

SUMMARY
Few therapies target the loss of tumor suppressor genes in cancer. We examine CRISPR-SpCas9 and RNA-interference loss-of-function screens to identify new therapeutic targets associated with genomic loss of tumor suppressor genes. The endosomal sorting complexes required for transport (ESCRT) ATPases VPS4A and VPS4B score as strong synthetic lethal dependencies. VPS4A is essential in cancers harboring loss of VPS4B adjacent to SMAD4 on chromosome 18q and VPS4B is required in tumors with co-deletion of VPS4A and CDH1 (E-cadherin) on chromosome 16q. We demonstrate that more than 30% of cancers selectively require VPS4A or VPS4B. VPS4A suppression in VPS4B-deficient cells selectively leads to ESCRT-III filament accumulation, cytokinesis defects, nuclear deformation, G2/M arrest, apoptosis, and potent tumor regression. CRISPR-SpCas9 screening and integrative genomic analysis reveal other ESCRT members, regulators of abscission, and interferon signaling as modifiers of VPS4A dependency. We describe a compendium of synthetic lethal vulnerabilities and nominate VPS4A and VPS4B as high-priority therapeutic targets for cancers with 18q or 16q loss.
In Brief
Neggers, Paolella, and colleagues identify the ATPases VPS4A and VPS4B as selective vulnerabilities and potential therapeutic targets in cancers harboring loss of chromosome 18q or 16q. In VPS4B-deficient cancers, VPS4A suppression leads to ESCRT-III dysfunction, nuclear deformation, and abscission defects. Moreover, ESCRT proteins and interferons can modulate dependency on VPS4A.
Graphical Abstract
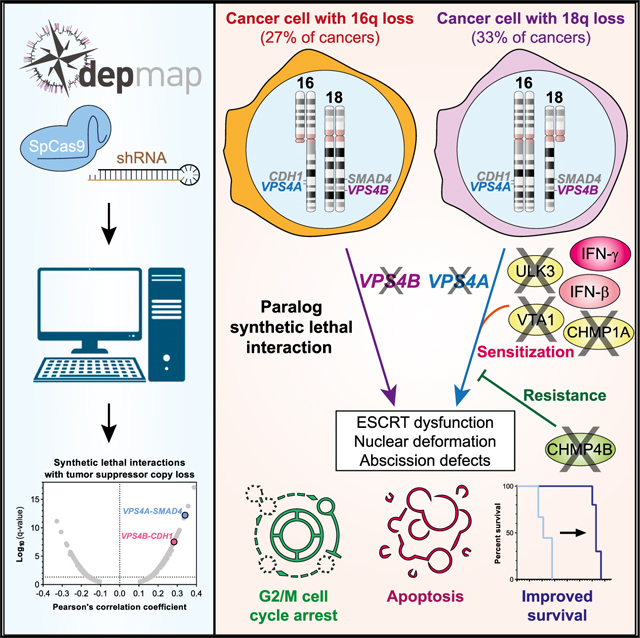
INTRODUCTION
Discovery of new biomarker-linked cancer therapeutic targets may enable drug development and lead to advances in clinical care. Somatic copy number alterations (CNAs) leading to loss of tumor suppressor gene (TSG) function constitute important driver events in tumorigenesis (Beroukhim et al., 2010; Fearon and Vogelstein, 1990). Unfortunately, few therapeutic options exist to target oncogenic processes evoked by tumor suppressor inactivation. However, developing drugs based on synthetic lethal interactions with common somatic CNAs represents a promising approach to attain cancer-selective therapeutics.
Synthetic lethality refers to the observation that, for certain gene pairs, inactivation of either gene is tolerated but combined loss-of-function of both genes results in decreased cell viability (Dobzhansky, 1946; Hartwell et al., 1997; Huang et al., 2020; Kaelin, 1999). Synthetic lethal relationships in cancer have been defined in different contexts. For example, BRCA1/2-mutant cancers harbor defects in homologous recombination DNA repair and are particularly sensitive to inhibition of the poly(ADP-ribose) polymerase (PARP) DNA repair enzyme (Sonnenblick et al., 2015).
Synthetic lethal relationships are also observed among paralog genes for which dependency on one paralog is conferred by loss of a second functionally redundant paralog gene, as demonstrated between SMARCA2-SMARCA4, ARID1A-ARID1B, UBB-UBC, and MAGOH-MAGOHB (Helming et al., 2014; Hoffman et al., 2014; Tsherniak et al., 2017; Viswanathan et al., 2018). Such paralog synthetic lethality may arise when there is a concomitant loss of a driver TSG and a paralog passenger gene nearby, a phenomenon that has been termed “collateral lethality” (Dey et al., 2017; Muller et al., 2012). Besides synthetic lethality, we and others have described selective dependencies on genes that have themselves undergone copy number loss in cancer (Liu et al., 2015; McDonald et al., 2017; Nijhawan et al., 2012; Paolella et al., 2017). Because targeting synthetic lethal relationships in cancer may yield a wide therapeutic window of efficacy between tumor and normal cells, identification of pharmacologically tractable synthetic lethal targets remains a priority for oncology drug development programs.
To systematically define synthetic lethal vulnerabilities associated with genomic loss of established TSGs, we analyzed genome-scale CRISPR-SpCas9 and RNA-interference loss-of-function screening data from more than 600 cancer cell lines. We identified and prioritized 175 synthetic lethal interactions with one or more of 51 TSGs. In particular, we discovered that the paralog genes encoding vacuolar protein sorting 4 homolog A and B (VPS4A and VPS4B) are selective genetic vulnerabilities for tumors harboring genomic copy loss of SMAD4 or CDH1 because of co-deletion of VPS4B or VPS4A, respectively. VPS4B is located on the long arm (q) of chromosome 18, 12.3 Mb away from SMAD4, whereas VPS4A is located 0.476 Mb downstream of CDH1 (E-cadherin) on chromosome 16q. Codeletion of SMAD4 and VPS4B is commonly observed in pancreatic, colorectal, stomach, and renal cell carcinomas and, to a lesser extent, in cancers of the bile duct, lung, prostate, esophagus, uterus, cervix, and ovary (Kojima et al., 2007; Thiagalingam et al., 1996; Zack et al., 2013). Meanwhile, loss of CDH1 and VPS4A occurs in cancers of the stomach, breast, skin, colon, and prostate (Berx et al., 1996; Graff et al., 1995; Yoshiura et al., 1995; Zack et al., 2013).
VPS4A and B function as AAA ATPases, which are critical for the regulation of endosomal sorting complex required for transport (ESCRT), a multimeric protein complex essential for inverse membrane remodeling. The ESCRT machinery is involved in a range of cellular processes, including cytokinesis, membrane repair, autophagy, and endosomal processing (Schöneberg et al., 2017; Vietri et al., 2020). VPS4A/B form asymmetric hexameric complexes that are stabilized by the VTA1 cofactor (Obita et al., 2007; Scott et al., 2005a; Su et al., 2017; Sun et al., 2017; Vietri et al., 2020). Here, we demonstrate that suppression of VPS4A in tumors with reduced copy number of VPS4B leads to accumulation of ESCRT-III filaments, cytokinesis defects, nuclear membrane abnormalities and micronucleation, ultimately resulting in G2/M cell cycle arrest, apoptosis, and tumor regression. Furthermore, using a CRISPR-SpCas9 genome-scale modifier screen, we identified multiple genes that promote or suppress VPS4A dependency. Our findings reveal a critical role for the ESCRT pathway in cancer cell survival and nominate the VPS4 enzymes as promising synthetic lethal targets specific for tumors harboring the loss of VPS4B on chromosome 18q or the loss of VPS4A on chromosome 16q.
RESULTS
Discovery of Synthetic Lethal Interactions with Genomic Loss of Established Tumor Suppressors
To uncover synthetic lethal interactions with genomic loss of TSGs, we analyzed genome-scale RNA interference (RNAi) and CRISPR-SpCas9 cancer-dependency datasets (https://depmap.org/portal; McFarland et al., 2018; Meyers et al., 2017; Tsherniak et al., 2017). We focused our analysis on 51 commonly lost TSGs to identify synthetic lethal relationships that could be relevant to a large fraction of human cancers (Table S1). We correlated log2-normalized copy number calls for each of those tumor suppressors with normalized, gene-level CRISPR-SpCas9 (622 cell lines, 18,333 genes) and RNAi (669 cell lines, 16,905 genes) dependency scores (Figure 1A). Only 1.0% of CRISPR-SpCas9 (9,339/934,983) and 2.0% of RNAi (16,866/862,155) dependency-TSG interactions were significant at a 10% false discovery rate (FDR; Data S1). Interestingly, a sizeable fraction of significant CRISPR-SpCas9 (31.4%; 2,935/9,339) and RNAi correlations (7.1%; 1,222/16,866) represented cis correlations in which the dependency gene localized to the same chromosomal arm as the TSG (Figures S1A–S1C). Collateral (partial) loss of essential genes adjacent to TSGs frequently occurs, resulting in enhanced dependency on expression of those genes. Such cis-occurring gene dependencies have previously been described as “CYCLOPS” (copy-number alterations yielding cancer liabilities owing to partial loss) (Nijhawan et al., 2012; Paolella et al., 2017).
Figure 1. Discovery of Synthetic Lethal Interactions with Genomic Loss of Established Tumor Suppressors.

(A) Synthetic lethality analysis workflow. Copy number of 51 tumor suppressor genes across cancer cell lines was correlated with gene-dependency scores from CRISPR-SpCas9 or RNAi interference screens.
(B) q-q plot showing significant, positive Pearson’s correlations between gene dependency scores and tumor suppressor gene deletion. For each pair, the dependency gene is listed first, followed by the associated tumor suppressor gene. Highly significant correlations (FDR 10%, q < 0.1) are highlighted (beige) with emphasis on the VPS4A-SMAD4 and VPS4B-CDH1 synthetic lethal interactions (pink).
(C) Number of significant synthetic lethal interactions for each tumor suppressor gene that shows significant dependency-copy number Pearson’s correlation coefficient in both RNAi and CRISPR datasets (q < 0.1).
(D) Log-normalized q-values for all significant and positive correlating gene dependencies (q < 0.1) for the 10 most-correlated tumor suppressor genes.
See also Figures S1 and S10, Table S1, and Data S1.
To expand beyond CYCLOPS genes, we focused on gene dependencies located on a different chromosome (i.e., trans) (Figure 1A). We identified 3,974 CRISPR-SpCas9 (0.4%) and 14,862 RNAi (1.8%) significant trans correlations at a 10% FDR (Figures S1D–S1F; Data S1); of these, 342 were significant in both datasets (Table S1). Some synthetic lethal interactions were unique to one dataset, partially because of an incomplete overlap in the probed genes and screened cell lines between datasets.
To identify synthetic lethal interactions, we focused on positive correlations for which TSG loss (lower copy number) confers increased dependency (lower dependency score) in both RNAi and CRISPR datasets to uncover 175 significant interactions across 122 genes (Figures 1B–1D and S1G). Several paralog and collateral synthetic lethal interactions were highly significant in our analysis, including those previously reported for MAGOH-CDKN1B and PRMT5/WDR77 with CDKN2A (Figures 1B and 1D; Kryukov et al., 2016; Viswanathan et al., 2018) as well as multiple new interactions, including ones linked to the loss of KEAP1, STK11, ATM, TP53, CDH1, and SMAD4 (Figures 1B, 1D, and S1G; Table S1).
The ESCRT Enzymes VPS4A and VPS4B Are Paralog Synthetic Lethal Vulnerabilities in Cancers Harboring SMAD4 or CDH1 Loss
We observed a striking number of synthetic lethal interactions with SMAD4 deletion (Figures 1C and S1G), one of the most commonly lost tumor suppressor genes in human cancer. VPS4A scored as the strongest correlated gene dependency with copy loss of SMAD4/18q (Figures 1B–1D; Table S1). Notably, VPS4B, a paralog of VPS4A (Figure S2A), resides 12.3 Mb adjacent to SMAD4 on chromosome 18q (Figure 2A). Across cancer cell lines, VPS4B copy loss was frequent and strongly correlated with SMAD4 loss (Figure 2B; R2 = 0.615). Although SMAD4 was homozygously deleted in some cell lines, we did not observe homozygous loss of VPS4B. Interestingly, some VPS4A-dependent cell lines harbored normal SMAD4 copy number but showed focal loss of VPS4B, whereas some VPS4A-independent cell lines harbored loss of SMAD4 but retained normal copy number for VPS4B (Figures 2B and S2B). Furthermore, VPS4A dependency correlated better with VPS4B copy loss than with SMAD4 copy loss in both CRISPR-SpCas9 (R2 = 0.208 versus R2 = 0.116) and RNAi datasets (R2 = 0.118 versus R2 = 0.021) (Figure S2D, left panels). Moreover, of all VPS4A-dependent cancer cell lines, 63.9% (106/166) demonstrated at least partial genomic loss of VPS4B (Figure S2E; Table S1). These results suggest that dependency on VPS4A is driven by loss of VPS4B, rather than SMAD4.
Figure 2. The ESCRT Enzymes VPS4A and VPS4B Are Paralog Synthetic Lethal Vulnerabilities in Cancers Harboring SMAD4 or CDH1 Loss.

(A) Genomic location of SMAD4 and VPS4B on human chromosome 18.
(B) Scatterplot showing SMAD4 (x axis) and VPS4B (y axis) log2-normalized relative copy numbers in 1,657 cancer cell lines and linear regression with 95% confidence interval (blue line) and Pearson’s coefficient.
(C) Illustration of ESCRT- and VPS4-mediated reverse topology membrane remodeling.
(D) Volcano plot of Pearson’s correlation coefficients between CRISPR-SpCas9 gene dependency scores and SMAD4 copy number (x axis) and the log10-normalized q-value for each of these correlations (y axis) across 622 cancer cell lines from the Dependency Map (19Q3). Significant dependencies are colored. Horizontal line: 10% FDR (q < 0.1).
(E) Smoothed histogram showing the distribution of CHMP4B dependency scores from the CRISPR-SpCas9 Dependency Map dataset (19Q3) across cancer cell lines. The number of dependent cell lines (CRISPR score less than —0.5, orange line) over the total number of probed cell lines is shown in the top left corner. Red line at —1: CRISPR score for a set of highly essential genes. Black line at 0: CRISPR score of negative control sgRNAs.
(F) Boxplot of CRISPR-SpCas9 CHMP4B gene dependency scores across cancer cell lines with neutral or reduced VPS4B or VPS4A copy number (Log2 relative copy number < 0.66) in the Dependency Map. ns, not significant; ****p < 0.0001.
(G) Frequency of VPS4A-dependent cancer cell lines by tumor lineage in the Dependency Map 19Q3 dataset.
(H) Summary of VPS4B copy number alterations in TCGA Pan-Cancer Atlas patient samples categorized by tumor type. Orange dots denote patient samples with strong VPS4B loss (less than −0.75).
See also Figures S2–S4 and Table S1.
We next evaluated whether a reciprocal relationship existed between VPS4B dependency and VPS4A copy number. VPS4B dependency significantly correlated with loss of the CDH1 tumor suppressor locus on chromosome 16q22.1 (Figure 1B; Table S1). VPS4A localizes only 476 kilobases downstream of the CDH1 TSG (Figure S2C), and VPS4A copy number strongly correlated with CDH1 copy number (Figure S2C; R2 = 0.846). As expected, dependency on VPS4B correlated with VPS4A copy number in both CRISPR-SpCas9 and RNAi datasets (Figures S2D and S2F), although that correlation was less profound than that between VPS4A dependency and VPS4B copy loss (compare Figures S2E and S2F; Table S1).
VPS4A and VPS4B encode 49-kDa AAA ATPases that are 81% identical (Figure S2A). These two paralog proteins form multimeric complexes with the ESCRT machinery to regulate reverse topology membrane remodeling and fission across many cellular processes (Vietri et al., 2020; Wollert et al., 2009; Figure 2C). CHMP4B is a core filament-forming ESCRT-III protein that is essential for ESCRT-mediated membrane remodeling (McCullough et al., 2018). Although VPS4A dependency scores positively correlated with SMAD4/VPS4B copy number, we observed a strong anticorrelation between CHMP4B dependency and SMAD4 copy number (Figure 2D; Table S1). Although CHMP4B is strongly essential for proliferation and survival in cells with euploid VPS4B copy number, cells that harbor loss of VPS4B are less sensitive to CHMP4B knockout (Figures 2E and 2F; Table S1). Interestingly, no clear relationship between CHMP4B dependency scores and VPS4A/CDH1 copy number was observed (Figures 2F and S3A).
Correlation of VPS4A and VPS4B CRISPR dependency scores with CRISPR dependency scores for all other genes highlighted that VPS4A and VPS4B are co-essential with other specialized ESCRT genes, such as CHMP1A, VTA1, and IST1 (Figures S3B–S3D; Table S1). Conversely, dependency on CHMP4Banticorrelated with VPS4A/VPS4B dependency (Figures S3B–S3D; Table S1). Like CHMP4B dependency (Figure 2F), CHMP1A dependency demonstrated significant interaction with VPS4B copy number loss but not with VPS4A copy loss (Figure S3D; Table S1). This could possibly be explained by the less-pronounced and less-prominent occurrence of VPS4A copy loss across cancer cell lines (Figures S2C and S2F) or may indicate a paralog-specific interaction between VPS4B expression and dependency on the CHMP1A and CHMP4B ESCRT-III proteins. In aggregate, our synthetic lethal analysis highlights a critical role for the ESCRT pathway in maintaining cancer cell survival.
VPS4A and VPS4B Undergo Frequent Copy Loss across Both Adult and Pediatric Cancer Types
Across datasets, 22.7% (142/624) of cancer cell lines screened by CRISPR-SpCas9 and 10.0% (55/546) of those screened by RNAi depend on VPS4A for proliferation and survival (Figure S4A, top panel), with more than 40% of pancreatic cancer and pediatric rhabdomyosarcoma (RMS) cancer cell lines demonstrating robust dependence (Figure 2G; Table S1). Additionally, VPS4A was essential in a substantial fraction of bladder, bile duct, lung, ovarian, colon, and esophageal cancer cell lines (Figure 2G). For VPS4B, 12.5% (78/624) of cancer cell lines screened by CRISPR-SpCas9 and 20.9% (146/700) of those screened by RNAi were dependent on VPS4B (Figure S4A, bottom panel), with more than 25% of ovarian, breast, pancreatic, liver, gastric, and bile duct cancer cell lines demonstrating strong dependency (Figure S4B; Table S1).
To confirm the relevance of those findings beyond cancer cell lines, we examined the frequency of VPS4A/B copy loss in patient tumor samples from The Cancer Genome Atlas (TCGA) pan-cancer copy number dataset (Taylor et al., 2018). In 10,712 adult cancers, VPS4B copy loss occurred in 33% (3,546/10,712) of cancers (Figure 2H; Table S1), including 47.5% of pancreatic ductal adenocarcinoma samples. Across the same TCGA dataset, VPS4A copy loss occurred in 27.1% of tumors, with common loss in ovarian, uterine, and sarcoma samples (Figure S4C; Table S1). Across TCGA samples, strong correlations between VPS4B and SMAD4 (R2 = 0.686) and VPS4A and CDH1 (R2 = 0.917) copy numbers were observed (Figure S4D; Table S1).
To expand our analysis to pediatric samples, we surveyed the Dana-Farber Cancer Institute (DFCI) patient targeted-sequencing database (Oncopanel/PROFILE) (Sanchez-Vega et al., 2018) by inferring VPS4B copy number from the neighboring BCL2 gene (~70 Kb) (Figure S4E). Notable VPS4B copy loss was observed in 40% of germ-cell tumors (6/15), 19% of osteosarcomas (6/31), and 9% of brain tumors (16/176) (Figure S4F). As pediatric RMS cell lines were the most enriched for VPS4A dependency (Figure 2G), we investigated the DFCI PROFILE RMS cohort and a published RMS cohort of pediatric patients (Chen et al., 2013) and observed that 5% (2/39; DFCI PROFILE RMS) and 16% (10/62; [Chen et al., 2013]) of RMS tumors harbored partial VPS4B copy loss, which was not subtype specific (Figures S4G and S4H).
Taken together, these data indicate that VPS4A and VPS4B copy loss occurs in many adult and pediatric tumor lineages and suggest that over a third of all human cancers may depend on VPS4A or VPS4B for survival. Given the robust synthetic lethal interaction between VPS4A and VPS4B/SMAD4 loss and the prominence of VPS4B/SMAD4 loss across cancers, we focused subsequent validation studies on VPS4A as a vulnerability in VPS4B-deficient cancers.
VPS4A Validates as a Strong Genetic Dependency in Cancer Cells with Copy Loss of VPS4B
To confirm whether VPS4A inactivation can selectively kill cells with copy loss of VPS4B, we examined whether cancer cells with partial VPS4B copy loss (VPS4Bloss cells) were more sensitive to VPS4A ablation than were cells without VPS4B CNAs (VPS4Bneutral cells). We first evaluated the effect of CRISPR-SpCas9-mediated VPS4A knockout on viability of eight VPS4Bneutral and 10 VPS4Bloss cell lines 7 days after lentiviral transduction. As expected, cell viability was significantly decreased in VPS4Bloss cells infected with any of the three single-guide RNAs (sgRNAs) targeting VPS4A (Figures 3A and S5A; Table S1). We next evaluated three VPS4A short hairpin RNAs (shRNAs) and their paired C9–11 seed controls (Buehler et al., 2012) for their ability to selectively suppress VPS4A expression using a doxycycline-inducible RNAi system. shVPS4A-2 and its paired shSeed2 control were selected for further experiments based on optimal VPS4A knockdown and minimal off-target seed effects (Figures S5B and S5C). Using that pair, RNAi-mediated suppression of VPS4A profoundly reduced proliferation of VPS4Bloss, but not VPS4Bneutral, cancer cell lines (Figures 3B, 3C, and S5C).
Figure 3. Validation of VPS4A as a Dependency in Cancer Cells with Copy Loss of VPS4B.

(A) Seven-day viability assays from eight VPS4Bneutral and 10 VPS4Bloss cell lines stably transduced with CRISPR-SpCas9. Each dot represents the mean viability effect (y axis) of cells infected with the indicated sgRNA (n = 3). Black bars indicate the mean cell viability effect of all three VPS4A sgRNAs. See Method Details. ****p < 0.0001.
(B) Seven-day viability assays from 2 VPS4Bneutral and 5 VPS4Bloss cell lines stably transduced with the shSeed2 control (orange) and shVPS4A-2 (blue) tetracycline-inducible RNAi reagents after treatment with 0.5 μM of doxycycline (dox; 0.222 μg/mL). Each dot represents a technical replicate and shows relative cell viability (y axis) compared with untreated cells. ns, not significant; ****p < 0.0001.
(C) Ten-day proliferation curve of VPS4Bloss SNU213 pancreatic cancer cells stably transduced with the tetracycline-inducible RNAi system for shSeed2 control (orange and brown) or shVPS4A-2 (blue). Cells were either grown in control or 1 mM dox (0.444 μg/mL) medium.
(D) In vivo subcutaneous tumor growth in immune-compromised NOG mice of VPS4Bloss SMSCTR rhabdomyosarcoma (top) and SNU213 pancreatic cancer cells (bottom) stably transduced with the SpCas9 endonuclease and either the shSeed2 control (orange) or the shVPS4A-2 (blue) tetracycline-inducible RNAi systems. ****p < 0.0001.
(E) Kaplan-Meier survival plot of NOG mice bearing subcutaneous SMSCTR (top) or SNU213 (bottom) xenografts described in (D). Crosses indicate censored mice. ****p < 0.0001.
(F) Digitized immunoblot for VPS4A, VPS4B, and Vinculin from SMSCTR (top) and SNU213 (bottom) xenograft tumors in NOG mice described in (D). SMSCTR, 7 days after randomization; SNU213, days 20~25 after treatment start for shSeed2 untreated or day 73 after treatment start for shVPS4A-2, +dox.
(G) Caspase 3/7 apoptosis activity (y axis) over time (x axis) in four cancer cell lines. Cells stably expressing SpCas9 were lentivirally transduced with a control sgRNA (orange) or an sgRNA targeting VPS4A (blue). Caspase 3/7 signal was normalized relative to time-matched uninfected cells. ****p < 0.0001.
(H) Stacked bar plots showing cell cycle distribution of ES2, KP4 (VPS4Bneutral, gray) and SMSCTR, 59M and JR (VPS4Bloss, black) cells using DAPI staining and EdU incorporation analyzed by flow cytometry 4 days after VPS4A ablation by CRISPR-SpCas9. *q < 0.05, **q < 0.01, ***q < 0.001, ****q < 0.0001.
We next investigated whether VPS4A suppression with the inducible RNAi system could impair the growth of established tumor xenografts using human VPS4Bloss SMSCTR (rhabdomyosarcoma) and SNU213 (pancreatic ductal adenocarcinoma) cancer cells. Induction of VPS4A suppression, but not of shSeed2 control, resulted in near-complete tumor regression (SMSCTR) or potent tumor growth inhibition (SNU213) and improved survival in both models (Figures 3D–3F, median survival SMSCTR, 30 versus 74 days; SNU213, 21 versus 63 days).
CRISPR-SpCas9-mediated disruption of VPS4A-induced apoptosis (caspase 3/7 activity) in RMS, head and neck, pancreatic, and ovarian cancer cell lines with VPS4Bloss loss (Figures 3G and S5D). In contrast, no apoptosis induction was observed in the VPS4Bneutral ovarian cancer cell line ES2 after VPS4A disruption (Figure S5E). We also observed consistent G2/M arrest upon VPS4A ablation in VPS4Bloss, but not in VPS4Bneutral, cancer cells (Figure 3H).
These data confirm that VPS4A is critical for proliferation and survival of cancer cells with genomic copy loss of VPS4B.
Altered VPS4B Expression Modulates VPS4A Dependency in Cancer Cells
In both the Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019) and TCGA (Taylor et al., 2018) datasets, VPS4B expression showed robust correlation with VPS4B copy number (Figures 4A, 4B, and S6A; R2 = 0.318 [CCLE], R2 = 0.172 [TCGA]), suggesting that gene dosage drives VPS4B expression and that VPS4Bloss cells express less VPS4B than VPS4Bneutral cells. Similar findings were obtained for VPS4A (Figures 4B, S6B, and S6C; R2 = 0.265 [CCLE], R2 = 0.325 [TCGA]). We next examined quantitative proteomics data from 374 cancer cell lines (Nusinow et al., 2020) and observed a significant decrease in VPS4B protein expression in VPS4Bloss cells (Figure 4C; R2 = 0.248). Significantly reduced VPS4B protein levels in VPS4Bloss cells were experimentally and independently confirmed by quantitative capillary-based immunodetection of VPS4B across 29 cancer cell lines (Figures 4D and 4E; R2 = 0.473).
Figure 4. Altered VPS4B Expression Modulates VPS4A Dependency in Cancer Cells.

(A) Linear regression with 95% confidence interval (blue line) and Pearson’s correlation between VPS4B RNA-seq expression (y axis) and VPS4B relative copy number (x axis) from 1,196 cancer cell lines in the CCLE.
(B) Histogram of Pearson’s correlation coefficients of correlating each gene’s mRNA expression level with its copy number for all 18,749 genes from the CCLE (orange histogram), with VPS4A and VPS4B indicated. The blue histogram shows a null distribution generated by correlating 18,749 random copy number-mRNA expression gene pairs.
(C) Linear regression correlation with 95% confidence interval (blue line) and Pearson’s correlation between VPS4B quantitative mass-spectrometry protein expression (y axis) and VPS4B relative copy number (x axis) from 374 cancer cell lines in the CCLE. y axis represents log2 protein expression of a cell line normalized to expression of the protein in a set of 10 reference cancer cell lines (zero as mean reference value).
(D) Digitized VPS4B and total protein immunoblot from 29 cancer cell lines (n = 14 VPS4Bneutral and 15 VPS4Bloss). Relative VPS4B copy number values from the CCLE are shown.
(E) Linear regression with 95% confidence interval (blue line) and Pearson’s correlation between quantified, normalized VPS4B protein levels from (D) and VPS4B relative copy number across cancer cell lines with neutral (gray) or reduced (black) VPS4B copy number. Violin plot with the average and standard deviation marked for normalized VPS4B protein level in VPS4Bneutral and VPS4Bloss cancer cells. ****p < 0.0001.
(F) VPS4B immunoblot from the parental RD-SpCas9 cancer cell line (VPS4Bneutral) and a mixture of two pools of four monoclonal RD-SpCas9 VPS4B−/− CRISPR-SpCas9 knockout cell lines.
(G) Cell viability of VPS4Bneutral RD-SpCas9 cells (left panel) and two pools of four monoclonal RD-SpCas9 VPS4B−/− cell lines, from (F) and Figure S6D. Each dot represents normalized cell viability from an individual assay well with the indicated sgRNAs (legend), and black bars indicate the mean of each group.
(H) VPS4B immunoblot from the VPS4Bloss JR-SpCas9 cancer cell line and a JR-SpCas9 cancer cell line overexpressing VPS4BWT fused to a V5 protein tag.
(I and J) Cell viability of VPS4Bloss JR-SpCas9 cancer cells (left) and JR-SpCas9 cells overexpressing the indicated controls or VPS4B (I) or VPS4A (J) cDNAs. ORF, open reading frame. Black bars indicate the mean of each group.
See also Figure S6.
To ascertain whether reduction in VPS4B expression sensitizes VPS4Bneutral cells to VPS4A depletion, we used CRISPR-SpCas9 to knockout VPS4B in the VPS4Bneutral non-dependent RMS cancer cell line RD (Figures 4F, S6D, and S6E). We mixed eight VPS4B−/− monoclonal cultures into two distinct pools of four clones and tested each pool’s tolerability to CRISPR-SpCas9-mediated knockout of VPS4A. Upon VPS4A ablation, both VPS4B−/− clone pools showed substantially reduced viability compared with negative controls, indicating that loss of VPS4B was enough to confer dependency on VPS4A in these cells (Figure 4G). Similar results were observed with the original polyclonal VPS4B knockout cultures (Figure S6F). VPS4B overexpression was sufficient to rescue polyclonal VPS4Bloss JR cells from VPS4A suppression (Figures 4H and 4I). Combined, these results demonstrate that VPS4B expression levels modulate dependency on VPS4A.
We next performed exogenous rescue experiments to evaluate the ability of wild-type and loss-of-function VPS4A alleles to rescue VPS4Bloss cancer cells from VPS4A dependency and to confirm the specificity of the VPS4A sgRNAs. We used sgVPS4A-2 and sgVPS4A-3, which targeted intron-exon junctions in VPS4A, to inactivate endogenously, but not exogenously expressed VPS4A variants in VPS4Bloss 59M ovarian cancer and JR rhabdomyosarcoma cells (Figure S6G). We attempted to stably express four different VPS4A variants: wild-type VPS4AWT; VPS4AL64A, which prevents binding of VPS4A’s microtubule interaction and trafficking (MIT) domain to ESCRT-III filaments (Scott et al., 2005a); VPS4AK173Q, which cannot bind ATP (Stuchell et al., 2004); or VPS4AE228Q, which cannot hydrolyze ATP (Scheuring et al., 2001; Tanaka et al., 2002). Stable expression of either ATP mutant (VPS4AK173Q or VPS4AE228Q) was not compatible with long-term cell culture (Figure S6H), suggesting dominant-negative functions for these mutants (Fujita et al., 2003) and indicating that impairment of ATP binding or hydrolysis functionally inactivates not only the mutant VPS4A protein but also co-expressed wild-type VPS4A or VPS4B proteins. Interestingly, both VPS4AWT and VPS4AL64A constructs could fully rescue cell viability upon disruption of endogenous VPS4A by CRISPR-SpCas9 in both JR and 59M cells (Figures 4J and S6I). Rescue by the VPS4AL64A mutant suggests that MIT domain interactions with ESCRT-III filaments are not required to rescue viability after VPS4A depletion.
VPS4A Suppression Leads to ESCRT-III Filament Accumulation, Deformed Nuclei, and Abscission Defects in VPS4Bloss Cancer Cells
We next sought to investigate how VPS4A suppression alters ESCRT function in insensitive (VPS4Bneutral) and sensitive (VPS4Bloss) cancer cell lines using immunofluorescence to study known ESCRT-dependent cellular processes (Figure 5A; Vietri et al., 2020). First, we visualized the core ESCRT-III subunit CHMP4B by confocal immunofluorescence imaging after suppression of VPS4A. After 6 days of VPS4A suppression (Figure 5B), we observed significantly increased formation of bright CHMP4B speckles in the cytoplasm and the nucleus of sensitive cell lines (PANC0403, SNU213, and 59M), but not in the VPS4Bneutral cell line KP4 (Figures 5C and 5D), suggesting that loss of VPS4A/B function leads to accumulation of CHMP4B filaments.
Figure 5. VPS4A Suppression Leads to ESCRT-III Filament Accumulation, Deformed Nuclei, and Abscission Defects in VPS4Bloss Cancer Cells.

(A) Known functions of the ESCRT machinery in membrane biology.
(B) Digitized immunoblot showing VPS4A and Vinculin protein levels in VPS4Bneutral and VPS4Bloss cancer cell lines with the dox-inducible shVPS4A-2 RNAi system after 5 days of treatment with control or 1 μM dox.
(C) Confocal immunofluorescence of CHMP4B in four cancer cell lines with the dox-inducible shVPS4A-2 RNAi system, from (B) imaged after 6 days of treatment. (grayscale, scale bars: 50 μm).
(D) Quantification of CHMP4B speckle formation in untreated (orange) and dox-treated (blue) cells from (C) on multiple random images (n = 3–9). ns, not significant; **q < 0.01, ****q < 0.0001.
(E) Confocal fluorescence imaging of DNA (DAPI, blue) of parental RD-SpCas9 cancer cells euploid for VPS4B copy and clone B2 RD-SpCas9 cancer cells with knockout of VPS4B (Figures 4F, 4G, and S6D). Scale bars: 50 mm. Nuclear surface size by CellProfiler. ****p < 0.0001.
(F) Confocal (immuno)fluorescence of the inner nuclear membrane protein Emerin (Alexa Fluor 561, red) and DNA (DAPI, blue) in four different cancer cell lines, from (B) after 6 days of treatment. Arrows: micronuclei. Scale bars: 50 μm.
(G) (Immuno)fluorescence of cytokinetic bridges and midbodies using tubulin (Alexa Fluor 488, green) and DNA (DAPI) in 3 different cancer cell lines after 4 days of induction of CRISPR-SpCas9-mediated disruption of an intergenic region (sgChr2–2) or VPS4A (sgVPS4A-1). Arrows: cytokinetic bridges.
(H) Quantification of cancer cells connected to neighboring cells by cytokinetic bridges. ns, not significant; **p < 0.01, ****p < 0.0001.
See also Figure S7.
Upon VPS4A suppression, we also noticed nuclear deformation and enlargement in VPS4Bloss cancer cells. This phenotype was also observed in the VPS4B−/− monoclonal RD cell lines we previously created (Figures 4F, 4G, and S6D), even without VPS4A suppression (Figure 5E). We visualized nuclear DNA (DAPI) and the inner nuclear membrane (Emerin staining) and observed that sustained VPS4A suppression gave rise to multilobed, fragmented nuclear structures, micronuclei, and multinucleation in VPS4Bloss pancreatic and ovarian cancer cells (Figure 5F). Three additional rhabdomyosarcoma lines (RD, JR, and SMSCTR) showed higher baseline CHMP4B expression with clear filament formation and some nuclear deformation and multinucleation (Figure S7A). After a 5-day suppression of VPS4A, we observed a slight increase in cytoplasmic CHMP4B speckles and nuclear deformation in VPS4Bloss JR (not significant; q = 0.06) and SMSCTR (significant; q = 0.005) cells, whereas no obvious changes were observed in the VPS4Bneutral RD cells (not significant; q = 0.86) (Figure S7B; 5% FDR).
Because the ESCRT machinery is required for cytoplasmic vesicle trafficking, we also investigated whether VPS4A suppression induced alterations in cytoplasmic membrane structures in VPS4Bloss SNU213 cancer cells. By immunofluorescence visualization of RAB7 (endosomes), LC3B (autophagosomes), and SEC61B (endoplasmic reticulum), we observed that sustained VPS4A suppression for 7 days induced small changes in endosomal and endoplasmic reticulum structures but did not significantly alter the amount or size of autophagosomes (Figures S7C and S7D).
Finally, we observed that cancer cells undergoing VPS4A suppression were still attached to other cells through long cytokinetic bridges, suggestive of abscission defects. Because the ESCRT and VPS4 machinery has a crucial role in cellular abscission, we investigated this phenotype further by immunofluorescence staining of tubulin 4 days after CRISPR-SpCas9-mediated knockout of VPS4A (Figure 5G). Cytokinetic bridging was evident upon visual inspection in VPS4Bloss cancer cells of multiple lineages, and quantification revealed a significantly increased fraction of VPS4Bloss cancer cells were still connected to a neighboring cell by a cytokinetic bridge (Figure 5H).
CRISPR-SpCas9 Screening for Modifiers of VPS4A Dependency Reveals an Important Role for ESCRT Proteins and the ULK3 Abscission Checkpoint Kinase
Because the ESCRT pathway is involved in several processes essential for cancer cell survival, we performed a genome-scale CRISPR-SpCas9 loss-of-function screen in combination with RNAi-mediated silencing of VPS4A to map suppressors and enhancers of cancer cell death elicited upon VPS4A suppression. For that purpose, we used the SNU213 pancreatic cancer cell line, which harbors VPS4B copy loss and is dependent on VPS4A (Figures 3A–3D and S5C). We transduced the Brunello sgRNA library (Doench et al., 2016) into SNU213 cells stably expressing SpCas9 and the dox-inducible shVPS4A-2 RNAi system and conducted the screen in the presence or absence of doxycycline (Figure 6A). Replicate guide and gene-level scores strongly correlated, indicating robust screening performance (Figure S8A–S8D).
Figure 6. CRISPR-SpCas9 Screening Reveals That ESCRT Proteins and the ULK3 Kinase Modify Sensitivity to VPS4A Suppression.

(A) CRISPR-SpCas9 loss-of-function screen in SNU213-SpCas9 pancreatic cancer cells to identify modifiers of VPS4A dependency.
(B) Volcano plot highlighting genes for which knockout altered cell viability of VPS4A-suppressed SNU213-SpCas9 cells. Each dot represents a gene. Difference in log2-normalized mean sgRNA abundance between untreated and dox-treated (VPS4A suppressed) cells (x axis) and the significance of the q-value of this difference (y axis) are shown. Significant genes that sensitized cells to VPS4A suppression are orange; genes promoting resistance to VPS4A suppression are green.
(C) Log2-normalized mean fold changes for individual sgRNA abundance (colored circles) in SNU213 cells for genes scoring as top hits in the differential analysis
(B) of untreated (orange) and dox-treated (blue) samples.
(D) Volcano plot, similar to (B), showing only genes related to the ESCRT machinery.
(E) Manually annotated protein network of the top 50 scoring genes causing sensitivity or resistance to VPS4A suppression from the screen (B). Grey connections indicate strength of interaction between proteins as predicted by STRING (https://string-db.org)
(F) Gene set-enrichment plot of statistical significance (x axis) of Metascape (https://metascape.org) summary gene sets mapping to the top 50 scoring genes from the screen.
SgRNAs enriched in doxycycline-treated (VPS4A-suppressed) cells indicate resistance genes for which knockout promotes cell survival in the presence of RNAi-mediated VPS4A suppression (Figure 6B, green points; Table S1). We identified Argonaute 2 (AGO2), an essential protein for RNAi-mediated gene silencing, as the most highly enriched gene, further validating the robustness of the screen (Figures 6B and 6C). sgRNAs targeting CHMP4B scored as the second most-enriched set of sgRNAs (Figures 6B–6E), supporting the notion that CHMP4B ESCRT-III filaments have a crucial role in mediating the mechanism of antiproliferation conferred by VPS4A suppression. sgRNAs targeting additional members of the ESCRT machinery were also significantly enriched and included sgRNAs targeting the ESCRT-I VPS28 and VPS37B, ESCRT-II SNF8, and ESCRT-III CHMP5 (Figure 6D). Other top-enriched target genes included the Itchy E3 ubiquitin ligase encoding ITCH, the endosome-associated and uncharacterized COMMD7 gene, the RNA G-quadruplex unfolding DEAH/RHA helicase DHX36, and the ETS family transcription factor ELF2 (Figures 6B, 6C, and 6E). We also observed enrichment of additional sgRNAs targeting various ribosomal and nucleoli genes, the endosome-associated COMMD2 and COMMD3 genes and various metabolism-related genes, such as GMPS, IMPDH2, and PGD (Figure 6E; Table S1).
We also observed strong depletion of sgRNAs targeting numerous genes in doxycycline-treated (VPS4A suppressed) cells relative to the untreated cells (VPS4A expressed), indicating genes for which knockout results in a synthetic lethal interaction or a selective enhancement of the antiproliferative effect of VPS4A-suppression (Figure 6B, orange points). Most notably, sgRNAs targeting the VPS4A/VPS4B complex cofactor VTA1, as well as the two accessory ESCRT-III filament genes CHMP1A and CHMP1B scored as strong sensitizers to VPS4A suppression (Figures 6B–6E). Next to VTA1, the ULK3 gene, which encodes an abscission checkpoint kinase, scored as the top synthetic lethal gene. Knockout of TIAL1, which encodes a splicing and apoptosis-related regulatory protein, and RUNX1, which encodes a transcriptional complex-core binding factor, as well as multiple members of other common pathways also scored as potent sensitization mechanisms (Figures 6B–6F).
We next validated whether targeted CHMP1A and CHMP4B ablation can modulate cancer dependency on VPS4A by transient transfection of SpCas9 and sgRNAs targeting those genes or controls into SNU213 cancer cells stably expressing the dox-inducible shVPS4A-2 system. Compared with the control, CRISPR-mediated polyclonal knockout of CHMP1A strongly sensitized SNU213 cells to VPS4A depletion, whereas polyclonal knockout of CHMP4B partially rescued cells from VPS4A depletion (Figures S8E–S8G), confirming the results of our modifier screen (Figure 6B).
Interferon Signaling and CHMP4B Expression Modulate VPS4A Dependency
Although VPS4A dependency correlates strongly with VPS4B copy number and expression (Figures 4F–4I, S2D, and S2E), not every cancer cell line showing VPS4B copy loss is sensitive to VPS4A suppression (Figures S2D and S2E). To search for additional biomarkers, we correlated VPS4A CRISPR dependency scores with gene-level RNA sequencing (RNA-seq) expression values across cancer cell lines (Figure 7A; Table S1). As expected, VPS4A dependency correlated with lower expression of genes located on 18q adjacent to VPS4B (Figure 7A, blue points). Gene-set enrichment analysis on the top 250 anticorrelated genes with VPS4A dependency indicated enrichment of the cellular response to viral infection, cytokine (interleukin) signaling, cell adhesion pathways, and cytoskeletal organization (Figure 7B; Table S1). Top enriched pathways were driven by interferon type 1 and 2 signaling pathways (Table S1), and complementary analysis of quantitative proteomic data (Figure S9A; Table S1) further identified a strong anticorrelation between VPS4A dependency and innate immune response genes, including type 1 interferon (α/β) and interleukin signaling (Figure S9B). This anticorrelation between VPS4A CRISPR dependency and the innate response against virus remained even after controlling for VPS4B loss (Figures S9C and S9D; Table S1), suggesting that increased expression of interferon response genes may enhance cellular sensitivity to VPS4A ablation. Consistent with this hypothesis, the VPS4Bloss cell lines PANC0403 and SNU213, but not the VPS4Bnormal cell line KP4, were sensitized to doxycycline-induced RNAi-mediated VPS4A suppression by co-treatment with interferon-γ (Figures 7C, S9E, and S9F). Interferon-β strongly sensitized SNU213 cells to VPS4A suppression but did not alter sensitivity of PANC0403 cells (Figures 7C and S9F). Notably, PANC0403 cells express lower levels of the type I IFNAR2 receptor and were overall less responsive to interferon-β (Figures S9F and S9G). Interestingly, these observed sensitization effects were specific to interferon because co-treatment of the same cells with the tubulin inhibitor paclitaxel or the Aurora kinase B inhibitor AZD2811 (Barasertib) did not sensitize cells to VPS4A ablation (Figure S9H). Mechanistically, interferon-β and, to a lesser extent, interferon-γ, activated canonical interferon signaling as evidenced by upregulation of interferon-stimulated gene 15 (ISG15) 2 days after treatment in SNU213 and KP4 cells (Figure S9I). Consistent with the lower interferon-β responsiveness of PANC0403, this induction was less pronounced in PANC0403 cells (Figure S9I). Interestingly, after 4-day treatment of SNU213 cells, interferon-β, but not interferon-γ, reduced VPS4A and VPS4B protein levels up to 30%–35% in a dose-dependent manner (Figure 7D), suggesting that type I signaling through interferon-β may diminish VPS4 protein levels and, therefore, sensitize cancer cells to VPS4A suppression, whereas interferon-γ might sensitize cancer cells to VPS4A suppression through an alternative mechanism.
Figure 7. Interferon Signaling and CHMP4B Expression Modulate VPS4A Dependency.

(A) Pearson’s correlation coefficients between gene mRNA expression and CRISPR-SpCas9 VPS4A dependency scores (x axis) and the log-normalized statistical significance (q value) of these interactions (y axis) across 619 CCLE cancer cell lines. Top negatively correlated genes are shown in orange, whereas genes localizing to chromosome 18q are shown in blue. Grey area, values outside 5% FDR q < 0.05.
(B) Gene set-enrichment plot showing statistical significance (x axis) of Metascape (https://metascape.org) summary gene sets mapped to the top 250 genes whose mRNA expression significantly anticorrelated with VPS4A CRISPR dependency scores, orange genes in (A). Gene sets associated with interferon signaling are highlighted (blue).
(C) Six-day cell viability of KP4 and SNU213 pancreatic cancer cell lines stably expressing the dox-inducible shVPS4A-2 RNAi system (see STAR Methods).
(D) Digitized immunoblot and quantification of VPS4A, VPS4B and total protein from SNU213-shVPS4A-2 cancer cells treated for 4 days with interferon-β (blue) or interferon-γ (orange-brown).
(E) Linear regression with 95% confidence interval (blue line) and Pearson’s correlation between prediction values from a 10-fold cross-validated multiple linear regression model (y axis) and observed VPS4A CRISPR dependency scores (x axis). The linear model uses normalized VPS4B, CHMP4B, ISG15, and ITCH mRNA expression values across 621 cancer cell lines to predict VPS4A dependency.
(F) As in (E) but for VPS4B CRISPR dependency scores (x axis).
We next investigated whether a multivariate model incorporating additional features along with VPS4B expression could yield an improved biomarker for VPS4A dependency. For this purpose, we generated a 10-fold cross-validated four-parameter linear model to predict VPS4A dependency by incorporating gene mRNA expression levels of VPS4B, CHMP4B, and ITCH, the two top-scoring modifiers of the CRISPR-VPS4A-dependency screen (Figure 6B), and the interferon response gene ISG15 (Figure S9I). This combined model significantly improved the correlation between predicted and observed VPS4A-dependency scores over models based on each gene independently or any combination of pairs or triplets of these four genes (Figures 7E and S9J; Table S1). This model also outperformed any univariate correlation between VPS4A dependency scores and other gene expression, copy number, or gene dependency features (Table S1). Applying a similar modeling strategy to the VPS4B-dependency prediction showed that addition of CHMP4B expression to VPS4A expression also strongly increased the predictive power over univariate models and correlations (Figures 7F and S9K; Table S1). In summary, these data indicate that the VPS4A-VPS4B synthetic lethal interaction is modified by expression levels of the associated VPS4 paralog and CHMP4B, with expression of interferon-response genes serving to further modulate VPS4A or VPS4B dependency.
DISCUSSION
We have performed a systematic analysis of genome-scale CRISPR-SpCas9 and RNAi screening data from the Cancer Dependency Map (https://www.depmap.org) and identified genetic vulnerabilities that correlate with copy number loss of one of 51 common TSGs. This compendium of synthetic lethal interactions for cancer nominates multiple known and novel targets for potential therapeutic development and further mechanistic study. We described a striking number of synthetic lethal interactions with copy loss of the SMAD4 tumor suppressor on chromosome 18q, one of the most frequent genomic alterations in human cancer. Most notably, we observed that a large subset of SMAD4-deficient cancer cells selectively requires expression of the ESCRT-related ATPase VPS4A for survival due to genomic loss of its paralog VPS4B, located 12.3 Mb downstream of SMAD4 on 18q. We further demonstrated that VPS4A suppression induces apoptosis and cell cycle arrest in in vitro cancer models with reduced copy number of VPS4B and results in profound in vivo tumor regression in subcutaneous cancer xenograft mouse models. Reciprocally, we observed that cancer cells with loss of CDH1 (encoding E-cadherin) on chromosome 16q show collateral loss of VPS4A, which sensitizes these cells to depletion of VPS4B. These results highlight dose-dependent relationships between ESCRT proteins and a critical role for the ESCRT machinery in maintaining cancer cell survival.
The VPS4A-VPS4B paralog dependency is an example of collateral synthetic lethality, where deletion of a neighboring bystander gene leads to cancer dependence on another related gene. Collateral lethality was first described for homozygous loss of ENO1 on chromosome 1p36.23 resulting in dependence on the paralog ENO2 in glioblastoma (Muller et al., 2012). This ENO1-ENO2 synthetic lethal relationship and a second recently described collateral lethality relationship between copy loss of ME2 (adjacent SMAD4) and dependence on its paralog gene ME3 (Dey et al., 2017) were not discovered in our combined analysis (Figures S10A and S10B). The VPS4A-VPS4B paralog dependency relationship has been previously reported in screening data without functional characterization or mechanistic study (McDonald et al., 2017; Viswanathan et al., 2018), and a recent complementary study demonstrated the VPS4A-VPS4B synthetic lethal interaction in a mouse xenograft model of colon cancer (Szymańska et al., 2020).
The SMAD4 tumor suppressor on chromosome 18q21.33 is lost in approximately 33% of human cancer, with particularly high rates of loss in pancreatic cancers (68%), colorectal (71%), and renal cell carcinomas (17%) (Zack et al., 2013). Given its proximity to SMAD4, VPS4B is often co-deleted with SMAD4, thereby sensitizing cells with 18q loss to VPS4A suppression. Conversely, VPS4A is adjacent to CDH1 and is also lost in other tumor types, including cancer lineages in which VPS4B is not commonly deleted, thus sensitizing those tumor cells to VPS4B depletion. Interestingly, we almost never observed complete genomic loss of either VPS4A or VPS4B, even though SMAD4 and CDH1 are sometimes lost completely. In aggregate, we estimate that more than one-third of cancers harbor a partial copy loss of VPS4A or VPS4B and that a diverse spectrum of tumors showing VPS4A or VPS4B loss will be sensitive to depletion or inhibition of the associated paralog.
In response to VPS4A suppression, we observed that cancer cells with genomic loss of VPS4B arrest in G2/M, accumulate CHMP4B-containing ESCRT-III filaments and demonstrate cytokinesis defects and nuclear deformation and micronucleation, ultimately leading to apoptosis. We also observed that VPS4 suppression leads to defects in endosomal and endoplasmic reticulum structure. Cytoplasmic vesicle trafficking is associated with surface receptor recycling and endogenous protein degradation, and disruption of the ESCRT machinery can trigger the accumulation of internalized receptors with their ligands that signal excessively from stalled endosomes resulting in spurious activation of EGFR, Hedgehog, Notch, Toll-like receptor, NF-κB, and many other signaling pathways (Alfred and Vaccari, 2016; Baldys and Raymond, 2009; Mamińska et al., 2016; Matusek et al., 2014). Moreover, VPS4 suppression has also been shown to cause defective mitotic spindle formation, disrupted endocytic and vesicular trafficking, impaired maturation of autophagosomes, increased cell-surface accumulation of receptor tyrosine kinases, defective plasma membrane repair, and even DNA damage (Bishop and Woodman, 2000; Lin et al., 2012; Mierzwa et al., 2017; Morita et al., 2010; Scheffer et al., 2014; Szymańska et al., 2020; Takahashi et al., 2018; Vietri et al., 2015; Zheng et al., 2012). Thus, VPS4 ablation has pleiotropic effects and may affect a multitude of cellular processes that could contribute to profound anticancer activity. Moreover, combined depletion of VPS4A and VPS4B was shown to cause cell-autonomous activation of NF-κB signaling and expression of immunomodulatory cytokines in colorectal cancer cells, leading to caspase-dependent apoptosis, RIPK1-mediated cell death, and potential activation of M1 macrophages in vitro (Szymańska et al., 2020). However, it remains to be determined whether this effect occurs in vivo. Future work will be required to investigate the precise mechanisms of tumor-cell intrinsic and extrinsic mechanisms of cell death in response to VPS4 inhibition.
CHMP4B is the main filament-forming ESCRT-III protein, which requires nucleation and activation to form multimeric filament structures (Christ et al., 2017). We observed an anticorrelation of CHMP4B dependency with VPS4B copy loss in genome-scale screening data (Figures 2E and 2F), suggesting that VPS4B-deficient cells harbor reduced fitness from CHMP4B accumulation and that depletion of CHMP4B may support the proliferation of these cells. In line with that hypothesis, CRISPR-SpCas9 screening for modifiers of VPS4A dependency revealed that knockout of CHMP4B conferred resistance to VPS4A suppression in VPS4B-deficient cells. Furthermore, integration of CHMP4B mRNA expression levels with VPS4B or VPS4A mRNA expression or gene copy number in a multiple linear model resulted in significantly improved prediction of VPS4A and VPS4B dependency, respectively (Table S1). Collectively, these data support the paradigm that cancer cells with increased levels of CHMP4B more strongly require VPS4 activity to maintain viability and that VPS4 suppression induces cell death in part because of excessive CHMP4B accumulation.
Cancer cell sensitivity to VPS4A suppression was also potently enhanced by disruption of regulators of the abscission checkpoint, including genes encoding the ULK3 kinase and the ESCRT-III proteins CHMP1A and CHMP1B. The abscission checkpoint is a genome protection mechanism that relies on Aurora B kinase (AURKB) and ESCRT-III subunits to delay abscission in response to chromosome mis-segregation to avoid DNA damage and aneuploidy. ULK3 is regulated by AURKB and binds to and phosphorylates ESCRT-III proteins, including CHMP1A, CHMP1B, CHMP2A, and IST1, resulting in inhibition of ESCRT-III polymerization and VPS4 activity (Carlton et al., 2012; Thoresen et al., 2014). Consequently, knockout of ULK3, CHMP1A, or CHMP1B would be expected to further disrupt the abscission checkpoint, leading to further impairment in cytokinesis beyond that observed with VPS4A suppression alone. Moreover, as CHMP1A and CHMP1B are regulatory ESCRT-III proteins with among the strongest affinity for VPS4 proteins (Scott et al., 2005a), knockout of these genes likely impairs recruitment of remaining VPS4 proteins to ESCRT-III filaments, enhancing the effect of VPS4 disruption on ESCRT-mediated processes. These findings suggest that inhibition of the ESCRT pathway and blockade of the abscission checkpoint could provide strategies to further enhance sensitivity of cancer cells to VPS4A suppression.
Integrative transcriptomic and proteomic analysis also identified a strong correlation between baseline interferon response gene expression and VPS4A dependency. Notably, the life cycle of many viruses requires the ESCRT/VPS4 pathway (Votteler and Sundquist, 2013), and interferon signaling has been shown to disrupt viral maturation and budding in part because of inhibition of VPS4/ESCRT function (Cabrera et al., 2019; Kuang et al., 2011; Pincetic et al., 2010). In particular, the ubiquitin-like protein interferon-stimulated gene 15 (ISG15) has been reported to be upregulated by the (type I) antiviral interferon response. ISG15 can bind the ESCRT-III subunits CHMP2A, CHMP4B, CHMP5, and CHMP6 to mask their MIT-interacting motif (MIM) domains to prevent the recruitment of VPS4 complexes (Kuang et al., 2011; Pincetic et al., 2010). Furthermore, cellular interferon response has also been shown to halt herpes simplex virus 1 maturation in mouse neurons by downregulation of VPS4 expression and induction of CHMP4B accumulation (Cabrera et al., 2019). Thus, we believe that baseline interferon response signaling in cancer cells may in part suppress VPS4/ESCRT function, thereby enhancing VPS4A dependency in the context of VPS4B loss. Indeed, when VPS4B-deficient cells were treated with interferon-β and interferon-γ to induce interferon signaling, we observed a marked sensitization of these cells to VPS4A depletion, which was not observed with chemotherapeutics. However, whereas interferon-β (interferon type I) strongly induced ISG15 levels and seemed to lower VPS4 protein levels directly, interferon-γ (interferon type II) did not, indicating that interferon-β and interferon-γ affect VPS4/ESCRT function through different mechanisms. Nevertheless, inclusion of the expression of the interferon response gene ISG15 and the immune-related E3 ubiquitin-protein ligase ITCH in a multiple linear model to predict VPS4A dependency improved the predictive power over models incorporating expression of VPS4B or CHMP4B alone or in combination. Thus, our results suggest that the modulation of inflammatory signaling in the context of VPS4 depletion forms a potential combinatorial therapeutic strategy for future consideration.
Finally, we have shown through mutant rescue experiments that the ATPase domain is critical for the function of VPS4A in mediating survival of cells with partial copy loss of VPS4B. Although VPS4A and B demonstrate 80.5% homology, the development of small molecules that differentially target VPS4A in cells with VPS4B loss or VPS4B in cells with VPS4A loss remains a tractable possibility because of small structural differences near the ATP-binding pocket (Inoue et al., 2008; Monroe et al., 2017; Scott et al., 2005b; Su et al., 2017). Moreover, combined inhibition of VPS4A and VPS4B may also prove effective and clinically tolerable given a potential therapeutic window arising from gene dosage alterations and differences in total VPS4A/B levels in tumor versus normal cells. Although currently no specific VPS4A/B inhibitor has been developed, non-specific inhibitors of AAA ATPases have been reported to bind VPS4 proteins (Pöhler et al., 2018; Zhang et al., 2016). Several key preclinical questions that will inform drug development remain to be addressed. For example, although our findings support functional redundancy of VPS4A and VPS4B, distinct functions of each paralog protein may also exist given the wide range of cellular processes regulated by the ESCRT machinery. Moreover, various studies using in vitro experiments or yeast cells (which normally express only a single VPS4 protein) have demonstrated that VPS4A and VPS4B could interact (Huttlin et al., 2015; Scheuring et al., 2001). However, the degree to which VPS4A and VPS4B cooperate and form functional homomeric versus heteromeric complexes in living human (cancer) cells remains to be elucidated.
Given the genomic biomarker prevalence and the potent synthetic lethal relationships demonstrated here, the development of small molecule inhibitors of VPS4 proteins may prove an important advance in the treatment of cancer.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for further information and for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew J. Aguirre (andrew_aguirre@dfci.harvard.edu).
Materials Availability
Plasmids and genetically engineered cancer cell lines that have been generated as part of this study are available upon request.
Data and Code Availability
Scripts and code were written for R, Python and CellProfiler and are available on request. The Public 19Q3 Broad Institute’s Cancer Dependency Map and Cancer Cell Line Encyclopedia datasets (https://depmap.org/portal) are available on fig share: https://doi.org/10.6084/m9.figshare.9170975.v1, https://doi.org/10.6084/m9.figshare.9201770.v3. A complementary dataset with CRISPR Cancer Dependency Scores from the Sanger Institute processed with the Broad Institute’s CERES pipeline is also available: https://doi.org/10.6084/m9.figshare.9116732.v1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
All parental cell lines were from validated sources and procured through the Broad Institute’s Dependency Map Project Cancer Cell Line Encyclopedia banks. SpCas9-expressing cell lines were obtained from the Broad Institute’s Genetic Perturbation Platform. All cell lines were originally obtained from authorized cell line banks including the American Type Culture Collection (ATCC), European Collection of Authenticated Cell Cultures (ECACC), Korean Cell Line Bank (KCLB), Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) and the Japanese Riken cell line bank. Cell lines were cultured in RPMI-1640 with L-glutamine and phenol red (Corning 10–040-CV) and 10% heat-inactivated fetal bovine serum (Sigma-Aldrich F4135) in addition to other supplements when indicated at 37°C and 5% CO2. Cell lines were initially thawed and expanded in their native, manufacturer recommended culture media supplemented with Penicillin-Streptomycin (Thermo-Fisher, 15140122). However, if the native media was not RPMI, cells were adapted and maintained in RPMI with 10% fetal bovine serum for all experiments after initial expansion. Cell lines were validated by STR profiling and tested for mycoplasma using the PCR-based Universal Mycoplasma Detection Kit (ATCC 30–1012K).
Parental cell lines
| Cell Line | Source | Catalog Number | Description |
|---|---|---|---|
|
| |||
| 59M | ECACC | 89081802 | High grade ovarian serous adenocarcinoma metastatic (ascites); female (65 years old) |
| CAL29 | DSMZ | ACC 515 | Urinary bladder transitional cell carcinoma primary; female (80 years old) |
| COV413A | Sigma-Aldrich (ECACC) | 07071905 | Ovarian epithelial-serous carcinoma metastatic (sigmoid colon); female |
| CW9019 | Academic | - | Alveolar rhabdomyosarcoma unknown |
| ES2 | ATCC | CRL-1978 | Ovarian clear cell adenocarcinoma primary; female (47 years old) |
| GSU | RIKEN | RCB2278 | Gastric carcinoma metastatic (ascites); male (37 years old) |
| HEK293T | ATCC | CRL-3216 | Embryonic kidney cells engineered to express the SV40 T-antigen embryonic kidney, female (fetus) |
| HUPT3 | DSMZ | ACC 259 | Pancreatic ductal adenocarcinoma, exocrine metastatic (ascites); male (66 years old) |
| JHOS2 | RIKEN | RCB1521 | High grade ovarian serous adenocarcinoma primary; female (45 years old) |
| JR (JR-1) | Academic | - | Alveolar rhabdomyosarcoma metastatic (lung); female (7 years old) |
| KP4 | RIKEN | RCB1005 | Pancreatic ductal adenocarcinoma, exocrine metastatic (ascites); male (50 years old) |
| MKN74 | JCRB | JCRB0255 | Gastric tubular adenocarcinoma metastatic (liver); male (37 years old) |
| NCIH747 | ATCC | CCL-252 | Cecum adenocarcinoma metastatic (common duct node); male (69 years old) |
| OVISE | JCRB | JCRB1043 | Ovarian clear cell adenocarcinoma metastatic (pelvis); female (40 years old) |
| OVK18 | RIKEN | RCB1903 | Ovarian endometrioid adenocarcinoma metastatic (ascites); female (49 years old) |
| PANC0403 | ATCC | CRL-2555 | Pancreatic ductal adenocarcinoma, exocrine primary; male (70 years old) |
| RD | ATCC | CCL-136 | Embryonal rhabdomyosarcoma Primary (pelvic mass); female (7 years old) |
| SMSCTR | Academic | - | Embryonal rhabdomyosarcoma primary (pelvic mass); male (1 year old) |
| SNU213 | KCLB | 00213 | Pancreatic ductal adenocarcinoma, exocrine primary; male (65 years old) |
| YAPC | DSMZ | ACC 382 | Pancreatic ductal adenocarcinoma, exocrine metastatic (ascites); Male (43 years old) |
| YD38 | KCLB | 60508 | Gingival squamous cell carcinoma primary (lower gingiva); male (67 years old) |
Engineered cancer cell lines
59M SpCas9-BlastR
59M SpCas9-BlastR-RFluciferase-RFP-shSeed (1–3)-HygroR
59M SpCas9-BlastR-RFluciferase-RFP-shVPS4A (1–3)-HygroR
59M SpCas9-BlastR-pLX_TRC313-VPS4A-WT-HygroR
59M SpCas9-BlastR-pLX_TRC313-VPS4A-L64A-HygroR
CAL29 SpCas9-BlastR
COV413A SpCas9-BlastR
COV413A SpCas9-BlastR-shSeed (1–3)-HygroR
COV413A SpCas9-BlastR-shVPS4A (1–3)-HygroR
CW9019 SpCas9-BlastR
ES2 SpCas9-BlastR
GSU SpCas9-BlastR
HUPT3 SpCas9-BlastR
JHOS2 SpCas9-BlastR
JR SpCas9-BlastR
JR SpCas9-BlastR-pLX_TRC313-VPS4B-HygroR
JR SpCas9-BlastR-pLX_TRC313-VPS4A-WT-HygroR
JR SpCas9-BlastR-pLX_TRC313-VPS4A-L64A-HygroR
KP4 shSeed (1–3)-HygroR
KP4 shVPS4A (1–3)-HygroR
MKN74 SpCas9-BlastR
NCIH7474 SpCas9-BlastR
OVISE SpCas9-BlastR
OVK18 SpCas9-BlastR
PANC0403 shSeed (1–3)-HygroR-RFluciferase-NeoR
PANC0403 shVPS4A (1–3)-HygroR-RFluciferase-NeoR
RD SpCas9-BlastR
RD SpCas9-BlastR-shSeed (1–3)-HygroR
RD SpCas9-BlastR-shVPS4A (1–3)-HygroR
RD SpCas9-BlastR-BRD003-VPS4B-PuroR-VPS4B−/−
SMSCTR SpCas9-BlastR
SMSCTR SpCas9-BlastR-shSeed (1–3)-HygroR
SMSCTR SpCas9-BlastR-shVPS4A (1–3)-HygroR
SNU213 SpCas9-BlastR
SNU213 SpCas9-BlastR-shSeed (1–3)-HygroR
SNU213 SpCas9-BlastR-shVPS4A (1–3)-HygroR
SNU213 SpCas9-BlastR-shSeed (1–3)-HygroR-RFluciferase-GFP
SNU213 SpCas9-BlastR-shVPS4A (1–3)-HygroR-RFluciferase-GFP
YAPC SpCas9-BlastR
YD38 SpCas9-BlastR
Subcutaneous mouse xenograft studies
Animal studies were done in accordance with Dana Farber Cancer Institute’s IACUC approved protocol (DFCI 16–015). Human rhabdomyosarcoma SMSCTR and pancreatic cancer SNU213 cells stably expressing SpCas9 and the shVPS4A-2 or shSeed2 tetracycline-inducible RNAi system were maintained in log phase growth in RPMI-1640 with 10% FBS and 300 μg/mL hygromycin. SNU213 cells also carried Renilla firefly luciferase coupled to GFP. Cell lines were confirmed as mycoplasma free and prepared for subcutaneous injection into immuno-deficient female CIEA NOG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) 8–10 weeks of age (Taconic labs). A total of 38 mice were injected once in the flank with 8*106 (SMSCTR) or 6.66*106 (SNU213) cells resuspended in 100 μL PBS without (SMSCTR) or with 50% growth-factor reduced, phenol-red free Matrigel (SNU213) (Corning 356231). Five mice were housed per cage and tumor size was monitored biweekly by caliper measurement after shaving. Then 3–5 weeks after injection, mice were randomized on a rolling basis to doxycycline containing diet (625 ppm) or control diet when tumors reached 200~300 mm3. Once tumors reached > 2000~4000 mm, mice were sacrificed, and tumors were harvested and stored at −80°C. Data was analyzed with GraphPad Prism using Bonferroni-corrected log-rank Mantel-Cox analysis for survival.
To assess on-target knockdown of VPS4A, one mouse for each treatment arm and for both shSeed2 and shVPS4A-2 tumors were selected (total of four mice) and sacrificed 7 days (SMSCTR) or at the end of treatment (SNU213). Tumors were harvested, weighted, and lysed in 15x tumor-weight in volume radioimmunoprecipitation assay lysis buffer (RIPA, Thermo Scientific, 89900) using 2 mL microcentrifuge tubes coupled to Precellys® Evolution bead-mill homogenization at 7,500 rpm for 3× 30 s. After homogenization, tubes were spun down at 4°C and the lysis supernatant was collected and stored at −20°C until immunoblotting. These experiments were repeated a second time using a cohort of eight female NRG mice (SMSCTR; NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ, 007799; The Jackson Laboratory) or ten female NOG Mice (SNU213; NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) (Taconic labs) 7–10 weeks of age, with two flank tumors per mouse. These achieved similar results.
METHOD DETAILS
Discovery of synthetic lethal interactions
To uncover synthetic lethal interactions with somatic CNAs of established TSGs, we analyzed and integrated data from pooled, genome-scale RNA interference (RNAi) and CRISPR-SpCas9 loss-of-function screening for effects on cell proliferation from over 600 well annotated cancer cell lines within the Broad’s Institute Cancer Dependency Map Public 19Q3 release (https://depmap.org/portal) (McFarland et al., 2018; Meyers et al., 2017; Tsherniak et al., 2017). We limited our analysis to 51 common TSGs (Table S1) and correlated Log2-normalized copy number calls for each of these tumor suppressors with normalized, gene-level CRISPR (622 cell lines, 18,333 genes) and RNAi (669 cell lines, 16,905 genes) dependency scores. These correlations were performed in R using the cor.test function. For each pair of tumor-suppressor gene and dependency gene, we calculated the Pearson’s correlation coefficient with its associated p value (F-test). We then applied a 10% false-discovery rate (FDR, Benjamini-Hochberg) using the p.adjust function in R. Gene chromosomal location information was obtained from the Atlas of Genetics and Cytogenetics in Oncology and Haematology (http://AtlasGeneticsOncology.org, downloaded June 2019). When the chromosomal arm-level location of a dependency gene was the same as the arm-level location of the correlated tumor suppressor gene, this interaction was classified as a cis interaction. If the chromosomal arm housing the dependency gene was different from the location of the tumor suppressor gene, the interaction was classified as a trans interaction instead (Data S1). For synthetic lethal interaction analysis, we only incorporated trans dependency genes that showed a positive correlation with copy loss of tumor suppressor genes. Significant synthetic lethal interactions were then cross-referenced between the RNAi and CRISPR datasets to obtain a list of highly confident synthetic lethal interactions (Table S1; Figure 1B). The resulting list was then analyzed further using Microsoft Excel 365 to counter the number of significant synthetic lethal interactions for each tumor suppressor and to generate a heatmap of these interactions. Finalized results were visualized with GraphPad Prism v8.3.0.
Illustrations and diagrams
Drawings detailing the chromosomal location of VPS4A/B, CDH1/SMAD4 and other genes (Figures 1A, 2A, S2C, and S4E), the function of ESCRT (Figures 2C and 5A) and the CRISPR modifier screen workflow (Figure 6A) were created in Adobe Illustrator v24.2.3 (2019, 64-Bit). The alignment of the human VPS4A and VPS4B protein sequences (Figure S2A) was first created using Geneious Prime v2019.1.1 and then edited in Adobe Illustrator.
Analysis of cancer patient copy number data
TCGA VPS4A/VPS4B copy number analysis
Copy number data from 10,712 TCGA patient samples (Sanchez-Vega et al., 2018; Taylor et al., 2018) were downloaded from the Pan-Cancer Atlas (https://gdc.cancer.gov/about-data/publications/pancan-aneuploidy and https://www.cbioportal.org/). Both GISTIC thresholded copy number calls (Mermel et al., 2011) and relative linear copy number calls were used to determine copy number status of VPS4B, VPS4A. Samples with GISTIC copy number values of “−1” (partial loss) or “−2” (deep deletions) or relative Log2 values below “−0.75” were called as at least partial copy loss.
Copy number analysis of DFCI Profile project
We analyzed DFCI’s database of all pediatric, adult pancreatic, adult ovarian, and adult sarcoma patient samples profiled using the OncoPanel targeted sequencing assay (Garcia et al., 2017; Sholl et al., 2016), in accordance with DFCI’s IRB approval. This data is available upon request from the Dana-Farber Cancer Institute or the AACR Project GENIE through a separate agreement. We chose samples with a known/annotated primary tumor type and over 20% histological tumor purity. Though VPS4B is not covered on OncoPanel, we looked for whether a neighboring gene’s copy number status could be used as a surrogate. We first used TCGA Pan-Cancer Atlas copy number calls (Cerami et al., 2012; Gao et al., 2013) to assess the positive and negative predictive values for 240 genes on chromosome 18 to predict concurrent deletion of VPS4B as both the prediction gene and VPS4B copy number status are known. In particular, we used a given gene’s “shallow deletion” copy call to infer whether the VPS4B copy call in that sample was also a “shallow deletion.” Of the chromosome 18 genes that were covered in the DFCI OncoPanel, BCL2 shallow deletion was the best predictor of VPS4B shallow deletion, with 99.7% positive predictive value and 99.9% negative predictive value (Figure S2E). In a similar way, we also assessed positive and negative predictive values for genes on chromosome 16 to infer VPS4A copy number and identified CDH1 as a surrogate marker for partial VPS4A loss in the DFCI OncoPanel.
Rhabdomyosarcoma VPS4B copy number analysis
Illumina whole exome and whole genome paired end sequences were downloaded from published RMS patient samples (Chen et al., 2013). . St. Jude RNaseq data was analyzed (https://github.com/jkobject/VPS4A) using star v2.6.1c, star fusion 1.6.0 and rsem v1.0 with gencode29 reference transcripts. We used GATK4 v4.0.8.0.(DePristo et al., 2011; Van der Auwera et al., 2013) to obtain relative copy number values, which were log2 transformed with a pseudo count of 1. Some patient samples were available multiple times in this dataset. These samples were often derived from multiple tissues, such as primary and metastatic tumor tissue or from patient-derived xenografts. For these cases, VPS4B copy number calls were highly concordant across samples and therefore we removed additional patient samples from the analysis to prevent double counting. Genomics data for the St. Jude Children’s Research Hospital Rhabdomyosarcoma profiling is available upon request through a separate agreement with St. Jude Children’s Research Hospital or through the European Genome-phenome Archive (EGAS00001000256).
Genetic engineering of cancer cell lines
CRISPR-SpCas9 and shRNA validation experiments were performed using lentiviral transduction. Lentiviral transduction was also used to generate stable cell lines expressing SpCas9 (pLEX_311-Cas9v2, EF1a-Cas9v2 SV40-BlastR) or the tetracycline-inducible RNAi systems (shSeed control and shVPS4A targeting systems, Cellecta). Lentiviral particles carrying the genetic material for these constructs were created by co-transfection into HEK293T cells under Geneticin selection with a packaging (psPAX2; Addgene 12260) and VSV-G envelope plasmid (pMD2.G; Addgene 12259) following standard protocol. Cell lines stably transduced with the CRISPR-SpCas9 endonuclease were selected for lentiviral integration and cultured with 1~4 μg/mL Blasticidin S due to the presence of a Blasticidin resistance cassette. Cells stably expressing the shVPS4A or shSeed control RNAi systems were selected and maintained with 300 μg/mL Hygromycin B.
CRISPR-based cell viability assays
Assay design and cell line optimization
For Figure 3A, CellTiter-Glo viability assays were performed with stably expressing SpCas9 cell lines in 96 well plates. Cells were seeded and infected with sgRNA expressing lentivirus in wells on day 0, and selected with puromycin 24 hours later. Cell titer-Glo viability was read out 7 days after plating and infection. Prior to CellTiter-Glo® viability assays, all cell lines were individually optimized for the assay including titrating both cell seeding density and volume of virus used for infection. All lentiviral preps for each sgRNA described below were batch controlled and titrated on three different cell lines representing cell lines with low, medium, and high lentiviral transduction efficiency. Optimal cell seeding densities and viral volumes for infections were then used for all subsequent viability assays using CRISPR.
For Figures S8E and S8F, 1,100,000 SNU213 cells stably expressing SpCas9 were transiently transfected with the indicated sgRNAs (0.66 μg sgRNA plasmid) using Amaxa 2b nucleofection (Program L-023) in 100 μL Buffer L (Lonza). Cells were immediately plated in 6-well plates containing 2 mL RPMI supplemented with 10% FBS and without any antibiotics. Cells were then cultured for 2 days undisturbed before they were harvested using trypsin detachment (0.25% in EDTA, 500 μL per well, 1500 μL RPMI-10% FBS). After collection, cells were counted and plated in equal volumes in T25 flasks. Cells were then grown for another seven days, during which the cells were harvested on day 3, 5 and 7 using trypsin detachment to count the number of viable cells using Trypan blue exclusion and a Vi-CELL XR Cell Viability Analyzer (Beckman Coulter). Remaining cells were replated in T25 and T75 flask until the end of the experiment. At the end, cells were collected for gDNA using the QiaAMP DNA Mini kit (QIAGEN, 51304). according to the manufacturer’s instructions with addition of Rnase A (QIAGEN, 19101). 10 ng of gDNA was then subjected to PCR using CloneAmp HiFi PCR Premix (Takara) following protocol (30 cycles) to amplify the genomic CHMP1A (primers: Fwd 5′-TGAGCTCCACTTGCCTTTCA, Rev 5′-CAGAGGATGCTTGGTGACGT) and CHMP4B (primers: Fwd 5′-CTAGAACCTCACCCTGTGCC, Rev 5′-CATTCCGGGACTAGCACTGC) sgRNA target sites to validate knockout. Amplified gDNA was then purified using the Monarch® PCR & DNA Cleanup Kit (New England Biolabs T1030S) according to the instructions and subjected to Sanger sequencing (Genewiz) using the CHMP1A-seq 5′-AGAATATGGCCCAGGTGACC and CHMP4B-seq: 5′ CGGGTGGACACGTACATGTT primers. The resulting sequences were then analyzed in TIDE. Primers were obtained as single-stranded, standard-desalted DNA from Integrated DNA Technologies.
sgRNAs design and rationale
For viability assays, we used 3 negative control guides (sgLacZ, sgChr2 and sgAAVS1), 3 positive control guides targeting pan-essential genes (sgPOLR2D, sgSF3B1 and sgKIF11), and 3 guides targeting VPS4A (sgVPS4A-1, sgVPS4A-2 and sgVPS4A-3). For negative control guides, we designed two “cutting control” guides that allow CRISPR-SpCas9 to cleave safe regions of the human genome to control for effects of DNA double strand breaks. For sgChr2, the sgRNA targets a gene desert on chromosome 2, which is also the least copy number altered chromosome across cancer (Beroukhim et al., 2010). For sgAAVS1, the sgRNA targets the safe harbor AAVS1 integration locus which is an intronic region in PPP1R12C. sgLacZ represents a non-targeting sequence not found in the human genome. All sgRNA were cloned into a 3rd generation lentiviral vector (Lentiguide-Puro, Addgene 52963) encoding for a human U6 promotor followed by the SpCas9 crRNA backbone sequence and a puro-resistance marker behind the human EF-1a promoter. Lentiviral particles were generated by transfection of the sgRNA plasmid with a packaging (psPAX2; Addgene 12260) and VSV-G envelope plasmid (pMD2.G; Addgene 12259) into HEK293T cells under Geneticin selection following standard protocol. For validation of the effect of CHMP1A and CHMP4B knockout on cell viability of SNU213 cells undergoing VPS4A suppression, sgRNAs were cloned into the non-lentiviral BPK1520 vector (Addgene 65777), which encodes a human U6 promoter followed by the SpCas9 crRNA backbone sequence. Type IIS BsmBI-mediated cloning was used to clone sgRNAs targeting sequences into both vectors (See Addgene for protocols). sgRNA target sequences with cloning adapters were obtained from Integrated DNA technologies (IDT) as single stranded standard-desalted dsDNA oligos. The 20 bp targeting sequences for each SpCas9 sgRNA were:
sgLacZ: 5′-AACGGCGGATTGACCGTAAT (negative control)
sgChr2: 5′-GGTGTGCGTATGAAGCAGTG (negative control)
sgAAVS1: 5′-AGGGAGACATCCGTCGGAGA (negative control)
sgPOLR2D: 5′-AGAGACTGCTGAGGAGTCCA (essential gene control)
sgSF3B1: 5′-AAGGGTATCCGCCAACACAG (essential gene control)
sgKIF11: 5′-CAGTATAGACACCACAGTTG (essential gene control)
sgVPS4A-1: 5′-ACTCACACTTGATAGCGTGG
sgVPS4A-2: 5′-GGGCCGCACGAAGTACCTGG (intron/exon, also for ORF rescue)
sgVPS4A-3: 5′-ATTGTTATTCCCCACCCCTG (intron/exon, also for ORF rescue)
sgCHMP1A: 5′-GAACCTGGACGTCCATACAT
sgCHMP4B: 5′-TCGATGGCACAAGCCATGAA
Viability assay data quality control
For Figure 3A, each assay was required to meet specific quality control metrics. There were 10 unique conditions, one for each of the 9 guides described above, plus a no infection control. There were 6 replicate wells per sgRNA infection, 3 were selected with puromycin and 3 were not. For quality control of raw luminescence from CellTiter-Glo®, we required infection efficiency (puro/no puro selection for each sgRNA) to be at least 80%, and all replicate wells had to be within 2 standard deviations of the mean for that sgRNA infection. Viability reduction from cutting controls, corresponding to DNA double strand breaks, was to be no more than 30% of the non-targeting sgLacZ. We also required SpCas9 activity from each cell line to be greater than 50%, determined by the percent viability reduction of the average of the 3 pan-essential genes (sgPOLR2D, sgSF3B1 and sgKIF11) to negative controls (sgLacZ, sgChr2 and sgAAVS1).
Data normalization
We sought to normalize and scale the viability data in a manner comparable to the DepMap dependency scores (CERES for CRISPR (Meyers et al., 2017) and DEMETER2 for RNAi (McFarland et al., 2018)). Viability scores were normalized on a scale from 0 (the average effect of negative sgRNA cutting controls) to −1 (the average effect of knockout from 3 different pan-essential genes). We first calculated the distance of each well to the average of the two-cutting control sgRNAs (sgChr2 and sgAAVS1).
For each assay well, we then scaled these values from 0 to −1. 0 represents the average viability effect of the cutting controls and −1 represents the average viability effect knockout of the 3 pan-essential genes run in the assay.
Scaling the cell viability effect in this way allows for one to compare across cells lines that have differential responses to “off target” effects of CRISPR e.g., DNA double strand breaks, and differential Cas9 activity when cell lines exhibit differences in maximum number of cells killed by pan essential gene ablation.
Doxycycline induced RNAi reagents
VPS4A targeting shRNA sequences were selected from project DRIVE (McDonald et al., 2017) and cloned into the pRSITEP-U6Tet(shRNA)-EF1-TetRep-2A-Puro vector (Cellecta #SVSHU6TEP-L) for doxycycline inducible shRNA expression. Negative control shRNA seed sequences were generated for each on-target shRNA. Seed sequences contain mutations in base pair positions 9–11 of the shRNA that are intended to remove on-target knockdown, but retain the same seed sequence (bp positions 2–8) and off-target effects (Buehler et al., 2012). The used shRNA target sequences are provided below:
shVPS4A-1: 5′-GCAAGAAGCCAGTCAAAGAGA
shSeed-1: 5′-GCAAGAAGCCTCACAAAGAGA
shVPS4A-2: 5′-CGAGAAGCTGAAGGATTATTT
shSeed-2: 5′-CGAGAAGCTGTTCGATTATTT
shVPS4A-3: 5′-GCCGAGAAGCTGAAGGATTAT
shSeed-3: 5′-GCCGAGAAGCACTAGGATTAT
RNAi-based cell viability assays
For Figure 3B, cell lines stably expressing doxycycline-inducible shVPS4A-2 or sequenced match shSeed-2 control were plated in 96-well plates in culture medium with 300 μg/mL Hygromycin B and in the presence or absence of 1 μM doxycycline. Cells were cultured for 7 days and then assayed for cell number using CellTiter-Glo® luminescence. Relative cell viability was calculated by dividing the doxycycline condition luminescence values by the no doxycycline treatment for each cell line. The culture media was refreshed every 2–3 days to replenish the antibiotics. For Figure 3C, regularly passaged SNU213 cells stably expressing the inducible RNAi system and CRISPR-SpCas9 were harvested with trypsin and counted using a Vi-CELL XR and trypan blue exclusion (Beckman Coulter). Then, 125.000 cells were plated in T25 flask with 6 mL of L-glutamine containing RPMI-1640 medium supplemented with 10% FBS, 300 μg/mL hygromycin B, 2 μg/mL blasticidin S and with the presence or absence of 1 μM doxycycline. Cells were then cultured at 37°C, 5% CO2 and harvested every 2–3 days with trypsin and counted using a Vi-CELL XR and trypan blue exclusion (Beckman Coulter). All remaining cells were replated in fresh culture medium supplemented with antibiotics in new T25 or T75 flasks as the experiment proceeded over a period of 10 days.
Long-term colony formation assays
Cell lines stably expressing doxycycline inducible shVPS4A or seed matched control RNAi reagents were plated in 24 well plates in triplicate with or without 1 μM doxycycline. Three different plating densities (18,000, 9,000 or 4,500 cells/well) were used to determine the optimal plating density. Plates with optimal density were selected as the plating density that generated negative control wells that reached confluence after 14 days of plating. For staining, 24 well plates were fixed with 10% buffered formalin 15 min, washed deionized water, stained with 0.1% crystal violet for 20 min, and washed with deionized water again. For quantification, crystal violet dye was extracted using 1 mL of 10% acetic acid for 20 min, diluted 4-fold with water and 50 mL were plated in triplicate in 96 well plates. Absorbance was quantified at 590 nm.
Immuno-based detection of proteins
Immunoblots were carried out on RIPA-generated lysates following either standard wet-transfer protocols imaged using LI-COR fluorescent secondary antibodies on an Odyssey CLx Imager (LI-COR Biosciences) or by automated capillary-based detection of chemiluminescent signal generated by HRP-conjugated secondary antibodies with a Wes system (Protein Simple, Compass v3.1.7).
Western blotting
Whole cell protein lysates were collected in cold RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) supplemented with cOmplete, Mini Protease Inhibitor Cocktail Tablets (Roche). Cell extracts were cleared by spinning at max speed in tabletop centrifuge at 4°C. Protein concentrations were quantified using BCA Protein Assay Kit (Pierce) and diluted to equivalent concentrations. Lysates were run on NuPAGE 4%–12% Bis-Tris Protein Gels and transferred to PVDF membranes. LI-COR fluorescent secondary antibodies were used to detect proteins using an Odyssey CLx Imager (LI-COR).
Protein Simple capillary-based detection
Cell lysates were prepared similarly using cold RIPA buffer supplemented with proteinase inhibitors. Protein concentrations were determined using the BCA Protein Assay Kit (Pierce). Samples were then diluted to 0.125 mg/mL total protein and prepared according to the instructions of the Protein Simple Wes System. Briefly, lysates were denatured by 5 min boiling at 95°C in sample buffer with 1% SDS and 40 mM DTT. 3–5 μL of denatured sample was then separated and detected using chemiluminescence generated by anti-mouse and anti-rabbit secondary antibodies conjugated to horseradish peroxidase using standard settings and volumes for the 12–230 kDa 25-capillary separation module used with a Wes System (SM-W004; ProteinSimple). For total protein quantification, the total protein detection module (DM-TP01) was used. To normalize signals to total protein stains, the Compass Software v.3.1.7 (Protein Simple) was used to quantify the peak area of total protein (Dropped lines, 115 KDa width). Peak area signals for VPS4A and VPS4B were quantified using Compass Software with a Gaussian fit and a width of 9 kDa. VPS4A and VPS4B peak area signals for a single sample were then divided by the corresponding peak area of the total protein stain for that sample. The total protein normalized VPS4A/VPS4B signals in treated samples were then normalized to the total protein normalized signal of the untreated samples.
Antibody dilutions
| Target | Species | Clonality | Western | Protein Simple | Vendor | Catalog # |
|---|---|---|---|---|---|---|
|
| ||||||
| VPS4A | Mouse | Monoclonal | 1:500 | 1:20 | Santa Cruz Biotechnology | sc-133122 |
| VPS4B | Rabbit | Monoclonal | 1:1000 | 1:33 | Abcam | ab137027 |
| V5 | Rabbit | Monoclonal | 1:1000 | 1:50 | Cell Signaling Technology | 13202S |
| GAPDH | Rabbit | Monoclonal | 1:1000 | - | Cell Signaling Technology | 2118 |
| Vinculin | Rabbit | Monoclonal | - | 1:75 | Abcam | ab129002 |
| ISG15 | Mouse | Monoclonal | 1:10 | Santa-Cruz | sc-166755 | |
| anti-mouse-IRDye 680LT | Goat | NA | 1:10000 | - | LI-COR Biosciences | 926–68020 |
| Anti-rabbit-IRDye 800CW | Goat | NA | 1:10000 | - | LI-COR Biosciences | 926–32211 |
| Anti-mouse-HRP | Goat | NA | - | undiluted | Protein Simple | DM-002 |
| anti-rabbit-HRP | Goat | NA | - | undiluted | Protein Simple | DM-001 |
| Total protein-HRP | - | NA | - | undiluted | Protein Simple | DM-TP01 |
Caspase 3/7 apoptosis assay using IncuCyte
Cell seeding
Stable SpCas9 expressing cells were plated and infected in a manner similar to CRISPR Cell-Titer Glo viability assay described above. Six replicate wells per sgRNA were seeded in clear bottom 96-well plates with EMEM media supplemented with 10% Fetal Bovine Serum and 1x Penicillin - Streptomycin - Glutamine. Standard RPMI-1640 contains riboflavin which can generate fluorescent background with caspase 3/7 signal in the Incucyte assay and was therefore not used. On the same day as cell seeding, cells were infected with sgRNAs expression vectors.
Antibiotic and caspase dye treatment
24 hours after cell plating, three of the six replicates received fresh media with 1 mg/mL of puromycin and three of the six replicates received fresh media without puromycin. All media conditions contained 5 mM of IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (catalog #: 4440). Media selection was performed in the dark due to the light-sensitive nature of the apoptosis reagent. Following selection, the plate was transferred into the IncuCyte® S3 Live-Cell Analysis System (catalog #4647) for imaging. Phase contrast images and green fluorescent channel images were captured using the 10x objective magnification every two hours for a total of 46 time-points. For each well, four images containing both phase contrast and green channel data were obtained.
IncuCyte data analysis
Using the IncuCyte® S3 Analysis System software, cell confluence over time was quantified along with the total area of green (apoptosis positive) objects in mm2/well. Computer generated masks for confluence and green area, trained on a sample set of images across time points and confluency levels, were manually checked for accuracy. The ruleset generated by the training image set was then applied to all images and all time points. Each metric was averaged over the four quadrants per well. First, the green object area metric for each well was divided by the confluence metric for each well, yielding a quantitation of the percent field-of-view positive for apoptosis. These values for each well at each time point were subsequently normalized to well average time-matched no-infection control, no-puromycin condition, which represent unperturbed cell growth. Standard error was computed and plotted using the 3 resulting values per condition, each representing a single well of a 96 well assay plate.
W = well
T = time point
G = total green object area
p = phase contrast confluence
N = no-infection control well average
Cell cycle analysis by flow cytometry
Cell lines stably expressing SpCas9 cells were plated in 6-well plates and infected with sgRNA expressing vectors. Cells were selected with puromycin 24 hours after infection. Four days after infection, cells were labeled with EdU for 1–3 hours and stained with the Click-iT Plus EdU Flow Cytometry Assay Kit following instructions (ThermoFisher, Catalog #: C10632). Cells were co-stained with DAPI and then analyzed by flow cytometry and analyzed with FlowJo v10.
Apoptosis analysis by AnnexinV flow cytometry
Cas9 stable cell lines were plated and infected with the indicated sgRNAs in 6 well plates (cell plating range 2e5 to 5e5 cells per well). Cells were selected with puromycin 24 hours after plating and infection and assayed 5 days after infection by flow cytometry. Inactivation of pan essential gene SF3B1 was used as a positive control for apoptosis induction. Cells were stained using the BD Phar-Mingen FITC Annexin V Apoptosis Detection Kit (catalog #: 556547) according to the manufacturer’s specifications and analyzed by flow cytometry with FlowJo version 10.
Quantification of VPS4B protein levels
VPS4B protein levels were examined using two methods. The first approach compared VPS4B quantitative proteomics across a subset of 375 cancer cell lines (Nusinow et al., 2020) from the CCLE (data available on https://gygi.med.harvard.edu/publications/ccle). For this dataset, tandem mass tagged (TMT) signal-to-noise values from MS3 scans were exported and paired with their MS2 peptide identities. Filtered TMT values were summed and normalized for loading within a ten-plex. Normalized protein abundance values were log2-transformed and mean protein expression per cell line was centered at 0. The second approach involved quantification of VPS4B and total protein across 29 cancer cell line lysates by Protein Simple capillary-based immunoblotting. VPS4B protein level was calculated by extracting VPS4B luminescent peak signal intensity, which were normalized by dividing with the total protein peak signal intensities.
Generation of isogenic VPS4B−/− cell lines
VPS4Aneutral RD cells, stably expressing SpCas9, were infected with a lentivirus expressing sgRNA targeting the sixth exon of VPS4B (sgVPS4B: 5′-CCACTTAGAAACAAGATCAG) using lentiGuide-Puro. As this vector contains a puromycin resistance cassette, cells were selected with 2 μg/mL puromycin. Due to the variable enzymatic activity of SpCas9 across single cells, the infected cells were serially diluted into clear bottom 96-well plates and examined for the presence of single cells. Wells containing single cells were expanded. Sixteen of the resulting clonal populations were interrogated for VPS4B knockout by western blot. DNA extracts from the isogenic cell clones were Sanger sequenced and presence of indels were assessed by the TIDE method of deconvolution (https://tide.nki.nl/ [Brinkman et al., 2014]) using VPS4B exon 6 targeting primers (VPS4B-For: 5′-GCCTAATCATGTTTCAGGTACAGA, VPS4B-Rev: 5′-GGCAAGAGAACACCTTGGAG). Cell lines that were both null by western blot and contained ≥ 80% indels by TIDEseq were selected for further experimentation and pooled into 2 groups of 4 to mitigate the effects of clonal variation.
Overexpression of VPS4B
A pLX_TRC313 ORF expression vector containing VPS4B was procured from the Broad Genetic Perturbation Platform (https://portals.broadinstitute.org/gpp/public/). For VPS4B overexpression, the VPS4Bloss JR cell line stably expressing SpCas9 was infected with pLX_TRC313-VPS4B lentiviral particles and selected with 200 ug/mL hygromycin. Cells were expanded and examined for increased VPS4B expression by western blot. Cells were then placed in 7-day Cell-Titer Glo viability assays as described above in the “CRISPR based cell viability assays with CellTiter-glo” Methods section.
Rescue by VPS4A mutant constructs
A Gateway pDONR223 VPS4A vector was procured from the Broad Genetic Perturbation Platform. Three mutations in VPS4A reported to alter function were selected from the literature to interrogate their ability to rescue cell viability in VPS4A-dependent cell lines following endogenous VPS4A inactivation. VPS4AL64A was reported to prevent MIT domain binding of ESCRT-III filament CHMP1B without disrupting MIT domain folding (Scott et al., 2005a), whereas VPS4AK173Q exhibited dominant negative activity that abrogates ATP binding (Stuchell et al., 2004). Lastly we engineered a VPS4AE228Q mutant that prevents ATP hydrolysis (Scheuring et al., 2001; Tanaka et al., 2002). Primers for site-directed mutagenesis were designed using the NEBasechanger tool (http://nebasechanger.neb.com/) and site-directed mutagenesis was performed with the Q5® Site-Directed Mutagenesis kit (catalog #: E0554S). Following confirmation by Sanger sequencing, we Gateway cloned the mutant constructs on the pDONR223 construct, as well as a wild-type VPS4A construct, into the pLX_TRC313 expression vector. Following confirmation by Sanger sequencing, lentivirus was generated in HEK293T cells. The SpCas9-expressing cell lines JR and 59M were transduced with the mutant expression lentivirus in 6 well dishes. 24 hours after transduction, the culture media was replaced with media containing 200μg/mL hygromycin B. The growth kinetics of the 59M cultures were tracked by repeated cell counts using a Vi-CELL XR (Beckman-Coulter).
Immunostaining and confocal microscopy
Immunostaining was performed on cells plated in 8-well chamber slides and grown for 5–6 days. Cells were fixed and permeabilized using standard paraformaldehyde and triton-based protocols. Immunostaining was performed with validated primary antibodies with Alexa Fluor-conjugated secondary antibodies. DNA was visualized with DAPI and images were obtained with either an upright epifluorescence microscope, or a Nikon Eclipse Ti inverted microscope equipped with a Yokogawa Life Sciences CSU-W1 spinning disc confocal system. Images were quantified using CellProfiler v3.1.9 and ImageJ where indicated.
For plating, cells were seeded into 8-well chamber slides (either Nunc Lab-Tek II Chamber Slides, or Ibidi tissue culture treated μ-Slide 8-Well). For Lab-Tek slides, chambers were coated with 1:50 dilutions of collagen I (Corning Collagen I, catalog #: 354249) and laminin (Sigma, catalog #: L2020) in 1x PBS for 1–3 hours at 37C. Depending on the cell line, cells were seeded at a range of densities from 5,000 to 30,000 cells per chamber. For experiments with doxycycline induced RNAi, cells were first plated in small T25 flasks in the presence or absence of 1 μM doxycycline and treated for 4–5 days (culture media refreshed once) and then harvested using 0.25% trypsin. Collected cells were then plated into chamber slides which were incubated for another 1–2 days, while remaining cells were harvested for VPS4A immunoblot analysis (Figure 5B). For CRISPR-based gene inactivation, SpCas9 stable cell lines were first seeded and infected in 6-well plates. Media was changed 24 hours later and selected with puromycin for an additional 24 hours. Selected cells were then trypsinized from 6-wells plates and moved to chamber slides in media that lacked puromycin and cultured for an additional 3 days.
After incubation, cells were washed with 1x PBS and fixed with fresh 4% paraformaldehyde in 1x PBS for 15 minutes. Fixation was quenched with two washes of 0.2M glycine in 1x PBS. Cells were permeabilized with 0.1~0.2% (v/v) Triton X-100 in 1x PBS for 10~15 minutes at room temperature and blocked with blocking buffer (1% (w/v) bovine serum albumin (BSA) in 1x PBS, or 10% normal goat serum (w/v) in 1x PBS). All primary antibodies were diluted in blocking buffer and incubated overnight at 4C (see below for antibody sources and dilutions). Cells were washed three times with 1x PBS and stained with Alexa Fluor conjugated secondary antibodies (Molecular Probes, ThermoFisher) diluted 1:500 ~1:1000 in blocking buffer. Cells were counterstained with 2~5 μg/mL DAPI in 1x PBS. Cells plated in Nunc Lab-Tek II plates were then incubated for 20 minutes and washed twice with deionized water, and coverslipped with ProLong Gold Antifade Mountant (ThermoFisher). Images of cellular abscission were collected with an upright epifluorescence microscope; other images were captured on a Nikon Eclipse Ti inverted microscope with a Yokogawa Life Sciences CSU-W1 spinning disc confocal.
Antibodies used for immunofluorescence
| Target | Species | Clonality | Dilution | Vendor | Lot | Catalog # |
|---|---|---|---|---|---|---|
|
| ||||||
| CHMP4B | Rabbit | Polyclonal | 4 μg/mL | Proteintech | 13683–1-AP | |
| Emerin | Mouse | Clone 0203 | 0.5 μg/mL | Novus Biologicals | 2602 | NBP2–52877 |
| RAB7 | Rabbit | Clone D95F2 | Cell Signaling Technology | 1 | #9367S | |
| LC3B | Rabbit | Clone D11 | 0.5 μg/mL | Cell Signaling Technology | 13 | #3868P |
| SEC61B | Rabbit | Polyclonal | 1 μg/mL | Sigma-Aldrich | A113820 | HPA049407 |
| Tubulin | Mouse | Clone DM1A | Sigma-Aldrich | T6199 | ||
| anti-rabbit Alexa Fluor 488 | Goat | Polyclonal | 2 μg/mL | ThermoFisher Scientific | 2018309 | A-11008 |
| anti-mouse Alexa Fluor 568 | Goat | Polyclonal | 4 μg/mL | ThermoFisher Scientific | 2014175 | A-11004 |
Quantification of cellular immunostaining
Confocal fluorescence images of CHMP4B speckles and other punctate stains of LC3B, RAB7 and SEC61B were analyzed and quantified after control and doxycycline treatment using a custom image analysis pipeline in CellProfiler v3.1.9 (Kamentsky et al., 2011). Briefly, cells and background were identified using background controlled nuclear (DAPI) and cytoplasmic (CHMP4B-alexa fluor 488/561 or Cellmask Deep Red Stain - ThermoFisher Scientific - H32721) immunofluorescent signals. Fluorescent signals above an adaptive background-controlled threshold were quantified and counted as speckles of > 3 μm and assigned to background or cellular areas. Cell speckle counts were then log2-normalized after adding a value of 1. Data was plotted using GraphPad Prism v8.3.0 and statistical significance was determined using GraphPad’s inbuilt ANOVA test with false-discovery correction using the corrected method of Benjamini and Yekutieli.
CRISPR VPS4A dependency modifier screen
SNU213 pancreatic cancer cells stably transduced with the CRISPR-SpCas9 endonuclease and the shVPS4A-2 inducible RNAi system were infected with a genome-scale Brunello lentiviral sgRNA library (Addgene - 73179). To prepare for infection, SNU213 cells were passaged and upscaled to 300 × 106 cells at 37◦C, 5% CO2 for 1.5 weeks in T75, T175 and then 500 cm2 bioassay plates (Nunc Nunclon Delta Treated Square BioAssay Dish, ThermoFisher Scientific - 166508) with standardized screening medium; L-glutamine containing RPMI-1640 medium supplemented with 10% FBS (Sigma-Aldrich - F4135), 2 μg/mL blasticidin S (SpCas9 selection; GIBCO ThermoFisher Scientific - A1113903) and 300 μg/mL hygromycin B (RNAi system selection; GIBCO ThermoFisher Scientific- 10687010). Cells were harvested with trypsin and counted using a Vi-CELL XR and trypan blue exclusion (Beckman Coulter).
For infection, cells were diluted to 1 × 106 cells / mL in 300 mL L-glutamine containing RPMI-1640 medium without antibiotics. Polybrene (MilliPore-Sigma - TR-1003-G) was added to a final concentration of 8 μg/mL, followed by 16 mL of previously titrated Brunello lentiviral particles to obtain an multiplicity of infection (MOI) of 0.4 and a coverage of 1.500 cells / sgRNA. The cell suspen- sion was mixed by manual pipetting and then plated into twelve 12-well plates at 2 ×106 cells per well. Plates were spinfected for 1.5 hours at 750 x g, 35°C and then further incubated overnight at 37°C, 5% CO2. The following morning, all infected cells were collected by trypsinization and combined into 1 pool. Cells were then diluted in standardized screening medium supplemented with 2 μg/mL puromycin (GIBCO ThermoFisher Scientific - A1113803) and plated in twelve 500 cm2 bioassay plates with 120 mL of cell suspension per plate. Cells were selected with puromycin for 5 days to allow for CRISPR mutagenesis.
Following counting, all cells were harvested by trypsin collection and counted. Then, 40 × 106 cells were plated for each treatment arm in four 500 cm2 bioassay plates (10 × 106 cells per plate, ~500x coverage) with 120 mL medium per plate. Each treatment arm was replicated (4 plates per replicate, 16 plates total) and cells were treated with either standardized screening medium with 2 μg/mL puromycin or standardized screening medium with 2 μM doxycycline and 2 μg/mL puromycin for a period of 2 weeks. During this period, the medium was refreshed every 3 days and cells were monitored and passaged to maintain 40 × 106 cells per replicate.
After treatment, surviving cells were collected by mild trypsinization and collected by centrifugation at 500 x g, supernatant was removed and cell pellets were frozen at −80°C. Genomic DNA from cell pellets (at least 30 million cells per replicate) was purified using silica-membrane-based nucleic acid extraction with the QiaAMP DNA Mini kit (QiaGen 51304). For this purpose, cell pellets for each replicate were suspended in PBS at a concentration of 25 × 106 cells/mL and divided into 1.5 mL tubes containing 5 × 106 cells each. These were then processed according to the QiaAMP DNA Mini kit protocol with 2 modifications. During the proteinase K incubation step, 1 mg/mL RNase A (QiaGen 19101) was added to degrade contaminating cellular RNA. For gDNA elution, spin columns were incubated with 125 μL elution buffer at 56°C for 1 hour before elution by centrifugation, this step was repeated once and both 125 μL fractions were combined. Following gDNA extraction, gDNA concentrations were measured using a NanoDrop 8000 (ThermoFisher Scientific ND-8000-GL).
To determine the sgRNA sequences present in the gDNA of surviving cells, a total of 240 μg of gDNA for each replicate was subjected to PCR amplification using primers with illumina P5 and P7 adapters. Each PCR reaction was performed in 100 μL using 10 μg of input gDNA. PCR was carried out over 28 cycles using the ExTaq hot-start DNA polymerase (Clontech RR001C). Amplified sgRNAs were purified using the AMPure XP magnetic bead purification system (Beckman Coulter, A63880). Amplified products were sequenced by next-generation single short-read 50-cycle Illumina-based sequencing on a HiSeq 2500. Individual sgRNA read counts were sample normalized to read counts per million, adjusted by a value of 1 and then log2 transformed. Log2-normalized sgRNA scores were then compared to the plasmid input library to determine sgRNA fold changes. Fold change scores were then sorted and collapsed into a single gene score. Gene-level scores between screening arms were then compared to identify genes that promote resistance or enhanced sensitivity to VPS4A suppression using the STARS v1.3 algorithm as a python script (Doench et al., 2016). For this purpose, a threshold of 50% was used with a 1000x null distribution to determine statistical significance values. The analysis was performed in both a negative (depletion) and positive (enrichment) direction and the lowest FDR q-value of these two directions was taken for each gene. Q-values below 0.05 were regarded as significant. The top 50 most significant genes were then clustered and visualized using functional associations predicted with STRING v11.0 (https://string-db.org). Clusters were grouped into functional groups by manual inspection. Integrated gene-set enrichment analysis was performed on the top 50 significant genes using metascape’s human-standardized express analysis (GO, Reactome, KEGG, CORUM gene sets) (https://metascape.org, update 2019–08-14). Data was visualized using Graphpad Prism v8.3.0.
Correlations with VPS4A and VPS4B dependency
CRISPR VPS4A or VPS4B dependency scores across 622 cell lines from the Broad Institute’s public 19Q3 dependency map were correlated with gene expression, copy number, proteomics, and other CRISPR dependency scores from the Broad Institute’s 19Q3 Dependency Map/CCLE release. Pearson’s correlations were performed in R using the cor.test function. P values were then corrected for false discovery using the Benjamini-Hochberg method of the p.adjust function in R and q-values were -log10-normalized. The results were plotted using GraphPad Prism v8.3.0. Some of the results were used for gene-set enrichment analysis. For this purpose, symbols for the top significantly (5% FDR) correlating genes were uploaded to Metascape (https://metascape.org, update 2019–08-14) and analyzed for Homo sapiens by incorporating GO, KEGG and Reactome gene sets.
For the multiple linear models, VPS4A and VPS4B CRISPR dependency scores together with RNaseq-determined mRNA expression values for the indicated genes were binned into 10 equal-sized groups of cell lines. A multiple linear model was then trained in R using the specified features using the in-built lm function. For this purpose, the model was first trained on 9 of the bins and then utilized to predict the last bin. This process was repeated 10 times to predict all 10 bins (10-fold cross validation) and all prediction scores were then collected and correlated with the real observed values using Pearson correlation. The results of the 4-parameter multiple linear models were plotted using GraphPad Prism v8.3.0 and statistics for each of the models were extracted and saved into a table using R.
Synergy with interferon or chemotherapeutics
To determine whether interferon treatment cooperates with VPS4A suppression, pancreatic cancer cell lines (KP4, PANC0403 and SNU213) stably expressing the doxycycline-inducible RNAi system against VPS4A (shVPS4A-2) were plated in white-walled 96-well plates at 100~400 cells per well in 100 μL of 10% FBS-supplemented RPMI-1640 with L-glutamine with or without 1 μM doxycycline. Plates were incubated at 37°C, 5% CO2 for 3 days. A 9-point log10 titration of a stock solution (5 μg/mL) of purified recombinant human interferon-β1 or interferon-γ (PeproTech 300–02BC; 300–02) dissolved in 1% BSA containing PBS with 0.3% Tween-20 or a titration of a stock solution of Paclitaxel and AZD2811 (Selleck Chemicals S1150; S1147) in DMSO was then added to the cells using a D300e Digital Dispenser (Tecan) using T8+ and D4+ dispense cassette heads (Hewlett-Packard). At the same time, the doxycycline was refreshed. Cells were then incubated for an additional 3 days at 37°C, 5% CO2. Afterward, 100 μL of premixed CellTiter-Glo (Promega) reagent was added to the wells to lyse cells by shaking the plates at 500 RPM for 10 minutes at room temperature. Luminescence for each well was then measured using an Envision plate reader (Perkin Elmer) to measure cell viability using the ATP-based readout. Luminescence signal for each well was normalized to the average signal from 6 wells treated without interferon for each cell line. These normalized values were then visualized using GraphPad Prism 8.3.0 fitted with a four-parameter log-based non-linear dose-response curve. For each dose point, 3 replicate wells were used, and the experiment was repeated once after two weeks (total of 6 values for each dose point). The 6-day timing of the assay was optimized to reach 50% inhibition in cell viability through VPS4A suppression with doxycycline treatment in the PANC0403 and SNU213 cell lines. Doxycycline was refreshed at day 3 to maintain stringent VPS4A suppression during the experiment.
Effect of interferon on ISG15 and VPS4
To determine whether interferon treatment induces ISG15 protein, KP4 (18,000 cells/well), PANC0403 (18,000 cells/well) and SNU213 (32,000 cells/well) cells stably expressing the tetracycline-inducible shVPS4A-2 RNAi system were plated in duplicate in 6-well plates in 2 mL RPMI supplemented with 10% FBS and 300 μg/mL Hygromycin B with or without 1 μM doxycyline. Cells were incubated for 3 days before the cell culture medium was refreshed. During refreshment, 0, 0.5 or 5 ng/mL of purified recombinant human interferon-β1 or interferon-γ (PeproTech 300–02BC; 300–02) dissolved in 1% BSA containing PBS (stocks 10 ng/mL) was added to the cells. Cells were then incubated for another 2 days and then collected for protein lysates by washing with PBS. After washing, cells were scraped in ice-cold RIPA buffer supplemented with proteinase inhibitors (150 μL / well, two wells per condition). After scraping, RIPA cell lysates were incubated in 1.5 mL microcentrifuge tubes on ice for 30 mins before spinning down to remove cell debris at 20,000 x g for 30 minutes at 4°C.
To determine whether interferon treatment affects VPS4A and VPS4B protein levels, 40,000 SNU213 cells stably expressing the tetracycline-inducible shVPS4A-2 RNAi system were plated in 6-well plates with RPMI supplemented with 10% FBS and a titration of purified recombinant human interferon-β1 or interferon-γ (PeproTech 300–02BC; 300–02). Cells were incubated for two days and then the medium containing interferon was refreshed. Cells were incubated for another two days and then washed with PBS. Following washing, cells were harvested by scraping in ice-cold RIPA buffer supplemented with proteinase inhibitors (150 μL / well, two wells per condition). After scraping, RIPA cell lysates were incubated in 1.5 mL microcentrifuge tubes on ice for 30 mins before spinning down to remove cell debris at 20,000 x g for 30 minutes at 4°C. Finally, for both experiments, protein content in the clear protein lysates (supernatant) were quantified using the BCA Protein Assay Kit (Pierce). Lysates were blotted using capillary-based western blotting and total protein stains were used for VPS4A and VPS4B normalization, which were normalized to the average signal of untreated cells.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data was analyzed using R and python code or using in-built statistical tools in GraphPad Prims v8.3.0. Description and details of the statistical tests are described below and in the method details for some of the individual experiments.
Statistical tests and supplementary information
| Figure | Statistical test |
|---|---|
|
| |
| Figures 1B and 1D | Two-sided p values were calculated using a t-distribution and adjusted using a Benjamini-Hochberg false-discovery rate to obtain q-values (10% FDR). |
| Figure 2D | t distribution (df = n—2) with Benjamini-Hochberg false-discovery rate to obtain q-values (10% FDR). |
| Figure 2F | Two-tailed ANOVA Kruskal-Wallis non-parametric test with Dunn’s correction. Boxes indicate 25th and 75th percentiles with median. Whiskers indicate 10th and 90th percentiles, outliers are shown as circles. |
| Figure 2G | A score < −0.5 for CRISPR-SpCas9 (blue) or RNAi (orange) was used to classify dependency. For each lineage, the number of dependent cell lines over the total number of screened cell lines is shown. |
| Figure 2H | Values show log2-normalized VPS4B copy number relative to the mean sample ploidy. Bars show mean VPS4B copy number ± standard deviation. |
| Figure 3A | Two-tailed unpaired t test on the grand mean of all sgRNAs in the VPS4Bneutral and VPS4Bloss groups. Results from at least two independent experiments are shown. Viability scores are normalized on a scale from 0 (negative controls) to −1 (essential gene controls). |
| Figure 3B | Boxplots indicate 25th-75th percentiles with median and whiskers indicate maximal outlier values. Results show a representative experiment that was repeated at least once. Two-way repeated-measures ANOVA with Sidak’s multiple comparisons test. |
| Figure 3C | Icons represent mean ± standard deviation of 2 independent experiments. Each dot represents a single measurement and each line tracks tumor volume (y axis) of an individual mouse tumor over time (x axis). |
| Figures 3D and 3E | Bonferroni-corrected log-rank Mantel-Cox analysis. For 3D, each dot represents a single measurement and each line tracks tumor volume (y axis) of an individual mouse tumor over time (x axis). |
| Figure 3G | Repeated-measures two-way ANOVA. Dots and errors bars represent means ± standard error of a single experiment using the average of 4 images per well from 3 different wells. |
| Figure 3H | Significance was determined by two-tailed unpaired t tests between each cell cycle stage (G1, G2/M or S) for sgChr2–2 and the individual VPS4A sgRNAs for each cell line individually using false discovery correction by two-stage step-up method from Benjamini, Krieger and Yekutieli. Each dot represents an individual technical replicate of cells treated with control sgRNA (sgChr2–2, orange) or VPS4A targeting sgRNAs shown only for G1 as a reference and to indicate variation between repeats. Stacked bars represent the mean for each sgRNA (n:3) with standard deviation (error bars). |
| Figure 4E, right panel | Two-tailed unpaired Student’s t test with Welch’s correction. VPS4B loss thresholded at Log2 relative copy number < 0.66. |
| Figures 4G, 4I, and 4J | Each dot represents the normalized cell viability from an individual well. Horizontal black bars indicate the mean of each group. For all panels ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 using an unpaired t test comparing the mean viability effect of the negative control sgRNAs to the indicated sgRNA treatment. |
| Figure 5C | Images show grayscale values from a single experiment. |
| Figure 5D | Two-tailed Brown-Forsythe ANOVA with corrected Benjamini-Yekutieli false-discovery rate (FDR). KP4 untreated (100 cells), +dox (67 cells), PANC0403 untreated (311 cells), +dox (395 cells), SNU213 untreated (113 cells), +dox (85 cells), 59M untreated (81 cells), +dox (48 cells). |
| Figure 5E | Two-tailed unpaired t test with Welch’s correction. Volcano plots show total spread, and he median (black bar) with 25%–75% percentiles (dotted lines) are shown. |
| Figure 5F | Images from a single experiment. |
| Figure 5G | Images from a single experiment. |
| Figure 5H | Two-tailed Fisher’s Exact test with Bonferroni correction. Images (n: 9–17) from panel 5G were quantified manually using ImageJ. |
| Figure 6B | The STARS v1.3 algorithm was used to determine significance. For this purpose, a threshold of 50% was used with a 1000x null distribution to determine statistical significance values. The analysis was performed in both a negative (depletion) and positive (enrichment) direction and the lowest FDR q-value of these two directions was taken for each gene. Q-values below 0.05 were regarded as significant. |
| Figure 6C | Log2-normalized mean fold changes are quantified using individual sgRNA abundance at the end of the screen over sgRNA abundance as quantified in the original plasmid DNA stock. Each colored circle represents a different sgRNA targeting the indicated gene (mean of both replicates). Volcano plots show total spread, and the median (black bar) with 25–75% percentiles (dotted lines) are shown. |
| Figure 6F | Numbers after gene set names indicate the number of top 50 screening hits in that set divided by the total amount of genes in the gene set. |
| Figure 7A | Two-sided p values were calculated using a t-distribution and adjusted using a Benjamini-Hochberg false-discovery rate to obtain q-values (5% FDR). |
| Figure 7B | Numbers after gene set names indicate the number of anticorrelated genes that are part of that set divided by the total amount of genes in that gene set. |
| Figure 7C | Each dot represents the mean of 2 experiments performed in triplicate, with error bars indicating standard deviation. |
| Figure 7D | Mean, relative and normalized protein quantification values with standard deviation are plotted in bar graphs (untreated average signal: dashed horizontal line). The experiment was performed twice. |
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| VPS4A (Mouse monoclonal) | Santa Cruz Biotechnology | sc-133122; RRID:AB_2304400 |
| VPS4B (Rabbit monoclonal) | Abcam | ab137027 |
| V5 (Rabbit monoclonal) | Cell Signaling Technology | 13202S; RRID:AB_2687461 |
| GAPDH (Rabbit monoclonal) | Cell Signaling Technology | 2118; RRID:AB_561053 |
| Vinculin (Rabbit monoclonal) | Abcam | ab129002; RRID:AB_11144129 |
| ISG15 (Mouse monoclonal) | Santa-Cruz | sc-166755; RRID:AB_2126308 |
| CHMP4B (Rabbit polyclonal) | Proteintech | 13683-1-AP; RRID:AB_2877971 |
| Emerin (Mouse monoclonal) | Novus Biologicals | NBP2-52877 |
| RAB7 (Rabbit monoclonal) | Cell Signaling Technology | 9367S; RRID:AB_1904103 |
| LC3B (Rabbit monoclonal) | Cell Signaling Technology | 3868P; RRID:AB_2137707 |
| SEC61B (Rabbit polyclonal) | Sigma-Aldrich | HPA049407; RRID:AB_2680750 |
| Tubulin (Mouse monoclonal) | Sigma-Aldrich | T6199; RRID:AB_477583 |
| anti-rabbit-Alexa Fluor 488 (Goat polyclonal) | ThermoFisher Scientific | A-11008; RRID:AB_143165 |
| anti-mouse-Alexa Fluor 568 (Goat polyclonal) | ThermoFisher Scientific | A-11004; RRID:AB_2534072 |
| Anti-mouse-IRDye 680LT (Goat) | LI-COR Biosciences | 926-68020; RRID:AB_10706161 |
| Anti-rabbit-IRDye 800CW (Goat) | LI-COR Biosciences | 926-32211; RRID:AB_621843 |
| Anti-mouse-HRP (Goat) | Protein Simple | DM-002 |
| Anti-rabbit-HRP (Goat) | Protein Simple | DM-001 |
| Total protein-HRP | Protein Simple | DM-TP01 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| E.Coli (DH5α K12) | New England Biolabs | C2987I |
| E.Coli (Stable K12) | New England Biolabs | C3040H |
| E. Coli (One Shot Stbl3 Chemically Competent) | ThermoFisher Scientific | C737303 |
|
| ||
| Chemicals Peptides and Recombinant Proteins | ||
|
| ||
| Puromycin dihydrochloride | Life Technologies | A1113803 |
| Blasticidin S | Life Technologies | A1113903 |
| Hygromycin B | Life Technologies | 10687010 |
| Doxycycline hydrochloride | Sigma-Aldrich | D3447 |
| Geneticin (G-418) | Fisher Scientific | 10131035 |
| Polybrene (Hexadimethrine Bromide) | MilliPore-Sigma | TR-1003-G |
| Gentian Violet (Crystal Violet) | Sigma-Aldrich | C0775 |
| Paraformaldehyde | VWR International | 100504-858 |
| Normal Goat Serum (pure) | Sigma-Aldrich | G9023 |
| 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) | Sigma Aldrich | D9542 |
| Interferon-β1 | PeproTech | 300-02BC |
| Interferon-γ | PeproTech | 300-02 |
| Paclitaxel | Selleck Chemicals | S1150 |
| AZD-2811 (Barasertib) | Selleck Chemicals | S1147 |
|
| ||
| Deposited Data | ||
|
| ||
| Public 19Q3 Cancer Dependency Map – RNAi (https://figshare.com/articles/DEMETER_2_Combined_RNAi/9170975/1) | Broad Institute | https://depmap.org/portal |
| Public 19Q3 Cancer Dependency Map – CRISPR (https://figshare.com/articles/DepMap_19Q3_Public/9201770/3) | Broad Institute | https://depmap.org/portal |
| Public 19Q3 Cancer Cell Line Encyclopedia (https://figshare.com/articles/DepMap_19Q3_Public/9201770/3) | Broad Institute | https://depmap.org/portal |
| Project Score – Sanger Cancer Dependency Map CERES (https://figshare.com/articles/Project_SCORE_processed_with_CERES/9116732/1) | Sanger Institute and Broad Institute | https://depmap.sanger.ac.uk/ |
| Dana-Farber Cancer Institute PROFILE/ OncoPanel (Separate agreement with the Dana-Farber Cancer Institute) | Dana-Farber Cancer Institute | AACR Project GENIE |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| 59M | ECACC | 89081802 |
| CAL29 | DSMZ | ACC 515 |
| COV413A | ECACC Sigma-Aldrich | 07071905 |
| CW9019 | Academic Source | N/A |
| ES2 | ATCC | CRL-1978 |
| GSU | RIKEN | RCB2278 |
| HEK293T | ATCC | CRL-3216 |
| HUPT3 | DSMZ | ACC 259 |
| JHOS2 | RIKEN | RCB1521 |
| JR (JR-1) | Academic Source | N/A |
| KP4 | RIKEN | RCB1005 |
| MKN74 | JCRB | JCRB0255 |
| NCIH747 | ATCC | CCL-252 |
| OVISE | JCRB | JCRB1043 |
| OVK18 | RIKEN | RCB1903 |
| PANC0403 | ATCC | CRL-2555 |
| RD | ATCC | CCL-136 |
| SMSCTR | Academic Source | N/A |
| SNU213 | KCLB | 00213 |
| YAPC | DSMZ | ACC 382 |
| YD38 | KCLB | 60508 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mice - NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac (Female) | Taconic | NOG-F |
| Mice - NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ (Female) | Jackson Laboratory | 007799 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLEX_311Cas9v2 (pXPR_BRD111) | Addgene | 78166 |
| BPK1520 | Addgene | 65777 |
| lentiGuide-Puro (pXPR_BRD003) | Addgene | 52963 |
| psPAX2 | Addgene | 12260 |
| pMD2.G | Addgene | 12259 |
| pRSITEP-U6Tet-(shRNA)-EF1-TetRep-2A-Puro | Cellecta | SVSHU6TEP-L |
| pLX313-Renilla luciferase (pLX_TRC313_Rfluc) | Addgene | 118016 |
| pDONR223 | Broad Institute | pDONR223 |
| Brunello CRISPR-SpCas9 sgRNA library | Addgene | 73179 |
|
| ||
| Software and Algorithms | ||
|
| ||
| GraphPad Prism (https://www.graphpad.com/) | GraphPad Software | Version 8.3.0 |
| Excel and Word 365 | Microsoft | Version 2009 |
| R for Windows with various freely available packages (https://cloud.r-project.org/) | R Foundation | Version 4.0.2 |
| Python for Windows with Pandas, Numpy, Scipy, Statsmodels (https://www.python.org/downloads/windows/) | Python Software Foundation | Version 2.7 |
| Genome Analysis Toolkit https://gatk.broadinstitute.org/hc/en-us | Broad Institute | Version 4.0.8.0 |
| Star | Cold Spring Harbor Laboratory | Version 2.6.1c |
| Star fusion | Broad Institute | Version 1.6.0 |
| Rsem | University of Wisconsin-Madison | Version 1.0 |
| NIS-Elements (Confocal) | Nikon Instruments | Version 4.0 |
| ImageJ (with Java) (https://imagej.nih.gov/ij/) | National Institutes of Health | Version 1.8.0_172 |
| CellProfiler (https://cellprofiler.org/) | Broad Institute Carpenter Lab | Version 3.1.9 |
| IncuCyte® S3 Software | Essen BioScience | Version 2018B |
| Adobe Illustrator | Adobe | Version 24.2.3 |
| FlowJo (https://www.flowjo.com/solutions/flowjo/downloads) | BD Life Sciences | Version 10.4 |
| Geneious Prime (https://www.geneious.com/prime/) | Biomatters | Version 2019.1.1 |
| NEBaseChanger (http://nebasechanger.neb.com/) | New England Biolabs | Version 2019 |
| TIDE (https://tide.nki.nl/) | Netherlands Cancer Institute | Version 3.2.0 |
| STARS (https://portals.broadinstitute.org/gpp/public/software/index) | John Doench | Version 1.3 |
| STRING (https://string-db.org) | String Consortium | Version 11.0 |
| Metascape (https://metascape.org) | Metascape Team | Update 2019–08-14 |
| Empiria Studio Software (https://www.licor.com/bio/empiria-studio/) | LI-COR | N/A |
| Compass for Simple Western (https://www.proteinsimple.com) | Protein Simple | Version 3.1.7 |
|
| ||
| Other | ||
|
| ||
| Cancer Cell Line Encyclopedia Quantitative Proteomics (https://gygi.med.harvard.edu/publications/ccle) | Harvard Medical School Steve Gygi Lab | https://gygi.med.harvard.edu |
| The Cancer Genome Atlas 2018 PanCancer Dataset (https://www.cbioportal.org/) | National Cancer Institute | https://portal.gdc.cancer.gov/ |
| Rhabdomyosarcoma profiling (St. Jude) (Separate agreement with St. Jude Children’s hospital) | St. Jude Children’s Research Hospital | EGAS00001000256 |
| Atlas of Genetics and Cytogenetics in Oncology (http://AtlasGeneticsOncology.org) | Atlas of Genetics and Cytogenetics in Oncology, Haematology | http://AtlasGeneticsOncology.org |
Highlights.
Analysis of synthetic lethal interactions with tumor suppressor gene loss in cancer
VPS4A and VPS4B form a synthetic lethal pair in SMAD4- or CDH1-deleted cancers
VPS4A ablation causes abscission defects, nuclear deformation, and apoptosis
Dependency on VPS4A is modulated by other ESCRT proteins and interferon signaling
ACKNOWLEDGMENTS
This work was funded in part by the Slim Initiative in Genomic Medicine for the Americas (Carlos Slim Foundation) (T.R.G., JB, and F.V.), the Walter and Marina Bornhorst family (T.R.G.), the Lustgarten Foundation (A.J.A. and B.M.W.), the Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research (A.J.A. and B.M.W.), the Doris Duke Charitable Foundation (A.J.A.), the Pancreatic Cancer Action Network (A.J.A.), Team Sciarappa Strong (Jimmy Fund Walk), HL Snyder Foundation (W.C.H.), St. Baldrick’s Foundation Robert J. Arceci Innovation Award (K.S.), PMC Team Eradicate (K.S.), the Rally Foundation for Childhood Cancer (N.V.D.), St. Baldrick’s Foundation (N.V.D. and A.D.D.), the Damon Runyon Foundation for Cancer Research DRG-2384-19 (J.E.N.) and DRG-24-18 (A.D.D.), DOD CDMRP W81XWH-19-1-0281 (A.L.H.), American Cancer Society MRSG-18-202-01-TBG (A.L.H.), and NIH-NCI K08 CA218420-02 (A.J.A.), P50 CA127003 (A.J.A. and W.C.H.), U01 CA224146 (A.J.A. and W.C.H.), U01 CA176058 (W.C.H.), and R35 CA210030 (K.S.).
DECLARATION OF INTERESTS
F.V. and B.R.P. have received research support for this work from Novo Ventures. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, and Merck & Co., Inc. and has research funding from Mirati Therapeutics and Deerfield, Inc. that is unrelated to this work. B.M.W. consults for BioLineRx, Celgene, G1 Therapeutics, and GRAIL and receives unrelated funding from Celgene and Eli Lilly. A.T. consults for Tango Therapeutics. W.C.H. consults for ThermoFisher, Solvasta Ventures, MPM Capital, KSQ Therapeutics, iTeos, Tyra Biosciences, Frontier Medicine, and Paraxel. K.S. has previously consulted for Novartis and Rigel Pharmaceuticals and receives funding through the Novartis/Dana-Farber Cancer Institute Drug Discovery Program unrelated to this project. T.R.G. is a consultant to GlaxoSmithKline, a founder of Sherlock Biosciences, and was formerly a consultant and equity holder in Foundation Medicine, acquired by Roche. T.R.G. receives research funding unrelated to this project from Bayer Healthcare. J.E.N., B.R.P., F.V., and A.J.A. are inventors on pending patent applications related to this work that are owned by the Broad Institute of MIT and Harvard and the Dana-Farber Cancer Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108493.
REFERENCES
- Alfred V, and Vaccari T. (2016). When membranes need an ESCRT: endosomal sorting and membrane remodelling in health and disease. Swiss Med. Wkly 146, w14347. [DOI] [PubMed] [Google Scholar]
- Baldys A, and Raymond JR (2009). Critical role of ESCRT machinery in EGFR recycling. Biochemistry 48, 9321–9323. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx G, Cleton-Jansen AM, Strumane K, de Leeuw WJ, Nollet F, van Roy F, and Cornelisse C. (1996). E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 13, 1919–1925. [PubMed] [Google Scholar]
- Bishop N, and Woodman P. (2000). ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell 11, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B. (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler E, Chen Y-C, and Martin S. (2012). C911: A bench-level control for sequence specific siRNA off-target effects. PLoS ONE 7, e51942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera JR, Manivanh R, North BJ, and Leib DA (2019). The ESCRT-related ATPase Vps4 is modulated by interferon during herpes simplex virus 1 infection. MBio 10, e02567–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JG, Caballe A, Agromayor M, Kloc M, and Martin-Serrano J. (2012). ESCRT-III governs the Aurora B–mediated abscission checkpoint through CHMP4C. Science 336, 220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, et al. ; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project (2013). Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ L, Raiborg C, Wenzel EM, Campsteijn C, and Stenmark H. (2017). Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci 42, 42–56. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A frame-work for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey P, Baddour J, Muller F, Wu CC, Wang H, Liao W-T, Lan Z, Chen A, Gutschner T, Kang Y, et al. (2017). Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 542, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. (1946). Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 31, 269–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, and Vogelstein B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Fujita H, Yamanaka M, Imamura K, Tanaka Y, Nara A, Yoshimori T, Yokota S, and Himeno M. (2003). A dominant negative form of the AAA ATPase SKD1/VPS4 impairs membrane trafficking out of endosomal/lysosomal compartments: class E vps phenotype in mammalian cells. J. Cell Sci 116, 401–414. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, Ligon AH, Sholl LM, Kuo FC, MacConaill LE, et al. (2017). Validation of oncopanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med 141, 751–758. [DOI] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, and Baylin SB (1995). E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 55, 5195–5199. [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med 20, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, et al. (2014). Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 111, 3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Garraway LA, Ashworth A, and Weber B. (2020). Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov 19, 23–38, Published online November 11, 2019. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex Network: a systematic exploration of the human interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Kamikubo H, Kataoka M, Kato R, Yoshimori T, Wakatsuki S, and Kawasaki M. (2008). Nucleotide-dependent conformational changes and assembly of the AAA ATPase SKD1/VPS4B. Traffic 9, 2180–2189. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr. (1999). Choosing anticancer drug targets in the postgenomic era. J. Clin. Invest 104, 1503–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray M-A, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, and Carpenter AE (2011). Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27, 1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim H-G, Schoeb TR, Grizzle WE, and Klug CA (2007). Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 67, 8121–8130. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Z, Seo EJ, and Leis J. (2011). Mechanism of inhibition of retrovirus release from cells by interferon-induced gene ISG15. J. Virol 85, 7153–7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HH, Li X, Chen J-L, Sun X, Cooper FN, Chen Y-R, Zhang W, Chung Y, Li A, Cheng C-T, et al. (2012). Identification of an AAA ATPase VPS4B-dependent pathway that modulates epidermal growth factor receptor abundance and signaling during hypoxia. Mol. Cell. Biol 32, 1124–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang X, Han C, Wan G, Huang X, Ivan C, Jiang D, Rodriguez-Aguayo C, Lopez-Berestein G, Rao PH, et al. (2015). TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520, 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamińska A, Bartosik A, Banach-OrƗowska M, Pilecka I, Jastrzębski K, Zdżalik-Bielecka D, Castanon I, Poulain M, Neyen C, Wolińska-NizioƗ L, et al. (2016). ESCRT proteins restrict constitutive NF-κB signaling by trafficking cytokine receptors. Sci. Signal 9, ra8. [DOI] [PubMed] [Google Scholar]
- Matusek T, Wendler F, Polés S, Pizette S, D’Angelo G, Fürthauer M, and Thérond PP (2014). The ESCRT machinery regulates the secretion and long-range activity of Hedgehog. Nature 516, 99–103. [DOI] [PubMed] [Google Scholar]
- McCullough J, Frost A, and Sundquist WI (2018). Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu. Rev. Cell Dev. Biol 34, 85–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. (2017). Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592.e10. [DOI] [PubMed] [Google Scholar]
- McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, et al. (2018). Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun 9, 4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G. (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa BE, Chiaruttini N, Redondo-Morata L, von Filseck JM, König J, Larios J, Poser I, Müller-Reichert T, Scheuring S, Roux A, and Gerlich DW (2017). Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat. Cell Biol 19, 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe N, Han H, Shen PS, Sundquist WI, and Hill CP (2017). Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 6, e24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Colf LA, Karren MA, Sandrin V, Rodesch CK, and Sundquist WI (2010). Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc. Natl. Acad. Sci. USA 107, 12889–12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al. (2012). Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, et al. (2012). Cancer vulnerabilities unveiled by genomic loss. Cell 150, 842–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jané-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, et al. (2020). Quantitative proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, and Williams RL (2007). Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739. [DOI] [PubMed] [Google Scholar]
- Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, et al. (2017). Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. eLife 6, e23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincetic A, Kuang Z, Seo EJ, and Leis J. (2010). The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol 84, 4725–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöhler R, Krahn JH, van den Boom J, Dobrynin G, Kaschani F, Eggenweiler H-M, Zenke FT, Kaiser M, and Meyer H. (2018). A non-competitive inhibitor of VCP/p97 and VPS4 reveals conserved allosteric circuits in type I and II AAA ATPases. Angew. Chem. Int. Ed. Engl 57, 1576–1580. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. ; Cancer Genome Atlas Research Network (2018). Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, and Jaiswal JK (2014). Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun 5, 5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuring S, Röhricht RA, Schöning-Burkhardt B, Beyer A, Müller S, Abts HF, and Köhrer K. (2001). Mammalian cells express two VPS4 proteins both of which are involved in intracellular protein trafficking. J. Mol. Biol 312, 469–480. [DOI] [PubMed] [Google Scholar]
- Schöneberg J, Lee I-H, Iwasa JH, and Hurley JH (2017). Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol 18, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Gaspar J, Stuchell-Brereton MD, Alam SL, Skalicky JJ, and Sundquist WI (2005a). Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc. Natl. Acad. Sci. USA 102, 13813–13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Chung H-Y, Gonciarz-Swiatek M, Hill GC, Whitby FG, Gaspar J, Holton JM, Viswanathan R, Ghaffarian S, Hill CP, and Sundquist WI (2005b). Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP, et al. (2016). Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenblick A, de Azambuja E, Azim HA Jr., and Piccart M. (2015). An update on PARP inhibitors–moving to the adjuvant setting. Nat. Rev. Clin. Oncol 12, 27–41. [DOI] [PubMed] [Google Scholar]
- Stuchell MD, Garrus JE, Müller B, Stray KM, Ghaffarian S, McKinnon R, Kräusslich H-G, Morham SG, and Sundquist WI (2004). The human endosomal sorting complex required for transport (ESCRT-I) and its role in HIV-1 budding. J. Biol. Chem 279, 36059–36071. [DOI] [PubMed] [Google Scholar]
- Su M, Guo EZ, Ding X, Li Y, Tarrasch JT, Brooks CL 3rd, Xu Z, and Skiniotis G. (2017). Mechanism of Vps4 hexamer function revealed by cryo-EM. Sci. Adv 3, e1700325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Li L, Yang F, Wang X, Fan F, Yang M, Chen C, Li X, Wang H-W, and Sui S-F (2017). Cryo-EM structures of the ATP-bound Vps4E233Q hexamer and its complex with Vta1 at near-atomic resolution. Nat. Commun 8, 16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymańska E, Nowak P, Kolmus K, Cybulska M, Goryca K, Derezińska-WoƗek E, Szumera-Ciećkiewicz A, Brewińska-Olchowik M, Grochowska A, Piwocka K, et al. (2020). Synthetic lethality between VPS4A and VPS4B triggers an inflammatory response in colorectal cancer. EMBO Mol. Med 12, e10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, Serfass JM, Chen L, Gebru M, Chen C, et al. (2018). An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun 9, 2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Fujita H, Katoh H, Mori K, and Negishi M. (2002). Vps4-A (vacuolar protein sorting 4-A) is a binding partner for a novel Rho family GTPase, Rnd2. Biochem. J 365, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. ; Cancer Genome Atlas Research Network (2018). Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33, 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, et al. (1996). Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet 13, 343–346. [DOI] [PubMed] [Google Scholar]
- Thoresen SB, Campsteijn C, Vietri M, Schink KO, Liestøl K, Andersen JS, Raiborg C, and Stenmark H. (2014). ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat. Cell Biol 16, 550–560. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, and Stenmark H. (2015). Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522, 231–235. [DOI] [PubMed] [Google Scholar]
- Vietri M, Radulovic M, and Stenmark H. (2020). The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol 21, 25–42. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Nogueira MF, Buss CG, Krill-Burger JM, Wawer MJ, Malolepsza E, Berger AC, Choi PS, Shih J, Taylor AM, et al. (2018). Genome-scale analysis identifies paralog lethality as a vulnerability of chromosome 1p loss in cancer. Nat. Genet 50, 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J, and Sundquist WI (2013). Virus budding and the ESCRT pathway. Cell Host Microbe 14, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Yang D, Ren X, Lee HH, Im YJ, and Hurley JH (2009). The ESCRT machinery at a glance. J. Cell Sci 122, 2163–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, and Hirohashi S. (1995). Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc. Natl. Acad. Sci. USA 92, 7416–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng C-Z, Wala J, Mermel CH, et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li W, Chu M, Chen H, Yu H, Fang C, Sun N, Wang Q, Luo T, Luo K, et al. (2016). The AAA ATPase Vps4 plays important roles in Candida albicans hyphal formation and is inhibited by DBeQ. Mycopathologia 181, 329–339. [DOI] [PubMed] [Google Scholar]
- Zheng Z-Y, Cheng C-M, Fu X-R, Chen L-Y, Xu L, Terrillon S, Wong ST, Bar-Sagi D, Songyang Z, and Chang EC (2012). CHMP6 and VPS4A mediate the recycling of Ras to the plasma membrane to promote growth factor signaling. Oncogene 31, 4630–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts and code were written for R, Python and CellProfiler and are available on request. The Public 19Q3 Broad Institute’s Cancer Dependency Map and Cancer Cell Line Encyclopedia datasets (https://depmap.org/portal) are available on fig share: https://doi.org/10.6084/m9.figshare.9170975.v1, https://doi.org/10.6084/m9.figshare.9201770.v3. A complementary dataset with CRISPR Cancer Dependency Scores from the Sanger Institute processed with the Broad Institute’s CERES pipeline is also available: https://doi.org/10.6084/m9.figshare.9116732.v1.