

Abstract
Limited information is available about epigenetic mechanisms by which cigarette smoke enhances the initiation and progression of lung cancer. To examine this issue, A549 and Calu-6 lung cancer cells were cultured in normal media with or without tobacco smoke condensate (TSC) under clinically relevant exposure conditions. Ten-day TSC exposure dramatically increased the tumorigenicity of lung cancer cells in nude mice. Microarray and quantitative reverse transcription-PCR (RT-PCR) experiments revealed that this phenomenon coincided with diminished expression of Dickkopf-1 (Dkk-1). Western blot, chromatin immunoprecipitation, methylation-specific PCR, and pyrosequencing experiments showed that repression of Dkk-1 coincided with decreased H4K16Ac, increased H3K27me3, and recruitment of SirT1, EZH2, SUZ12, and Bmi1 without DNA hypermethylation within the Dkk-1 promoter despite prolonged TSCexposures. Removal of TSCfrom culture media resulted in loss of promoter-associated polycomb repressor complexes and reexpression of Dkk-1. siRNA-mediated knockdown of EZH2 and SirT1 partially abrogated TSC-mediated inhibition of Dkk-1 expression. Western blot and quantitative RT-PCR array experiments showed that TSC exposure as well as knockdown of Dkk-1 activated Wnt signaling and significantly up-regulated Wnt5a in lung cancer cells. Knockdown of Dkk-1 recapitulated the dramatic protumorigenic effects of TSC exposure in C alu-6 cells. Despite the transient nature of Dkk-1 repression following TSCexposure in vitro, Dkk-1 remained silenced in tumor xenografts derived from TSC t-reated C alu-6 cells. Collectively, these data provide evidence that cigarette smoke directly engages polycomb machinery to activate a signaling network implicated in maintenance of cancer stem cells.
Introduction
Lung cancer ranks among the most lethal malignancies worldwide, with an incidence exceeding 1,000,000 cases per year; the vast majority of these neoplasms are directly attributable to cigarette smoking. Mounting evidence indicates that aberrant expression/activity of epigenetic regulators of gene expression contributes significantly to pulmonary carcinogenesis (1). For instance, increased DNA methyltransferase expression coincides with hypermethylation of tumor suppressor genes and diminished survival of lung cancer patients (2, 3). Aberrant expression of various histone deacetylases correlates with advanced stage of disease and adverse outcome in these individuals (4, 5).
Recently, polycomb group (PcG) proteins, which regulate stem cell pluripotency, have emerged as critical mediators of aberrant gene silencing in cancer cells (6). Stem cell PcG targets are significantly more likely to exhibit cancer-specific promoter hypermethylation than non-polycomb targets (7). Furthermore, the polycomb-mediated H3K27me3 mark is associated with promoters of all tumor suppressor genes that are hypermethylated in cancer cells (8, 9). The majority of lung cancers exhibit disruption of Rb-mediated regulation of E2F, and a number of PcG genes including EZH2, EED, and SUZ12, which mediate H3K27 trimethylation, are potential E2F targets (10). Overexpression of Bmi1 correlates with methylation-mediated silencing of p16 in lung cancer cells (11). Interestingly, loss of p16 activity in primary epithelial cells results in up-regulation of EZH2 and SUZ12 and targeting of the respective PcG proteins as well as DNA methyltransferases to HOXA9, a gene frequently silenced by promoter hypermethylation in cancer cells (12). The fact that pulmonary squamous cell carcinomas and their precursor lesions exhibit increased expression of Bmi1 and EZH2 (13) suggests that dysregulation of PcG complexes occurs early during lung cancer development.
Whereas the link between cigarette smoking and lung cancer risk is well established, the epigenetic mechanisms by which tobacco smoke initiates and maintains the malignant phenotype of lung cancer cells remain poorly understood. Interestingly, despite the fact that tobacco carcinogens mediate inactivation of numerous tumor suppressor genes and induce expression of synuclein-γ via epigenetic mechanisms lung cancer cells (14, 15), the relevance of cigarette smoking as to overall prognosis of lung cancer patients remains controversial (16, 17). The present study was undertaken to ascertain if cigarette smoke induces epigenetic alterations, which directly enhance the malignant phenotype of lung cancer cells.
Materials and Methods
Cell lines and treatment conditions.
Calu-6, A549, H226, SK-LU-1, H841, SW480, and DLD-1 cancer lines were obtained from American Type Culture Collection. Primary normal human small airway epithelial cells (SAEC) were obtained from Lonza, Inc., and cultured per vendor instructions. Tobacco smoke condensates (TSC) derived from Kentucky Reference 1R4F research blend cigarettes (University of Kentucky) were prepared as described (18) and generously provided by Wanda L. Fields (R.J. Reynolds, Inc., Winston-Salem, NC) at a concentration of 9 puffs, 10 mg tar/mL in DMSO. All cancer lines were maintained in RPMI medium supplemented with 10% FCS, 10 mmol/L of glutamic acid, and penicillin/streptomycin.
For smoke exposure experiments, cells were cultured in 10-cm dishes in appropriate normal media (NM) with or without TSC (0.0005, 0.001, or 0.003 puff/mL). Medium was changed daily with the addition of fresh TSC. Cells were subcultured as necessary. Recombinant human Dickkopf-1 (rhDkk-1) was obtained from R&D Systems and used at a concentration of 300 ng/mL for 90 min according to vendor instructions.
Proliferation assays.
Cells (1 × 105) were plated per well in six-well plates and cultured in NM with or without TSC. Triplicate wells were harvested and counted by trypan blue exclusion techniques.
RNA isolation and microarray analysis.
Total RNA was isolated from A549 or Calu-6 cells cultured for 5 d in NM with or without TSC (0.0005, 0.001, or 0.003 puff/mL) using the RNeasy minikit (Qiagen). Gene expression profiles were analyzed using Gene-Chip Human Genome U133 2.0 plus Arrays (Affymetrix) according to the vendor’s instructions.
Real-time quantitative reverse transcription-PCR.
Real-time quantitative reverse transcription-PCR (RT-PCR) was done as described (19) using Dkk-1, EZH2, SirT1, and β-actin primers obtained from Applied Biosystems.
Methylation-specific PCR and pyrosequencing.
The previously predicted CpG island within the Dkk-1 promoter region (20) was confirmed using the online CpG island search engine.1 Genomic DNA was isolated from TSC-treated or control cells using the Qiagen DNeasy kit. Bisulfite modification of DNA was done using the Qiagen Epifect kit. Bisulfite-modified DNA was amplified using methylation-specific primers and PCR conditions as described (20). PCR products were resolved by 2% agarose gel electrophoresis and visualized using ethidium bromide techniques.
For pyrosequencing, PCR primer sequences (F) 5ʹ-GGGTGAAGAGTGTTAAAGGTT-3ʹ and (R) biotin-5ʹTTGAGTTTTTTTGAGATGATGGTTT3ʹ and sequencing primers 5ʹ-TTTTTTTATGTATATAAA-3ʹ and 5ʹ-TTGTGTTTTTTGTAGTTA-3ʹ were designed to amplify methylated as well as unmethylated bisulfite-modified DNA using the Biotage Assay Design software. Dispensation order pyrosequencing reactions and data analysis were done using the Pyromark MD pyrosequencer with software provided by Biotage. For Wnt RT-PCR arrays, 96-well focused Wnt signaling RT-PCR arrays (PAH-043C) were obtained from SuperArray. One microgram of RNA from control Calu-6 cells, Calu-6 cells cultured in 0.003 puff/mL of TSC, or Calu-6 cells transfected with shRNA targeting Dkk-1 was used to generate cDNA using the iScript RT kit (Bio-Rad). The entire cDNA reaction was diluted and distributed among the 96 wells. Reactions were done with RT2 SYBR Green/ROX PCR Master Mix in an ABI 7900HT Fast Real-Time PCR machine. Data were analyzed using the web-based Superarray portal.2
Immunoblot.
Total cell proteins were extracted using the Cell Signaling lysis buffer (Cell Signaling technology) supplemented with 1× protease inhibitor (Roche, Inc.) and 1 mmol/L phenylmethylsulfonyl fluoride. Cell lysates were resolved on Bis-Tris polyacrylamide gels (Invitrogen), transferred to nitrocellulose membranes, and incubated for 1 h at room temperature with the following antibodies: Dkk-1 and β-actin (Santa Cruz Biotechnology), EZH2 (BD Biosciences), and LRP-6, phospo-LRP-6, Dvl-2, SirT1, phospho-c-jun NH2-terminal kinase (JNK), and JNK (Cell Signaling Technology). Immunoblot signals were detected using appropriate horse-radish peroxidase–conjugated secondary antibodies and SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology). Membranes were stripped and reprobed as necessary using Restore stripping buffer (Pierce).
Chromatin immunoprecipitation.
Chromatin immunoprecipitation was done as previously described (21) with minor modifications. Briefly, DNA-protein complexes were cross-linked with formaldehyde at a final concentration of 1% for 15 min. Immune complexes were formed with either nonspecific IgG or chromatin immunoprecipitation grade antibodies recognizing H3K4me3 (Abcam), H3K9me3, H3K27me3, H4K16Ac, Suz12, Bmi1 (Millipore), EZH2 (BD Biosciences), RNA polymerase II (Santa Cruz Biotechnology), or SirT1 (Cell Signaling Technology and Sigma-Aldrich). DNA was eluted and purified from complexes, followed by PCR amplification of the Dkk-1 promoter using primers and conditions as described (20).
Luciferase promoter-reporter transient transfection experiments.
The T-cell factor responsive vector TOPflash and the mutant negative control vector FOPflash were purchased from Millipore. Cells (1 × 104) were plated per well in 24-well plates and cultured in NM with or without TSC for 5 d, followed by transient transfection of TOPflash or FOPflash vectors using Lipofectin. Forty-eight hours later, cells were lysed and assayed for luciferase activity using the Promega dual luciferase reporter assay according to the manufacturer’s instruction. Renilla luciferase activity was used to normalize intersample variability.
siRNA and shRNA knockdown.
Calu-6 cells were transiently transfected with siRNAs targeting EZH2 and SirT1 or sham siRNA sequences (Ambion) using Oligofectamine (Invitrogen). Target gene knockdown was confirmed by RT-PCR and Western blot techniques. In addition, Calu-6 cells were transfected with shRNA vectors targeting Dkk-1 and sham shRNA constructs (Origene) using Lipofectin (Invitrogen). Stable transfectants were isolated under puromycin (1 µg/mL) selection. Clonal populations exhibiting knockdown of Dkk-1 comparable to TSC-treated Calu-6 cells were expanded, with Dkk-1 knockdown confirmed by RT-PCR and Western blot techniques.
Murine xenograft experiments.
Calu-6 and A549 cells cultured in NM with or without TSC (0.003 puff/mL) for 10 d or Calu-6 cells stably transfected with shRNA targeting Dkk-1 or control vectors were suspended in PBS at a concentration of 1 × 106 cells/100 µL. Athymic nude mice were inoculated s.c. in the left flank with TSC-treated or Dkk-1 knockdown cells and in the right flank with control cells. Four weeks later, mice were euthanized and evaluated for percent tumor take and tumor mass. Xenograft tissues were harvested immediately after CO2 euthanasia. Twenty-five animals in the TSC-treated and 20 animals in the Dkk-1 knockdown cohorts were included in at least two independent experiments. All animal procedures were approved by the National Cancer Institute Animal Care and Use Committee and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Results
TSC enhances the tumorigenicity of lung cancer cells.
A series of experiments were initiated to examine the effects of tobacco smoke on the epigenome and malignant phenotype of Calu-6 and A549 lung cancer cells. These cells were chosen for study due to their marked responses to chromatin remodeling agents suggestive of epigenomic plasticity potentially inducible by environmental carcinogens (22–24) and strikingly different tumorigenic potentials; Calu-6 cells exhibit low tumorigenicity, whereas A549 cells are highly tumorigenic in nude mice when inoculated s.c. in PBS.
Extrapolation of data pertaining to cotinin levels in saliva and systemic circulation of active smokers (25), as well as nicotine content in the condensates, suggested that 0.001 to 0.003 puff/mL approximated exposure conditions in active smokers (~1 pack per day). Preliminary experiments indicated that TSC mediated modest growth inhibitory effects in A549 and Calu-6 cells (Fig. 1A). Interestingly, TSC exposure dramatically increased the tumorigenicity of Calu-6 cells (Fig. 1B). For example, only 24% of mice inoculated with control Calu-6 cells developed tumors; in contrast, 92% of mice inoculated with Calu-6 cells exposed to 0.003 puff/mL of TSC for 10 d before injection developed tumors (P = 0.000001, exact test). Furthermore, tumors arising from TSC-treated Calu-6 cells were significantly larger than those developing from control cells (439.8 ± 329.1 versus 75.2 ± 44.7 mg; P < 0.0001, two-tailed t test). Essentially 100% of mice inoculated with A549 cells developed tumors irrespective of preinjection treatment. Tumors arising from TSC-treated A549 cells tended to be somewhat larger than those developing from untreated cells (364 ± 333 versus 200 ± 329 mg; P = 0.25, two-tailed t test; data not shown).
Figure 1.

Effects of TSC in A549 and Calu-6 lung cancer cells. A, TSC-mediated effects on proliferation of cultured lung cancer cells. B, effects of TSC on the tumorigenicity of Calu-6 cells in nude mice. This effect was less pronounced in A549 cells that normally exhibit high tumorigenicity in nude mice (data not shown). C, quantitative RT-PCR analysis of Dkk-1 expression in lung cancer cell lines following 5-day TSC exposure. D, correlation of Dkk-1 mRNA copy numbers assessed by quantitative RT-PCR (top) and Dkk-1 protein levels in A549 and Calu-6 lung cancer cells detected by Western blot techniques (bottom).
TSC down-regulates Dkk-1 in lung cancer cells.
Affymetrix microarrays were used to examine if gene expression profiles in cultured lung cancer cells coincided with enhanced tumorigenicity following TSC exposure. Using criteria of ≥2-fold up-regulated/repressed relative to controls and P < 0.05, six genes were repressed, whereas 28 genes were up-regulated in a dose-dependent manner in Calu-6cells exposed to TSC (0.0005, 0.001, and 0.003 puff/mL). Using similar criteria, 219 genes were significantly repressed, whereas 111 genes were up-regulated in a dose-dependent manner in A549 cells following TSC exposure.3 Interestingly, Dkk-1, encoding a Wnt signaling antagonist frequently silenced by DNA methylation in cancer cells (20, 26), was down-regulated in a dose-dependent manner in both cell lines. Quantitative RT-PCR experiments (Fig. 1C) confirmed that TSC mediated dose-dependent decreases in Dkk-1 mRNA copy numbers in A549 and Calu-6 cells, which exhibited relatively high Dkk-1 expression. This phenomenon was also observed in several additional lung cancer lines such as SK-LU-1 and H226, which exhibit somewhat lower basal levels of Dkk-1. No appreciable effects were observed in H1299, H460, or H841 cells expressing very low levels of Dkk-1. Diminished Dkk-1 expression coincided with reduced Dkk-1 protein levels in A549 and Calu-6 cells following TSC exposure (Fig. 1D).
TSC-mediated repression of Dkk-1 is not associated with promoter methylation.
Methylation-specific PCR and pyrosequencing experiments were done to ascertain if TSC exposure induced methylation of the Dkk-1 promoter in cultured lung cancer cells. Briefly, A549 and Calu-6 cells were treated continuously with NM ± TSC (0.003 puff/mL) for 5, 10, 15, 30, 45, and 60 days. Figure 2A depicts primer annealing sites for methylation-specific PCR and pyrosequencing relative to the transcription site in the Dkk-1 promoter region. Methylation-specific PCR analysis (Fig. 2B) confirmed that the Dkk-1 promoter is methylated in control DLD-1 colon cancer cells deficient for Dkk-1 expression, but not in SW480 colon cancer cells exhibiting high level of Dkk-1 expression (20). No appreciable increase in Dkk-1 promoter methylation was evident following methylation-specific PCR analysis of A549 or Calu-6 cells despite relatively prolonged TSC exposures. Pyrosequencing experiments (Fig. 2C) confirmed the results of methylation-specific PCR, suggesting that DNA methylation was not the primary mechanism by which TSC mediated repression of Dkk-1 in cultured lung cancer cells.
Figure 2.

Analysis of Dkk-1 promoter methylation in A549 and Calu-6 lung cancer cells following TSC exposure. A, schematic representation of a portion of the CpG island flanking the transcription start site within the Dkk-1 promoter. Thin black arrow, transcription start site. White and black arrows, positions of methylation-specific PCR and pyrosequencing primers, respectively; 18 CpG sites were analyzed by pyrosequencing. Striped arrows, chromatin immunoprecipitation primer sites. B, methylation-specific PCR analysis of Dkk-1 in A549 and Calu-6 cells cultured in NM ± TSC (0.003 puff/mL) for 5 to 60 d. DLD-1 and SW480 colon cancer cells served as positive and negative controls, respectively. C, pyrosequencing analysis confirming no significant increase in Dkk-1 promoter methylation in A549 and Calu-6 cells despite relatively prolonged TSC exposures.
TSC exposure decreases H4K16 acetylation and increases H3K27 trimethylation within the Dkk-1 promoter.
Chromatin immunoprecipitation techniques were used to further investigate epigenetic phenomena associated with TSC-mediated repression of Dkk-1 in lung cancer cells. Briefly, A549 and Calu-6 cells were cultured in NM ± TSC (0.003 puff/mL) for 10 days before chromatin immunoprecipitation analysis. SW480 and DLD-1 were used as controls for activation (H3K4me3 and H4K16Ac) and repression (H3K9me3 and H3K27me3) histone marks, respectively. Primers for chromatin immunoprecipitation analysis encompassed the transcription start site as well as a portion of the adjacent CpG island within the Dkk-1 promoter (Fig. 2A). As shown in Fig. 3A, chromatin immunoprecipitation analysis revealed no apparent changes in RNA polymerase II, H3K4me3, or H3K9me3 levels within the Dkk-1 promoter in lung cancer cells following TSC exposure. Interestingly, TSC exposure decreased H4K16Ac and increased H3K27me3 levels within the Dkk-1 promoter region in both lung cancer lines.
Figure 3.

Chromatin immunoprecipitation analysis of histone alterations coinciding with Dkk-1 expression in cultured lung cancer cells. A, chromatin immunoprecipitation analysis of the Dkk-1 promoter in A549 and Calu-6 cells cultured in NM ± TSC (0.003 puff/mL) for 10 d. SW480 and DLD1 colon cancer cells were used as controls for activation and repression marks, respectively. B, chromatin immunoprecipitation analysis of polycomb proteins recruited to the Dkk-1 promoter in cultured lung cancer cells following TSC exposure. C and D, chromatin immunoprecipitation analysis of the Dkk-1 promoter (C) and quantitative RT-PCR assessment of Dkk-1 expression in Calu-6 cells (D) before and after removal of TSC from culture media.
TSC exposure induces reversible recruitment of polycomb proteins to the Dkk-1 promoter in lung cancer cells.
Because H3K27me3 levels within the Dkk-1 promoter were increased in lung cancer cells following TSC exposure, additional chromatin immunoprecipitation experiments were done to ascertain if components of polycomb repressor complexes (6, 27) were also recruited to this promoter. Calu-6 cells were cultured in NM ± TSC for 10 days. Chromatin immunoprecipitation analysis (Fig. 3B) revealed a dose-dependent increase in H3K27me3, which coincided with recruitment of EZH2 and SUZ12 (components of the ‘‘initiation’’ polycomb repressor complex 2 that catalyze H3K27me3), as well as Bmi1, a member of ‘‘maintenance’’ polycomb repressor complex 1 (27). Additional analysis confirmed the dose-dependent deacetylation of H4K16, which coincided with recruitment of SirT1, a class III histone deacetylase that mediates H4K16 deacetylation (28). Consistent with previous data pertaining to polycomb-mediated transcriptional repression (29), no appreciable changes in RNA polymerase II levels within the Dkk-1 promoter were observed following TSC treatment.
Additional experiments were done to examine the stability of polycomb occupancy within the Dkk-1 promoter in TSC-treated lung cancer cells. Briefly, Calu-6 cells were cultured in NM ± TSC (0.003 puff/mL) for 10 days. Subcultures from TSC treated cells were allowed to proliferate in the absence of smoke for 5 or 10 days. Subsequent chromatin immunoprecipitation experiments (Fig. 3C) showed time-dependent reduction of H3K27me3 coinciding with diminished EZH2, SUZ12, and Bmi1 levels within the Dkk-1 promoter. A concomitant, progressive increase in H4K16Ac levels coincided with dissociation of SirT1 from the Dkk-1 promoter following cessation of TSC treatment. Real-time quantitative RT-PCR experiments with RNA isolated from cell cultures used for the chromatin immunoprecipitation experiments confirmed that the aforementioned histone modifications coincided with restoration of Dkk-1 expression in Calu-6 cells following removal of TSC from culture media (Fig. 3D).
Additional experiments were done to ascertain if TSC could mediate similar effects in normal respiratory epithelia. Briefly, SAEC were treated with TSC under conditions identical to those used for Calu-6 and A549 cells. Quantitative RT-PCR experiments revealed that TSC markedly decreased Dkk-1 mRNA copy numbers in SAEC. Western blot and chromatin immunoprecipitation experiments showed that despite relatively low levels of EZH2 and SirT1 in SAEC compared with cultured lung cancer cells, TSC-mediated repression of Dkk-1 in SAEC coincided with recruitment of EZH2, trimethylation of H3K27, and decreased acetylation of H4K16 within the Dkk-1 promoter (Supplementary Fig. S1).
siRNA knockdown of EZH2 and SirT1 abrogates TSC-mediated repression of Dkk-1.
Additional studies were done to further examine the role of polycomb complexes in the repression of Dkk-1 in lung cancer cells following TSC exposure. Briefly, parental Calu-6 cells or Calu-6 cells transfected with either sham siRNAs or siRNAs targeting EZH2 and/or SirT1 were cultured in NM with or without TSC (0.001 or 0.003 puff/mL) for 2 days. This abbreviated exposure duration was selected to minimize cumulative toxicities of transfections, target gene knockdown, and TSC exposures that might confound experimental results. Quantitative RT-PCR (data not shown) and Western blot experiments (Fig. 4A) showed no appreciable changes in EZH2 or SirT1 expression in control or transfected Calu-6 cells following TSC exposure. Additional analysis confirmed specific target gene knockdown in transfected cells (Fig. 4B). Quantitative RT-PCR analysis showed that knockdown of EZH2 increased Dkk-1 expression in untreated Calu-6 cells and partially abrogated TSC-mediated repression of Dkk-1 (Fig. 4C). Knockdown of SirT1 also seemed to somewhat increase Dkk-1 expression in untreated Calu-6 cells and partially abrogate TSC-mediated repression of Dkk-1 (Fig. 4C). The effects of SirT1 knockdown were less pronounced than those resulting from EZH2 depletion, possibly due to less efficient target gene inhibition [53% knockdown relative to sham control for siSirT1 versus 83% knockdown for siEZH2 by quantitative RT-PCR analysis (data not shown)]. Simultaneous knockdown of EZH2 and SirT1 also seemed to increase Dkk-1 expression and partially abrogate TSC-mediated repression of Dkk-1. This phenomenon was most likely mediated primarily by EZH2 knockdown (73%); the contribution of SirT1 knockdown (44%) in the combination knockdown experiment was not readily apparent. Correlative Western blot experiments showed that EZH2, SirT1, and combined EZH2/SirT1 knockdown modestly increased Dkk-1 protein levels in untreated Calu-6 cells and diminished the depletion of Dkk-1 in these cells following TSC exposure (Fig. 4D).
Figure 4.

Effects of EZH2 and SirT1 knockdown on TSC-mediated repression of Dkk-1 in cultured lung cancer cells. A, Western blot analysis of EZH2 or SirT1 protein levels in Calu-6 lung cancer cells following TSC exposure. B, Western blot analysis of EZH2 and SirT1 protein levels in Calu-6 cells transfected with siRNAs against EZH2 or SirT1 or with sham control sequences showing specific target gene knockdown. C, quantitative RT-PCR analysis of Dkk-1 expression in Calu-6 cells exhibiting siRNA-mediated knockdown of EZH2 and/or SirT1.D, Western blot analysis of Dkk-1 expression in untreated and TSC-exposed Calu-6 cells transfected with sham siRNAs or siRNAs targeting EZH2 and/or SirT1. Corresponding densitometry showing that knockdown of EZH2 and, to a lesser extent, SirT1 increases Dkk-1 protein levels in untreated Calu-6 cells and partially abrogates the depletion of Dkk-1 mediated by TSC.
Dkk-1 repression modulates noncanonical Wnt signaling in lung cancer cells.
A series of experiments were undertaken to ascertain if down-regulation of Dkk-1 enhances Wnt signaling in lung cancer cells. As shown in Fig. 5A, TSC treatment resulted in a modest, statistically insignificant increase in T-cell factor reporter activity reflective of canonical Wnt signaling (30, 31) in A549 and Calu-6 cells. Subsequent Western blot experiments revealed an ~2-fold increase in cyclin D levels but no appreciable alterations in β-catenin localization in these cell lines following TSC exposure (data not shown).
Figure 5.
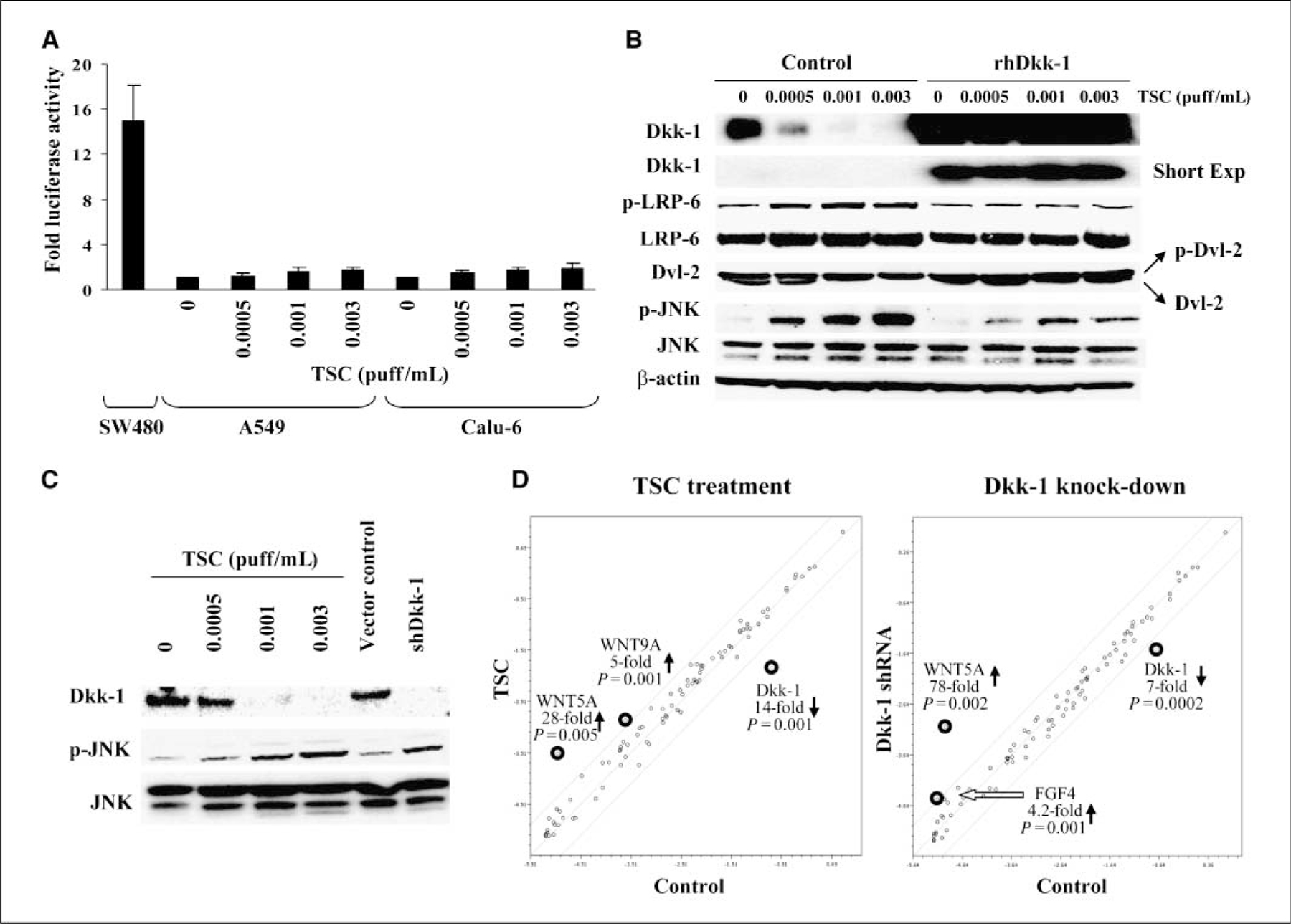
Analysis of Wnt signaling mediated by TSC or Dkk-1 knockdown in cultured lung cancer cells. A, TOP-flash promoter-reporter activity in A549 and Calu-6 cells following TSC exposure. Columns, mean of triplicate experiments measured as a ratio of TOP/FOP values, normalized to Renilla luciferase; bars, SD. B, Western blot analysis of Wnt signaling components in Calu-6 cells exposed to TSC in the presence or absence of exogenous recombinant Dkk-1 (rhDkk-1). C, Western blot analysis of Dkk-1, phospho-JNK, and total JNK levels in Calu-6 lung cancer cells exposed to TSC or stably transfected with shRNA targeting Dkk-1. D, focused quantitative RT-PCR analysis of Wnt signaling in Calu-6 lung cancer cells exposed to TSC (0.003 puff/mL) or stably transfected with shRNA silencing Dkk-1 . Results depicted are a summation of triplicate arrays. Lines above and below the center regression lines indicate 4-fold changes in gene expression.
Additional experiments were done to examine alternative Wnt pathways that might be modulated by inactivation of Dkk-1. Western blot analysis (Fig. 5B) revealed that TSC-mediated repression of Dkk-1 coincided with dose-dependent phosphorylation of LDL receptor related protein 6 (LRP-6) and dishevelled-2 (Dvl-2) indicative of activation of Wnt receptor complex, as well as JNK, a downstream effector of noncanonical Wnt signaling. Exogenous recombinant Dkk-1 abrogated Wnt receptor as well as JNK activation by TSC. Additional experiments revealed that shRNA-mediated knockdown of Dkk-1 enhanced phosphorylation of JNK in Calu-6 cells (Fig. 5C), suggesting that activation of JNK in these cells following TSC exposure was due, at least in part, to activation of Wnt signaling, rather than nonspecific responses to genotoxic stress. Subsequent quantitative RT-PCR array experiments confirmed that TSC exposure enhances Wnt signaling in lung cancer cells (Fig. 5D); interestingly, Wnt5a (which encodes a ligand implicated in noncanonical Wnt signaling; ref. 32) was markedly induced in TSC-treated Calu-6 cells as well as Calu-6 cells transfected with shRNA targeting Dkk-1. Taken together, these data suggest that repression of Dkk-1 contributes to activation of noncanonical Wnt signaling in lung cancer cells following TSC exposure.
Dkk-1 knockdown enhances the tumorigenicity of Calu-6 cells in nude mice.
Additional experiments were done to directly assess the relevance of Dkk-1 repression with respect to the tumorigenic potential of lung cancer cells mediated by TSC. Briefly, nude mice were inoculated s.c. with Calu-6 cells stably expressing a shRNA targeting Dkk-1 or a vector control. Western blot experiments (Fig. 5C) confirmed Dkk-1 knockdown. As shown in Fig. 6A, mice inoculated with Calu-6 cells exhibiting knockdown of Dkk-1 were considerably more likely to develop tumors relative to mice injected with vector control cells (18 of 20 versus 6 of 20; P = 0.0002, exact test). In addition, tumors arising from Dkk-1 knockdown cells were markedly larger than those derived from vector control cells (574.3 ± 410.1 versus 69.3 ± 33.4 mg; P = 0.00011, two-tailed t test). Quantitative RT-PCR analysis confirmed very low levels of Dkk-1 expression in Dkk-1 knockdown xenografts but not in those derived from vector control cells (Fig. 6B). Interestingly, additional quantitative RT-PCR and pyrosequencing analyses revealed persistently low levels of Dkk-1 expression without apparent promoter methylation in TSC-treated Calu-6 tumor xenografts, suggesting a strong in vivo selection pressure to maintain silencing of this tumor suppressor gene.
Figure 6.
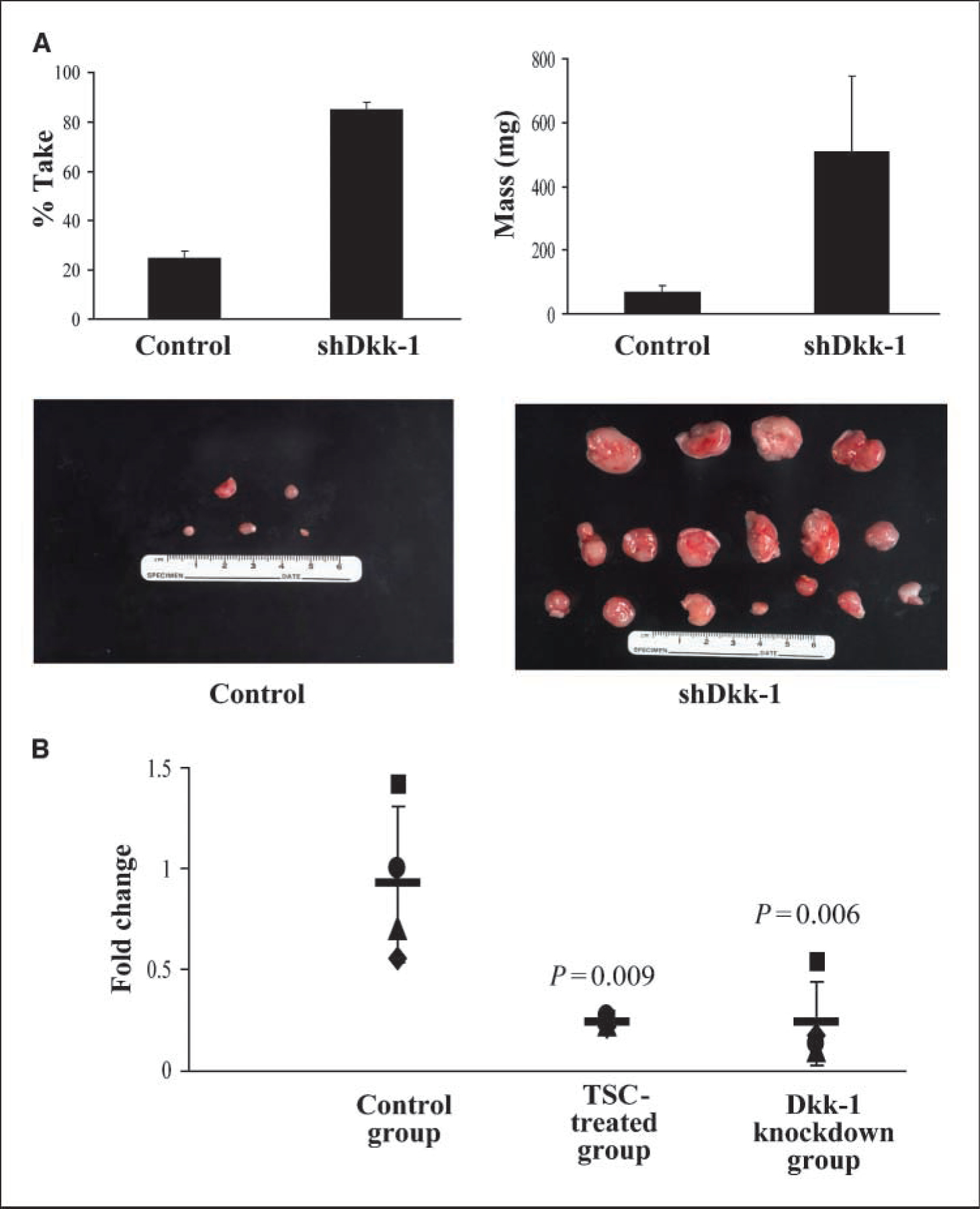
Effects of targeted inhibition of Dkk-1 expression on the tumorigenicity of Calu-6 lung cancer cells. A, percent take and mass of Calu-6 tumor xenografts following knockdown of Dkk-1 . Inhibition of Dkk-1 expression (Fig. 5C) dramatically increased the percent take as well as the mass of Calu-6 tumor xenografts. B, quantitative RT-PCR analysis showing reduced levels of Dkk-1 expression in xenografts derived from TSC-exposed and Dkk-1 knockdown Calu-6 cells relative to controls.
Discussion
Whereas the epidemiologic association between tobacco smoke and lung cancer is irrefutable, the epigenetic events contributing to the initiation and progression of this malignancy have yet to be fully elucidated. Pertinent to this study, ~30% of lung cancer patients continue to smoke following diagnosis (17, 33). Surprisingly, the effect of cigarette smoking on the lung cancer epigenome relative to cancer-specific mortality has not been firmly established.
Polycomb repressor complexes maintain pluripotency in germ cells by establishing a bivalent chromatin structure and silencing genes that regulate development and differentiation (6, 34). Several recent studies indicate that aberrant promoter DNA methylation in cancer cells frequently involves genes with relatively low transcriptional activity exhibiting bivalent histone marks; DNA demethylating agents and histone deacetylase inhibitors derepress these aberrantly silenced genes but do not revert them to fully euchromatic states (35, 36). Data presented in this article strongly suggest that tobacco smoke induces a bivalent chromatin structure within the Dkk-1 promoter, coinciding with polycomb-mediated repression of this Wnt antagonist in lung cancer cells. Whereas various components of Wnt signaling including Dkk-1 are known to be potential polycomb targets (8, 37), our data provide the first evidence of a major environmental carcinogen directly engaging polycomb machinery to acutely silence a tumor suppressor gene, thereby activating a signaling network implicated in stem cell renewal, embryogenesis, and cancer (31, 38). Our inability to completely abolish TSC-mediated repression of Dkk-1 using siRNAs targeting EZH2 and/or SirT1 may be attributable, at least in part, to inefficient target gene inhibition and/or the need to simultaneously deplete additional polycomb proteins such as EZH1, which complements EZH2 in maintaining H3K27 trimethylation and repression of polycomb target genes in embryonic stem cells (39).
Polycomb targets seem to be preferentially silenced by de novo methylation mechanisms during malignant transformation (7–9). Our data suggest that recruitment of polycomb repressor complexes precedes promoter-specific hypermethylation of Dkk-1 during pulmonary carcinogenesis (26). Our observations pertaining to repression of Dkk-1 in the absence of promoter methylation in cultured lung cancer cells despite prolonged TSC exposures are consistent with recent studies showing that polycomb-mediated gene silencing in cancer cells can occur independent of DNA methylation (40). Whereas the mechanisms underlying these phenomena have not been established, several recent reports have shown that EZH2 promotes formation of intrachromosomal and interchromosomal loops that mediate long-range transcriptional repression; intrachromosomal loops frequently involve genes with relatively low transcriptional activity that are ‘‘poised’’ for activation or further repression via DNA methylation (41, 42). Conceivably, chromatin looping contributes to polycomb-mediated repression of Dkk-1 in cells exposed to tobacco smoke.
The fact that knockdown of Dkk-1 seemed to be sufficient to recapitulate the effects of TSC exposure clearly suggests that epigenetic silencing of this Wnt antagonist enhances the tumorigenic potential of lung cancer cells. These data are consistent with recent studies indicating that Dkk-1 inhibits the tumorigenicity of colon and breast cancer cells (20, 43). Interestingly, TSC- and shRNA-mediated repression of Dkk-1 dramatically induced expression of Wnt5a , which encodes a noncanonical Wnt ligand critical for distal airway development (44). Although the effects of Wnt5a vary depending on tissue histology and receptor context, recent data indicate that this ligand activates a planar cell polarity pathway mediated via JNK, which confers a proinvasive phenotype in cancer cells (32). Of particular relevance in this regard, Huang and colleagues (45) observed that Wnt5a expression in non–small-cell lung cancers correlates with proliferation index, stromal expression of vascular endothelial growth factor A, and diminished patient survival.
Because Wnt signaling seems to be critical for maintenance of normal as well as cancer stem cells (46–48), it is not surprising that numerous Wnt antagonists are silenced via epigenetic mechanisms in lung cancers (26, 49). Data presented in this article clearly show that cigarette smoke can engage polycomb repressor complexes to mediate epigenetic silencing of Dkk-1, thereby enhancing the malignant phenotype of lung cancer cells. These observations provide a potential mechanism by which continued smoking could adversely affect survival in lung cancer patients and support further analysis of the epigenetic effects of tobacco smoke during pulmonary carcinogenesis.
Supplementary Material
Acknowledgments
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Full microarray results can be viewed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13309.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Xing J, Stewart DJ, Gu J, Lu C, Spitz MR, Wu X. Expression of methylation-related genes is associated with overall survival in patients with non-small cell lung cancer. Br J Cancer 2008;98:1716–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT, Wang YC. Alteration of DNA methyltransferases contributes to 5ʹCpG methylation and poor prognosis in lung cancer. Lung Cancer 2007;55:205–13. [DOI] [PubMed] [Google Scholar]
- 3.Brock MV, Hooker CM, Ota-Machida E, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med 2008;358:1118–28. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki H, Moriyama S, Nakashima Y, et al. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 2004;46:171–8. [DOI] [PubMed] [Google Scholar]
- 5.Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer 2004;112:26–32. [DOI] [PubMed] [Google Scholar]
- 6.Kanno R, Janakiraman H, Kanno M. Epigenetic regulator polycomb group protein complexes control cell fate and cancer. Cancer Sci 2008;99:1077–84. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Widschwendter M, Fiegl H, Egle D, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007;39:157–8. [DOI] [PubMed] [Google Scholar]
- 8.Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007;39:232–6. [DOI] [PubMed] [Google Scholar]
- 9.Ohm JE, McGarvey KM, Yu X, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007;39:237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003;22:5323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vonlanthen S, Heighway J, Altermatt HJ, et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer 2001;84:1372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reynolds PA, Sigaroudinia M, Zardo G, et al. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem 2006;281:24790–802. [DOI] [PubMed] [Google Scholar]
- 13.Breuer RH, Snijders PJ, Smit EF, et al. Increased expression of the EZH2 polycomb group gene in BMI-1-positive neoplastic cells during bronchial carcinogenesis. Neoplasia 2004;6:736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res 2006;66:1371–5. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Zhou Y, Boggs SE, Belinsky SA, Liu J. Cigarette smoke induces demethylation of prometastatic oncogene synuclein-γ in lung cancer cells by down-regulation of DNMT3B. Oncogene 2007;26:5900–10. [DOI] [PubMed] [Google Scholar]
- 16.Itaya T, Yamaoto N, Ando M, et al. Influence of histological type, smoking history and chemotherapy on survival after first-line therapy in patients with advanced non-small cell lung cancer. Cancer Sci 2007;98: 226–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W, Heist RS, Liu G, et al. Smoking cessation before diagnosis and survival in early stage non-small cell lung cancer patients. Lung Cancer 2006;53:375–80. [DOI] [PubMed] [Google Scholar]
- 18.Fields WR, Leonard RM, Odom PS, Nordskog BK, Ogden MW, Doolittle DJ. Gene expression in normal human bronchial epithelial (NHBE) cells following in vitro exposure to cigarette smoke condensate. Toxicol Sci 2005;86:84–91. [DOI] [PubMed] [Google Scholar]
- 19.Hong JA, Kang Y, Abdullaev Z, et al. Reciprocal binding of CTCF and BORIS to the NY-ESO-1 promoter coincides with derepression of this cancer-testis gene in lung cancer cells. Cancer Res 2005;65:7763–74. [DOI] [PubMed] [Google Scholar]
- 20.Aguilera O, Fraga MF, Ballestar E, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 2006;25: 4116–21. [DOI] [PubMed] [Google Scholar]
- 21.Kang Y, Hong JA, Chen GA, Nguyen DM, Schrump DS. Dynamic transcriptional regulatory complexes including BORIS, CTCF and Sp1 modulate NY-ESO-1 expression in lung cancer cells. Oncogene 2007;26:4394–403. [DOI] [PubMed] [Google Scholar]
- 22.Schrump DS, Hong JA, Nguyen DM. Utilization of chromatin remodeling agents for lung cancer therapy. Cancer J 2007;13:56–64. [DOI] [PubMed] [Google Scholar]
- 23.Lotem J, Sachs L. Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene 2006;25:7663–72. [DOI] [PubMed] [Google Scholar]
- 24.Dolinoy DC, Jirtle RL. Environmental epigenomics in human health and disease. Environ Mol Mutagen 2008; 49:4–8. [DOI] [PubMed] [Google Scholar]
- 25.Dhar P. Measuring tobacco smoke exposure: quantifying nicotine/cotinine concentration in biological samples by colorimetry, chromatography and immunoassay methods. J Pharm Biomed Anal 2004; 35:155–68. [DOI] [PubMed] [Google Scholar]
- 26.Licchesi JD, Westra WH, Hooker CM, Machida EO, Baylin SB, Herman JG. Epigenetic alteration of Wnt pathway antagonists in progressive glandular neoplasia of the lung. Carcinogenesis 2008;29:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz YB, Pirrotta V. Polycomb complexes and epigenetic states. Curr Opin Cell Biol 2008;20:266–73. [DOI] [PubMed] [Google Scholar]
- 28.Vaquero A, Sternglanz R, Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007;26:5505–20. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 2007;8:9–22. [DOI] [PubMed] [Google Scholar]
- 30.Katoh M, Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res 2007;13:4042–5. [DOI] [PubMed] [Google Scholar]
- 31.Kestler HA, Kuhl M. From individual Wnt pathways towards a Wnt signalling network. Philos Trans R Soc Lond B Biol Sci 2008;363:1333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pukrop T, Binder C. The complex pathways of Wnt 5a in cancer progression. J Mol Med 2008;86:259–66. [DOI] [PubMed] [Google Scholar]
- 33.Cox LS, Africano NL, Tercyak KP, Taylor KL. Nicotine dependence treatment for patients with cancer. Cancer 2003;98:632–44. [DOI] [PubMed] [Google Scholar]
- 34.Ku M, Koche RP, Rheinbay E, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 2008;4: e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez J, Munoz M, Vives L, Frangou CG, Groudine M, Peinado MA. Bivalent domains enforce transcriptional memory of DNA methylated genes in cancer cells. Proc Natl Acad Sci U S A 2008;105: 19809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGarvey KM, Van NL, Cope L, et al. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res 2008;68: 5753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006;20:1123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008;8:387–98. [DOI] [PubMed] [Google Scholar]
- 39.Shen X, Liu Y, Hsu YJ, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008;32:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 2008;40: 741–50. [DOI] [PubMed] [Google Scholar]
- 41.Tiwari VK, McGarvey KM, Licchesi JD, et al. PcG proteins, DNA methylation, and gene repression by chromatin looping. PLoS Biol 2008;6:2911–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiwari VK, Cope L, McGarvey KM, Ohm JE, Baylin SB. A novel 6C assay uncovers Polycomb-mediated higher order chromatin conformations. Genome Res 2008;18: 1171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett 2008;269:67–77. [DOI] [PubMed] [Google Scholar]
- 44.Li C, Hu L, Xiao J, et al. Wnt5a regulates Shh and Fgf10 signaling during lung development. Dev Biol 2005; 287:86–97. [DOI] [PubMed] [Google Scholar]
- 45.Huang CL, Liu D, Nakano J, et al. Wnt5a expression is associated with the tumor proliferation and the stromal vascular endothelial growth factor-an expression in non-small-cell lung cancer. J Clin Oncol 2005; 23:8765–73. [DOI] [PubMed] [Google Scholar]
- 46.Nusse R. Wnt signaling and stem cell control. Cell Res 2008;18:523–7. [DOI] [PubMed] [Google Scholar]
- 47.Malanchi I, Peinado H, Kassen D, et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 2008;452:650–3. [DOI] [PubMed] [Google Scholar]
- 48.Malanchi I, Huelsken J. Cancer stem cells: never Wnt away from the niche. Curr Opin Oncol 2009;21:41–6. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki M, Shigematsu H, Nakajima T, et al. Synchronous alterations of Wnt and epidermal growth factor receptor signaling pathways through aberrant methylation and mutation in non small cell lung cancer. Clin Cancer Res 2007;13:6087–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.