

Abstract
Pneumonia is the leading cause of morbidity and mortality in children worldwide. The ten valent pneumococcal vaccine (PCV10) was introduced in Pakistan's Expanded Program on Immunization (EPI) in 2012 as a 3 + 0 schedule without catchup immunization. Nasopharyngeal carriage is taken as a surrogate marker to measure the impact of pneumococcal vaccine on populations. Carriage surveys are necessary to monitor the persistence of Vaccine Type (VT) serotypes, the emergence of Non-Vaccine Type (NVT) serotypes, and their role in both transmission and disease. This article describes various troubleshooting measures which we undertook to adopt the protocol to our setting. We also used an innovative approach to describe various epidemiological parameters of vaccine effectiveness against carriage. It is important to publish these methods to allow for valid regional and temporal comparisons of results in different settings. Thus, in this article, we describe the following methods for isolating upper airway pneumococcal carriage:
-
•
Methods for the collection, transport, and storage of nasopharyngeal samples.
-
•
Methods for identification and serotyping of pneumococci.
-
•
Methods for estimation of the direct and indirect effects of pneumococcal vaccines on nasopharyngeal carriage.
Keywords: Streptococcus pneumoniae, Pneumococcal vaccines, Herd immunity, Nasopharyngeal carriage, Surveillance, Pakistan
Graphical abstract

Specifications Table
| Subject Area | Immunology and Microbiology |
| More specific subject area | Vaccine impact and serotype replacement |
| Method name | Detection of upper respiratory carriage of Streptococcus pneumoniae |
| Name and reference of original method | Satzke et al. Standard method for detecting upper respiratory carriage of Streptococcus pneumoniae: Updated recommendations from the World Health Organization Pneumococcal Carriage Working Group. Vaccine [1]. |
| Resource availability | n/a |
Method details
Study setting
Our study site was Matiari, which is a district in southern province of Sindh and has a total population of 0.7 million residing in more than 90,000 households across 1600 villages (2018 census). It is located around 182 km away from the Aga Khan University Hospital in Karachi. This study was carried out in two union councils (smallest administrative units), Khyber and Shah Alam Shah Jee Wasi, in the rural part of Matiari which had a cumulative population of around 88,739 with 15% of children under the age of 2 years (Fig. 1).
Fig. 1.

Map showing study area comprising of UCs Seekhat and Shah Alam.
Study design and participants
A time series/serial cross-sectional study was carried out from October 2014 to September 2018. A continuously updated line listing of all children under the age of 2 years formed the sampling frame of the study from which 15 children were randomly selected every week and enrolled after obtaining written informed consent from the guardian. Children with nose and throat abnormalities or with a serious illness requiring hospitalization were excluded from the study. Each child was enrolled only once. At the time of enrollment, a nasopharyngeal specimens was collected. Data on household demographics, recent clinical history including hospitalization and outpatient visits, exposure to household smoke and indoor air pollution was collected by study personnel on smartphones. A brief clinical exam including measurement of fever, respiratory rate, and observation for chest wall indrawing was also done. Data on vaccination was recorded based on verbal recall or vaccination cards where available. In case vaccination card was unavailable, the mothers were probed for PCV-10 vaccination by confirming the age at which their child got vaccinated and site where it was administered. It is a common practice by vaccinators that PCV-10 is usually injected in left leg and the pentavalent vaccine in the right leg. The Expanded Program on Immunization by Government of Pakistan offered a 3 + 0 schedule with doses at 6, 10 and 14 weeks of age.
We performed a two monthly household surveillance to enumerate all births in the area. Thus, a line listing of all children eligible to receive the vaccine was available. To promote vaccine uptake in the community, we contacted the primary caretakers either on mobile phones or by making a physical visit on the due dates of the vaccination to encourage taking their children for vaccination. Transport was provided where needed. We identified low coverage areas and hired a social mobilization team to sensitize the communities to timely vaccinations. Vaccination in these communities was either through fixed centers where Lady Health Workers (LHWs) vaccinated the children coming to these centers or through outreach services by a male vaccinator in the uncovered areas (Fig. 2).
Fig. 2.

Collection of swab from nasopharynx.
Laboratory methods
Materials and instruments for nasopharyngeal swab collection
-
•
Nasopharyngeal Swabs (NP Swabs) Single with Ultrafine twisted wire, MW173P Transwab® Pernasal Amies Plain
-
•
Skim-milk Tryptone Glucose Glycerine (STGG) liquid medium,
-
a.
Tryptone soya broth, Cat # CM#0129b
-
b.
D-(+) – glucose Cat # g8270
-
c.
Skim milk powder, LP0031 BD-DIFCO REF: 232100
-
d.
Glycerol (btl/1ltr) BDH LAB, Cat # 1.04091.1000
-
•
Latex gloves
-
•
Disposal box
-
•
Gauge pieces
-
•
Red bags for waste disposal
Procedure
Nasopharyngeal specimens were collected by trained staff as per the established the World Health Organization's (WHO) consensus methods for the collection and transport of specimens [1].
-
1.
The infant or young child was held securely preferably in a lying position with head tipped slightly backwards and the study worker gently inserted the flocked swab into one of the nostrils directly backwards (not upwards), along the floor of the nasal passage until it reached the posterior wall of the nasopharynx located around two-thirds the distance from the nostril to the ear lobe.
-
2.
We gently rotated the swab for 5–10 s to loosen the epithelial cells. In cases where an obstruction was encountered, we took the swab from the other nostril. Care was taken to avoid touching the tongue and the cheeks with the swab or hand.
-
3.
We removed the swab slowly and placed into the viral transport media by inserting it at least ½ inch below the surface of the media. We clipped the swab handle to fit the transport medium tube and reattached the cap securely.
-
4.
We labelled the specimen appropriately, placed it in biohazard bag with completed requisition, and sent it to the laboratory.
Transport of NP swabs
-
1.
The NP samples collected from the Matiari field site were sent to the Pediatrics Infectious Disease Research Lab (IDRL) within 8 h in a cool box (Coleman) at 2–8 °C along with the sample receiving form with complete patient and specimen information.
-
2.
On receiving the cool box (Coleman) at the IDRL Lab, we recorded the cold chain temperature, the specimen information on the sample tube and the sample receiving form in the lab book.
Materials used for pneumococcal culture and identification
-
1.
Todd Hewitt broth supplemented with 1% yeast extract (THY), Oxoid ™ CM0189.
-
2.
Rabbit serum: Gibco Cat # 16120-107 Gibco.
-
3.
5 µg gentamycin plate: Cat #: Blood Agar Base Cat # CM0271, Sheep Blood in-house, Gentamycin Sulphate salt: Sigma cat # G3632-1 G.
-
4.
10 µg gentamycin plate: Cat #: Blood Agar Base Cat # CM0271, Sheep Blood in-house, Gentamycin Sulphate salt: Sigma cat # G3632-1 G.
-
5.
Sheep Blood Agar (SBA), Oxoid ™ code CAT # CM 0271B.
-
6.
Optochin disk, Cat #: 5 µg Oxoid Code: DD-0001T.
-
7.
Blood Colistin Nalidixic Acid (BCNA) agar, Blood Agar Base Cat # CM0271, Nalidixic and Colistin: Cat # Oxoid SR70E, Sheep Blood in-house.
Procedure for performing a pneumococcal culture
-
1.
We vortexed the specimen at high speed for 10–20 s to disperse organisms from the swab tip.
-
2.
We then transferred 200 µl of the vortexed NP‐STGG sample to 5 ml of Todd Hewitt broth supplemented with 1% yeast extract (THY), Oxoid ™ CM0189 and 1 ml of rabbit serum.
-
3.
The remaining NP-STGG was aliquoted into two sterile cryovials, which were labelled with study ID and immediately frozen in upright position at ‐80 °C.
-
4.
We vortexed and incubated the THY at 37 °C in a 5% CO2 incubator for 6 h.
-
5.
After incubation, we vortexed the sample again and inoculated one loop (10 µl) of the THY on each of the following media: (a) Sheep Blood Agar (SBA), Ooid ™ code CAT # CM 0271B with Optochin disk, (b) Blood Colistin Nalidixic Acid (BCNA) agar, (c) 5 µg gentamycin, Remel ™ Blood Agar w/ Gentamicin and (d) 10 µg gentamycin, Remel ™ Blood Agar w/ Gentamicin. We incubated the plates at 37 °C in a 5% CO2‐incubator for 18–24 h.
Trouble shooting
Since isolation of pneumococcus in the presence of other gram-negative rods can sometimes be suppressed, we used multiple media for isolation. Growth on anyone was considered positive. In case of growth on multiple media, selection for next step was preferred in following order; SBA with Optochin, BCNA, 5 µg gentamycin, 10 µg gentamycin
Pneumococcal isolate detection and identification
-
1.
After incubation, we carefully examined the growth plates for typical small, grayish, and moist pneumococcal colonies, surrounded by a greenish zone of alpha‐hemolysis.
-
2.
We transferred suspected pneumococcal colonies (halo diameter >14 mm) to fresh SBA (with Optochin 5 ug disk) and incubated them for 18–24 h at 37 °C in a 5% CO2‐incubator.
-
3.
We then isolated pure pneumococcal colonies (halo diameter >14 mm), by gently scraping the growth using a sterile disposable loop into a labelled cryotube containing 1.0 ml of STGG medium. The cryotubes were stored immediately at ‐80 °C for pneumococcal serotyping using molecular techniques.
Troubleshooting
We performed a bile solubility test for S. pneumoniae isolates which were optochin resistant or with a smaller zone of inhibition (halo diameter of <14 mm) to complete our identification tests. (Fig. 3)
Fig. 3.

Bile solubility test- turbid (left tube) is negative. Clear (right tube) is positive.
Procedure for performing bile solubility tests
-
1.
We prepared a heavy suspension of pure culture by allowing the broth culture to age in the incubator for 18–24 h in a glass test tube containing 1 ml of 0.5% saline. We adjusted the turbidity of the uniform suspension to that of no. 1 McFarland standard: approximate cell count density = 3 × 10^8 cells) and divided it in two tubes (test and control) of 0.5 ml each.
-
2.
We added 0.5 ml of 2% sodium deoxycholate (bile salts) Oxoid ™ Cat # LP 0055 to the test tube and 0.5 ml of normal saline to the control tube. We vortexed both the tubes and incubated them in a 5% CO2‐incubator at 35 to 37 °C for up to 2 h.
-
3.
We observed a clearing of turbidity in the S. pneumoniae containing test tube compared to the control tube, while any other alpha‐hemolytic streptococci test tube remained turbid even after the 2 h incubation.
Troubleshooting
A partial clearing (partial solubility) was not considered positive for pneumococcal identification. Partially soluble strains with optochin zones of inhibition <14 mm was not considered pneumococci.
Antimicrobial susceptibility testing
We received around 15 samples from the field site every week. The samples were cultured and only the ones positive for pneumococcus were tested further for antibiotic sensitivity. Antimicrobial Susceptibility Testing was performed by using the Kirby–Bauer Method as per Clinical laboratory Standard Institute (CLSI) Guidelines.
Quality control (QC)
-
1.
Optochin Disc QC: Each new lot of optochin disks were tested with positive and negative controls. The growth of S. pneumoniae strain ATCC 49619 was inhibited by optochin and the growth of Streptococcus mitis ATCC 49456 was not inhibited by optochin.
-
2.
STGG Sterility: We checked the sterility of the STGG medium by plating a loop full of a homogenized vial from each lot onto SBA and incubating the agar plate at 37 °C for 24 h. The presence of any growth on the plates was considered a failed sterility check and the entire lot was discarded.
-
3.
Todd Hewitt broth Sterility: We confirmed the sterility of the (THY) medium by plating a full loop of a homogenized vial from each lot onto SBA and incubating the agar plate at 37 °C for 24 h. The presence of any growth on the plates was considered a failed sterility check and the entire lot was discarded.
-
4.
Agar Plate Media Sterility: We randomly chose freshly prepared uninoculated plates and incubated them at 37 °C for 24 h for a sterility check. The presence of any growth on the plates was considered a failed sterility check and the entire lot was discarded.
-
5.
Bile Solubility test QC: Each new lot of sodium deoxycholate was tested with positive and negative QC strains. S. pneumoniae strain ATCC 49619 was the positive control and S. mitis strain ATCC 49456 was used as the negative control.
DNA extraction of streptococcus pneumoniae from isolates
Equipment used:
-
•
Micro centrifuge with refrigerating function (Hitachi, Chiyoda, Tokyo, Japan).
-
•
Biosafety Cabinet Class II type A2 (Stringer Baker, Sanford, Maine, USA).
-
•
Heating Block (VWR Digital Heat block, Radnor, Pennsylvania, USA).
-
•
Vortex mixer (LW Scientific, Lawrenceville, GA, USA).
-
•
−20 °C Freezer (Revco/ Thermo Electron Corporation, Waltham, Massachusetts, USA).
Consumables:
-
•
1.5 ml micro centrifuge tubes (sterile, DNase free, or PCR grade) (Thermo Scientific, Waltham, Massachusetts, USA).
-
•
70% ethanol (Merck Millipore, Burlington, Massachusetts, USA).
-
•
1 set of micropipettes (1–10 µl, 2–20 µl, 20–200 µl, and 100–1000 µl) (Gilson, Middleton, Wisconsin, USA).
-
•
Pre-sterilized filter tips (10 µl, 200 µl, and 1000 µl) (Ambion, Austin, Texas, USA).
Reagents:
TE Buffer (10 mM TrisHCl, pH 8.0, 1 mM EDTA) (Invitrogen, Carlsbad, California, USA)
We used crude boiling method for extraction of DNA. The biosafety cabinet was wiped with 70% ethanol and exposed to UV light at least for 15 min prior to use. We turned on the dry block heater and set at 100 °C. We then dispensed 1000 µl of TE Buffer into a 1.5 ml microcentrifuge tube. We took a loopful of pneumococcal colonies from the culture plates and emulsified the inoculum into the 1.5 ml tube, containing TE Buffer. The contents of the tube were vortexed for 10–20 s. Then we placed the tube on the dry block heater for10 min at 100 °C. After boiling, we centrifuged the tube at 10,000 rpm for 10 min. We carefully removed the supernatant without disturbing the pellet and dispensed it in a pre-labelled 1.5 ml fresh microcentrifuge tube for storage at −20 °C. A negative control containing only TE Buffer was also extracted without addition of any colonies following the same protocol. This served as Extraction Negative Control (ENC).
Sequential multiplex PCR for streptococcus pneumoniae sero-grouping /serotyping
Equipment used:
-
•
PCR thermo cycler (Eppendorf Master cycler Gradient, Hamburg, Germany).
-
•
General purpose PCR UV cabinet workstation (Grant Bio, Chelmsford, UK).
-
•
−20 °C Freezer (Revco/ Thermo Electron Corp, Waltham, Massachusetts, USA).
-
•
Heating Block (VWR Digital Heat block, Radnor, Pennsylvania, USA).
-
•
Vortex mixer (LW Scientific, Atlanta, Georgia, USA).
-
•
Minicentrifuge (Apollo Instrumentation, Roodepoort, South Africa).
-
•
Electrophoresis tank (Thermo Scientific A2, Waltham, Massachusetts, USA).
-
•
Power supply (Biometra, Gottingen, Germany).
-
•
Microwave oven (Dawlance Combi Grill, Karachi, Pakistan).
-
•
GEL-DOC XR Molecular imager with image lab software (Biorad Universal Hood II, Hercules, California, USA).
Consumables:
-
•
70% ethanol (Merck Millipore CAS 64-17-5, Burlington, Massachusetts, USA).
-
•
1.5 mL microcentrifuge tubes (sterile DNase free or PCR grade) (Thermo Scientific, Waltham, Massachusetts, USA).
-
•
Nuclease & pyrogen free, clear, non-sterile 0.2 ml PCR 8-tube strips with strip caps (Mbp inc, Toronto, Canada).
-
•
1 set of micropipettes (1–10 µl, 2–20 µl, 20–200 µl, and 100–1000 µl) (Gilson, Middleton, Wisconsin, USA).
-
•
Pre-sterilized filter tips (10 µl, 200 µl, and 1000 µl) (Ambion, Austin, Texas, USA).
-
•
Ice bucket/bench top cooler.
Reagents:
-
•
Sequential Multiplex Primers sets (forward and reverse) for PCR reactions 1–8 (Eurofins MWG operon, Luxembourg, Europe).
-
•
Qiagen Multiplex Kit (Qiagen cat # 206145, Hilden, Germany).
-
•
Positive control DNA extract (Serotype specific positive strains received from CDC).
-
•
PCR grade water (Invitrogen, Carlsbad, California, USA).
-
•
Agarose powder (OmniPur Agarose - CAS 9012-36-6- Calbiochem, San Diego, CA, USA).
-
•
100 bp DNA ladder (Promega G8291, Madison, Wisconsin, USA).
-
•
6x DNA loading dye (SIGMA, G-7654, Munich, Germany).
-
•
Sybr Green I Dye (SIGMA, S9430, Munich, Germany).
-
•
Trizma Base (SIGMA 1 kg, T6066, Munich, Germany).
-
•
EDTA (SIGMA E5134–500 G, Munich, Germany).
-
•
Glacial Acetic Acid (Merck Millipore CAS 64–19–7, Burlington, Massachusetts, USA).
We prepared following reagent stock / working solutions
-
(1)
EDTA, 0.5 M, pH 8.0 (100 ml): We dissolved 186 g EDTA in 800 ml of ddH2O and adjusted its pH to 8.0 with 10 M NaOH (~5 ml). We added ddH2O to 100 ml of this solution and mixed it well. The prepared solution was then autoclaved and stored at room temperature.
-
(2)
Sybr Green I staining solution: We mixed 120 µl of Sybr Green I dye in 1000 ml of 1X TAE buffer and stored it at 4°C in a dark colored (amber) bottle.
-
(3)
TAE (Tris/acetate/EDTA) electrophoresis buffer, 50 X stock solution: We dissolved 242 g Trizma base into 500 ml of ddH2O, followed by the addition of 57.1 ml of glacial acetic acid and 100 ml of 0.5 M EDTA pH 8.0 to 750 ml of ddH2O. We adjusted its pH to 8.0 and added ddH2O to make up the volume up to 1000 ml. It was mixed well on a stir plate, autoclaved, and stored at room temperature. We diluted the TAE stock solution 1X in H2O before use.
-
(4)
Agarose gel (2.5%): We added 2 g of electrophoresis-grade agarose to 100 ml of 1X TAE buffer in a 250 ml flask or bottle. We heated the solution in a microwave until clear and cooled it at room temperature for 3 to 5 min before pouring in the casting tray to solidify.
-
(5)
Primer working stock solution: We adjusted the primer working solution to 25 µM with PCR grade water from stock solution of 200 µM.
We deduced serotypes using the published sequential multiplex PCR assay and performed further confirmation by monoplex PCR [2,3].
-
1.
Prior to beginning the PCR, we filled the plate template worksheet, turned on heating block and set it at 56 °C.
-
2.
We wiped the PCR workstation with 70% ethanol and exposed to UV light at least for 15 min prior to use.
-
3.
DNA extracts and positive controls stored at −20 °C were allowed to thaw at room temperature. Once thawed they were placed on 56 °C for 10 to 15 min.
-
4.
In the PCR reaction assembly area, we gathered the reagents needed for the PCR reactions, including PCR master mix, primers, and PCR grade water. If the reagents were stored at −20 °C, we allowed them to thaw completely and vortexed/flicked and quick spin before use.
-
5.
Once thawed all the reagents were kept on ice or in bench top cooler. Sequential multiplex PCR reactions, based on one of the described schemes, were prepared in standard 25 µl reaction volumes in sterile 1.5 ml microcentrifuge tubes keeping the tubes on ice using primers and concentrations described below:
-
6.
We pipetted 23 µl of this master mix into each appropriate well of 96-well plate or individual tubes placed on cooler or ice according to plate template worksheet.
-
7.
We added 2 µl of template DNA to each appropriate well in a separate area away from PCR workstation that was designated for addition of DNA template specifically.
-
8.
We set up one extraction negative control, one PCR negative control and 4,5 serotype specific positive controls (depending on the multiplex PCR reaction) for all PCR assays.
-
9.
For the PCR Negative control: we added 2 µl of PCR grade water to a reaction well instead of DNA template.
-
10.
For Extraction Negative control (ENC): we added 2 µl of ENC that was extracted during boiling method to a reaction well instead of DNA template.
-
11.
For Positive control: we added 2 µl of DNA template that is known to contain the amplified sequence to a reaction well.
-
12.
The plate was quickly spun at 1000 rpm to bring down any droplets.
-
13.
The thermo cycler was programmed to the conditions described in the table below:
Tables 3–10. Protocol for Multiplex PCR Reaction.
Table 1.
Reagents used for master mix.
| Reagents: | Volume (µl) |
|---|---|
| PCR Grade Water | Varies |
| 2X PCR Buffer-Qiagen Multiplex PCR Kit (cat# 206145) | 12.5 |
| Forward Primer | Vary (refer tables below for PCR reactions 1–8) |
| Reverse Primer | Vary (refer tables below for PCR reactions 1–8) |
| Qiagen Q solution (cat # 206145), | 2.5 |
| Total Master Mix | 23 |
| DNA Template | 2 |
Table 2.
Thermal cycling parameter.
| (a)1x | 95 °C for 15 min |
| (b) 35x | 94 °C for 30 s |
| 54 °C for 90 s | |
| 72 °C for 60 s | |
| (c)1x | 72 °C for 10 min |
| (d) Hold | 4 °C |
Table 4.
Multiplex PCR Reaction 2.
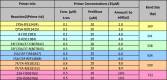 |
Table 5.
Multiplex PCR Reaction 3.
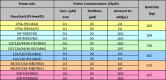 |
Table 6.
Multiplex PCR Reaction 4.
 |
Table 7.
Multiplex PCR Reaction 5.
 |
Table 8.
Multiplex PCR Reaction 6.
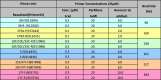 |
Table 9.
Multiplex PCR Reaction 7.
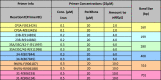 |
Table 11.
List of primers and working stock concentration used.
| Target | Primer orProbeName | Real-time Primers and ProbesNucleotide Sequence (5′ to 3′) | Working Stock Concentration (µM) |
|---|---|---|---|
| S. pneumoniaelytA | F373 (Forward Primer) | ACGCAATCTAGCAGATGAAGCA | 10 µM |
| R424 (Reverse Primer) | TCGTGCGTTTTAATTCCAGCT | 10 µM | |
| Pb400 (Probe) | 5′- d FAM-TGCCGAAAACGCTTGATACAGGGAG-BHQ-13′ | 5 µM |
Table 12.
List of reagents used.
| Reagents | Volume per reaction (µl) |
|---|---|
| PCR Grade Water |
|
|
|
|
|
|
|
|
|
|
|
|
|
Table 13.
Thermal cycling parameters.
| Thermal Cycling Parameters | |
|---|---|
| 1 X | 50 °C for 2 min |
| 1 X | 95 °C for 10 min |
| 50 X | 95 °C for 15 s |
| 60 °C for 60 s | |
Table 14.
List of Oligonucleotide primers used for pneumococcal serotype deduction by conventional multiplex PCR.
| S.No | Description |
|---|---|
| 1 | PRIMERS S.Pneumo CPSA-FWD. |
| GCA GTA CAG TTT GTT GGA CTG ACC | |
| 2 | PRIMERS S.Pneumo CPSA-REV. |
| GAA TAT TTT CAT TAT CAG TCC CAG TC | |
| 3 | PRIMERS S.Pneumo 1-FWD. |
| CTC TAT AGA ATG GAG TAT ATA AAC TAT GGT TA | |
| 4 | PRIMERS S.Pneumo 1-REV. |
| CCA AAG AAA ATA CTA ACA TTA TCA CAA TAT TGG C | |
| 5 | PRIMERS S.Pneumo 2-FWD. |
| TAT CCC AGT TCA ATA TTT CTC CAC TAC ACC | |
| 6 | PRIMERS S.Pneumo 2-REV. |
| ACA CAA AAT ATA GGC AGA GAG AGA CTA CT | |
| 7 | PRIMERS S.Pneumo 3-FWD. |
| ATG GTG TGA TTT CTC CTA GAT TGG AAA GTA G | |
| 8 | PRIMERS S.Pneumo 3-REV. |
| CTT CTC CAA TTG CTT ACC AAG TGC AAT AAC G | |
| 9 | PRIMERS S.Pneumo 4-FWD. |
| CTG TTA CTT GTT CTG GAC TCT CGA TAA TTG G | |
| 10 | PRIMERS S.Pneumo 4-REV. |
| GCC CAC TCC TGT TAA AAT CCT ACC CGC ATT G | |
| 11 | PRIMERS S.Pneumo 5-FWD. |
| ATA CCT ACA CAA CTT CTG ATT ATG CCT TTG TG | |
| 12 | PRIMERS S.Pneumo 5-REV. |
| GCT CGA TAA ACA TAA TCA ATA TTT GAA AAA GTA TG | |
| 13 | PRIMERS S.Pneumo 6A/B/C/D-FWD. |
| AAT TTG TAT TTT ATT CAT GCC TAT ATC TGG | |
| 14 | PRIMERS S.Pneumo 6A/B/C/D-REV. |
| TTA GCG GAG ATA ATT TAA AAT GAT GAC TA | |
| 15 | PRIMERS S.Pneumo 6C/6D-FWD. |
| CAT TTT AGT GAA GTT GGC GGT GGA GTT | |
| 16 | PRIMERS S.Pneumo 6C/6D-REV. |
| AGC TTC GAA GCC CAT ACT CTT CAA TTA | |
| 17 | PRIMERS S.Pneumo 7C/B/40-FWD. |
| CTA TCT CAG TCA TCT ATT GTT AAA GTT TAC GAC GGG A | |
| 18 | PRIMERS S.Pneumo 7C/B/40-REV. |
| GAA CAT AGA TGT TGA GAC ATC TTT TGT AAT TTC | |
| 19 | PRIMERS S.Pneumo 7F/A-FWD. |
| TCC AAA CTA TTA CAG TGG GAA TTA CGG | |
| 20 | PRIMERS S.Pneumo 7F/A-REV. |
| ATA GGA ATT GAG ATT GCC AAA GCG AC | |
| 21 | PRIMERS S.Pneumo 8-FWD. |
| GAA ACG AAA CTG TCA GAG CAT TTA CAT | |
| 22 | PRIMERS S.Pneumo 8-REV. |
| CTA TAG ATA CTA GTA GAG CTG TTC TAG TCT | |
| 23 | PRIMERS S.Pneumo 9 N/L-FWD. |
| GAA CTG AAT AAG TCA GAT TTA ATC AGC | |
| 24 | PRIMERS S.Pneumo 9 N/L-REV. |
| ACC AAG ATC TGA CGG GCT AAT CAA T | |
| 25 | PRIMERS S.Pneumo 9 V/A-FWD. |
| GGG TTC AAA G TC AGA CAG TG A ATC TTA A | |
| 26 | PRIMERS S.Pneumo 9 V/A-REV. |
| CCA TGA ATG A AA TCA ACA TT G TCA GTA GC | |
| 27 | PRIMERS S.Pneumo 10A-FWD. |
| GGT GTA GAT TTA CCA TTA GTG TCG GCA GAC | |
| 28 | PRIMERS S.Pneumo 10A-REV. |
| GAA TTT CTT TAA GAT TCG GAT ATT TCT C | |
| 29 | PRIMERS S.Pneumo 10F/C/33C-FWD. |
| GGA GTT TAT CGG TAG TGC TCA TTT TAG CA | |
| 30 | PRIMERS S.Pneumo 10F/C/33C-REV. |
| CTA ACA AAT TCG CAA CAC GAG GCA ACA | |
| 31 | PRIMERS S.Pneumo 11A/D-FWD. |
| GGA CAT GTT CAG GTG ATT TCC CAA TAT AGT G | |
| 32 | PRIMERS S.Pneumo 11A/D-REV. |
| GAT TAT GAG TGT AAT TTA TTC CAA CTT CTC CC | |
| 33 | PRIMERS S.Pneumo 12F/A/44/46-FWD. |
| GCA ACA AAC GGC GTG AAA GTA GTT G | |
| 34 | PRIMERS S.Pneumo 12F/A/44/46-REV. |
| CAA GAT GAA TAT CAC TAC CAA TAA CAA AAC | |
| 35 | PRIMERS S.Pneumo 13-FWD. |
| TAC TAA GGT AAT CTC TGG AAA TCG AAA GG | |
| 36 | PRIMERS S.Pneumo 13-REV. |
| CTC ATG CAT TTT ATT AAC CG C TTT TTG TTC | |
| 37 | PRIMERS S.Pneumo 14-FWD. |
| GAA ATG TTA CTT GGC GCA GGT GTC AGA ATT | |
| 38 | PRIMERS S.Pneumo 14-REV. |
| GCC AAT ACT TCT TAG TCT CTC AGA TGA AT | |
| 39 | PRIMERS S.Pneumo 15A/F-FWD. |
| ATT AGT ACA GCT GGA ATA TCT CTT C | |
| 40 | PRIMERS S.Pneumo 15A/F-REV. |
| GAT CTA GTG AAC GTA CTA TTC CAA AC | |
| 41 | PRIMERS S.Pneumo 15B/C-FWD. |
| TTG GAA TTT AAT TAG TGG CTT ACC TA | |
| 42 | PRIMERS S.Pneumo 15B/C-REV. |
| CAT CCG CTT ATT AAT TGA AGT AAT CTG AAC C | |
| 43 | PRIMERS S.Pneumo 16F-FWD. |
| GAA TTT TTC AGG CGT GGG TGT TAA AAG | |
| 44 | PRIMERS S.Pneumo 16F-REV. |
| CAG CAT ATA GCA CCG CTA AGC AAA TA | |
| 45 | PRIMERS S.Pneumo 17F-FWD. |
| TTC GTG ATG ATA ATT CCA ATG ATC AAA CAA GAG | |
| 46 | PRIMERS S.Pneumo 17F-REV. |
| GAT GTA ACA AAT TTG TAG CGA CTA AGG TCT GC | |
| 47 | PRIMERS S.Pneumo 18A/B/C/F-FWD. |
| CTT AAT AGC TCT CAT TAT TCT TTT AAG CC | |
| 48 | PRIMERS S.Pneumo 18A/B/C/F-REV. |
| TTA TCT GTA AAC CAT ATC AGC ATC TGA AAC | |
| 49 | PRIMERS S.Pneumo 19A-FWD. |
| GAG AGA TTC ATA ATC TTG CAC TTA GCC A | |
| 50 | PRIMERS S.Pneumo 19A-REV. |
| CAT AAT AGC TAC AAA TGA CTC ATC GCC | |
| 51 | PRIMERS S.Pneumo 19F-FWD. |
| GTT AAG ATT GCT GAT CGA TTA ATT GAT ATC C | |
| 52 | PRIMERS S.Pneumo 19F-REV. |
| GTA ATA TGT CTT TAG GGC GTT TAT GGC GAT AG | |
| 53 | PRIMERS S.Pneumo 20-FWD. |
| GAG CAA GAG TTT TTC ACC TGA CAG CGA GAA G | |
| 54 | PRIMERS S.Pneumo 20-REV. |
| CTA AAT TCC TGT AAT TTA GCT AAA ACT CTT ATC | |
| 55 | PRIMERS S.Pneumo 21-FWD. |
| CTA TGG TTA TTT CAA CTC AAT CGT CAC C | |
| 56 | PRIMERS S.Pneumo 21-REV. |
| GGC AAA CTC AGA CAT AGT ATA GCA TAG | |
| 57 | PRIMERS S.Pneumo 22/F/A-FWD. |
| GAG TAT AGC CAG ATT ATG GCA GTT TTA TTG TC | |
| 58 | PRIMERS S.Pneumo 22/F/A-REV. |
| CTC CAG CAC TTG CGC TGG AAA CAA CAG ACA AC | |
| 59 | PRIMERS S.Pneumo 23A-FWD. |
| TAT TCT AGC AAG TGA CGA AGA TGC G | |
| 60 | PRIMERS S.Pneumo 23A-REV. |
| CCA ACA TGC TTA AAA ACG CTG CTT TAC | |
| 61 | PRIMERS S.Pneumo 23B-FWD. |
| CCA CAA TTA G CG CTA TAT TCA TTC AAT CG | |
| 62 | PRIMERS S.Pneumo 23B-REV. |
| GTC CAC GCT GAA TAA AAT GAA GCT CCG | |
| 63 | PRIMERS S.Pneumo 23F-FWD. |
| GTA ACA GTT GCT GTA GAG GGA ATT GGC TTT TC | |
| 64 | PRIMERS S.Pneumo 23F-REV. |
| CAC AAC ACC TAA CAC TCG ATG GCT ATA TGA TTC | |
| 65 | PRIMERS S.Pneumo 24A/B/F-FWD. |
| GCT CCC TGC TAT TGT AAT CTT TAA AGA G | |
| 66 | PRIMERS S.Pneumo 24A/B/F-REV. |
| GTG TCT TTT ATT GAC TTT ATC ATA GGT CGG | |
| 67 | PRIMERS S.Pneumo 31-FWD. |
| GGA AGT TTT CAA GGA TAT GAT AGT GGT GC | |
| 68 | PRIMERS S.Pneumo 31-REV. |
| CCG AAT ATA TTC AAT ATA TTC CTA CTC | |
| 69 | PRIMERS S.Pneumo 33F/A/37-FWD. |
| GAA GGC AAT CAA TGT GAT TGT GTC GCG | |
| 70 | PRIMERS S.Pneumo 33F/A37-REV. |
| CTT CAA AAT GAA GAT TAT AGT ACC CTT CTA C | |
| 71 | PRIMERS S.Pneumo 34-FWD. |
| GCT TTT GTA AGA GGA GAT TAT TTT CAC CCA AC | |
| 72 | PRIMERS S.Pneumo 34-REV. |
| CAA TCC GAC TAA GTC TTC AGT AAA CTT TAC | |
| 73 | PRIMERS S.Pneumo 35A/C/42-FWD. |
| ATT ACG ACT CCT TAT GTG ACG CGC ATA | |
| 74 | PRIMERS S.Pneumo 35A/C/42-REV. |
| CCA ATC CCA AGA TAT ATG CAA CTA GGT T | |
| 75 | PRIMERS S.Pneumo 35B-FWD. |
| GAT AAG TCT GTT GTG GAG ACT TAA AAA GAA TG | |
| 76 | PRIMERS S.Pneumo 35B-REV. |
| CTT TCC AGA TAA TTA CAG GTA TTC CTG AAG CAA G | |
| 77 | PRIMERS S.Pneumo 35F/47F-FWD. |
| GAA CAT AGT CGC TAT TGT ATT TTA TTT AAA GCA A | |
| 78 | PRIMERS S.Pneumo 35F/47F-REV. |
| GAC TAG GAG CAT TAT TCC TAG AGC GAG TAA ACC | |
| 79 | PRIMERS S.Pneumo 38/25F/25A-FWD. |
| CGT TCT TTT ATC TCA CTG TAT AGT ATC TTT ATG | |
| 80 | PRIMERS S.Pneumo 38/25F/25A-REV. |
| ATG TTT GAA TTA AAG CTA ACG TAA CAA TCC | |
| 81 | PRIMERS S.Pneumo 39-FWD. |
| TCA TTG TAT TAA CCC TAT GCT TTA TTG GTG | |
| 82 | PRIMERS S.Pneumo 39-REV. |
| GAG TAT CTC CAT TGT ATT GAA ATC TAC CAA |
Table 3.
Multiplex PCR Reaction 1.
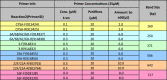 |
Table 10.
Multiplex PCR Reaction 8.
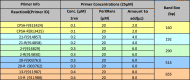 |
Agarose gel electrophoresis
PCR products (10 µl) were run on 2.5% agarose gels to determine band sizes using positive controls. A positive control for each serotype and a 100 bp ladder molecular size marker was included on each gel.
-
1.
We added 1X TAE buffer to the electrophoresis tank and placed the gel cassette containing the solidified 2% agarose gel into the tank.
-
2.
We spun the PCR plate or tubes at 500 x g to ensure all liquid is at the bottom and mixed 10 µl of PCR reaction with 2 µl of 6X loading dye.
-
3.
We pipetted the DNA/loading dye mixture into the wells and loaded 6µl of 100 bp DNA ladder after every 6 isolates.
-
4.
We ran the gel at 90–120 Vs for 30–45 min or until the bromophenol blue dye band was halfway down the gel.
-
5.
We placed the gel in Sybr green staining solution for 15–20 min.
-
6.
We visualized the gel under a UV light and labelled the wells for interpretation.
-
7.
Each reaction gave two bands, i.e., species-specific positive control (cpsA, although some were cpsA negative) and a serotype-specific band.
-
8.
The remainder of the amplicon was stored at −20°C, if necessary.
We matched the band sizes on agarose gels with those of positive controls before assigning a putative serotype. The PCR reaction was setup sequentially, if a strain was negative for any of the serotypes included in reaction 1 then we proceeded to reaction 2 and so forth until a serotype was determined.
In case of positivity of any isolate for particular serotype, a monoplex reaction was run for confirmation. For the monoplex confirmation of serotypes, a protocol similar to multiplex serotyping PCR was followed with 2 µl of the extracted DNA directly used in the PCR reaction. We included the cpsA gene in each monoplex reaction to act as an internal positive control. The PCR master mix consisted of 0.1 ul of 25 µM cpsA primers, 25 uM working stock of serotype specific forward and reverse primers, 12.5 ul of 2X Qiagen multiplex PCR buffer, 2.5 ul Qiagen Q solution (cat # 206145), and nuclease free water for making up the volume up to 25 ul. Amplification was carried out in an Eppendorf Master Cycler Gradient with initial denaturation at 95 °C for 15 min, 35 cycles of 94 °C for 30 s, 54 °C for 90 s, and 72 °C for 60 s, finally at 72 °C for 10 min for final extension.
If no serotype was identified on the sequential multiplex PCRs, then the strain was labelled as not typeable by our sequential multiplex PCR scheme. CpsA negative non typeable were confirmed with a lytA Real time PCR as described by Carvalho MDa et al. [4].
Confirmation of cpsA negative nontypeables by lytA gene real time PCR
We performed lytA real time PCR for the confirmation of S.pneumoniae from nontypeable isolates using TaqMan assay. The assay was based on amplification of the lytA target gene, using primers and TaqMan probe specific for lytA gene of S.pneumoniae. The lytA gene, encoding autolysin, is a single copy gene present in all encapsulated and nontypeable S.pneumoniae.
Equipment:
-
•
Applied Biosystems. 7500/7500 Fast Real-Time PCR System (Thermo Scientific, Waltham, Massachusetts, USA) or,
-
•
QIAGEN Rotor-Gene Q (Corbett Rotor-Gene 6000, Hilden, Germany).
-
•
General purpose PCR UV cabinet workstation (Grant Bio, Chelmsford, UK).
-
•
−20 °C Freezer (Revco/ Thermo Electron Corp, Waltham, Massachusetts, USA).
-
•
Heating Block (VWR Digital Heat block, Radnor, Pennsylvania, USA).
-
•
Minicentrifuge (Apollo Instrumentation, Roodepoort, South Africa).
-
•
Vortex mixer (LW Scientific, Lawrenceville, GA, USA).
Consumables:
-
•
70% ethanol (Merck Millipore CAS 64-17-5, Burlington, Massachusetts, USA).
-
•
1.5 ml microcentrifuge tubes (sterile, DNase free, or PCR grade) (Thermo Scientific, Waltham, Massachusetts, USA).
-
•
MicroAmp™ Optical 8-Tube Strip, 0.2 mL (Cat # 4316567, Thermo Fisher Scientific, Waltham, Massachusetts, USA).
-
•
MicroAmp™ Optical 8-Cap Strips (Catalog number: 4323032, Thermo Fisher Scientific, Waltham, Massachusetts, USA).
-
•
1 set of micropipettes (1–10 µl, 2–20 µl, 20–200 µl, and 100–1000 µl) (Gilson, Middleton, Wisconsin, USA).
-
•
Pre-sterilized filter tips (10 µl, 200 µl, and 1000 µl) (Ambion, Austin, Texas, USA).
-
•
Ice bucket/bench top cooler.
Reagents:
-
•
ABI TaqMan Universal PCR Master Mix 2X (Part No. 4304437, Thermo Fisher Scientific, Waltham, Massachusetts, USA).
-
•
Primers and dual-labelled hydrolysis probes (Eurofins MWG operon, Luxembourg, Europe).
-
•
Positive control DNA extract.
-
•
DNA extracts from nontypeable isolates.
-
•
PCR grade water (Invitrogen, Carlsbad, California, USA).
Note: TaqMan Master Mix was stored at 2–4 °C. Stock and working solutions of primers and probes were stored at −20 °C
Preparation of primers and probe working stocks
We diluted primers and probe stock vial as per manufacturer's instruction and working stocks were set at the concentrations mentioned below in the table.
Real-time PCR protocol
-
1.
We wiped the workplace and PCR hood with 70% ethanol and exposed it to UV light at least for 15 min prior to use.
-
2.
We removed DNA templates and positive control DNA from −20 °C to allow them to thaw completely at room temperature. Once thawed, we placed them on 56 °C for 10 to 15 min.
-
3.
We set up one Non-template control (NTC) and one Positive control for each assay that was run. NTC contained all reagents except for template DNA; instead, 2 ul of sterile, PCR-grade water was added to the NTC reaction wells. Positive control reaction was performed using DNA from known positive isolates.
-
4.
In the PCR reaction assembly area, we gathered reagents needed for the PCR reactions, including PCR master mix, primers, and PCR grade water. We thawed all reagents stored at −20 °C, and vortex/flicked and quick spin each tube before use.
-
5.
For each extraction to be tested, the master mix contained following reagents:
-
6.
We pipetted 23 ul of master mix into each appropriate well of individual optical strip tubes placed on cooler or ice according to the plate template worksheet.
-
7.
We then added 2 ul of template DNA and positive control DNA to each appropriate well.
-
8.
We then spun the PCR strip tubes at 500 x g for a few seconds to bring down any droplets and to mix them well. We transported the strip tubes directly to the amplification area and placed it in the real-time PCR machine.
-
9.
We set the cycling parameters as suggested for the primers and probes according to the table below:
Data analysis and interpretation
We recorded the cycle threshold (Ct) value for each well. The Ct value was set according to the amplification cycle at which the fluorescence of the sample exceeded the threshold value. As with any experiment, results for the unknowns could not be evaluated without first assessing the results for the controls. NTCs gave a value of “No Ct” and produced amplification curves that were straight lines near zero. Positive controls gave appropriate Ct values and amplifications plots of the positive controls and any unknown specimens that generated curves were all sigmoidal. When determining whether to call any nontypeable isolate positive or negative for S.pneumoniae, we set the cutoff value at 30. We considered isolates with Ct ≤ 30 showing smooth and sigmoidal amplification curve as positive for S.pneumoniae but nontypeables. Whereas isolates with Ct > 30 showing curves that were straight lines near zero were reported negative for S.pneumoniae.
Trouble Shooting Guide:
A failed test (i.e., positive control(s) reported to be negative) were due to any of the following reasons:
-
1.
Equipment not working properly
-
2.
Expired reagents
-
3.
Incorrectly made-up primers and probes
-
4.
Incorrect primer sequence
-
5.
Pipetting error
-
6.
Plate not sealed tightly with caps
-
7.
If negative control came up positive, then water may be contaminated or otherwise one or all the reagents may be contaminated. In this situation, we discarded the reagents.
-
8.
In the event of a failed test i.e., positive control yielding no target, the test was repeated with a new control and/or reagent.
If cross contamination was suspected because of the Ct values in known negative controls or NTCs then:
-
(a)
We cleaned the workplace with 70% ethanol.
-
(b)
The reagents used in the assay may have been contaminated. We made new aliquots of the reagents including primers, probes, master mix and water.
-
(c)
We limited the amount of positive control DNA in the plate and made sure the pipettes were sterile.
Resolution of serogroup 6A/6B/6C/6D into serotypes by conventional PCR
Resolution of sero-group 6A/B/C/D into individual serotypes, 6A, 6B, 6C, and 6D was done as per the methods described by Ping et al. with slight modifications [5].
We prepared a 25 µl PCR reaction mix with 1 µl of 25 µM working stocks of forward and reverse primers each, 12.5 ul of 2X Qiagen multiplex PCR buffer, 2.5 ul of Qiagen Q solution (cat # 206145), and 6ul of nuclease free water, with 2 µl of DNA template added to the reaction mixture. We conducted PCR amplification using a Eppendorf Master Cycler Gradient with the following thermal profile: initial denaturation at 95 °C for 15 min, followed by 35 cycles of 94 °C for 30 s, 66.2 °C for 60 s, and 72 °C for 60 s, then a final extension step at 72 °C for 10 min. The amplified PCR products were separated on 2.5% (w/v) agarose gel electrophoresis, stained with 1X SYBR -green staining solution, and documented under a Biorad Gel Doc imager.
Quality control
A positive control for each serotype included in the PCR reaction(s) was run on each gel to ensure that bands are assigned to the correct serotype.
Troubleshooting
For unclear serotyping results, as part of our Internal Quality Assessment, we repeated the assay for confirmation and double checked them with microbiological identification, including bile solubility and optochin sensitivity assays and confirmatory monoplex using CDC controls and negative control extraction.
-
(1)
Statistical analysis
Vaccine Type (VT) carriage was defined as isolation of any of the 10 serotypes included in PCV10 (serotypes 1, 4, 5, 6B, 7F, 9 V, 14, 18C, 19F, 23F). Non-vaccine type (NVT) carriage was defined as presence of all other serotypes. PCV13 serotypes were defined as any of the PCV10 serotypes plus serotypes 3, 6A and 19A. A child was categorized as fully vaccinated if he/she had received all three doses of PCV10 (either card verified or verbal). We described carriage rates by vaccination status and study year. A study year ran from October to the September of next year.
Measures of direct, indirect, total, and overall effects were calculated using a modified Halloran Model described in Fig. 4. Logistic regression analysis was performed to identify predictors of colonization with a PCV10 serotype. All variables with a p-value less than 0.25 in the bivariate analysis were used to build a multivariable model. A backward selection procedure was used to derive a parsimonious model for retaining only variables significant at p-value ≤ 0.05.
Fig. 4.

Sequential multiplex PCR.
Statistical analysis was done using STATA version 15.0.
Direct effect was calculated as 1– (VT carriage rate in children who received all three doses /VT carriage rate in children who received zero dose), indirect effect was defined as 1- (VT carriage rate in children who received zero dose /26.7%), total effect was defined as 1-(VT carriage rate in children who received all three doses/26.7%) and overall effect was defined as 1- (carriage rate in the study population / 26.7%) (Fig. 5).
Fig. 5.

Schematic representation of direct, indirect, total, and overall effect in two populations (modified from Halloran 2011).
Ethics
Ethical approval was obtained from Aga Khan University's Ethical Review Committee (3181-Ped-ERC-14) in 2014.
Supplementary material and/or additional information
n/a.
Declaration of Competimg Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We would like to acknowledge all the children and their parents who agreed to take part in this study. We would also like to acknowledge all the field and lab staff who contributed to the successful implementation of the project.
Footnotes
Direct Submission or Co-Submission
Co-submissions are papers that have been submitted alongside an original research paper accepted for publication by another Elsevier journal.
Co-Submission
Co-Submission with the journal “Vaccine”
Manuscript number JVAC-d-20–01,605. Accepted for publication.
References
- 1.Satzke C., Turner P., Virolainen-Julkunen A., Adrian P.V., Antonio M., Hare K.M. Standard method for detecting upper respiratory carriage of Streptococcus pneumoniae: updated recommendations from the World Health Organization pneumococcal carriage working group. Vaccine. 2013;32:165–179. doi: 10.1016/j.vaccine.2013.08.062. [DOI] [PubMed] [Google Scholar]
- 2.da Gloria Carvalho M., Pimenta F.C., Jackson D., Roundtree A., Ahmad Y., Millar E.V. Revisiting pneumococcal carriage by use of broth enrichment and PCR techniques for enhanced detection of carriage and serotypes. J. Clin. Microbiol. 2010;48:1611–1618. doi: 10.1128/JCM.02243-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pai R., Gertz R.E., Beall B. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J. Clin. Microbiol. 2006;44:124–131. doi: 10.1128/JCM.44.1.124-131.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carvalho Mda G., Tondella M.L., McCaustland K., Weidlich L., McGee L., Mayer L.W. Evaluation and improvement of real-time PCR assays targeting lytA, ply, and psaA genes for detection of pneumococcal DNA. J. Clin. Microbiol. 2007;45:2460–2466. doi: 10.1128/JCM.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin P., Xiao M., Kong F., Oftadeh S., Zhou F., Liu C. Simple, accurate, serotype-specific PCR assay to differentiate Streptococcus pneumoniae serotypes 6A, 6B, and 6C. J. Clin. Microbiol. 2009;47:2470–2474. doi: 10.1128/JCM.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]