatacept in CTLA‐4 deficiency and LRBA deficiency, JAK inhibitors (baricitinib, ruxolitinib) in STAT‐related disorders and tocilizumab in STAT‐3 GOF [81, 82].
Diseases with EBV‐related lymphoproliferation
EBV infection, if not adequately controlled by the immune system, can result in polyclonal B cell activation and proliferation and potentially in the development of lymphoma, as the infectious agent is able to interfere with cellular apoptosis and express proteins that induce B cell activation [83].
An exuberant EBV‐associated lymphoproliferation is described in different PIDs, in most cases depending on defects of the cellular immune response. In particular, it is reported in patients with defects in T cell proliferative response to mitogens, in defective early T cell receptor (TCR) signaling or co‐stimulatory molecules (Table 4) [84, 85, 86, 87, 88, 89, 90, 91, 92, 93]. All the diseases mentioned cause persistent or recurrent lymphadenopathies triggered by an uncontrolled EBV infection and prolonged EBV viremia, and are associated with an increased risk of developing EBV‐associated lymphoid malignancies. Although most of these conditions are caused by molecular defects selectively impairing the immune response against EBV, in disorders depending upon a non‐selective immune defect patients can experience also severe infections caused by other pathogens [84]. Indeed, disorders of T cell proliferative response and signaling, including RAS guanyl‐releasing protein 1 (RASGRP1), CTP synthase 1 (CTPS1) and interleukin‐2‐inducible T cell kinase (ITK) deficiencies, are associated with an increased risk of other severe viral infections [caused by cytomegalovirus, herpes simplex virus, varicella‐zoster virus, human papillomavirus (HPV) and others], bacterial and fungal infections and virus‐associated neoplasms (i.e. HPV‐related) [84, 86]. Two other peculiar conditions featured by non‐EBV selective immune impairment are represented by X‐linked immunodeficiency with magnesium defect, EBV infection and neoplasia (XMEN) disease and serine/threonine kinase 4 (STK4) deficiency. XMEN is a rare PID caused by a deficiency of the magnesium transporter 1 (MAGT1) gene. It presents in males with recurrent sinopulmonary infections, otitis and chronic lymphadenopathy, often accompanied by splenomegaly. In the disease, sharing common clinical features with ALPS, the lymphoproliferation can be both EBV‐related and EBV‐independent and carries also a significant risk of developing lymphoid neoplasms [85, 94]. In STK4 deficiency, a PID featured by lymphopenia mainly affecting T CD4 cells, patients experience recurrent infections with increased susceptibility to EBV and potential development of both benign lymphoproliferation and lymphoma. The role of STK4 in the surveillance against the development of neoplasms underlies the elevated risk of lymphomas and leukemia in these patients [88]. The clinical relevance of EBV‐related disorders points out the importance of performing a PCR analysis for EBV DNA in patients with features of immunodeficiency or immune dysregulation and lymphoproliferation, as well as the necessity of an EBV encoding region (EBER) in‐situ hybridization on the histological samples from these patients [95]. Finally, patients with defective killing function, as reported in X‐linked proliferative syndrome 1 (XLP1) and XLP2 or patients with disorders involving check‐points of the cellular cycle, have enhanced susceptibility to the development of EBV‐associated hemophagocytic lymphohistiocytosis (HLH) (Table 4) [96, 97, 98]. Among these, XLP1 is of particular interest, as its complex clinical picture is not limited to EBV‐related HLH (in which the presence of lymphadenopathy is not the prominent feature), but includes hypogammaglobulinemia with immune dysregulation, EBV‐related lymphoproliferation and increased susceptibility to the development of lymphoid malignancies, particularly NHL [99, 100] (Table 1).
TABLE 4.
Disorders associated with increased susceptibility to EBV‐related lymphadenopathy
| Disease | Molecular defect | Other clinical/laboratory features and implications |
|---|---|---|
| RASGRP1 deficiency [84, 86] | Defective T cell proliferative response to mitogens, reduced expression of CTPS1 | Susceptibility to CMV, HPV, HSV, bacterial and fungal infections |
| CTPS1 deficiency [84, 89] | Defective T cell proliferation after activation of TCR. No deficit in effector function | Recurrent bacterial and viral infections |
| ITK deficiency [84, 90] | Defective TCR signaling, with impaired expansion end effector function of CD8 cells | Increased risk of virus‐associated neoplasia [i.e HPV associated] |
| XMEN [84, 85, 94] | MAGT1 deficiency causes impaired TCR signaling and defective NK2GD expression on CD8 and NK cells, responsible for a defective killing of EBV‐infected cells | Recurrent sinopulmonary infections, otitis, splenomegaly. ↑Peripheral B cells, inverted CD4/CD8 ratio |
| CD70 deficiency [84, 91, 93] | Defective CD70/CD27 interaction, important in T cell survival and effector function, including the expansion against EBV | Highly selective for EBV infections Hypogammaglobulinemia |
| CD27 deficiency [84, 92, 93] | Defective CD70/CD27 interaction, important in T cell survival and effector function, including the expansion against EBV | Highly selective for EBV infections Potential EBV‐associated HLH |
| CD137 deficiency [84, 87] | TNFRSF9 mutations cause Impaired expansion of EBV‐specific T cells, for the absence of co‐stimulatory activity mediated by CD137 | Highly selective for EBV infections. Chronic EBV viremia. Potential EBV‐associated HLH |
| STK4 deficiency [88] | STK4 mutations lead to enhanced T cell apoptosis and reduced T cell proliferation. STK4 is also a regulator of cellular checkpoints | Recurrent infections. Reduced levels of T cells. Hepatomegaly, splenomegaly |
| XLP1 [84, 99, 100] | Mutations in the SH2DIA gene, causing a defective killing of EBV‐infected cells by T and NK cells | EBV‐associated HLH. Hypogammaglobulinemia |
| XLP2 [84, 96] | XIAP mutations, causing enhanced T cell apoptosis and inflammasome dysregulation | EBV or CMV‐associated HLH. Other inflammatory disorders [IBD] |
| FAAP24 deficiency [97] | FAAP24 mutation causes altered DNA repair and ineffective checkpoint response | EBV‐associated HLH |
Abbreviations: CTPS1 = CTP synthase 1; EBV = Epstein–Barr virus; FAAP24 = Fanconi anemia‐associated protein 24; HL = Hodgkin’s lymphoma; HLH = hemophagocytic lymphohistiocytosis; ITK = interleukin‐2‐inducible T cell kinase; MAGT1 = magnesium transporter 1; NHL = non‐Hodgkin’s lymphoma; NK = natural killer; NK2GD = natural killer group 2 member; DRASGRP1 = RAS guanyl‐releasing protein 1; SH2DIA = SH2 domain‐containing protein 1A; STAT = signal transduced and activator of transcription; STK4 = serine/threonine kinase 4; TNFRSF9 = tumor necrosis factor receptor subfamily 9; XIAP = X‐linked inhibitor of apoptosis; XMEN = X‐linked immunodeficiency with magnesium defect, EBV infection and neoplasia. XLP = X‐linked proliferative syndrome.
Autoinflammatory disorders
Autoinflammatory disorders are a wide spectrum of diseases featured by an uncontrolled inflammatory response, caused by defects in the structure and function of the inflammasome. Their most recognized clinical presentation is represented by recurrent episodes of fever accompanied by other signs or symptoms, which may include lymphadenopathy [101]. Differently from the other diseases discussed in this review, autoinflammatory disorders are not associated with the development of lymphoid malignancies, but the clinical phenotype is often difficult to interpret. Lymphadenopathy represents a prominent clinical feature of the periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome and hyper‐IgD syndrome (HIDS), although it is rarely also described in patients with other autoinflammatory disorders (Table 5] [102, 103, 104, 105, 106, 107].
TABLE 5.
Genetic background of autoinflammatory disorders associated with lymphadenopathies and clinical implications
| Disease | Molecular defect | Other clinical features and implications | Laboratory findings |
|---|---|---|---|
| PFAPA syndrome [106, 108] | Unknown | Periodic episodes of fever (duration 3‐5 days, recurrence 21‐28 days). Pharyngitis, apthosis, possible abdominal pain | ↑ Inflammatory biomarkers during the febrile episodes |
| HIDS [107, 109] | Mutation of MVK causes accumulation of mevalonic acid and reduced production of isoprenoids | Periodic fever (duration 3–8 days, recurrence every 2–8 weeks), abdominal pain arthralgia/arthritis, cutaneous rash, splenomegaly, hepatomegaly, vomiting, serositis, apthosis | ↑ Inflammatory biomarkers during the febrile episodes, serum cytokines, serum IgD |
| ↓ MVK activity | |||
| FMF [103] | Mutation of MEFV causes the production of dysfunctional protein pyrin, with consequent activation of the inflammasome resulting in an enhanced inflammatory response | Periodic fever (duration 1–3 days, variable recurrence), abdominal pain arthralgia/arthritis, cutaneous rash, arthralgia/arthritis, serositis | ↑ Inflammatory biomarkers during the febrile episodes, serum cytokines |
| TRAPS [102] | Mutations of TNFRSF1A cause reduced serum TNFR (with enhanced TNF‐induced response), defective autophagy, and increased damage by reactive oxygen species | Periodic fever (duration 7–21 days, 2–4 episodes/year), cutaneous rash, abdominal pain conjunctivitis, ocular pain, myalgia, arthralgia/arthritis | ↑ Inflammatory biomarkers during the febrile episodes, serum cytokines. ↓ Serum TNFR |
| Blau syndrome [104] | NOD2 mutation, causing activation of NF‐kB | Granulomatous arthritis, uveitis, dermatitis. Less common: sialoadenitis, vasculitis, erythema nodosum, neuropathies, nephritis, hepatic and splenic granulomas, interstitial lung disease | ↑ Inflammatory biomarkers (chronic elevation) |
| DADA‐2 [105] | CECR1 mutation causes deficient activity of ADA‐2, resulting in imbalanced monocyte differentiation, increased activity of neutrophils | Vasculitis (polyarteritis nodosa), cutaneous rash, livedo reticularis, musculoskeletal and renal involvement, susceptibility to stroke Immune dysregulation, splenomegaly, hepatomegaly. Increased risk of sinopulmonary infections and herpesvirus infections | ↑ Inflammatory biomarkers |
| ↓ Memory B cells, terminally differentiated B cells; plasma cells | |||
| Hypogammaglobulinemia (↓IgM) | |||
| Disease | Molecular defect | Other clinical features and implications | Laboratory findings |
| PFAPA syndrome [106, 108] | Unknown | Periodic episodes of fever (duration 3–5 days, recurrence 21–28 days). Pharyngitis, apthosis, possible abdominal pain | ↑ Inflammatory biomarkers during the febrile episodes |
| HIDS [107, 109] | Mutation of MVK causes accumulation of mevalonic acid and reduced production of isoprenoids | Periodic fever (duration 3–8 days, recurrence every 2–8 weeks), abdominal pain arthralgia/arthritis, cutaneous rash, splenomegaly, hepatomegaly, vomiting, serositis, apthosis | ↑ Inflammatory biomarkers during the febrile episodes, serum cytokines, serum IgD |
| ↓ MVK activity |
Abbreviations: ADA‐2 = adenosine deaminase‐2 ;CECR1 = cat eye syndrome chromosome region 1; DADA‐2 = adenosine deaminase‐2 deficiency; FMF = familial Mediterranean fever; HIDS = hyper‐immunoglobulin (Ig)D syndrome; MEFV = Mediterranean fever; MVK = mevalonate kinase; NF‐kB = nuclear factor kappa B; NOD2 = nucleotide‐binding oligomerization domain 2; PFAPA = periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis; TNFR = tumour necrosis factor receptor; TNFRSF1A = tumour necrosis factor receptor superfamily member 1A; TRAPS = tumour necrosis factor receptor‐associated periodic syndrome.
PFAPA syndrome is the most common autoinflammatory disorder of childhood [101], with a mean age at onset of 2–4 years, and presents with recurrent episodes of fever with a duration of 3–6 days, accompanied by cervical lymphadenopathy, pharyngitis and apthosis [106]. Laboratory examinations are non‐specific, but typically show elevation of inflammatory markers during the febrile episodes, with normalization in the intercritic period [106]. Given this clinical phenotype, patients are usually referred to immunologists, infectious disease specialists or otolaryngologists [108]. However, in a subgroup of patients recurrent fever and lymphadenopathy can be the leading signs of the disease, thus justifying the request of a hematological consultation. The absence of an identified causative mutation contributes to complicating the diagnostic approach to the disease.
HIDS, caused by mutations of the mevalonate kinase (MVK) gene, usually shows onset during the first year of life, and most of the patients present to clinical attention before the fifth year of life. Patients with HIDS experience recurrent episodes of fever occurring every 2–8 weeks with a duration of 3–7 days, associated with a wide spectrum of symptoms such as abdominal pain, cutaneous rash, arthralgia or arthritis [109]. In most patients, during the febrile episodes lymphadenopathy is present, mainly in the cervical region, and can be accompanied by splenomegaly and hepatomegaly [107]. Differently from PFAPA syndrome, which usually resolves in scholar age, a significant percentage of patients with HIDS continue to experience febrile episodes and the associated signs and symptoms after adolescent age [109]. During the episodes, an elevation of inflammatory markers, including serum amyloid protein, is observed, while the levels of serum cytokines (IL‐1β, IL‐6) also remain elevated in many cases in the intercritic phase. Elevated levels of IgD are highly suggestive for HIDS but can be absent in some patients, thus not representing a sensible biomarker [107].
CONCLUSIONS AND DIAGNOSTIC IMPLICATIONS
During past decades, following the rapid advances in the knowledge of the molecular and immunological mechanisms underlying the pathogenesis of the immune diseases, new clinical entities at the crossroad of immunodeficiency, immune dysregulation and autoinflammation have been disclosed and some well‐known diseases have been reclassified. This makes the diagnostic process of these conditions a great challenge, due to the need to dissect the prevalent pathogenic aspects among the above‐mentioned diseases. In this work, we provide an overview of the most relevant PIDs associated with lymphadenopathy, including diseases with immune dysregulation and autoinflammatory disorders, summarizing their pathogenic aspects and clinical features. Beyond the genetic analysis, an extensive approach focused upon the recognition of the clinical signs and immunological features of the single condition (after a wide critical reasoning to exclude infectious, oncological and hematological diseases) can significantly help in the formulation of the diagnosis. Together with the known classic red flags for PIDs (high susceptibility to infections, mainly towards opportunistic pathogens or restricted to the same ones, the evidence of autoimmunity or other signs of immune dysregulation), some peculiar manifestations such as severe eczema, a periodic disease course or the finding of GLILD on computerized tomography (CT) scan are relevant warning signs, helping in the diagnostic process. Therefore, with the purpose of guiding the clinician towards a diagnostic assessment, we elaborated an algorithm focusing upon the main clinical signs at presentation (Figure 4). Although most of the described conditions are associated with an increased risk of infections, a peculiar infectious pattern can help in driving the diagnostic process. Indeed, the presence of severe herpesvirus infection is common in disorders featured by a combined immune defect (i.e. APDS), while mucocutaneous candidiasis suggests the diagnosis of STAT‐1 GOF, thus highlighting the importance of performing complete microbiological investigations (serological tastings, blood cultures, PCR for viral genomes and analysis of histological samples) in these patients. The finding of eczema and enteropathy in association with lymphadenopathy is strongly suggestive of the diagnosis of IPEX or other Tregopathies (CTLA‐4 deficiency, LATAI), while the simultaneous presence of eczema and diffuse lymphoproliferation in the newborn can lead to the suspect diagnostic of OS, particularly when it is associated with peripheral eosinophilia [110]. Also, the identification of autoimmunity can provide important indications for the diagnostic process. Indeed, CVID, ALPS and APDS frequently present with autoimmune cytopenias, while arthritis is more commonly evidenced in patients with PKCD and RALD (also in the context of an SLE‐like clinical phenotype), and autoimmune endocrinopathies are one of the hallmarks of IPEX syndrome and related disorders. A periodical clinical course featured by the recurrence of lymphadenopathies, fever and other symptoms (pharyngitis, serositis and others) should represent a warning sign for the diagnosis of an autoinflammatory disorder. Finally, in patients with respiratory involvement, the evidence of GLILD supports the diagnostic suspect of CVID, CTLA‐4 deficiency, LATAI or STAT‐3 GOF.
FIGURE 4.

Diagnostic approach to lymphadenopathies in patients with immune disorders. The figure shows an approach to the diagnosis of immune diseases (immunodeficiencies, immune dysregulation and autoinflammatory disorders) associated with lymphoproliferation. The approach is based on the identification of specific clinical signs, including features of autoimmunity (arthritis, cytopenia, endocrinopathy), the finding of eczema, specific infectious patterns, periodic disease course and respiratory involvement, in the form of granulomatous lymphocytic interstitial lung disease (GLILD)
Despite the significant overlap evidenced in the clinical and immunological pictures of immunodeficiencies and immune dysregulation syndromes associated with lymphoproliferation, an adequate clinical assessment could guide the diagnostic approach and execution of the appropriate genetic investigations. Further research will help in more clearly defining the clinical and immunological features associated with different PIDs, leading to the formulation of genotype–phenotype correlations and giving a basis to provide disease‐specific follow‐up and therapeutic indications. In conclusion, due to the rarity and multi‐faceted complexity of these conditions, deep critical reasoning of the clinician first observing the patient and the close collaboration with a multi‐disciplinary group experienced in immunodeficiency and autoinflammation are necessary to provide an adequate diagnostic approach.
CONFLICT OF INTERESTS
The authors declare that they have no financial disclosures or conflicts of interest.
AUTHOR CONTRIBUTIONS
Giorgio Costagliola wrote the manuscript, which was critically revised by Rita Costagliola. Both authors contributed to manuscript revisions and read and approved the submitted version.
ACKNOWLEDGEMENTS
Not applicable.
Costagliola G, Consolini R. Lymphadenopathy at the crossroad between immunodeficiency and autoinflammation: An intriguing challenge. Clin Exp Immunol. 2021;205:288–305. 10.1111/cei.13620
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1.Routes JM, Verbsky JW. Immunodeficiency presenting as an undiagnosed disease. Pediatr Clin North Am. 2017;64:27–37. [DOI] [PubMed] [Google Scholar]
- 2.Sánchez‐Ramón S, Bermúdez A, González‐Granado LI, Rodríguez‐Gallego C, Sastre A, Soler‐Palacín P. Primary and secondary immunodeficiency diseases in oncohaematology: warning signs, diagnosis, and management. Front Immunol. 2019;10:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riaz IB, Faridi W, Patnaik MM, Abraham RS. A systematic review on predisposition to lymphoid (B and T cell) neoplasias in patients with primary immunodeficiencies and immune dysregulatory disorders (inborn errors of immunity). Front Immunol. 2019;10:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al‐Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2018;38:129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. 2016;138:957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. 2014;5:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho HE, Cunningham‐Rundles C. Non‐infectious complications of common variable immunodeficiency: updated clinical spectrum, sequelae, and insights to pathogenesis. Front Immunol. 2020;11:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gathmann B, Mahlaoui N, Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134:116–26. [DOI] [PubMed] [Google Scholar]
- 9.Maglione PJ. Autoimmune and lymphoproliferative complications of common variable immunodeficiency. Curr Allergy Asthma Rep. 2016;16:19. [DOI] [PubMed] [Google Scholar]
- 10.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 2008;112:277–86. [DOI] [PubMed] [Google Scholar]
- 11.Yakaboski E, Fuleihan RL, Sullivan KE, Cunningham‐Rundles C, Feuille E. Lymphoproliferative disease in CVID: a report of types and frequencies from a US patient registry. J Clin Immunol. 2020;40:524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 2008;111:77–85. [DOI] [PubMed] [Google Scholar]
- 13.Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. 2016;53:575–90. [DOI] [PubMed] [Google Scholar]
- 14.Christiansen M, Offersen R, Jensen JMB, Petersen MS, Larsen CS, Mogensen TH. Identification of novel genetic variants in CVID patients with autoimmunity, autoinflammation, or malignancy. Front Immunol. 2019;10:3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss‐of‐function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol. 2018;142:1285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ardeniz O, Cunningham‐Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol. 2009;133:198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonilla FA, Barlan I, Chapel H, Costa‐Carvalho BT, Cunningham‐Rundles C, de la Morena MT, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4:38–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sánchez‐Ramón S, Radigan L, Yu JE, Bard S, Cunningham‐Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128:314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiaee F, Azizi G, Rafiemanesh H, Zainaldain H, Sadaat Rizvi F, Alizadeh M, et al. Malignancy in common variable immunodeficiency: a systematic review and meta‐analysis. Expert Rev Clin Immunol. 2019;15:1105–13. [DOI] [PubMed] [Google Scholar]
- 20.Cunningham‐Rundles C. Common variable immune deficiency: case studies. Blood 2019;134:1787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.da Silva SP, Resnick E, Lucas M, Lortan J, Patel S, Cunningham‐Rundles C, et al. Lymphoid proliferations of indeterminate malignant potential arising in adults with common variable immunodeficiency disorders: unusual case studies and immunohistological review in the light of possible causative events. J Clin Immunol. 2011;31:784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Resnick ES, Moshier EL, Godbold JH, Cunningham‐Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012;119:1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol. 2020. [DOI] [PubMed] [Google Scholar]
- 24.Bagheri Y, Vosughi A, Azizi G, Yazdani R, kiaee F, Hafezi N, et al. Comparison of clinical and immunological features and mortality in common variable immunodeficiency and agammaglobulinemia patients. Immunol Lett. 2019;210:55–62. [DOI] [PubMed] [Google Scholar]
- 25.Shillitoe B, Gennery A. X‐linked agammaglobulinaemia: outcomes in the modern era. Clin Immunol. 2017;183:54–62. [DOI] [PubMed] [Google Scholar]
- 26.Staines Boone AT, Torres Martínez MG, López Herrera G, de Leija Portilla JO, Espinosa Padilla SE, Espinosa Rosales FJ, et al. Gastric adenocarcinoma in the context of X‐linked agammaglobulinemia: case report and review of the literature. J Clin Immunol. 2014;34:134–7. [DOI] [PubMed] [Google Scholar]
- 27.Kavli B, Otterlei M, Slupphaug G, Krokan HE. Uracil in DNA – general mutagen, but normal intermediate in acquired immunity. DNA Repair. 2007;6:505–16. [DOI] [PubMed] [Google Scholar]
- 28.Leavis H, Zwerina J, Manger B, Fritsch‐Stork RDE. Novel developments in primary immunodeficiencies (PID) – a rheumatological perspective. Curr Rheumatol Rep. 2019;21:55. [DOI] [PubMed] [Google Scholar]
- 29.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cirillo E, Giardino G, Gallo V, D'Assante R, Grasso F, Romano R, et al. Severe combined immunodeficiency–an update. Ann NY Acad Sci. 2015;1356:90–106. [DOI] [PubMed] [Google Scholar]
- 31.Aleman K, Noordzij JG, de Groot R , van Dongen JJ , Hartwig NG. Reviewing Omenn syndrome. Eur J Pediatr. 2001;160:718–25. [DOI] [PubMed] [Google Scholar]
- 32.Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: current status and future perspectives. Front Pediatr. 2019;7:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villa A, Notarangelo LD. RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev. 2019;287:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358:2030–8. [DOI] [PubMed] [Google Scholar]
- 35.Neven B, Pérot P, Bruneau J, Pasquet M, Ramirez M, Diana JS, et al. Cutaneous and visceral chronic granulomatous disease triggered by a rubella virus vaccine strain in children with primary immunodeficiencies. Clin Infect Dis. 2017;64:83–6. [DOI] [PubMed] [Google Scholar]
- 36.Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I, et al. PROMIDISα: a T‐cell receptor α signature associated with immunodeficiencies caused by V(D)J recombination defects. J Allergy Clin Immunol. 2019;143:325–34.e2. [DOI] [PubMed] [Google Scholar]
- 37.Turvey SE, Durandy A, Fischer A, Fung S‐Y, Geha RS, Gewies A, et al. The CARD11‐BCL10‐MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol. 2014;134:276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arjunaraja S, Nosé BD, Sukumar G, Lott NM, Dalgard CL, Snow AL. Intrinsic plasma cell differentiation defects in B cell expansion with NF‐κB and T Cell anergy patient B cells. Front Immunol. 2017;8:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, et al. Human interleukin‐2 receptor β mutations associated with defects in immunity and peripheral tolerance. J Exp Med. 2019;216:1311–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castro CN, Rosenzwajg M, Carapito R, Shahrooei M, Konantz M, Khan A, et al. NCKAP1L defects lead to a novel syndrome combining immunodeficiency, lymphoproliferation, and hyperinflammation. J Exp Med. 2020;217:e20192275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh A, Joshi V, Jindal AK, Mathew B, Rawat A. An updated review on activated PI3 kinase delta syndrome (APDS). Genes Dis. 2020;7:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunes‐Santos CJ, Uzel G, Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. 2019;143:1676–87. [DOI] [PubMed] [Google Scholar]
- 43.Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3‐kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol. 2017;139:597–606.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol. 2016;16:702–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tessarin G, Rossi S, Baronio M, Gazzurelli L, Colpani M, Benvenuto A, et al. Activated phosphoinositide 3‐kinase delta syndrome 1: clinical and immunological data from an Italian cohort of patients. J Clin Med. 2020;9:3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eissing M, Ripken L, Schreibelt G, Westdorp H, Ligtenberg M, Netea‐Maier R, et al. PTEN hamartoma tumor syndrome and immune dysregulation. Transl Oncol. 2019;12:361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henrickson SE, Jongco AM, Thomsen KF, Garabedian EK, Thomsen IP. Noninfectious manifestations and complications of chronic granulomatous disease. J Pediatric Infect Dis Soc. 2018;7:S18–s24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shcherbina A, Candotti F, Rosen FS, Remold‐O’Donnell E. High incidence of lymphomas in a subgroup of Wiskott–Aldrich syndrome patients. Br J Haematol. 2003;121:529–30. [DOI] [PubMed] [Google Scholar]
- 49.Chrzanowska KH, Gregorek H, Dembowska‐Bagińska B, Kalina MA, Digweed M. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis. 2012;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arora H, Chacon AH, Choudhary S, McLeod MP, Meshkov L, Nouri K, et al. Bloom syndrome. Int J Dermatol. 2014;53:798–802. [DOI] [PubMed] [Google Scholar]
- 51.Rothblum‐Oviatt C, Wright J, Lefton‐Greif MA, McGrath‐Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. 2016;11:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vakkilainen S, Taskinen M, Mäkitie O. Immunodeficiency in cartilage‐hair hypoplasia: Pathogenesis, clinical course and management. Scand J Immunol. 2020;92:e12913. [DOI] [PubMed] [Google Scholar]
- 53.Calvo KR, Price S, Braylan RC, Oliveira JB, Lenardo M, Fleisher TA, et al. JMML and RALD (Ras‐associated autoimmune leukoproliferative disorder): common genetic etiology yet clinically distinct entities. Blood 2015;125:2753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neven Q, Boulanger C, Bruwier A, de Ville de Goyet M, Meyts I, Moens L, et al. Clinical spectrum of ras‐associated autoimmune leukoproliferative disorder (RALD). J Clin Immunol. 2021;41:51–8. [DOI] [PubMed] [Google Scholar]
- 55.Matson DR, Yang DT. Autoimmune lymphoproliferative syndrome: an overview. Arch Pathol Lab Med. 2020;144:245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bride K, Teachey D. Autoimmune lymphoproliferative syndrome: more than a FAScinating disease. F1000Res. 2017;6:1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Molnár E, Radwan N, Kovács G, Andrikovics H, Henriquez F, Zarafov A, et al. Key diagnostic markers for autoimmune lymphoproliferative syndrome with molecular genetic diagnosis. Blood. 2020;136:1933–45. [DOI] [PubMed] [Google Scholar]
- 58.Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler‐Stevenson M, Strober W, et al. Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol. 1998;153:1541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen‐Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 2001;98:194–200. [DOI] [PubMed] [Google Scholar]
- 60.Stremenova Spegarova J, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood 2020;136:1055–66. [DOI] [PubMed] [Google Scholar]
- 61.Salzer E, Santos‐Valente E, Keller B, Warnatz K, Boztug K. Protein kinase C δ: a gatekeeper of immune homeostasis. J Clin Immunol. 2016;36:631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li P, Huang P, Yang Y, Hao M, Peng H, Li F. Updated understanding of autoimmune lymphoproliferative syndrome (ALPS). Clin Rev Allergy Immunol. 2016;50:55–63. [DOI] [PubMed] [Google Scholar]
- 63.Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: monogenic diseases resulting in regulatory T‐cell deficiency. J Allergy Clin Immunol. 2018;142:1679–95. [DOI] [PubMed] [Google Scholar]
- 64.Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. Immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome: a systematic review. Autoimmun Rev. 2020;19:102526. [DOI] [PubMed] [Google Scholar]
- 65.Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover‐Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome. Front Immunol. 2018;9:2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA‐4 (CD152) mutations. Clin Exp Immunol. 2017;190:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T‐lymphocyte antigen 4‐insufficient subjects. J Allergy Clin Immunol. 2018;142:1932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of Phenotypes Associated with Mutations in LRBA. J Clin Immunol. 2016;36:33–45. [DOI] [PubMed] [Google Scholar]
- 69.Cagdas D, Halaçlı SO, Tan Ç, Lo B, Çetinkaya PG, Esenboğa S, et al. A spectrum of clinical findings from ALPS to CVID: several novel LRBA defects. J Clin Immunol. 2019;39:726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jamee M, Hosseinzadeh S, Sharifinejad N, Zaki‐Dizaji M, Matloubi M, Hasani M, et al. Comprehensive comparison between 222 CTLA‐4 haploinsufficiency and 212 LRBA deficiency patients: a systematic review. Clin Exp Immunol. 2021. doi: 10.1111/cei.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Mannurita SC, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146:248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X‐linked‐like syndrome, and defective IL‐10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119:482–7. [DOI] [PubMed] [Google Scholar]
- 73.Afzali B, Grönholm J, Vandrovcova J, O'Brien C, Sun H‐W, Vanderleyden I, et al. BACH2 immunodeficiency illustrates an association between super‐enhancers and haploinsufficiency. Nat Immunol. 2017;18:813–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lorenzini T, Dotta L, Giacomelli M, Vairo D, Badolato R. STAT mutations as program switchers: turning primary immunodeficiencies into autoimmune diseases. J Leukoc Biol. 2017;101:29–38. [DOI] [PubMed] [Google Scholar]
- 75.Milner JD, Vogel TP, Forbes L, Ma CA, Stray‐Pedersen A, Niemela JE, et al. Early‐onset lymphoproliferation and autoimmunity caused by germline STAT3 gain‐of‐function mutations. Blood 2015;125:591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, et al. STAT3 gain‐of‐function mutations associated with autoimmune lymphoproliferative syndrome like disease deregulate lymphocyte apoptosis and can be targeted by BH3 mimetic compounds. Clin Immunol. 2017;181:32–42. [DOI] [PubMed] [Google Scholar]
- 77.Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich P‐S, et al. Clinical aspects of STAT3 Gain‐of‐function germline mutations: a systematic review. J Allergy Clin Immunol Pract. 2019;7:1958–69.e9. [DOI] [PubMed] [Google Scholar]
- 78.Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain‐of‐function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016;127:3154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Henrickson SE, Dolan JG, Forbes LR, Vargas‐Hernández A, Nishimura S, Okada S, et al. Gain‐of‐function STAT1 mutation with familial lymphadenopathy and Hodgkin lymphoma. Front Pediatr. 2019;7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hadjadj J, Castro CN, Tusseau M, Stolzenberg M‐C, Mazerolles F, Aladjidi N, et al. Early‐onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. 2020;11:5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delmonte OM, Castagnoli R, Calzoni E, Notarangelo LD. Inborn errors of immunity with immune dysregulation: from bench to bedside. Front Pediatr. 2019;7:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Autoimmune Disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436–40. [DOI] [PubMed] [Google Scholar]
- 83.Saha A, Robertson ES. Impact of EBV essential nuclear protein EBNA‐3C on B‐cell proliferation and apoptosis. Future Microbiol. 2013;8:323–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Latour S, Fischer A. Signaling pathways involved in the T‐cell‐mediated immunity against Epstein–Barr virus: lessons from genetic diseases. Immunol Rev. 2019;291:174–89. [DOI] [PubMed] [Google Scholar]
- 85.Ravell JC, Matsuda‐Lennikov M, Chauvin SD, Zou J, Biancalana M, Deeb SJ, et al. Defective glycosylation and multisystem abnormalities characterize the primary immunodeficiency XMEN disease. J Clin Invest. 2020;130:507–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Somekh I, Marquardt B, Liu Y, Rohlfs M, Hollizeck S, Karakukcu M, et al. Novel mutations in RASGRP1 are associated with immunodeficiency, immune dysregulation, and EBV‐induced lymphoma. J Clin Immunol. 2018;38:699–710. [DOI] [PubMed] [Google Scholar]
- 87.Alosaimi MF, Hoenig M, Jaber F, Platt CD, Jones J, Wallace J, et al. Immunodeficiency and EBV‐induced lymphoproliferation caused by 4–1BB deficiency. J Allergy Clin Immunol. 2019;144:574–83.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schipp C, Schlütermann D, Hönscheid A, Nabhani S, Höll J, Oommen PT, et al. EBV negative lymphoma and autoimmune lymphoproliferative syndrome like phenotype extend the clinical spectrum of primary immunodeficiency caused by STK4 deficiency. Front Immunol. 2018;9:2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature 2014;510:288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ghosh S, Drexler I, Bhatia S, Adler H, Gennery AR, Borkhardt A. Interleukin‐2‐inducible T‐cell kinase deficiency‐new patients, new insight? Front Immunol. 2018;9:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70–CD27 pathway in immunity to Epstein–Barr virus infection. J Exp Med. 2017;214:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alkhairy OK, Perez‐Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol. 2015;136:703–12.e10. [DOI] [PubMed] [Google Scholar]
- 93.Ghosh S, Köstel Bal S, Edwards ESJ, Pillay B, Jiménez Heredia R, Erol Cipe F, et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood 2020;136:2638–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ravell JC, Chauvin SD, He T, Lenardo M. An update on XMEN disease. J Clin Immunol. 2020;40:671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weiss LM, Chen YY. EBER in situ hybridization for Epstein–Barr virus. Methods Mol Biol. 2013;999:223–30. [DOI] [PubMed] [Google Scholar]
- 96.Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol. 2014;34:772–9. [DOI] [PubMed] [Google Scholar]
- 97.Daschkey S, Bienemann K, Schuster V, Kreth HW, Linka RM, Hönscheid A, et al. Fatal lymphoproliferative disease in two siblings lacking functional FAAP24. J Clin Immunol. 2016;36:684–92. [DOI] [PubMed] [Google Scholar]
- 98.Aguilar C, Latour S. X‐linked inhibitor of apoptosis protein deficiency: more than an X‐linked lymphoproliferative syndrome. J Clin Immunol. 2015;35:331–8. [DOI] [PubMed] [Google Scholar]
- 99.Shadur B, Abuzaitoun O, NaserEddin A, Even‐Or E, Zaidman I, Stepensky P. Management of XLP‐1 and ITK deficiency: The challenges posed by PID with an unpredictable spectrum of disease manifestations. Clin Immunol. 2019;198:39–45. [DOI] [PubMed] [Google Scholar]
- 100.Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol. 2011;127:1329‐41.e2; quiz 42‐3. [DOI] [PubMed] [Google Scholar]
- 101.Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019;78:1025–32. [DOI] [PubMed] [Google Scholar]
- 102.Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF receptor‐associated periodic syndrome (TRAPS): current perspectives. Int J Mol Sci. 2020;21:3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alghamdi M. Familial Mediterranean fever, review of the literature. Clin Rheumatol. 2017;36:1707–13. [DOI] [PubMed] [Google Scholar]
- 104.Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto‐inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014;12:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caorsi R, Penco F, Schena F, Gattorno M. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol Online J. 2016;14:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Batu ED. Periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome: main features and an algorithm for clinical practice. Rheumatol Int. 2019;39:957–970. [DOI] [PubMed] [Google Scholar]
- 107.Favier LA, Schulert GS. Mevalonate kinase deficiency: current perspectives. Appl Clin Genet. 2016;9:101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Costagliola G, Maiorino G, Consolini R. Periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome (PFAPA): a clinical challenge for primary care physicians and rheumatologists. Front Pediatr. 2019;7:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J. 2016;14:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Costagliola G, Marco SD, Comberiati P, D’Elios S, Petashvili N, Di Cicco ME, et al. Practical approach to children presenting with eosinophila and hypereosinophilia. Curr Pediatr Rev. 2020;16:81–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.