

Abstract
Worldwide honeybees (Apis mellifera L.) are one of the most widely kept domesticated animals, supporting domestic and commercial livelihoods through the production of honey and wax, as well as in the delivery of pollination services to crops. Quantifying which plant species are foraged upon by honeybees provides insights into their nutritional status as well as patterns of landscape scale habitat utilization. Here we outline a rapid and reproducible methodology for identifying environmental DNA (eDNA) originating principally from pollen grains suspended within honey. The process is based on a DNA extraction incorporating vacuum filtration prior to universal eukaryotic internal transcribed spacer 2 region (ITS2) amplicon generation, sequencing and identification. To provide a pre-cursor to sequence phylotyping, we outline systems for error-corrected processing amplicon sequence variant abundance tables that removes chimeras. This methodology underpins the new UK National Honey Monitoring Scheme.
-
•
We compare the efficacy and speed of centrifugation and filtration systems for removing pollen from honey samples as a precursor to plant DNA barcoding.
-
•
We introduce the ‘HONEYPI’ informatics pipeline, an open access resource implemented in python 2.7, to ensure long-term reproducibility during the process of amplicon sequence variant classification.
Keywords: Amplicon sequence variants (ASV), Illumina generated PhiX control library, ITS2, Internal transcribed spacer 2, MiSeq platform, Naive bayesian classifier, Vacuum filtration
Graphical abstract
Specifications table
| Subject Area: | Biochemistry, Genetics and Molecular Biology |
| More specific subject area: | Extraction and metabarcode analysis of plant DNA extracted from UK honey |
| Method name: | DNeasy PowerPlant Pro Kit (Qiagen, Hilden, Germany) |
| Name and reference of original method: | DNeasy PowerPlant Pro Kit (Qiagen, Hilden, Germany) |
| Resource availability: | HONEYPI’ pipeline implemented in python 2.7 and is open access (https://github.com/hsgweon/honeypi). |
Methods details
Background
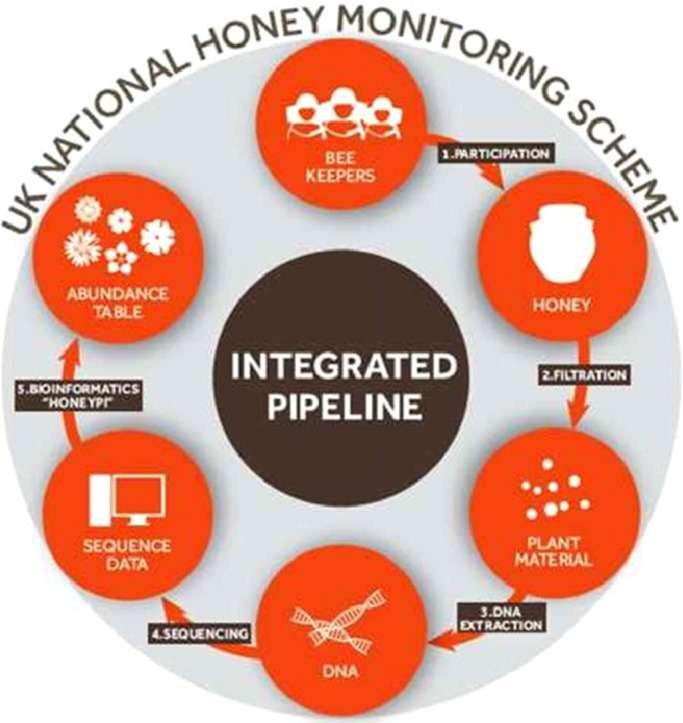
Honeybees are central place foragers typically travelling several kilometres from their hives [1,2]. As such, they readily integrate significant amounts of information on the landscape-scale floral resource available to honeybees as well as other generalist insect pollinators [3]. Additionally, their honey provides information on environmental contaminants, such as pesticides, to which bees are exposed when foraging in agricultural systems [4]. Information on foraging preferences is also critical for the parametrization of dynamic honeybee colony models [5], as well as quantifying their contribution to crop pollination services [6] and/or competitive interaction with wild pollinators [7]. In the UK, beekeeping is rapidly growing with over 29,000 beekeepers managing around 126,000 colonies [8]. This popularity has provided an opportunity for the rapid acquisition, using controlled methodologies, of large quantities of honey samples suitable for assessing foraging preferences and is currently implemented by the UK National Honey Monitoring Scheme which collected >800 honey samples across Great Britain in 2020 alone (https://honey-monitoring.ac.uk/).
Historically, microscopy has been used to determine the species of plants fed upon by bees, through the identification of pollen grains either in honey (Melissopalynology), or collected directly from foraging honeybees returning to hives [9,10]. However, both the need for specialist knowledge and significant processing time makes such methods inappropriate for processing large numbers of samples. The key to the success of such a citizen science national monitoring scheme will be the development and operational deployment of sophisticated protocols for barcoding and interpreting large volumes of honey samples. This methodological description outlines a simple pipeline for the application of these approaches, from the processing of the raw honey samples to the final step of species level phylotyping of amplicon sequence variants (ASV). A schematic of this pipeline is presented in Fig. 1.
Fig. 1.

Schematic showing the methodological pipeline used to isolate and identify the botanical origin of DNA contained within honey samples.
Extraction of plant material from honey
Current methodologies for the extraction of plant material (mainly pollen) from honey are done through either centrifugation [11], [12], [13], [14], filtration [15], or a combination of both [16], [17], [18] and depend upon downstream requirements- for example, when preparing high quality pollen for scanning electron microscopy. High speed or repeated centrifugation required to pellet buoyant palynomorphs can damage exine walls and there is always the risk of additional loss upon decanting [19]. Methods have been developed to reduce the potential negative impacts of centrifugation and include diluting honey in ethanol to reduce specific gravity for more efficient centrifugation [14] and the use of nested cell strainers as a gentle isolation method for large and fragile pollens [18]. However, the presence of ethanol and other chemicals can inhibit nucleic acid isolation therefore the investigation of an alternative approach which integrates these techniques was necessary. Consequently in this study, we compare the two methods (centrifugation or filtration for the isolation of pollen grains and associated plant DNA from honey dissolved in water) to establish a protocol that aims for optimal reproducibility and ease of scaling to large sample numbers.
Eight test samples were processed representing a range of UK honey types in terms of floral and geographical origin. From these samples, approximately 15 g of honey was weighed into a separate sterile 50 ml falcon tubes and diluted to 50 ml using molecular grade water. Diluted honey was then heated at 55 °C for 1 h, with occasional mixing in order to thoroughly dissolve and equally disperse any plant material. Where wax from capped honeycomb was suspended on the top of diluted honey, this layer was carefully removed and discarded using a clean spatula, as it was found to inhibit efficient DNA extraction. This procedure was carried out in duplicate and later centrifuged or filtered using the procedures outlined below.
Prior to centrifugation, diluted honey was loaded into 50 ml Beckman coulter centrifuge tubes. Samples were spun for 30 min at 15,000 x g in a Beckman Aventi Centrifuge with JA-20 rotor (Beckman Coulter Life Sciences, Indianapolis, USA). This isolation method was compared with replicate dilutions individually filtered using a reusable bottle top vacuum filtration system (Nalgene), fitted with 47 mm diameter mixed cellulose esters (MCE) membrane filters with a pore size of 1.2 µm (Millipore, Massachusetts, USA).
Total DNA was extracted from either the pellet or half a filter using the DNeasy PowerPlant Pro Kit (Qiagen, Hilden, Germany), with the following additions to the manufacturer's recommended protocol. To account for the small size of pollen grains, approx. 0.25 g of ≤ 106 µm autoclaved, acid washed glass beads (Merck, Darmstadt, Germany) were added to the PowerBead tubes- already containing 2.38 mm metal beads. Individual filters were sliced into smaller fragments using sterile dissection scissors and placed into the PowerBead tubes. To ensure complete cellular lysis, filters were immersed in 410 µl Bead Solution, 40 µl Phenolic Separation Solution (PSS) and 5 µl of proteinase K solution (20 mg/ml) and incubated at 60 °C for 1 h. After the addition of Solution SL and RNase A Solution (Step 2 of the manufacturer's protocol) tissue homogenization was undertaken for 1 min at speed setting 5.5 K using a Fastprep 24 tissue disrupter (MP Biomedicals, Solon, Ohio, USA). Samples were centrifuged at 13,000 x g for 3 min and the lysate transferred to a clean 2 ml microcentrifuge tube, 250 µl of Solution IR was added and the manufacturer's recommended protocol followed. Finally, due to the presence of PCR inhibitors associated with honey samples an additional wash of 500 µl, 97% ethanol was employed prior to a drying spin of 3 min (13,000 x g) and sample elution using Solution EB. Resultant DNA was quantified using a Nanodrop One spectrophotometer (Thermo scientific, Waltham, MA, USA) and extractions normalised to a concentration of ~10 ng/µl.
Amplicon generation and sequencing
Approximately 20 ng of extracted DNA template was used for plant DNA barcoding. Amplification was undertaken in a 50 µl reaction containing 0.5 µl Q5 High Fidelity Polymerase (New England Biolabs, Hitchin, UK),5X buffer, 1 µl 10 mM dNTP Mix, molecular grade water and 50 mM of a sample-unique, barcode-primer combination to allow for separation of sequences [20]. Primers were based on the universal eukaryotic internal transcribed spacer 2 region (herein, ITS2) and optimised for pollen analysis using Illumina MiSeq v3 chemistry [21]. Amplification included an initial denaturation at 98 °C for 30 s followed by 37 cycles of denaturation at 98 °C for 10 s; annealing at 49 °C for 20 s and elongation at 72 °C for 25 s. This was completed by a final extension step at 72 °C for 2 min. Amplicons were normalised using SequalPrep Normalisation Plate Kit, 96-well (Invitrogen, Carlsbad, CA), gel purified and quantified using Qubit high sensitivity dsDNA Assay kit (Invitrogen, Carlsbad, CA). The resultant amplicon library was sequenced at a concentration of 5.4 pM with a 0.6 pM addition of an Illumina generated PhiX control library. Sequencing was performed on an Illumina MiSeq platform using MiSeq Reagent Kit v3 (Illumina Inc., San Diego, USA).
HONEYPI bioinformatics pipeline
To ensure long-term reproducibility, we developed the HONEYPI pipeline implemented in python 2.7 and is open access (https://github.com/hsgweon/honeypi).The HONEYPI pipeline is divided into several parts as follows: (1) the raw amplicon sequences are quality filtered and adapters removed using TrimGalore v.0.6.4 (https://github.com/FelixKrueger/TrimGalore); (2) DADA2 pipeline is subsequently used to generate an Amplicon Sequence Variant (ASV) abundance table containing chimera-removed, high-quality error-corrected sequences [22]. (3). For each ASV, conserved regions flanking ITS2 are removed with ITSx v.1.1b [23]; and (4) resulting sequences taxonomically classified using the naive Bayesian classifier [24] against our in-house ITS2 database. This database was created by first downloading a total of 1958,909 sequences from NCBI on 25 March 2020 using the query “internal transcribed spacer [All Fields] AND 10:10,000[SLEN]”. These downloaded sequences were de-replicated with VSEARCH v.2.13.7 [25] to produce a sub-set of 1411,443 sequences. Of these sequences ITS2 regions were retrieved using ITSx [23] which removed and flanking conserved regions. Sequences shorter than 100 bps and those classified as non-eukaryotes were then removed, and from the resulting ITS2 (966,676 sequences) a RDP compatible training database was created using RDP Tools [24]. Unless stated otherwise, default parameters were used for the steps listed. Since HONEYPI uses ASVs rather than clusters of sequences for classification, it allows combining of ASV tables, i.e. data from two or more separate sequencing runs can be merged without re-clustering sequences.
Molecular statistics
All statistical analysis was performed using the statistical program R v.3.6.2 [26]. After quality filtering 1589,399 sequences remained. In order to identify taxonomically similar units, amplicon sequence variants were phylotyped (taxa identified as taxonomically the same) at the species level using the function aggregate_taxa in R package phyloseq v.1.30.0 [27] (Fig. 2). Taxa unassignable at the Kingdom/Phylum level and Non-Angiosperm taxa (Fungi, Metazoa, Chlorophyta) were considered erroneous or non-relevant to this study and therefore removed from the analysis. Additionally, to account for sequence bias samples with < 20, 000 sequences were removed from analysis and data was rarefied to an even depth of 20,159, using ‘Phyloseq’ function rarefy_even_depth. This rarefaction cut-off of 20,159 was considered to be the point at which samples had reached their asymptote based upon rarefaction curves log series rarecurve in R package ‘Vegan’ v.2.5–6 [28]. From this data set conventional descriptive community ecology metrics can then be performed. For the purposes of comparing filtration and centrifuge methods for DNA extraction from honey, we derive Fishers [alpha] log-series diversity index using the vegan package in R. Samples from the two methods were compared using the Welch t-statistic which allows for unequal variances between treatments but does assume a normal distribution.
Fig. 2.

Proportional relative abundance of rarefied sequences obtained from 8 test commercial honey samples. Sequences were identified to lowest possible taxonomic rank using the HONEYPI pipeline, then grouped to genus level (phylotyped). Taxonomic profiles were compared to show differences found between filtration and centrifugation pollen isolation prior to DNA extraction. The two methods are largely comparable, when looking at the dominant taxa from which samples are comprised.
Method validation
Samples that had undergone filtration contained a significantly higher Fishers diversity of plant species when compared to those extracted using the centrifugation approach (Welch t27= 2.58, p = 0.02) (Fig. 3). Filtered samples were in general highly reproducible, reduced sample variance suggesting that this method is reliable for a large datasets such as those produced by the National Honey Monitoring Scheme. Further, this methodology has the advantage of being both affordable and easy to scale up in terms of sample numbers through the use of multiple filter units. Differences between the two extraction methodologies can be explained by natural variation in viscosity between honey samples making DNA extraction post centrifugation significantly less reproducible within our system.
Fig. 3.

Fishers [alpha] log series diversity index (B) determined from 8 test commercial honey samples comparing the alternative extraction methods of filtration and centrifugation.
Application to support an eDNA national monitoring scheme
In conclusion, we have successfully integrated a series of modified existing DNA extraction and barcoding methodologies, and combined them with an innovative bioinformatics pipeline to provide a practical and highly efficient processing chain viable for the large scale determination of pollen amplicon variant sequences derived from honey samples. The application of this integrated methodology underpins a highly successful, mass participation citizen scheme national monitoring scheme – the UK National Honey Monitoring Scheme (https://honey-monitoring.ac.uk/) - which processes and reports on approximately 800 samples a year. This system enables a large-scale spatially explicit data resource describing multi-year national scale patterns of floral resource utilization by honeybees supporting scientific research, conservation policy and the livelihoods of beekeepers.
Declaration of Competing Interest
Authors confirm that there are no conflicts of interest.
Acknowledgments
This research was funded by the Natural Environment Research Council (NERC) and the Biotechnology and Biological Sciences Research Council (BBSRC) under research program NE/N018125/1 LTS-M ASSIST – Achieving Sustainable Agricultural Systems (www.assist.ceh.ac.uk). We are indebted to Peter Martin, Emily Abbott (Hive and Keeper), Pam Hunter (BBKA), Ged Marshall (BFA) and others for their advice and support in testing this methodology.
References
- 1.Steffan-Dewenter I., Kuhn A. Honeybee foraging in differentially structured landscapes. Proc. Royal Soc. B. 2003;270:569–575. doi: 10.1098/rspb.2002.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visscher P.K. Foraging strategy of honeybee colonies in a temperate deciduous forest. Ecology. 1982;63:1790–1801. [Google Scholar]
- 3.Bansch S. Foraging of honey bees in agricultural landscapes with changing patterns of flower resources. Agric. Ecosyst. Environ. 2020;291 [Google Scholar]
- 4.Woodcock B.A. Neonicotinoid residues in UK honey despite European Union moratorium. PLOS ONE. 2018;13 doi: 10.1371/journal.pone.0189681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becher M.A. BEEHAVE: a systems model of honeybee colony dynamics and foraging to explore multifactorial causes of colony failure. J. Appl. Ecol. 2014;51:470–482. doi: 10.1111/1365-2664.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodcock B.A. Spill-over of pest control and pollination services into arable crops. Agric. Ecosyst. Environ. 2016;231:15–23. [Google Scholar]
- 7.Herbertsson L. Competition between managed honeybees and wild bumblebees depends on landscape context. Basic Appl. Ecol. 2016;17:609–616. [Google Scholar]
- 8.Downing E., Sutherland N. The House of Commons Library; London, UK: 2017. Commons Library Debate Pack - The UK bee Population. [Google Scholar]
- 9.Smart M.D. A comparison of honey bee-collected pollen from working agricultural lands using light microscopy and ITS metabarcoding. Environ. Entomol. 2017;46:38–49. doi: 10.1093/ee/nvw159. [DOI] [PubMed] [Google Scholar]
- 10.Woodcock B.A. Country-specific effects of neonicotinoid pesticides on honeybees and wild bees. Science. 2017;356:1393–1395. doi: 10.1126/science.aaa1190. [DOI] [PubMed] [Google Scholar]
- 11.Soares S. Improving DNA isolation from honey for the botanical origin identification. Food Cont. 2015;48:130–136. [Google Scholar]
- 12.Torricelli M. In-house Validation of a DNA extraction protocol from honey and bee pollen and analysis in fast real-time PCR of commercial honey samples using a knowledge-based approach. Food Anal. Method. 2016;9:3439–3450. [Google Scholar]
- 13.de Vere N. Using DNA metabarcoding to investigate honey bee foraging reveals limited flower use despite high floral availability. Sci. Rep. 2017;7:42838. doi: 10.1038/srep42838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones G.D., Bryant J.V.M. The use of ETOH for the dilution of honey. Grana. 2004;43:174–182. [Google Scholar]
- 15.Lutier P.M., Vaissière B.E. An improved method for pollen analysis of honey. Rev. Palaeobot. Palynol. 1993;78:129–144. [Google Scholar]
- 16.Green O.R. A Manual of Practical Laboratory and Field Techniques in Palaeobiology. Springer; Netherlands, Dordrecht: 2001. Extraction techniques for palaeobotanical and palynological material; pp. 256–287. Green, O.R. [Google Scholar]
- 17.Lieux M.H. Acetolysis applied to microscopical honey analysis. Grana. 1980;19:57–61. [Google Scholar]
- 18.Urban M.A. Nested cell strainers: an alternative method of preparing palynomorphs and charcoal. Rev. Palaeobot. Palynol. 2018;253:101–109. [Google Scholar]
- 19.Traverse A. Springer; GermanyNetherlands: 2007. Paleopalynology. [Google Scholar]
- 20.Kozich J. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sickel W. Increased efficiency in identifying mixed pollen samples by meta-barcoding with a dual-indexing approach. Bmc Ecol. 2015;15:20. doi: 10.1186/s12898-015-0051-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callahan B.J. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bengtsson-Palme J. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Method Ecol. Evol. 2013;4:914–919. [Google Scholar]
- 24.Wang Q. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rognes T. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Core Development Team . R Foundation for Statistical Computing; Vienna, Austria: 2020. R: A language and Environment for Statistical Computing. [Google Scholar]
- 27.McMurdie P.J., Holmes S. Phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census Data. PLOS ONE. 2013;8:61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oksanen J., et al., vegan: Community Ecology Package. R package version 2.56 (2019).