

Abstract
Germline pathogenic variants in TP53 are associated with Li-Fraumeni syndrome (LFS), a cancer predisposition disorder inherited in an autosomal dominant pattern associated with high risk of malignancy, including early onset breast cancers, sarcomas, adrenocortical carcinomas, and brain tumors. Intense cancer surveillance for individuals with TP53 germline pathogenic variants is associated with reduced cancer-related mortality. Accurate and consistent classification of germline variants across clinical and research laboratories is important to ensure appropriate cancer surveillance recommendations. Here, we describe the work performed by the Clinical Genome Resource TP53 Variant Curation Expert Panel (ClinGen TP53 VCEP) focused on specifying the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines for germline variant classification to the TP53 gene. Specifications were developed for twenty ACMG/AMP criteria while nine were deemed not applicable. The original strength level for ten criteria was also adjusted due to current evidence. Use of TP53-specific guidelines and sharing of clinical data amongst experts and clinical laboratories led to a decrease in variants of uncertain significance from 28% to 12% compared with the original guidelines. The ClinGen TP53 VCEP recommends the use of these TP53-specific ACMG/AMP guidelines as the standard strategy for TP53 germline variant classification.
Keywords: TP53, variant curation, pathogenic variant, cancer
INTRODUCTION
The TP53 gene encodes p53, a protein with essential roles in genome stability and key cellular functions such as cell cycle, metabolism, apoptosis, senescence and differentiation (Lane, 1992; Zerdoumi et al., 2017). Germline pathogenic variants in TP53 occur in ~70% of individuals with Li-Fraumeni syndrome (LFS) (Varley, 2003), defined by patterns of personal and family history of certain early onset cancers, mainly pre-menopausal breast cancer, bone and soft tissue sarcomas, adrenocortical carcinomas and brain tumors. The cumulative incidence of cancer in LFS families is known to be high, with breast cancer risk reported to be higher than for BRCA1/2 carriers (Shin et al., 2020), and up to 100% risk of any cancer by age 70y for both males and females (Mai et al., 2016). This high penetrance and severe consequences of undiagnosed LFS has led to recommendations for intensive cancer surveillance and other clinical management strategies (Ballinger et al., 2017; Evans, Birch, Ramsden, Sharif, & Baser, 2006; Frebourg, Bajalica Lagercrantz, Oliveira, Magenheim, & Evans, 2020; Hanson et al., 2020; Kratz et al., 2017; Schon & Tischkowitz, 2018; Villani et al., 2016). Reported prevalence estimates range from 1:5,000 to 1:20,000 (Gonzalez, Noltner, et al., 2009; Lalloo et al., 2006), but recent studies suggest the frequency of germline TP53 suspected pathogenic variants may be higher (Amendola et al., 2015; de Andrade et al., 2019; de Andrade et al., 2017).
Germline TP53 genetic testing was initially recommended for individuals meeting Classic LFS (Li et al., 1988) and/or Chompret criteria, most recently revised in 2015 (Bougeard et al., 2015). Advances in sequencing technologies have greatly expanded the use of multi-gene panel testing, and even exome and genome sequencing, in the clinical setting. This has led to an increased number of variants of uncertain significance identified in TP53, including in cancer patients who do not meet LFS criteria, and individuals without cancer (Bittar et al., 2019). Given the significant, not only clinical but also emotional, challenges (Peters et al., 2016; Young et al., 2019) that come with an LFS diagnosis , it is essential to correctly assign TP53 germline variant pathogenicity.
The American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) variant curation guidelines are a series of generic criteria with varying levels of strength for and against pathogenicity, incorporating evidence from multiple data sources (Richards et al., 2015). As part of the directive of the Clinical Genome consortium (ClinGen: https://clinicalgenome.org), specifications of these guidelines for specific gene/disease pairs are developed and documented by a Variant Curation Expert Panel (VCEP), and have been completed for hereditary cancer genes including PTEN (Mester et al., 2018), CDH1 (Lee et al., 2018) and RUNX1(Luo et al., 2019).
Herein, we present the scientific rationale and recommendations of the ClinGen TP53 VCEP to adapt the ACMG/AMP guidelines for the classification of TP53 germline variants and present results from pilot testing of the finalized guidelines.
METHODS
The TP53 VCEP followed ClinGen standard operating procedures (see https://clinicalgenome.org/site/assets/files/3677/clingen_variant-curation_sopv1.pdf). The VCEP formed in 2015 by recruiting international TP53 experts knowledgeable in phenotype, molecular diagnosis and functional processes. Twenty-four members contributed to at least one of three different evidence type working groups: Population/Computational, Functional, and Clinical. Each group reviewed the ACMG/AMP TP53-specifications in detail, incorporated this with critical review of the relevant literature and analyses of relevant data to inform evidence weights, and came to consensus for each specification.
The VCEP members nominated variants for pilot testing the TP53-specific ACMG/AMP guidelines. Specifically, 23 variants were were chosen to cover varying molecular effects and the availability of data to assess the usability of different rule codes. Seven variants from the International Agency on Research in Cancer (IARC) TP53 Database (Bouaoun et al., 2016) were included for their rich phenotypic information, but had no prior variant classifications. Thirteen variants from the ClinVar database (Landrum et al., 2018) were used to balance the spectrum of suspected classifications (with annotation at time of study initiation ranging from benign to pathogenic). In addition to evidence available from public databases, case level evidence available from clinical laboratory databases was provided by relevant VCEP members to the biocurators, including information regarding cancer type(s) and family history, familial variant segregation, and de novo observations. Variant classifications (pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB) and benign (B)) were also provided by the nominating VCEP member, which are referred to as prior expert assertions. Of note, variants sourced from ClinVar had assertions submitted by laboratories with representation on our VCEP, so that laboratory’s assertion in 2017 was considered as the prior expert assertion. Each variant was independently curated by two of the collaborating five biocurators using the original ACMG/AMP guidelines and the TP53 specifications to test user interpretability. Only one of the five biocurators had prior experience with the rule specifications during development. The criteria combinations for a given classification tier were followed as originally proposed (Richards et al., 2015), with the additional combination of very strong plus supporting criteria reaching LP for the TP53-specific guidelines supported by the Tavtigian et al. Bayesian rule combination calculator (Tavtigian et al., 2018). The resulting classifications were compared between biocurators, against prior assertions by the nominating expert(s) or contributing laboratories, and with ClinVar assertions (Landrum et al., 2018) when possible. During this phase, the TP53-specific guidelines were refined, and the final draft and results of the application of the evidence codes to the pilot variants was presented to the ClinGen Sequence Variant Interpretation (SVI) Committee for approval.
RESULTS
TP53-specific variant curation criteria
Final TP53-specific ACMG/AMP guidelines are summarized in Table 1. Of the 28 original criteria, nine (PM3, PM4, PP2, PP4, PP5, BP1, BP3, BP5, and BP6) were excluded due to either limited data to support use of a rule code, irrelevance to TP53, or to avoid redundancy with another criterion specification. Additional details are shown in Supplementary Table S1. The remaining 19 criteria were modified by detailing the content and/or changing the strength level. Rationale for criteria specification are explained below.
Table 1.
Summary of the TP53-specific ACMG/AMP criteria developed by the ClinGen TP53 Variant Curation Expert Panel
| Original ACMG/AMP guidelines | TP53 specifications | |
|---|---|---|
| Rule code | Criteria description | |
| PVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LOF is a known mechanism of disease | Use SVI-approved decision tree to determine the strength of this criterion (refer to Abou Tayoun et al. for more details). |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change |
Use original description with the following additions: PS1:
|
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history | Use SVI-approved scoring system to determine the strength of this criterion (refer to Table 2 for more details) |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product |
PS3: transactivation assays in yeast demonstrate a low functioning allele (≤20% activity) AND there is evidence of dominant negative effect and loss-of-function OR there is a second assay showing low function (colony formation assays, apoptosis assays, tetramer assays, knock-in mouse models and growth suppression assays). PS3_Moderate: transactivation assays in yeast demonstrate a partially functioning allele (>20% and ≤75% activity) AND there is evidence of dominant negative effect and loss-of-function OR there is a second assay showing low function (colony formation assays, apoptosis assays, tetramer assays, knock-in mouse models and growth suppression assays). PS3_Moderate: there is no data available from transactivation assays in yeast BUT there is evidence of dominant negative effect and loss-of-function AND there is a second assay showing low function (colony formation assays, apoptosis assays, tetramer assays, knock-in mouse models and growth suppression assays). Refer to Figure 1 for more details. |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls | Use SVI-approved scoring system to determine the strength of this criterion (refer to Table 3 for more details). This criterion cannot be applied when a variant also meets BA1 or BS1. Refrain from considering probands who have another pathogenic variant(s) in a highly penetrant cancer gene(s) that is a logical cause for presentation. Caveat: Please be mindful of the risk of clonal hematopoieses of indeterminate potential with TP53 variants (Coffee et al., 2017; Weitzel et al., 2017). One should take care to ensure that probands have germline and not mosaic somatic TP53 variants. |
| PM1 | Located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation |
Located in a mutational hotspot defined as:
|
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium | PM2_Supporting: absent from population databases (gnomAD (most up-to-date non-cancer dataset) is the preferred population database at this time http://gnomad.broadinstitute.org). |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant | Excluded (refer to Supplementary Table S1 for more details). |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a nonrepeat region or stop-loss variants | Excluded (refer to Supplementary Table S1 for more details). |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before |
PM5: novel missense change at an amino acid residue where at least two other different missense changes determined to be pathogenic by the TP53 VCEP have been seen before. PM5_Supporting: novel missense change at an amino acid residue where a different missense change determined to be pathogenic by the TP53 VCEP has been seen before. Both criteria require the following additions:
|
| PM6 | Assumed de novo, but without confirmation of paternity and maternity | Use SVI-approved scoring system to determine the strength of this criterion (refer to Table 2 for more details). |
| PP1 | Co-segregation with disease in multiple affected family members in a gene definitively known to cause the disease |
PP1: co-segregation with disease is observed in 3–4 meioses in one family. PP1_Moderate: co-segregation with disease is observed in 5–6 meioses in one family. PP1_Strong: co-segregation with disease is observed >7 meioses in >1 family. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease | Excluded (refer to Supplementary Table S1 for more details). |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) |
PP3: Use original description with the following additions:
|
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | Excluded (refer to Supplementary Table S1 for more details). |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation | Excluded (refer to Supplementary Table S1 for more details). |
| BA1 | Allele frequency is >5% in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium | Allele frequency is ≥0.1% in a non-founder population with a minimum of five alleles (gnomAD (most up-to-date non-cancer dataset)) is the preferred population database at this time http://gnomad.broadinstitute.org). |
| BS1 | Allele frequency is greater than expected for disorder | Allele frequency is ≥0.03% and <0.1% in a non-founder population with a minimum of five alleles (gnomAD (most up-to-date non-cancer dataset) is the preferred population database at this time http://gnomad.broadinstitute.org). |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age |
BS2: observed in a single dataset in ≥8 females, who have reached at least 60 years of age without cancer (i.e. cancer diagnoses after age 60 are ignored). BS2_Supporting: observed in a single dataset in 2–7 females, who have reached at least 60 years of age without cancer. Caveat: Be mindful of the risk of clonal hematopoiesis of indeterminate potential with TP53 variants (Coffee et al., 2017; Weitzel et al., 2017). Individuals with mosaic somatic TP53 variants should not be included as evidence for BS2. |
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing |
|
| BS4 | Lack of segregation in affected members of a family | The variant segregates to opposite side of the family meeting LFS criteria, or the variant is present in >3 living unaffected individuals (at least two of three should be female) above 55 years of age. |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | Excluded (refer to Supplementary Table S1 for more details). |
| BP2 | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern | Variant is observed in trans with a TP53 pathogenic variant (phase confirmed), or there are three or more observations with a TP53 pathogenic variant when phase is unknown (at least two different TP53 pathogenic variants). The other observed pathogenic variants must have been classified using the TP53-specific guidelines. |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function | Excluded (refer to Supplementary Table S1 for more details). |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) |
Same rule description with the following additions:
|
| BP5 | Variant found in a case with an alternate molecular basis for disease | Excluded (refer to Supplementary Table S1 for more details). |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation | Excluded (refer to Supplementary Table S1 for more details). |
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved |
Same description with the following additions:
|
Abbreviations: SVI = Sequence Variant Interpretation; VCEP = Variant Curation Expert Panel
Population/Computational Working Group
Population data
BS1 and BA1 are criteria against pathogenicity based on the frequency of a germline variant in healthy individuals. To define the TP53 variant frequency cutoff for BS1, we calculated the maximum credible population allele frequency as reported by Whiffin et al. (Whiffin et al., 2017). In order not to over-estimate the prevalence of LFS, we used values at the lower end of published estimates of germline pathogenic TP53 variants, namely 1 in 5,000 individuals (Lalloo et al., 2006). Similarly, 30% cancer risk was selected for penetrance in order to allow for the inclusion of hypomorphic allelles, based on the reported penetrance of the Brazilian founder c.1010G>A (p.R337H) variant for LFS malignancies(Achatz & Zambetti, 2016). Genetic heterogeneity was set at 1.0 as TP53 is the only LFS-associated gene. This resulted in BS1 at 0.03% (0.0003). BA1 was then defined as 0.1% (0.001) based on a lifetime penetrance estimate of 70%, holding allelic and genetic heterogeneity at 1.0, and then increasing the derived maximum credible population allele frequency of 0.01% (0.0001) by an order of magnitude to arrive at a cutoff of 0.1% (0.001). For both criteria, a minimum of five alleles in a given population was required to ensure a comparable cohort. The TP53 VCEP specifically recommends the use of the most up-to-date non-cancer dataset of the gnomAD database (Karczewski et al., 2019), as this is the largest control database currently publicly available, and to ignore frequencies in Ashkenazi Jewish and Finnish populations due to founder effects (Kaariainen, Muilu, Perola, & Kristiansson, 2017; Shi et al., 2017).
The PM2 criterion uses absence in controls as evidence towards pathogenicity. Due to the overall rarity of TP53 germline variants (benign or pathogenic), the TP53 VCEP downgraded this criterion to supporting strength level. This criterion would not be applied to rare variants that do not fill additional pathogenic rule criteria and would otherwise meet a classification as a benign or likely benign variant.
PS4 requires case-control analyses, considered impractical for TP53 due to variant rarity and limited number of published studies. We instead developed a proband counting system to assign pathogenicity, based on the number of variant carriers meeting each of the existing clinical criteria for LFS; this is also the recommendation of the ClinGen SVI Committee. A likelihood ratio (LR) towards pathogenicity was calculated by dividing the proportion of carriers meeting classic LFS or Chompret 2015 criteria by the proportion of non-carriers meeting the same criteria, using data from individuals undergoing multigene panel testing at Ambry Genetics (Supplementary Figure S1). Results indicated that one proband with a variant meeting classic LFS or Chompret 2015 criteria would provide enough evidence to reach moderate or supporting (LRs=15.47 or 3.37 as per our analyses, respectively) strength level, respectively (Tavtigian et al., 2018). However, given the width of the confidence intervals, more reduced evidence weights were assigned using a point system based on the number of probands meeting classic LFS or Chompret 2015 (Table 2). Additionally, it was decided that PS4 should not be applied if the variant also meets the population rules BS1 or BA1, to avoid coincidental accumulation of proband counts for common variants, following the approach previously developed for PTEN (Mester et al., 2018).
Table 2.
Point system created for the specifications of the PS4 rule*
Classic LFS
|
PS4 Evidence Strength | # of Points Required |
| PS4 | 4 or more points | |
| PS4_Moderate | 2–3 points | |
| PS4_Supporting | 1 point |
Not to be used if a variant also meets BS1 or BA1 rules.
The BS2 criterion uses the presence of a variant in unaffected adults as evidence against pathogenicity. Two independent datasets (Ambry Genetics and GeneDx) were used to calculate how many observations of cancer-free adults at age 60 equated to strong evidence against pathogenicity. The LR towards pathogenicity was calculated by comparing the proportion of these individuals with a known TP53 pathogenic variant versus individuals without a TP53 pathogenic variant (Supplementary Table S2). The LRs estimated for observation of a variant in one healthy individual were 0.66 (Ambry dataset) and 0.28 (GeneDx dataset). Selecting the more conservative LR of 0.66, two to seven healthy individuals were considered necessary to apply BS2_Supporting (LRs ranging from 0.44 to 0.06), and eight or more to apply BS2 (LRs lower than 0.04) (Tavtigian et al., 2018). Given that the dataset used for this analysis included mostly females, and the associated risk of pre-menopausal breast cancer (Mai et al., 2016) for female TP53 carriers elevates their cancer risk compared with male TP53 carriers, it was stipulated that this criterion should be applied to female carriers only.
Computational and predictive data
PP3 and BP4 are commonly used criteria related to predictions using bioinformatic tools. Based on previous findings (Fortuno, James, Young, et al., 2018), an optimized version of Align-GVGD (Mathe et al., 2006) combined with BayesDel (Feng, 2017) (both included in the IARC TP53 Database R20 (Bouaoun et al., 2016)) were selected for bioinformatic prediction of TP53 missense variant pathogenicity. To assess prediction of effects on splicing, we suggest the use of a metapredictor, such as SpliceAI (Jaganathan et al., 2019) or VarSEAK (https://varseak.bio/).
The BP7 criterion for silent variants was expanded to specify the use of a metapredictor to exclude the use of this code for variants predicted to have any effects on splicing.
The strength level for PM5, which relies on previous observations of pathogenic variants at a given location as evidence of pathogenicity for other variants at the same location, was assessed as follows: the 493 non-functional variants from yeast transactivation assays (Kato et al., 2003) were assessed for the number of additional variants at the same amino acid residue that were also reported as non-functional, and compared to the number of functional or super trans variants (N=1239) at that position to generate a LR towards pathogenicity (Supplementary Table S3). Results indicated PM5 was applicable if two other pathogenic variants have been seen at a given amino acid residue (LR=6.46), but should be downgraded to supporting strength level if only one other pathogenic variant has been seen at the same residue (LR=2.90) (Tavtigian et al., 2018). Further, the following restrictions were added: (i) known P/LP variants must be based on classifications using the TP53-specific guidelines; (ii) variants using this rule must have equal or higher Grantham score (Grantham, 1974) than at least one pathogenic variant observed at that codon; (iii) this criterion cannot be used for any variant for which PM1 has been applied; (iv) splicing effects are excluded based on bioinformatic evidence from a metapredictor.
PS1 may be applied as strong strength if effects on splicing due to the nucleotide change are excluded using splicing assay data, or downgraded to PS1_Moderate if only bioinformatic predictions are available as evidence against aberrant splicing. The comparative variant must have been classified as P/LP using the TP53-specific guidelines.
PVS1 is the only original criterion with a very strong strength level for pathogenicity, which applies to loss of function variants. The ClinGen SVI Committee has published further guidance on this rule (Abou Tayoun et al., 2018), which the TP53 VCEP has agreed to follow, including applying relative strengths for different types of null variants based on characteristics, such as variant type and location.
Functional Working Group
Functional data
There are multiple mechanisms by which p53 function may be altered. To date, there are only three published systematic functional studies of p53 missense variants, which measure relevant disease mechanisms: transactivation activity (Kato et al., 2003), loss of function (LOF) (Giacomelli et al., 2018; Kotler et al., 2018) and dominant-negative effect (DNE) (Giacomelli et al., 2018). To elaborate the specifications of the functional-related criteria PS3 and BS3, and in agreement with SVI recommendations for application of the functional criteria (Brnich et al., 2019), we first assessed the relative performance of these assays as positive and negative predictors of variant pathogenicity (i.e., clinical calibration) using the Matthews correlation coefficient; clinical reference sets were assumed pathogenic missense variants (present in classic LFS probands in the IARC TP53 Database R19 and absent in controls, N=52) and assumed benign missense variants (in controls from non-cancer gnomAD v2.1.1 or FLOSSIES (https://whi.color.com/) at a frequency higher than 0.0001 and not found in patients, N=31) (Supplementary Table S4). In addition, we also calculated LRs towards pathogenicity for different combinations of functional results to assist application of strength levels (Supplementary Table S5). Following these results, a conservative decision tree considering availability and relative performance of the different assays is shown in Figure 1. Of note, this decision tree also considers other non-systematic functional assays, especially when those show conflicting evidence, as noted.
Figure 1.
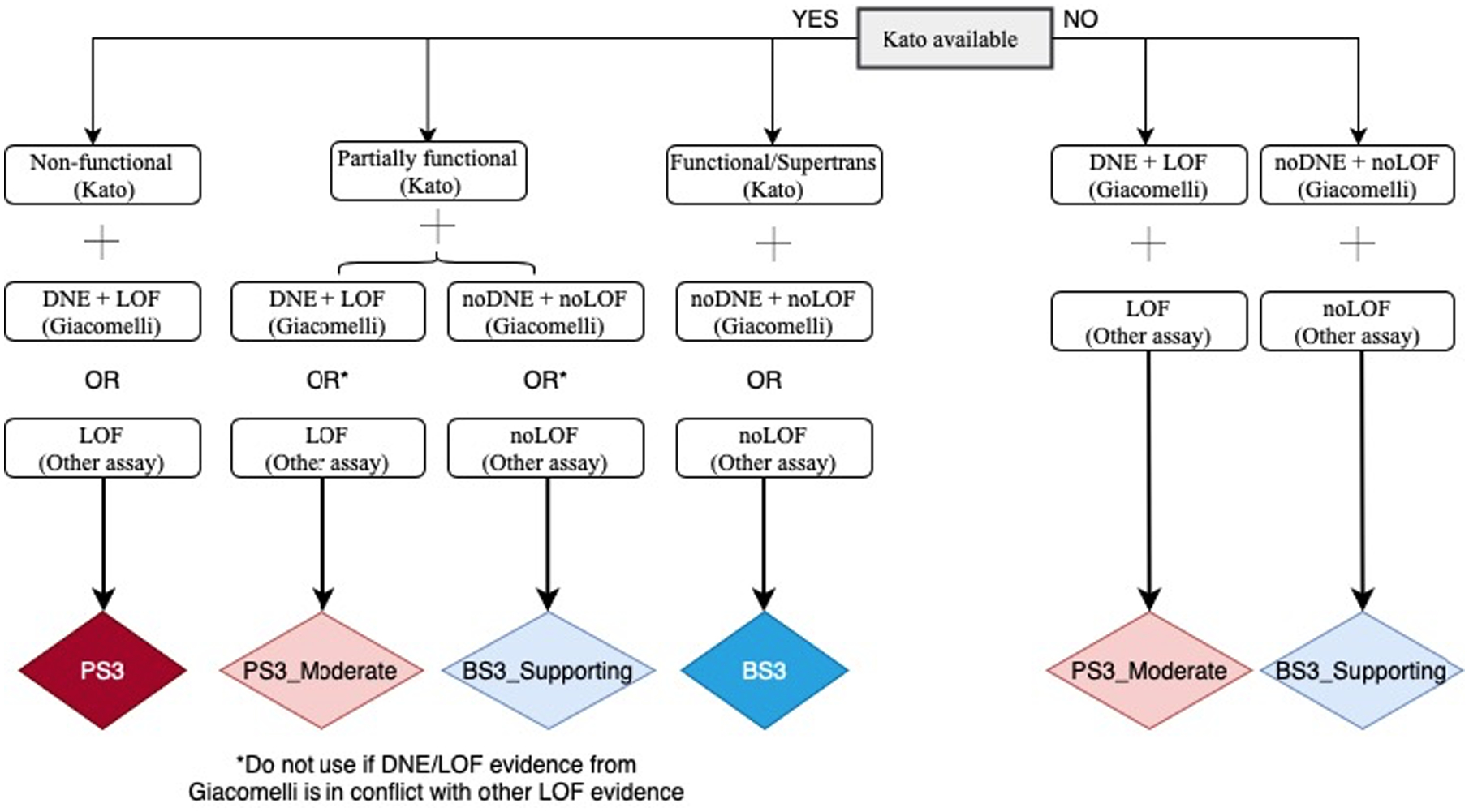
Flow chart for the specifications of PS3 and BS3 criteria.
Non-functional (Kato) = median transactivation activity ≤20%; Partially functional (Kato) = median transactivation activity >20 and ≤75%; Functional/Supertrans (Kato) = median transactivation activity >75%; DNE+LOF (Giacomelli) = p53WTNutlin3 Z-score ≥ 0.61 and Etoposide Z-score ≤ −0.21 = noDNE+noLOF (Giacomelli) = p53WTNutlin3 Z-score < 0.61 and Etoposide Z-score > −0.21. Other assays are available in IARC TP53 Database or original publications, and include in vitro growth assays in H1299 human cells from Kotler et al., (2018) with RFS score > −1.0 for LOF and RFS score < −1.0 for noLOF; or colony formation assays, growth suppression assays, apoptosis assays, tetramer assays, knock-in mouse models.
* If a variant does not match any of the possibilities shown, it is considered to have “no evidence to review” and no functional criterion can be applied.
Abbreviations: DNE = Dominant-negative effect; LOF = Loss-of-function.
Hotspot data
The second part of the PM1 criterion related to protein regions was considered not applicable, as there is no known functional domain without benign variation in TP53 given the evidence available. However, there are several well-described hotspots in TP53, occurring at amino acid positions 175, 245, 248, 249, 273, and 282 (Bouaoun et al., 2016; Fortuno, Pesaran, et al., 2019), for which PM1 is applicable. Additionally, analyses of tumor DNA sequencing have identified a large number of TP53 variants as somatic hotspots, with information available at cancerhotspots.org (Chang et al., 2016; Chang et al., 2018). Following published recommendations from the ClinGen Germline/Somatic Variant Curation Subcommittee (Walsh et al., 2018), it was specified that the PM1 criterion can also apply to somatically detected hotspots with ≥10 occurrences in cancerhotspots.org. This information is annotated in the IARC TP53 Database R20 (Bouaoun et al., 2016).
Clinical Working Group
Segregation data
The original ACMG/AMP criteria use segregation as supporting strength for pathogenicity (PP1), allowing for stronger evidence if there is more segregation data, or a strong strength of evidence against pathogenicity when there is lack of segregation (BS4). Given the wide spectrum of cancer types that have been reported for TP53 carriers (Caron et al., 2016; Kratz et al., 2017; Olivier, Hollstein, & Hainaut, 2010), the criteria were specified based only on the number of meioses of any LFS-associated cancer type, and number of families reported. Based on the gradations created previously for PTEN, the resulting TP53 specifications for PP1 are detailed in Table 1.
For BS4, which uses lack of segregation as evidence against pathogenicity, we considered potential issues due to the high de novo rate reported for TP53 (Gonzalez, Buzin, et al., 2009), and specified use under these two scenarios: the variant segregates to the side of the family that does not meet LFS criteria, or the variant is present in three or more living unaffected individuals (where at least two of three are female) above 55 years of age (age specification consistent with Chompret 2015 criteria).
De novo data
The TP53 VCEP provides guidance for assigning the strength for the original de novo PS2 and PM6 criteria that should be based on the type of cancer and its relevance to the TP53 spectrum. This was accomplished using a point system incorporating parentage and proband cancer type which also allows for combination of points if there are multiple de novo reports of the same variant (Table 3).
Table 3.
Point systems created for the specifications of the PS2 and PM6 criteria and TP53-associated cancers with different strength levels*
Strong LFS criteria
|
|
||
Moderate LFS criteria
|
|
||
| Total points required to assign the following rule codes | |||
| PM6_Supporting | PS2_Moderate or PM6 | PS2 or PM6_Strong | PS2_VeryStrong or PM6_VeryStrong |
| 0.5 | 1 | 2 | 4 |
If there are multiple reports of de novo probands, the points for each de novo observation can be summed. If the proband has multiple cancers, only the strongest associated LFS cancer should be used.
Cis/trans testing data
The use of BP2, which uses observation with another pathogenic variant as evidence against pathogenicity, was specified as follows: variant is observed in trans with a TP53 pathogenic variant (phase confirmed), or there are three or more observations with a TP53 pathogenic variant when phase is unknown. In this scenario, the variant must be seen with at least two different TP53 pathogenic variants (as specified by the TP53 VCEP).
Testing of the TP53 specifications on a pilot set of variants
There were 43 variants used for pilot testing. Classifications were compared between biocurators using the original ACMG/AMP and the TP53-specific guidelines, the existing assertions in ClinVar, and prior assertions by experts (Table 4). Of the 42 pilot TP53 variants recorded in ClinVar in September 2019, 16 (38%) were annotated as conflicting (see Table 4). Between-biocurator consistency of variant classification was high: 72.1% when using the original ACMG/AMP guidelines, and 81.4% using TP53-specific guidelines (Table 4). There were fewer VUS using TP53-specific guidelines compared with the original guidelines (5/43 (12%) vs 12/43 (28%)) (Figure 2). Using the TP53-specific guidelines and sharing clinical data amongst experts and clinical laboratories, the majority of VUS were downgraded to LB (13/16; 81%), two moved to LP (2/16; 12.5%), and one remained at VUS (1/16; 6%).
Table 4. Variant classified during the pilot testing phase using the TP53-specific ACMG/AMP guidelines.
All variants were annotated in relation to the transcript NM_000546.5 and protein NP_000537.3.
| TP53 variant | ClinVar ID | ClinVar Classifications (September, 2019)* | Prior Variant Classifications^ | Dual Biocurator Classifications | TP53-specific ACMG/AMP Final Classifications | Final Criteria Applied | |
|---|---|---|---|---|---|---|---|
| Original ACMG/AMP Guidelines# | TP53-specific ACMG/AMP Guidelines% | ||||||
| c.1079G>C p.(Gly360Ala) | 142003 | B/LB | LB | B/VUS | LB | LB | BS1, BS3_Supporting, BP4 |
| c.883C>T p.(Pro295Ser) | 428862 | LB | LB | VUS | LB | VUS | BP4 |
| c.206C>G p.(Ala69Gly) | 230112 | LB | LB | VUS | VUS | VUS | PM2_Supporting, BP4 |
| c.1093C>T p.(His365Tyr) | 80708 | VUS | VUS | VUS | LB | LB | BS3, BP4 |
| c.403T>G p.(Cys135Gly) | 376563 | VUS | VUS | LP | VUS | LP | PS3, PM2_Supporting, PP3_Moderate |
| c.1000G>C p.(Gly334Arg) | 182969 | VUS | VUS | VUS | VUS | VUS | BS3, PP3, PS4_Supporting |
| c.532C>G p.(His178Asp) | 482223 | LP | NA | LP/P | LP/P | LP | PS3, PM2_supporting, PM6 PP3_Moderate |
| c.538G>A p.(Glu180Lys) | 245711 | LP | P | P | VUS | LP | PM2_Supporting, PP3, PS4_Supporting, PM6, PS3_Moderate |
| c.431A>T p.(Gln144Leu) | 376647 | LP | NA | VUS | VUS | VUS | PM2_Supporting, PS4_Supporting, BS3_Supporting |
| c.578A>C p.(His193Pro) | 376612 | LP | NA | P | P | P | PS3, PM6, PM2_Supporting, PP3_Moderate, PS4_Supporting |
| c.743G>T p.(Arg248Leu) | 230253 | LP | P | P/LP | LP | P | PS3, PM1, PM2_Supporting, PP3_Moderate, PS4_Supporting |
| c.97–1G>A | 638853 | LP | P | P | LP/P | P | PVS1_Strong, PM2_Supporting, PS4_Supporting |
| c.537T>A p.(His179Gln) | 406578 | P/LP | NA | P | P | LP | PM6, PS3, PM2_Supporting, PP3 |
| c.517G>A p.(Val173Met) | 233951 | P/LP | LP | P | LP/P | P | PS2, PS3, PM2_Supporting, PS4_Supporting, PP3 |
| c.743G>A p.(Arg248Gln) | 12356 | P/LP | P | P | LP | P | PS3, PM1, PP3, PS4, PS2 |
| c.818G>A p.(Arg273His) | 12366 | P/LP | P | P | P | P | PS3, PS2, PS4, PM1, PP3 |
| c.1031T>C p.(Leu344Pro) | 12375 | P | LP | P/LP | LP | P | PS3, PM2_Supporting, PP3_Moderate, PS4_Moderate |
| c.659A>G p.(Tyr220Cys) | 127819 | P | P | P | P | P | PS3, PP1_strong, PM6, PP3_Moderate, PS4_Moderate |
| c.742C>T p.(Arg248Trp) | 12347 | P | P | P | P | P | PS2, PS3, PP1_Strong, PM1, PS4, PP3_Moderate |
| c.993+1delG | 428898 | P | P | P | P | P | PVS1, PM2_Supporting, PS4_Supporting, PP1_Moderate |
| c.892G>T p.(Glu298*) | 93323 | P | P | P | P | P | PVS1, PM2_Supporting, PS4_Supporting |
| c.372C>A p.(Cys124*) | 458537 | P | NA | P | P | P | PVS1, PM6, PM2_Supporting |
| c.488A>G p.(Tyr163Cys) | 127814 | P | NA | LP/P | P | P | PS3, PS2_Moderate, PM2_Supporting, PP3_Moderate, PS4_Supporting |
| c.455C>T p.(Pro152Leu) | 142766 | P | P | P | LP/P | P | PS3, PP3_Moderate, PS4, PP1 |
| c.919+1G>A | 633606 | P | P | P | P | P | PVS1_Strong, PM6_Supporting, PM2_Supporting |
| c.733G>A p.(Gly245Ser) | 12365 | P | P | P | P | P | PS3, PM1, PP3, PS4 |
| c.869G>A p.(Arg290His) | 127825 | Conflicting | VUS | VUS | VUS | B | BS3, BP4, BS1, BS2_Supporting |
| c.847C>T p.(Arg283Cys) | 127824 | Conflicting | VUS | VUS | VUS | VUS | BS3, PP3 |
| c.1040C>A p.(Ala347Asp) | 43587 | Conflicting | P | P | LP/P | LP | PS3, PM2_Supporting, PP1_Moderate, PS4_Supporting |
| c.1120G>C p.(Gly374Arg) | 230269 | Conflicting | LB/VUS | LB/VUS | LB | LB | BS3, BP4 |
| c.1096T>G p.(Ser366Ala) | 135360 | Conflicting | LB/VUS | LB/VUS | LB | LB | BS3, BP4 |
| c.935C>G p.(Thr312Ser) | 141102 | Conflicting | LB/VUS | B/LB | B/LB | B | BS1, BS3, BP4 |
| c.892G>A p.(Glu298Lys) | 141483 | Conflicting | LB | LB | LB | LB | BP4, BS3 |
| c.704A>G p.(Asn235Ser) | 127821 | Conflicting | LB/VUS | VUS | B | B | BS1, BS3, BS4, BP4, BP2, BS2_Supporting |
| c.245C>T p.(Pro82Leu) | 182946 | Conflicting | LB/VUS | VUS | LB | LB | BS3, BP4 |
| c.28G>A p.(Val10Ile) | 127806 | Conflicting | LB/VUS | LB | LB/VUS | LB | BS3, BP4 |
| c.21T>A p.(Asp7Glu) | 140782 | Conflicting | LB | VUS | VUS | LB | PM2_Supporting, BS3, BP4 |
| c.145G>A p.(Asp49Asn) | 186363 | Conflicting | LB | VUS | LB | LB | BP4, BS3 |
| c.641A>G p.(His214Arg) | 376615 | Conflicting | LP | LP/P | LP | LP | PS3, PM2_Supporting, PS4_Supporting |
| c.217G>A p.(Val73Met) | 142386 | Conflicting | LB | VUS | LB/VUS | B | BS1, BS3, BP4 |
| c.139C>T p.(Pro47Ser) | 43588 | Conflicting | B | B/LB | B | B | BA1, BS3, BP4, BS2 |
| c.319T>C p.(Tyr107His) | 140786 | Conflicting | LB/VUS | LB/VUS | B | B | BA1, BP4, BS3_Supporting, BS2_Supporting |
| c.428T>C p.(Val143Ala) | 792574 | NA | NA | LP/P | LP | LP | PS3, PM6, PM2_Supporting, PP3 |
Abbreviations: B = Benign, LB = Likely benign, VUS = Variant of uncertain significance, LP = Likely pathogenic, P = Pathogenic, Conflicting = Conflicting interpretations of pathogenicity, NA = Not Available.
ClinVar assertions were documented in September 2019. Conflicting variants were those that spanned different clinically relevant classifications.
Assertions provided by VCEP members with variant nominations. Some variants were nominated by multiple members; some with conflicting assertions. Variants listed as “NA” were not nominated by a VCEP member.
Variants were assessed by two curators using the original ACMG/AMP guidelines and any curation assertions conflicts are noted.
Additional rule specifications after pilot testing are largely responsible for differences in classifications between the Biocurator TP53-specified Classifications and the Final TP53 VCEP Classifications.
Figure 2.
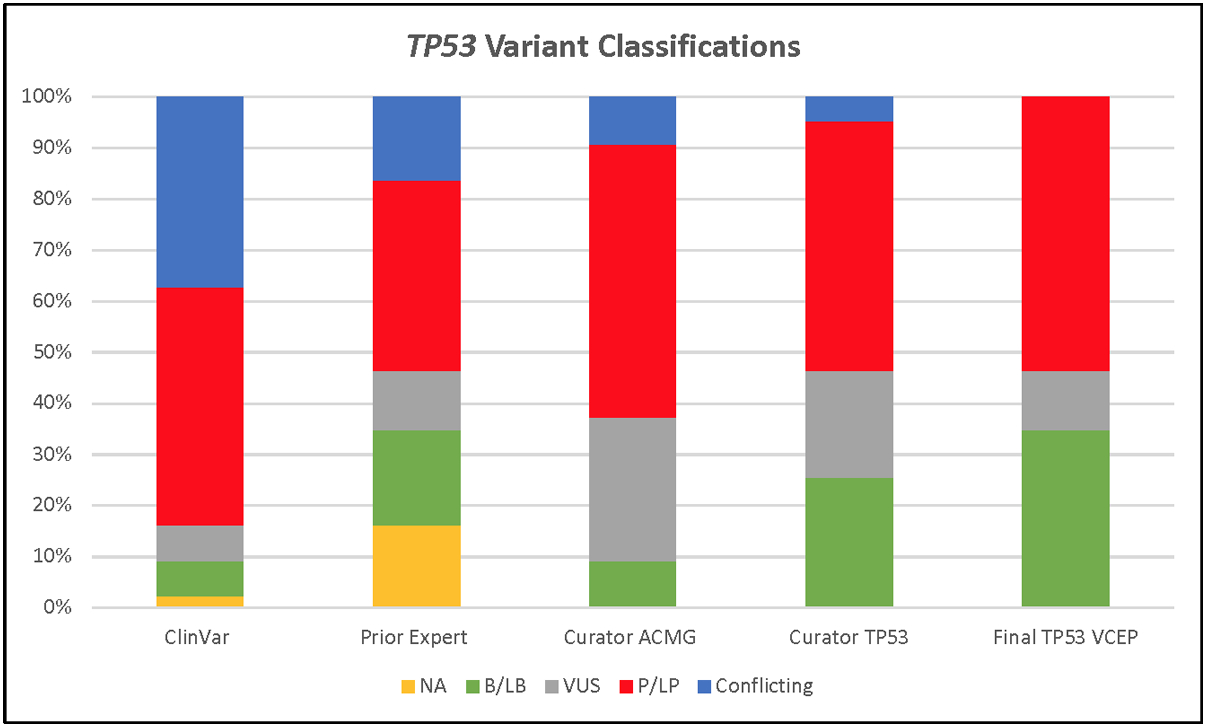
Variant classifications for 43 pilot TP53 variants in ClinVar, from the nominating expert(s), biocurators using the original ACMG/AMP guidelines, and the TP53-specific guidelines.
Abbreviations: B = Benign, LB = Likely benign, VUS = Variant of uncertain significance, LP = Likely pathogenic, P = Pathogenic, Conflicting = Clinically relevant conflicting interpretations of pathogenicity, and NA = Not Available.
DISCUSSION
The task of the TP53 VCEP was to specify the ACMG/AMP guidelines to assist with the clinical classification of germline variants in the TP53 gene. There is a rapidly growing number of individuals without personal or family history consistent with LFS who have presumed germline TP53 pathogenic variants (Batalini et al., 2019; Fortuno, James, & Spurdle, 2018), and cancer surveillance for people with TP53 pathogenic germline variants is time intensive and emotionally stressful. Given that TP53 is also on the ACMG Secondary Findings medically actionable list for return of results (Kalia et al., 2017), this work will also be important to help streamline variant curation and hopefully decrease variant classification discrepancies between laboratories.
Our pilot study demonstrated that the TP53-specific guidelines decreased the number of VUS compared with the original guidelines and increased the number of variants classified as not clinically relevant. Our study also showed strong intra-biocurator consistency, suggesting that the criteria appear to be straightforward to interpret; this should allow for fewer conflicting variant calls between laboratories. For example, the criteria related to population data can be applied more often, bioinformatic and functional data can now be applied more broadly.
One of the benefits of curating TP53 variants was the wealth of existing knowledge, including data readily available in public databases. The IARC TP53 database (Bouaoun et al., 2016) is a rich source of data that was helpful in assessing pathogenicity codes, including PS1, PS2, PS4, PM5, PM6, and PP1. The existing data on p53 mouse models searchable by knockout variant of interest was also useful for adapting functional rule codes. Additionally, the TP53 VCEP benefited from data sharing amongst participating researchers, clinicians, and clinical laboratories. VCEP members added clinical information pertaining to additional probands to those identified in the literature, to increase the number of pathogenic codes applicable to certain variants and to shift classifications from VUS to LP. Laboratory members also shared data from their large hereditary cancer panels that were helpful in defining and using the benign codes BS2 and BP2.
Consistent with the ClinGen VCEP process, all of the TP53 classified variants have been submitted to the ClinVar database (Landrum et al., 2018) with a summary of the classification process. More details about the evidence used to classify each variant is available through a link to the public access ClinGen Evidence Repository (https://erepo.clinicalgenome.org/evrepo/). The TP53 VCEP will continue to meet monthly to curate additional variants and submit them to ClinVar. Variants previously classified as LP or VUS will be reviewed at least every two years in the event new evidence has emerged, while variants classified as LB will be reviewed when new evidence is available or when requested by the public via the ClinGen website (www.clinicalgenome.org). It is also anticipated that these specifications will be updated and reviewed as needed. For example, in the first iteration of these specifications, we suggested the use of MaxEntScan (Yeo & Burge, 2004) and Human Splicing Finder (HSF) (Desmet et al., 2009), to predict variant spliceogenicity. However, HSF is no longer freely available. As a consequence, we conducted comparisons of predictions using other tools and now suggest the use of a metapredictor that captures multiple spliceogenic mechanisms. This follows advice of the SVI Committee. More definitive studies are warranted to calibrate the strength of predictions of splicing algorithms, and so future iterations of these specifications may provide further details on the use of a specific splicing predictor. The specifications may also consider additional evidence types to improve TP53 variant classification, such as the relationship between somatic and germline counts reported to be positively correlated only for pathogenic variants (Fortuno, Cipponi, et al., 2019) or HER2+ breast tumor pathology as a positive predictor of TP53 variant pathogenicity (Fortuno et al., 2020). The most up to date version of the ClinGen TP53 VCEP specifications is available at https://clinicalgenome.org/affiliation/50013/.
Supplementary Material
ACKNOWLEDGEMENTS
The ClinGen TP53 Variant Curation Expert Panel thanks the ClinGen PTEN Variant Curation Expert Panel and the Executive Committee of the Hereditary Cancer Clinical Domain Working Group for sharing their experience with developing criteria specifications. We also thank Steven Harrison, Leslie Biesecker and the remainder of the ClinGen Sequence Variant Interpretation Working Group for their guidance. Finally, thank present and past members of the TP53 VCEP, Maria Isabel Achatz, Rebecca Bassett, Jeffrey Bissonnette, David Goldgar, Sharisse Jimenez, Chimene Kesserwan, Deb Ritter, Leighton Telling, and Mackenzie Trapp.
FUNDING
This Expert Panel is funded by National Human Genome Research & National Cancer Institutes (1U41HG006834, 1U01HG007437, 1U01HG007436, HHSN261200800001E, U41HG009650, 1U41HG00964). The work of C.F. was supported by a University of Queensland (UQ) International Scholarship from the UQ School of Medicine. The work of M.F., K.C.A., and S.A.S. was supported by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health. The work of A.B.S. was supported by Australian National Health and Medical Research funding (ID1061779, ID1161589). The work of D.G.E. was supported by the NIHR Manchester Biomedical Research Centre (IS-BRC-1215-20007). The work of T.P.S. was supported by NIH-NCI K08CA234394 and R01CA242218.
Footnotes
CONFLICTS OF INTEREST
The following authors have no conflicts of interest to disclose: K.L., M.O., K.C.A., S.B., A.K.M.F., M.F., B.A.S., L.W., L.Z. The following authors have made extensive contributions to the TP53 literature and have previously published assertions on TP53 variants: C.F., P.L.M., L.D.A., D.G.E., P.J., K.M., T.P.S., A.B.S., S.A.S. The following authors are an employee, trainee or consultant for a commercial laboratory that offers genetic testing for TP53: T.P., R.H., K.M., S.E.P. J.M. is an employee of GeneDx/BioReference Laboratories, Inc./OPKO Health and has a salary as the only disclosure. The PERCH software, for which B.J.F. is the inventor, has been non-exclusively licensed to Ambry Genetics Corporation for their clinical genetic testing services and research. B.J.F. also reports funding and sponsorship to his institution on his behalf from Pfizer Inc. and Regeneron Genetics Center LLC.
DATA AVAILABILITY STATEMENT
Most of the data that support the findings of this study are openly available in the IARC TP53 Database at https://p53.iarc.fr (version R20, July 2019). Other data supporting the findings of this study are not publicly available due to private or ethical restrictions. The publicly available Clinical Genome Resource (ClinGen) Evidence Repository contains all of the curated evidence for the variants submitted to ClinVar (e.g., https://erepo.clinicalgenome.org/evrepo/ui/interpretation/7b4332f9-03a6-43f1-afde-508d91bd92d5).
REFERENCES
- Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, & Harrison SM (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat, 39(11), 1517–1524. doi: 10.1002/humu.23626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achatz MI, & Zambetti GP (2016). The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harb Perspect Med, 6(12). doi: 10.1101/cshperspect.a026195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, … Jarvik GP (2015). Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res, 25(3), 305–315. doi: 10.1101/gr.183483.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger ML, Best A, Mai PL, Khincha PP, Loud JT, Peters JA, … Savage SA (2017). Baseline Surveillance in Li-Fraumeni Syndrome Using Whole-Body Magnetic Resonance Imaging: A Meta-analysis. JAMA Oncol. doi: 10.1001/jamaoncol.2017.1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batalini F, Peacock EG, Stobie L, Robertson A, Garber J, Weitzel JN, & Tung NM (2019). Li-Fraumeni syndrome: not a straightforward diagnosis anymore-the interpretation of pathogenic variants of low allele frequency and the differences between germline PVs, mosaicism, and clonal hematopoiesis. Breast Cancer Res, 21(1), 107. doi: 10.1186/s13058-019-1193-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, & Harrison SM (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med, 20(12), 1687–1688. doi: 10.1038/gim.2018.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittar CM, Vieira IA, Sabato CS, Andreis TF, Alemar B, Artigalas O, … Ashton-Prolla P (2019). TP53 variants of uncertain significance: increasing challenges in variant interpretation and genetic counseling. Fam Cancer, 18(4), 451–456. doi: 10.1007/s10689-019-00140-w [DOI] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, & Olivier M (2016). TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat, 37(9), 865–876. doi: 10.1002/humu.23035 [DOI] [PubMed] [Google Scholar]
- Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, … Frebourg T (2015). Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol, 33(21), 2345–2352. doi: 10.1200/JCO.2014.59.5728 [DOI] [PubMed] [Google Scholar]
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, … Berg JS (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med, 12(1), 3. doi: 10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron O, Frébourg T, Benusiglio PR, Faivre L, Dugast C, Bonadona V, … Brugieres L (2016). The 2016 Li-Fraumeni syndrome cancer spectrum. Journal of Clinical Oncology, 34(15_suppl), 1535–1535. doi: 10.1200/JCO.2016.34.15_suppl.1535 [DOI] [Google Scholar]
- Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, … Taylor BS (2016). Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol, 34(2), 155–163. doi: 10.1038/nbt.3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, … Taylor BS (2018). Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov, 8(2), 174–183. doi: 10.1158/2159-8290.CD-17-0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B, Cox HC, Kidd J, Sizemore S, Brown K, Manley S, & Mancini-DiNardo D (2017). Detection of somatic variants in peripheral blood lymphocytes using a next generation sequencing multigene pan cancer panel. Cancer Genet, 211, 5–8. doi: 10.1016/j.cancergen.2017.01.002 [DOI] [PubMed] [Google Scholar]
- de Andrade KC, Frone MN, Wegman-Ostrosky T, Khincha PP, Kim J, Amadou A, … Achatz MI (2019). Variable population prevalence estimates of germline TP53 variants: A gnomAD-based analysis. Hum Mutat, 40(1), 97–105. doi: 10.1002/humu.23673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade KC, Mirabello L, Stewart DR, Karlins E, Koster R, Wang M, … Achatz MI (2017). Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum Mutat, 38(12), 1723–1730. doi: 10.1002/humu.23320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, & Beroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res, 37(9), e67. doi: 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Birch JM, Ramsden RT, Sharif S, & Baser ME (2006). Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet, 43(4), 289–294. doi: 10.1136/jmg.2005.036319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng BJ (2017). PERCH: A Unified Framework for Disease Gene Prioritization. Hum Mutat, 38(3), 243–251. doi: 10.1002/humu.23158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuno C, Cipponi A, Ballinger ML, Tavtigian SV, Olivier M, Ruparel V, … James PA (2019). A quantitative model to predict pathogenicity of missense variants in the TP53 gene. Hum Mutat, 40(6), 788–800. doi: 10.1002/humu.23739 [DOI] [PubMed] [Google Scholar]
- Fortuno C, James PA, & Spurdle AB (2018). Current review of TP53 pathogenic germline variants in breast cancer patients outside Li-Fraumeni syndrome. Hum Mutat, 39(12), 1764–1773. doi: 10.1002/humu.23656 [DOI] [PubMed] [Google Scholar]
- Fortuno C, James PA, Young EL, Feng B, Olivier M, Pesaran T, … Spurdle AB (2018). Improved, ACMG-Compliant, in silico prediction of pathogenicity for missense substitutions encoded by TP53 variants. Hum Mutat. doi: 10.1002/humu.23553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuno C, Mester J, Pesaran T, Weitzel JN, Dolinsky J, Yussuf A, … Spurdle AB (2020). Suggested application of HER2+ breast tumor phenotype for germline TP53 variant classification within ACMG/AMP guidelines. Hum Mutat. doi: 10.1002/humu.24060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuno C, Pesaran T, Dolinsky J, Yussuf A, McGoldrick K, Kho PF, … Spurdle AB (2019). p53 major hotspot variants are associated with poorer prognostic features in hereditary cancer patients. Cancer Genet, 235–236, 21–27. doi: 10.1016/j.cancergen.2019.05.002 [DOI] [PubMed] [Google Scholar]
- Frebourg T, Bajalica Lagercrantz S, Oliveira C, Magenheim R, & Evans DG (2020). Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet. doi: 10.1038/s41431-020-0638-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, … Hahn WC (2018). Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet, 50(10), 1381–1387. doi: 10.1038/s41588-018-0204-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez KD, Buzin CH, Noltner KA, Gu D, Li W, Malkin D, & Sommer SS (2009). High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet, 46(10), 689–693. doi: 10.1136/jmg.2008.058958 [DOI] [PubMed] [Google Scholar]
- Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, … Weitzel JN (2009). Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol, 27(8), 1250–1256. doi: 10.1200/JCO.2008.16.6959 [DOI] [PubMed] [Google Scholar]
- Grantham R (1974). Amino acid difference formula to help explain protein evolution. Science, 185(4154), 862–864. doi: 10.1126/science.185.4154.862 [DOI] [PubMed] [Google Scholar]
- Hanson H, Brady AF, Crawford G, Eeles RA, Gibson S, Jorgensen M, … Evans DG (2020). UKCGG Consensus Group guidelines for the management of patients with constitutional TP53 pathogenic variants. J Med Genet. doi: 10.1136/jmedgenet-2020-106876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, … Farh KK (2019). Predicting Splicing from Primary Sequence with Deep Learning. Cell, 176(3), 535–548.e524. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Kaariainen H, Muilu J, Perola M, & Kristiansson K (2017). Genetics in an isolated population like Finland: a different basis for genomic medicine? J Community Genet, 8(4), 319–326. doi: 10.1007/s12687-017-0318-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, … Miller DT (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med, 19(2), 249–255. doi: 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, … MacArthur DG (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv, 531210. doi: 10.1101/531210 [DOI] [Google Scholar]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, & Ishioka C (2003). Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A, 100(14), 8424–8429. doi: 10.1073/pnas.1431692100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, … Segal E (2018). A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol Cell, 71(1), 178–190 e178. doi: 10.1016/j.molcel.2018.06.012 [DOI] [PubMed] [Google Scholar]
- Kratz CP, Achatz MI, Brugieres L, Frebourg T, Garber JE, Greer MC, … Malkin D (2017). Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res, 23(11), e38–e45. doi: 10.1158/1078-0432.ccr-17-0408 [DOI] [PubMed] [Google Scholar]
- Lalloo F, Varley J, Moran A, Ellis D, O’Dair L, Pharoah P, … Evans DG (2006). BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur J Cancer, 42(8), 1143–1150. doi: 10.1016/j.ejca.2005.11.032 [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, … Maglott DR (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res, 46(D1), D1062–D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP (1992). Cancer. p53, guardian of the genome. Nature, 358(6381), 15–16. doi: 10.1038/358015a0 [DOI] [PubMed] [Google Scholar]
- Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, … Karam R (2018). Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat, 39(11), 1553–1568. doi: 10.1002/humu.23650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FP, Fraumeni JF Jr., Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, & Miller RW (1988). A cancer family syndrome in twenty-four kindreds. Cancer Res, 48(18), 5358–5362. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3409256 [PubMed] [Google Scholar]
- Luo X, Feurstein S, Mohan S, Porter CC, Jackson SA, Keel S, … Godley LA (2019). ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv, 3(20), 2962–2979. doi: 10.1182/bloodadvances.2019000644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, … Savage SA (2016). Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer, 122(23), 3673–3681. doi: 10.1002/cncr.30248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, & Tavtigian SV (2006). Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res, 34(5), 1317–1325. doi: 10.1093/nar/gkj518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester JL, Ghosh R, Pesaran T, Huether R, Karam R, Hruska KS, … Eng C (2018). Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum Mutat, 39(11), 1581–1592. doi: 10.1002/humu.23636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, & Hainaut P (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol, 2(1), a001008. doi: 10.1101/cshperspect.a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JA, Kenen R, Bremer R, Givens S, Savage SA, & Mai PL (2016). Easing the Burden: Describing the Role of Social, Emotional and Spiritual Support in Research Families with Li-Fraumeni Syndrome. J Genet Couns, 25(3), 529–542. doi: 10.1007/s10897-015-9905-x [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon K, & Tischkowitz M (2018). Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res Treat, 167(2), 417–423. doi: 10.1007/s10549-017-4531-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Webb BD, Birch AH, Elkhoury L, McCarthy J, Cai X, … Kornreich R (2017). Comprehensive population screening in the Ashkenazi Jewish population for recurrent disease-causing variants. Clin Genet, 91(4), 599–604. doi: 10.1111/cge.12834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SJ, Dodd-Eaton EB, Peng G, Bojadzieva J, Chen J, Amos CI, … Wang W (2020). Penetrance of Different Cancer Types in Families with Li-Fraumeni Syndrome: A Validation Study Using Multicenter Cohorts. Cancer Res, 80(2), 354–360. doi: 10.1158/0008-5472.Can-19-0728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, & Biesecker LG (2018). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. doi: 10.1038/gim.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley JM (2003). Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat, 21(3), 313–320. doi: 10.1002/humu.10185 [DOI] [PubMed] [Google Scholar]
- Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, … Malkin D (2016). Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol, 17(9), 1295–1305. doi: 10.1016/S1470-2045(16)30249-2 [DOI] [PubMed] [Google Scholar]
- Walsh MF, Ritter DI, Kesserwan C, Sonkin D, Chakravarty D, Chao E, … Plon SE (2018). Integrating somatic variant data and biomarkers for germline variant classification in cancer predisposition genes. Hum Mutat, 39(11), 1542–1552. doi: 10.1002/humu.23640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzel JN, Chao EC, Nehoray B, Van Tongeren LR, LaDuca H, Blazer KR, … Jasperson K (2017). Somatic TP53 variants frequently confound germ-line testing results. Genet Med. doi: 10.1038/gim.2017.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, … Ware JS (2017). Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. doi: 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, & Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol, 11(2–3), 377–394. doi: 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Young JL, Pantaleao A, Zaspel L, Bayer J, Peters JA, Khincha PP, … Werner-Lin A (2019). Couples coping with screening burden and diagnostic uncertainty in Li-Fraumeni syndrome: Connection versus independence. J Psychosoc Oncol, 37(2), 178–193. doi: 10.1080/07347332.2018.1543376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerdoumi Y, Lanos R, Raad S, Flaman JM, Bougeard G, Frebourg T, & Tournier I (2017). Germline TP53 mutations result into a constitutive defect of p53 DNA binding and transcriptional response to DNA damage. Hum Mol Genet, 26(14), 2591–2602. doi: 10.1093/hmg/ddx106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Most of the data that support the findings of this study are openly available in the IARC TP53 Database at https://p53.iarc.fr (version R20, July 2019). Other data supporting the findings of this study are not publicly available due to private or ethical restrictions. The publicly available Clinical Genome Resource (ClinGen) Evidence Repository contains all of the curated evidence for the variants submitted to ClinVar (e.g., https://erepo.clinicalgenome.org/evrepo/ui/interpretation/7b4332f9-03a6-43f1-afde-508d91bd92d5).