

Abstract
Despite being widely heralded following their discovery, the effectiveness and clinical utility of antidepressants has been questioned, in part due to the release of several decades of regulatory trial data. Upon investigation, contemporary regulatory trials of antidepressants have demonstrated a nearly identical effect size (0.3) for the past 40 years, regardless of placebo response or attempts to improve trial design.
In this review, we examine the historical methods of antidepressant trials and re-evaluate regulatory trial data over time and according to drug class (SSRIs, SNRIs, and atypicals) with the addition of two classes of antidepressants not previously analyzed: tricyclics used as active comparators and the recently-approved NMDA receptor antagonist, esketamine. We show that among these five classes of antidepressants there were no significant differences between effect sizes or percent symptom reduction. We suggest that within the context of a regulatory trial of antidepressants, effect sizes will remain modest (~0.3) regardless of class or novel drug mechanism, possibly due to regulatory changes to trial design and conduct following the Kefauver-Harris Act of 1962.
We comment that the regulatory double-blind, parallel, placebo-controlled trial model is an artificial creation for a narrow purpose—designed to demonstrate simple superiority over placebo and to determine basic safety. We should be cautious of stretching trial results beyond their limited capacity to inform clinical practice as trials are not representative of real-world patients or medication management practices. There is a substantial need to develop more realistic models to evaluate the clinical utility of antidepressants.
Keywords: clinical trials, antidepressants, regulatory trials, effect size, methodology
Introduction
Historically, depression has been conceptualized as an episodic and self-limiting ailment for which there are many types of interventions and treatments. These include religion-based therapy, professional psychotherapy, exercise, diet, and acupuncture to name a few.1 Given this wide array of traditional and alternative approaches to depression treatment, questions have been raised regarding the purported effectiveness of pharmacological treatments and whether such effects are specific or nonspecific. Moreover, if antidepressants do possess specific psychopharmacological effects, the question remains whether antidepressant classes are different from one another in terms of treatment efficacy.
These concerns are in part rooted in the history of the development of antidepressants. Many of the early antidepressants were discovered and evaluated using relatively simple clinical observations. These early trials were conducted by interested parties such as pharmaceutical companies and supported in part by medical enthusiasts. Needless to say, these early methods of discovery and evaluation provided a favorable climate for putative compounds, potentially exaggerating their effect. Although it is important to note that several drugs developed over the past 60 years have virtually disappeared from clinical practice due to safety concerns (for example MAO inhibitors and tricyclic antidepressants). Because of this, they never faced the scrutiny of modern-day double-blind placebo-controlled trials.
However, there is a persistent and nostalgic perception that these early antidepressants had better therapeutic effects compared to the antidepressants in use currently. Specifically, it is often purported that smaller effect sizes seen in more recent antidepressant trials suggest that these antidepressants are not as effective.
In this manuscript we propose that the current antidepressant clinical trial methodology implemented after the Kefauver-Harris Act has had a revolutionary effect on the outcome of antidepressant trials. Specifically, the methodology implemented by world-wide regulatory agencies, particularly the randomized, double-blind, placebo-controlled, parallel design of the US FDA, has systematically achieved two goals: a relatively stable but modest effect size among antidepressant trials and furthermore, minimized apparent differences in therapeutic outcomes among various classes of antidepressants.
To explore this phenomenon, we will first review the clinical trial methodology used in antidepressant studies prior to the close involvement of regulatory agencies. Following this, we will review the efficacy trial data for five classes of antidepressants (tricyclics, SSRIs, SNRIs, atypicals, and an NMDA receptor antagonist) limiting our search to only directly-sourced data from regulatory trials submitted to and reviewed by the US FDA.
We will demonstrate that data from modern antidepressants, including tricyclics used as active comparators, show almost identical efficacy using tightly regulated clinical trial methodology. Following this we will describe the factors that may have contributed to this phenomenon—in other words, the potentially equalizing and minimizing effects of regulatory trial methodology. Lastly, we will review potential pitfalls in the design of future antidepressant trials.
As it stands, it appears that antidepressants are not curative for most depressed patients. This concern was especially poignant after a large trial, the Sequenced Treatment Alternatives to Relieve Depression (STAR*D),2 reported surprisingly modest effect sizes for patients treated with antidepressants. However, the clinical reality is that antidepressants are widely used (with one out of every eight U.S. citizens over the age of 12 taking antidepressants in a given month)3 and as such, patients who would never be included in clinical trials represent a large part of the treatment population.4 It is important to note, trials such as the STAR*D designed to assess antidepressant efficacy in more “real world” contexts are still not representative of patients in clinical practice and the discerning application of antidepressants by experienced clinicians. Dr. Peter Kramer5 explores this issue more comprehensively, highlighting how antidepressants are used effectively in clinical settings and how this application differs from the assigned treatment in clinical trials. It appears that these discrepancies between trials and practice are not fully understood or appreciated. Thus, the larger aim of this review is to highlight the ways in which modern regulatory trials diverge from real-world psychopharmacology, resulting in their lack of clinical relevance.
Early History of Antidepressant Trials
In 1952, the psychoactive effects of iproniazid were discovered serendipitously among listless and depressed patients being treated for tuberculosis. The descriptions of these patients ‘dancing in the halls’ is legendary.6 Dr. William Furst conducted one of the early case series of iproniazid and based on his reports of 100 depressed patients said that “approximately three out of every four endogenously depressed patients [given iproniazid] respond as well, if not better, than with electroshock therapy”.7 As shown in Table 1 below, in one of the earliest studies of iproniazid’s effects, Loomer et al8 summarized that 70% (12/17) of depressed patients treated with iproniazid had a favorable response, 18% (3/17) of depressed patients had an unfavorable response and 12% (2/17) of depressed patients had no response.
Table 1. Summary of Treatment Effects for Depressed Patients Treated with Iproniazid as Reported in Loomer et al8.
| Type of ResponSe | Number of Patients |
Per Cent of PatientS |
||
| No Response | 2 | 12% | ||
| Favorable Response | 12 | 70% | ||
| Unfavorable Response | 3 | 18% |
These findings spurred the development of over six MAO inhibitors used for the treatment of depression. With estimates of half to three-quarters of patients experiencing sudden and dramatic relief of depression symptoms with iproniazid, expectations for antidepressant potency soared as new compounds were discovered.
One iconic drug amongst the early MAOI antidepressants was phenelzine, which was featured in many of the early depression trials of the 1950s. Dr. Furst9 originally characterized its effectiveness with outpatients as approximately as effective as iproniazid (with 50–69% of patients achieving full remission). In an additional study, Arnow10 found that approximately 66% of depressed patients among 580 case records achieved remission and 85% had a favorable response according to the overseeing practitioner. Furthermore, improvement was found in 55–70% of depressed patients treated with phenelzine and psychotherapy in a study of 50 in and outpatients.11 However, it is important to note that none of these early trials were placebo-controlled.
In 1955, shortly after the boom in MAO inhibitor development, a Swiss psychiatrist by the name of Ronald Kuhn was searching for another tricyclic-based sedative (or “antipsychotic”) and incidentally discovered the antidepressant effects of imipramine. Kuhn seemed to have discovered imipramine by simply giving the drug to depressed patients and observing their response to it, as described in his elegant case histories and descriptions that are illustrative to this day.12 The other tricyclic antidepressants were also essentially discovered by direct observation of depressed patients after receiving investigational compounds. Via this method of direct observation by a physician, several tricyclic antidepressants were developed and became the mainstay of depression treatments for almost three decades.
The demand for controlled studies, including placebo control, grew as more promising compounds were discovered. Many of these early controlled studies continued to support a robust effect size for tricyclic antidepressants. Citing over 30 references of studies examining the effects of imipramine, Klerman and Cole13 concluded that “among the large number of clinical reports, there is fairly general agreement that significant improvement can be expected from the use of imipramine in 60 to 80% of various types of depression.” Some of these studies are summarized in Table 2 below.
Table 2. Summary of Treatment Effects from Placebo-Controlled Trials of Depression Patients Treated with Imipramine from Klerman and Cole13.
| Authors | Diagnosis | Imipramine Dose (mg/day) | No. of Patients | Control Dose (mg/day) | Evaluation | Drug-Placebo Difference | ||||||||||||
| Imipramine | ||||||||||||||||||
| Improved | UNimproved | Improved | UNimproved | |||||||||||||||
| Abraham et al.3 | Psychotic and neurotic | 100 | 36 | 12 | 7 | 10 | Inert substance | 2 wk | Yes | |||||||||
| Ball and Kiloh27 | Psychotic | 250 | 20 | 7 | 6 | 22 | Inert substances | 4 wk | Yes | |||||||||
| Ball and Kiloh27 | Neurotic | 250 | 13 | 9 | 4 | 16 | Inert substances | 4 wk | Yes | |||||||||
| Daneman | Neurotic | 200 | 73 | 21 | 16 | 85 | Atropine 1.4 mg | 4 wk | Yes | |||||||||
| Uhlenhuth and Park | Neurotic | 150 | 9 | 13 | 6 | 14 | Atropine 0.6 mg | 2 wk | Yes | |||||||||
Importantly, most of the depression trials of the 1950s used some version of clinician impression to classify responder rates rather than quantifying symptoms or characterizing depression severity over time. However, depression trial endpoints changed in the 1960s when Max Hamilton developed and published a new rating scale (HAM-D) using prevalent symptoms seen in depressed patients at the time.14 Observing his melancholic inpatients being treated for severe depressive episodes, Hamilton developed a set of common symptoms to evaluate the severity of the illness. With the use of the 17–22 item multidimensional HAM-D scale it was thought that depression severity could be quantified using a standardized composite score. Shortly after its development, the HAM-D became the gold standard for measuring depressive symptoms in trials of antidepressants.
Influential trials of the 1980s replicated the robust results of depression trials in the 60s and 70s. In 1985, Rickels et al15 published the results of a placebo-controlled study of alprazolam, amitriptyline, and doxepin and reported that after 6 weeks of treatment, antidepressant treatment reduced HAM-D total symptoms by 42–49% as compared to 28% symptom reduction with placebo. As can be seen in Table 3 below, Rickels et al16 later reported that among depressed patients who were given adequate doses of study medications, the response rate was as follows: 68% with imipramine, 62% with alprazolam, 40% with diazepam and 32% with placebo.
Table 3. Summary of Treatment Effects From A Placebo-Controlled Study of Alprazolam, Amitriptyline, and Doxepin as Reported in Rickels et al,15 Percent Improved Patients*.
| Medication Intake, Capsules per Day |
ImIpramIne Hydrochloride |
Alprazolam | Diazepam | Placebo | P | |||||
| ⩾6 (n = 89) | 68 | 62 | 40 | 32 | .02 | |||||
| <6 (n = 58) | 86 | 73 | 55 | 60 | NS |
*Greater than 50% decrease in Hamilton Rating Scale for Depression score. NS indicates not significant.
These trials of early antidepressants led to this embedded expectation among psycho-pharmacologists that approximately one-third of patients on placebo would have a therapeutic effect compared to two-thirds of patients on antidepressants. However, among contemporary antidepressants, efficacy estimates have fallen short of these expectations. Although there is no definitive explanation for this decline, we suggest that the advent of modern, regulatory RCT models have dramatically altered the methods for assessing drug efficacy, possibly lowering the ceiling for antidepressant effects as measured in these trials. Next we explore the historical context of these major changes in trial design.
Evolution of the Regulatory RCT for Antidepressants
The period between 1962 and the 1980s saw a permanent change in the landscape for the discovery and development of new antidepressants (and pharmacological agents in general). Most notably, the clinical trial model radically evolved after 1962 as regulatory oversight increased following the passage of the Kefauver-Harris Drug Amendment Act.17 Although it was originally intended only to address issues of safety of new medications reaching the market, the Act eventually came to include a requirement of proof of efficacy.18 In order to achieve this aim, stringent, randomized, double-blind, placebo-controlled trials (RCTs) were developed to assess putative new drugs, including antidepressants.
Evaluation of new antidepressants came under the purview of the US FDA and similar regulatory agencies worldwide. Suspicion regarding fraud and lack of transparency led the US FDA to take on an antagonistic role with the pharmaceutical companies as well as clinical pharmacologists, which extended to the development of newer antidepressants. As a result, the previously collegial atmosphere between pharmaceutical companies and psycho-pharmacologists changed dramatically. Data analysis submitted by the sponsors of these trials could not serve as proof of efficacy out of fear of mishandling and so the FDA staff and scientists (statisticians in particular) were authorized and charged with auditing the collection practices of original data and conducting independent data analysis on these data.
As has occurred for over 70 years, statisticians and scientists have held firm the belief that physicians are impressionistic and prone to sentimentality—not critical enough in their thinking when it comes to their own experiences and patient reports.19,20 In order to overcome such diathesis among physicians, the FDA statisticians designed much stricter criteria for data collection and analysis. For example, they ruled out endpoints using impressionistic tools such as overall clinical impression; tools that had previously led to the actual discovery of earlier psychotropic agents and antidepressants.
Importantly, around the same time as FDA regulatory clinical trial models were being developed the American Psychiatric Association (APA) was formulating the third revision of the Diagnostic and Statistical Manual for Mental Disorders (DSM-III). This revised manual would include a complex, symptom-based diagnostic model of major depression (MDD). Following the publication of the DSM-III, the psychiatric community adopted this diagnostic understanding and over time major depressive disorder came to be thought of as a circumscribed and unique syndrome. This new understanding of major depression was highly influential in shaping patient selection practices for depression trials.
In this context of revising the psychiatric nomenclature, the community of scientists and psychiatrists were tasked with choosing and appropriate standard dependent measure for regulatory trials. The HAM-D scale was the gold standard in depression trials at the time and therefore served as a convenient solution to the problem of impressionistic endpoints. Without consideration of the disconnect between the HAM-D and the APA’s syndromal construction of major depression, the US FDA accepted the HAM-D total score as the default measure in trials of antidepressants. Therefore, outpatients fitting the APA’s new diagnostic criteria for depression were recruited for regulatory trials and antidepressant effects were measured on a scale designed to detect symptoms of the iconic melancholic inpatients of the 1960s.
Another major change occurred around 2006, when it was brought to the attention of trial developers that the HAM-D was incongruent with the definitions of Major Depression put forth by the American Psychiatric Association. An alternative scale called the Montgomery-Asberg Depression Rating Scale (MADRS) had been developed based on population surveys and focused on the psychological attributes of MDD, which more closely matched the APA’s description of the condition.21 The HAM-D was thought to be too heavily weighted for physical symptoms that did not match the modern depressed population for which modern antidepressants were being developed. The US FDA regulatory agency accepted the MADRS as a standard endpoint for antidepressant trials and it has been used for the past decade and a half.
Additionally, the FDA statisticians took almost a decade to decide on the statistical models to be used and so in total it took over 15 years from the passing of legislation to designing and instituting the double-blind placebo-controlled randomized clinical trial for the approval of any newer antidepressants. Thus, the first of the antidepressants to be approved under these new set of rules were zimelidine and nomifensine, neither of which continued to be used because of severe toxicity and the results of these first regulatory clinical trials have been lost in antiquity. However, another half a dozen antidepressants were approved using this regulatory clinical trial model over the next decade. It is with these first modern antidepressants that the impact of regulatory trial design becomes evident.
Earlier Analysis; a First Investigation of the Effects of Regulatory Trials
In the early 2000s, concern in the field over investigator bias and the safety of placebo use in depression studies with suicidal patients prompted our earlier investigation of data exclusively sourced from FDA regulatory trials. Once we obtained and published the data from the US FDA via the Freedom of Information Act, it became obvious that antidepressant effects fell drastically short of what was expected based on historical trials. The difference of two-thirds of depressed patients responding to antidepressants compared to one-third responding to placebo seen in earlier reports were supplanted with modest antidepressant-placebo differences of about 10% (40% of symptoms reduced with antidepressants and 30% with placebo).22 An entire cottage industry erupted out of this revelation, with the singular intent to reduce and control the increasing placebo response.
When we repeated our analysis nearly 15 years later and analyzed the regulatory trial data for 6 new antidepressants approved after 2001, it was clear that despite efforts to reduce the placebo response, efficacy profiles had not changed substantially.23 As a measure of the antidepressant-placebo difference, the mean effect size of modern antidepressants in the context of regulatory clinical trials was around 0.3, corresponding still to approximately 10% more reduction of depression symptoms with antidepressant compared with placebo.
Given the considerable effort that had gone into improving the design and conduct of antidepressant trials (including the increasingly scrupulous monitoring of site raters using ‘unbiased external raters’), many investigators have been perplexed by the consistently modest placebo-subtracted treatment effects seen in these trials. However, the fact of the matter is that efforts to improve on trial design have not brought about much improvement in treatment effect sizes. If there has been any improvement at all it has been in the rate of trial success, which can only be attributed to the increase in trial size allowing for greater statistical power and consistency of outcomes.
Furthermore, what these data suggest is that the efficacy profiles of almost all modern antidepressants are essentially equivalent to one another. This fact is unexpected and counterintuitive to the prevailing assumption in the field that some antidepressants are better than others. Interestingly, data from published studies continue to suggest that there may be differences in the efficacy profile of some antidepressants compared to others24 although it is important to note that the vast majority of the trials included in this analysis were underpowered25 and likely suffered from a myriad of methodological flaws including publication bias26 which may have impacted these findings. Nonetheless, it is curious how studies based on non-regulatory trials could show differences between antidepressants and their classes while FDA regulated trials show flattened differences if any at all.
And the question remains, were the early antidepressants (including tricyclics) truly just more potent drugs than the current classes of antidepressants? Reviews of published data suggest that this may not be true,27 however this question has not been examined in the isolated context of regulatory trials. Perhaps novel antidepressant classes like the highly anticipated ketamine-based drugs would perform better?
Current Analysis
To shed light on these lingering questions, we undertook the current analysis presenting the available efficacy trial data from FDA regulatory trials of antidepressants. Using this dataset, we aimed to test the assumption that the tricyclics used in pre-regulatory clinical trials were more potent drugs. By presenting data from tricyclics used as active comparators in these regulatory trials, we sought to evaluate if tricyclics outperform modern antidepressants under parallel trial conditions. We also opted to include the available efficacy data from the trial submitted for the approval of the novel antidepressant esketamine to provide a comprehensive and updated dataset of all approved antidepressants for the treatment of major depression.
Selection of Programs/Trials
We included only acute, parallel-group, double-blind, placebo-controlled trials for investigational antidepressants approved after registering an NDA program with the US FDA. Trials were included if they enrolled adult patients with a primary diagnosis of Major Depressive Disorder (MDD) and if the trials were analyzed in the Medical and Statistical Review of Efficacy by an FDA examiner.
We excluded data from treatment arms of investigational antidepressants at dosing levels not approved by the FDA. We also excluded depression trials enrolling only geriatric (>65 years old) patients, children (<18 years old), and inpatients, as well as relapse prevention or maintenance studies due to incomparability of data from these designs. Data from treatment arms evaluating tricyclic antidepressants as active comparators were included in this analysis for the aforementioned reasons.
Data Analysis
All statistics were calculated with IBM Statistical Package for the Social Sciences (SPSS). ANOVA tests were used to compare symptom reduction with drug and placebo, drug-placebo difference in symptom reduction, and effect sizes between the classes of antidepressants. Regression values were calculated for the data in Figure 1 examining drug-placebo difference in symptom reduction and effect sizes over time.
Figure 1.
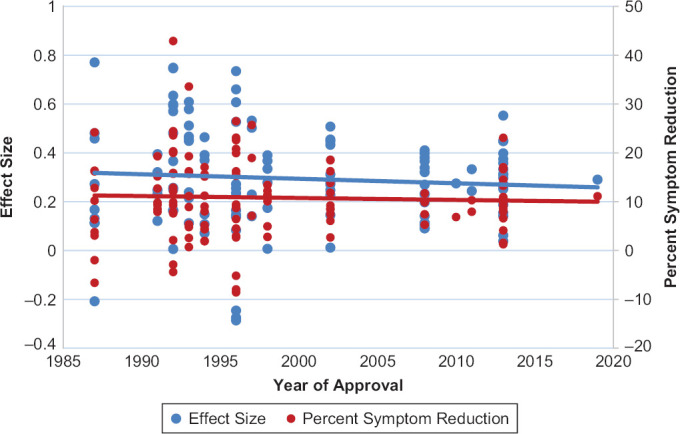
Scatterplot Evaluating Effect Sizes and Placebo-Subtracted Percent Symptom Reduction from 135 Treatment Arms Evaluating Investigational and Active Comparator Antidepressants Over Time (from 1987–2019).
Efficacy Data from FDA Statistical Review
The Medical and Statistical Reviews conducted by the FDA contain the published results of efficacy analysis along with treatment group raw baseline and change scores when available. We encountered several statistical methods for handling missing data from patient dropout in the reporting and analysis of this efficacy data. These methods included Observed Cases analysis, Analysis of Covariance (ANCOVA), and Last Observation Carried Forward (LOCF). Since data from LOCF analysis was available for all trials, we decided to use data (primary efficacy measure scores, p-values, and patient numbers) from these LOCF statistical computation tables.
Measures
Effect Size and Percent Symptom Reduction: The methods for calculating effect sizes using this dataset have been described extensively in previous publications.23 As an additional measure of treatment response and to address situations where data were insufficient to calculate a standardized effect size, we calculated the magnitude of symptom reduction as a percent (using change score over baseline for drug and placebo arms). We then subtracted the placebo response magnitude from the drug response magnitude to generate an estimated treatment effect for each individual treatment arm.
Dataset
After review of the FDA database for NDA registrations approved between 1987 and 2019, we identified a total of 17 adult depression approval programs for inclusion in this current analysis.
The investigational antidepressants (year of approval) were: fluoxetine hydrochloride (1987), sertraline hydrochloride (1991), paroxetine hydrochloride (1992), venlafaxine hydrochloride (1993), nefazodone hydrochloride (1994), mirtazapine (1996), bupropion hydrochloride SR (1996), venlafaxine hydrochloride ER (1997), citalopram (1998), escitalopram oxalate (2002), duloxetine hydrochloride (2002), desvenlafaxine succinate (2008), trazodone hydrochloride ER (2010), vilazodone hydrochloride (2011), levomilnacipran hydrochloride (2013), vortioxetine hydrobromide (2013), and esketamine (2019).
These programs cited a total of 126 efficacy evaluation trials with enough data available in the NDA report to perform this current analysis. From these trials, we excluded 40 trials after applying our inclusion/exclusion criteria: 6 geriatric population, 22 uncontrolled, 4 inpatient studies, 4 relapse-prevention design, and 4 unapproved dose trials. After exclusion: 86 registration trials were examined in this analysis.
These 86 trials provided data for 140 treatment arms evaluating an investigational antidepressant: 24 utilized an ineffective dose of the investigational antidepressant leaving 116 treatment arms for analysis. Of the 86 trials, 19 provided sufficient data from active comparators of a tricyclic antidepressant (either imipramine or amitriptyline) providing an additional 19 treatment arms for analysis, with a total of 135 treatment arms in the dataset. Table 4 reports the data collected from the selected trials and treatment arms, organized by antidepressant class.
Table 4. Efficacy Data from 86 trials of SSRIs, SNRIs, Atypicals, and Glutamate Antagonists Along with Data from Trials using tricyclics as Active Comparators.
| Program (Year) Protocol Number Dosing Schedule/Duration (weeks) | Placebo Baseline/Change Score (% Response) [N Patients] | Investigational Antidepressant Baseline/Change Score (% Response) [N Patients] | Effect Size | |||
| A. Serotonin Selective Reuptake Inhibitors (SSRIs) | ||||||
| Fluoxetine (1987) | ||||||
| 19Flex/4 | 28.2/−5.5 (19.5%) [24] | 28.6/−12.5 (43.7%) [22] | 0.77 | |||
| 27Flex/6 | 28.2/−8.4 (29.8%) [163] | 27.5/−11.0 (40.0%) [181] IMIP: 28.2/−12.0 (42.6%) [181] |
0.27 --- |
|||
| 25Flex/4 | 25.8/−8.8 (34.1%) [24] | 26.2/−7.2 (27.5%) [18] | −0.21 | |||
| 62−AFix/6 | 17.4/−5.8 (33.3%) [56] | 16.8/−6.2 (36.9%) [105] 16.6/−6.0 (36.1%) [103] 17.2/−5.4 (31.4%) [100] |
0.13
0.11 0.11 |
|||
| 62−BFix/6 | 24.3/−5.7 (23.4%) [48] | 24.7/−9.8 (39.6%) [97] 24.1/−9.6 (39.8%) [97] 24.2/−7.2 (29.8%) [103] |
0.48
0.46 0.17 |
|||
| Sertraline (1991) | ||||||
| 103Fix/6 | 25.3/−7.6 (30.0%) [86] | 24.8/−10.6 (42.7%) [90] 24.9/−9.8 (39.4%) [89] 25.7/−9.9 (38.5%) [82] |
0.32
0.25 0.23 |
|||
| 104Flex/8 | 23.4/−8.2 (35.0%) [141] | 23.3/−11.7 (50.2%) [142] AMIT: 23.2/−12.6 (54.3%) [144] |
0.39 --- |
|||
| 315Flex/8 | 22.2/−6.7 (30.2%) [73] | 23.1/−8.8 (38.1%) [76] AMIT: 23.5/−9.4 (40.0%) [70] |
0.12--- | |||
| Paroxetine (1992) | ||||||
| 01–001Flex/6 | 27.4/−10.5 (38.3%) [24] | 28.0/−13.5 (48.2%) [24] | 0.37 | |||
| 02−001Flex/6 | 25.9/−6.8 (26.3%) [53] | 26.6/−12.3 (46.2%) [51] | 0.57 | |||
| 02/002Flex/6 | 24.9/−5.8 (23.3%) [34] | 25.0/−10.9 (43.6%) [36] | 0.60 | |||
| 02/003Flex/6 | 28.9/−7.2 (24.9%) [33] | 28.6/−9.7 (33.9%) [33] | 0.25 | |||
| 02−004Flex/4 | 27.3/−7.2 (26.4%) [38] | 28.9/−12.2 (42.2%) [36] | 0.75 | |||
| 03−005Flex/6 | 26.8/−4.0 (14.9%) [42] | 26.1/−10.0 (38.3%) [40] IMIP: 27.4/−15.6 (56.9%) [37] |
0.60 --- |
|||
| 03−006Flex/6 | 28.7/−3.0 (10.5%) [38] | 29.7/−9.1 (30.6%) [39] IMIP: 27.9/−7.3 (26.2%) [39] |
0.75--- | |||
| 03−001Flex/4 | 24.8/−4.7 (19.0%) [38] | 24.9/−10.8 (42.4%) [40] IMIP: 24.8/−7.7 (31.1%) [40] |
0.63--- | |||
| 03−002Flex/4 | 25.6/−6.2 (24.2%) [40] | 24.9/−8.0 (32.1%) [40] IMIP: 24.4/−7.8 (32.0%) [40] |
0.26--- | |||
| 03−003Flex/4 | 27.0/−9.2 (34.1%) [42] | 25.7/−9.3 (36.2%) [39] IMIP: 26.3/−7.8 (29.7%) [41] |
0.01--- | |||
| 03−004Flex/4 | 27.0/−6.7 (24.8%) [37] | 27.6/−10.4 (37.7%) [37] IMIP: 26.5/−5.8 (21.9%) [40] |
0.48--- | |||
| 09Fix/12/HAM−D total | --/−8.2 (--%) [51] | --/−10.6 (--%) [104] --/−9.00 (--%) [99] --/−9.4 (--%) [100] |
0.16
0.16 0.16 |
|||
| Citalopram (1998) | ||||||
| 85AFlex/4 | 33.7/−9.6 (28.5%) [78] | 33.5/−12.9 (38.5%) [82] | 0.33 | |||
| 86141Flex/6 | 21.0/−4.9 (23.3) [50] | 22.2/−6.3 (28.4%) [97] | 0.17 | |||
| 89303Fix/6 | 23.7/−10.6 (44.7%) [64] | 23.0/−13.3 (57.8%) [61] | 0.28 | |||
| 91206Fix/6 | 24.6/−9.3 (37.8%) [124] | 24.4/−12.2 (50.0%) [120] 24.5/−12.1 (49.4%) [110] |
0.39
0.37 |
|||
| 89306Fix/6 | 33.1/−16.0 (48.3%) [88] | 31.3/−16.0 (51.1%) [97] | 0.01 | |||
| Escitalopram (2002) | ||||||
| MD 01Fix/8 | 29.5/−9.4 (31.9%) [119] | 28.0/−12.8 (45.7%) [118] 28.9/−13.9 (48.1%) [125] |
0.45
0.51 |
|||
| MD 02Flex/8 | 28.8/−11.2 (38.9%) [125] | 28.7/−12.9 (45.0%) [124] | 0.15 | |||
| 99001Fix/8 | 28.7/−13.6 (47.4%) [189] | 29.2/−16.3 (55.8%) [188] | 0.28 | |||
| 99003Flex/8 | 28.7/−12.5 (43.6%) [154] | 29.0/−15.3 (52.8%) [155] | 0.31 | |||
| Vilazodone (2011) | ||||||
| GNSC-04DP-02Flex/8 | 30.7/−9.7 (31.6%) [199] | 30.8/−12.9 (41.9%) [198] | 0.33 | |||
| CLDA-07DP-02Fix/8 | 32.0/−10.8 (33.8%) [231] | 31.9/−13.3 (41.7%) [232] | 0.24 | |||
| Vortioxetine (2013) | ||||||
| 11492AFix/6 | 33.9/−14.5 (42.8%) [105] | 34.0/−20.2 (59.4%) [100] | 0.55 | |||
| 305Fix/8/HAM-D total | 32.7/−11.3 (35.6%) [139] | 33.1/−16.2 (48.9%) [139] | 0.40 | |||
| 13267AFix/8/MADRS | 31.5/−11.7 (37.1%) [158] | 31.8/−17.2 (54.1%) [149] 31.2/−18.8 (60.3%) [151] |
0.45
0.45 |
|||
| 315USFix/8/MADRS | 31.5/−12.8 (40.6%) [153] | 31.9/−14.3 (44.8%) [145] 32.0/−15.6 (48.8%) [147] |
0.14
0.26 |
|||
| 316USFix/8/MADRS | 32.0/−10.8 (33.8%) [155] | 32.2/−13.0 (40.4%) [154] 32.5/−14.4 (44.3%) [148] |
0.19
0.36 |
|||
| 11984AFix/8/MADRS | --/−14.8 (--%) [145] | --/−16.3 (--%) [151] | 0.15 | |||
| 317Fix/8/MADRS | 33.4/−12.9 (38.6%) [149] | 34.1/−13.7 (40.2%) [143] 33.6/−13.4 (39.9%) [142] |
0.06
0.04 |
|||
| B. Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs) | ||||||
| Venlafaxine (1993) | ||||||
| 600A-206Flex/4 | 28.6/−4.8 (16.8%) [47] | 28.2/−14.2 (50.4%) [46] | 0.58 | |||
| 600A-301Flex/6 | 24.6/−9.5 (38.6%) [78] | 25.4/−13.9 (54.7%) [64] IMIP: 24.2/−10.5 (43.4%) [71] |
0.61 --- |
|||
| 600A-302Flex/6 | 24.4/−8.9 (36.5%) [75] | 25.0/−11.9 (47.6%) [65] | 0.45 | |||
| 600A-303Flex/6 | 24.6/−9.9 (40.2%) [79] | 23.6/−10.1 (42.8%) [70] IMIP: 24.7/−10.1 (40.9%) [72] |
0.11--- | |||
| 600A-203Fix/6 | 25.3/−6.7 (26.5%) [92] | 26.0/−11.1 (42.7%) [77] 26.0/−11.9 (45.8%) [79] 24.9/−10.5 (42.2%) [75] |
0.45
0.51 0.47 |
|||
| 600A-313Fix/6 | 25.4/−9.5 (37.4%) [75] | 25.6/−10.9 (42.6%) [72] 25.6/−11.8 (46.1%) [77] |
0.21
0.24 |
|||
| Venlafaxine ER (1997) | ||||||
| 208Flex/12 | 24.6/−8.7 (35.4%) [91] | 24.4/−14.9 (61.1%) [85] | 0.50 | |||
| 209Flex/8 | 23.6/−6.8 (28.8%) [100] | 24.5/−11.7 (47.8%) [91] | 0.53 | |||
| 367Flex/8 | 26.6/−13.1 (49.2%) [81] | 26.5/−15.6 (58.9%) [83] -- /-- [85] |
0.14
0.23 |
|||
| Duloxetine (2002) | ||||||
| HMAQaFlex/8 | 20.6/−6.5 (31.6%) [57] | 19.6/−8.5 (43.4%) [56] | 0.27 | |||
| HMAQbFlex/8 | 20.0/−5.7 (28.5%) [55] | 19.9/−6.2 (31.2%) [61] | 0.01 | |||
| HMATaFix/85/29/2021 | 17.8/−4.3 (24.2%) [89] | 17.4/−5.5 (31.6%) [81] | 0.23 | |||
| HMATbFix/8 | 17.2/−4.2 (24.2%) [88] | 18.1/−7.7 (42.7%) [86] | 0.45 | |||
| HMBHaFix/9 | 21.1/−5.2 (24.5%) [115] | 21.5/−9.3 (43.0%) [121] | 0.43 | |||
| HMBHbFix/9 | 20.5/−7.2 (35.3%) [136] | 20.3/−8.9 (43.8%) [123] | 0.25 | |||
| Desvenlafaxine (2008) | ||||||
| 332Fix/9 | 23.0/−9.6 (41.7%) [150] | 23.4/−11.5 (49.2%) [150] 23.4/−11.0 (47.0%) [147] |
0.27
0.20 |
|||
| 333Fix/8 | 24.3/−10.8 (44.4%) [161] | 24.3/−13.2 (54.3%) [164] 24.4/−13.7 (56.2%) [158] |
0.32
0.37 |
|||
| 223Fix/8 | --/-- (--%) [78] | --/-- (--%) [63] --/-- (--%) [72] |
0.09
0.11 |
|||
| 306Fix/8 | --/−7.7 (--%) [118] | --/−10.5 (--%) [114] --/−9.6 (--%) [116] --/−10.5 (--%) [113] |
0.38
0.23 0.41 |
|||
| 308Fix/8 | --/−9.3 (--%) [124] | --/−12.6 (--%) [121] --/−12.1 (--%) [124] |
0.40
0.34 |
|||
| 304Flex/8 | --/-- (--%) [114] | --/-- (--%) [120] | 0.14 | |||
| 309Flex/8 | --/−12.5 (--%) [120] | --/−13.4 (--%) [117] | 0.11 | |||
| 317Flex/8 | --/−9.8 (--%) [125] | --/−10.5 (--%) [110] | 0.09 | |||
| 320Flex/8 | --/−7.5 (--%) [118] | --/−9.1 (--%) [117] | 0.23 | |||
| Levomilnacipran (2013) | ||||||
| MD-01Fix/8 | 35.6/−11.6 (32.6%) [175] | 36.0/−14.8 (41.1%) [176] 36.1/−15.6 (43.2%) [177] 36.0/−16.5 (45.8%) [176] |
0.25
0.31 0.37 |
|||
| MD-03Flex/8 | 35.2/−12.2 (33.8%) [214] | 35.0/−15.3 (43.7%) [215] | 0.27 | |||
| MD-10Flex/8 | 31.0/−11.3 (36.5%) [185] | 30.8/−14.6 (47.4%) [185] 31.2/−14.4 (46.2%) [187] |
0.31
0.30 |
|||
| F02695 LP2 02Flex/10 | 30.5/−14.5 (47.5%) [277] | 30.7/−18.7 (60.9%) [276] | 0.33 | |||
| C. Atypical Antidepressants | ||||||
| Nefazodone (1994) | ||||||
| 030A2-0007Fix/6 | 26.4/−9.8 (37.1%) [47] | 25.4/−10.7 (42.1%) [47] | 0.11 | |||
| 03A0A-003Fix/6 | 25.9/−6.8 (26.3%) [45] | 25.4/−11.0 (43.3%) [44] IMIP: 25.8/−10.8 (41.9%) [45] |
0.46 --- |
|||
| 03A0A-004AFix/6 | 23.5/−8.5 (36.2%) [77] | 23.6/−9.0 (38.1%) [76] | 0.07 | |||
| 03A0A-004BFix/6 | 25.0/−9.4 (37.6%) [80] | 25.4/−12.4 (48.8%) [78] | 0.37 | |||
| CN104-005Flex/8 | 23.5/−8.0 (34.0%) [90] | 24.4/−12.0 (49.2%) [86] IMIP: 24.3/−10.2 (42.0%) [83] |
0.39--- | |||
| CN104-006Flex/8 | 23.8/−8.9 (37.4%) [78] | 23.5/−10.0 (42.6%) [80] IMIP: 23.7/−11.0 (46.4%) [79] |
0.15--- | |||
| 030A2-0004/0005 | 24.0/−9.8 (40.8%) [70] | 23.4/−10.0 (42.7%) [74] IMIP: 24.1/−10.9 (45.2%) [73] |
--- --- |
|||
| Mirtazapine (1996) | ||||||
| 003-002Flex/6 | 24.7/−5.4 (21.9%) [44] | 24.2/−11.7 (48.3%) [44] | 0.73 | |||
| 003-003Flex/6 | 25.5/−8.8 (34.5%) [45] | 25.4/−10.4 (40.9%) [45] | 0.14 | |||
| 003-008Fix/6 | 25.8/−9.6 (37.2%) [28] | 26.0/−7.6 (29.2%) [30] 25.5/−7.3 (28.6%) [28] 25.3/−8.1 (32.0%) [30] |
−0.28 −0.29 −0.25 |
|||
| 003-020Flex/6 | 29.5/−4.8 (16.3%) [39] | 27.8/−10.3 (37.1%) [41] AMIT: 29.2/−11.5 (39.4%) [40] |
0.66 --- |
|||
| 003-021Flex/6 | 24.4/−9.5 (38.9%) [48] | 24.2/−11.7 (48.3%) [45] AMIT: 25.0/−13.6 (54.4%) [48] |
0.25 --- |
|||
| 003-022Flex/6 | 31.2/−9.0 (28.8%) [50] | 33.0/−16.1 (48.8%) [49] AMIT: 32.0/−14.1 (44.1%) [50] |
0.61 --- |
|||
| 003-024Flex/6 | 27.7/−7.7 (27.8%) [48] | 27.5/−12.1 (44.0%) [50] AMIT: 27.6/−13.9 (50.4%) [49] |
0.53--- | |||
| 85027Flex/4 | 26.2/−10.9 (41.6%) [61] | 26.4/−13.4 (50.8%) [64] | 0.23 | |||
| Bupropion SR (1996) | ||||||
| 203Fix/8 | 23.2/−8.1 (34.9%) [117] | 23.4/−10.2 (43.6%) [113] | 0.27 | |||
| 205Fix/8 | 23.4/−8.3 (35.5%) [116] | 23.6/−9.0 (38.1%) [111] 24.2/−9.3 (38.4%) [111] |
0.08
0.14 |
|||
| 212Fix/8 | 23.9/−9.8 (41.0%) [148] | 24.4/−11.1 (45.5%) [144] | 0.16 | |||
| Trazodone ER (2010) | ||||||
| 04ACL3-001Flex/8 | 22.4/−9.25 (41.3%) [206] | 23.2/−11.2 (48.2%) [206] | 0.27 | |||
| D. Glutamate (NMDA receptor) Antagonist | ||||||
| Esketamine (2019) | ||||||
| Study 1Flex/4 | 37.3/−15.8 (42.4%) [109] | 37.0/−19.8 (53.5%) [114] | 0.29 | |||
Italics: Tricyclic comparator (IMIP= imipramine, AMIT=amitriptyline).
Findings
As can be seen in Figure 1, the mean effect size and the mean symptom reduction among the four classes of antidepressants has not changed significantly over time (Effect size: R2 = 0.006, β = −0.002, p = 0.40; Symptom reduction: R2 = 0.002, β = −0.04, p = 0.66).
Table 4 presents the mean efficacy endpoints (symptom reduction, drug-placebo difference, and effect size) from the 87 trials and the results of the ANOVA test across groups. As can be seen in Table 5, there were no statistically significant differences between groups in the overall symptom reduction with the investigational antidepressant, the drug-placebo difference in symptom reduction, or the effect size.
Table 5. Comparison of Mean Symptom Reduction and Effect Size Across Four Classes of Antidepressants (Tricyclics, SSRIs, SNRIs, Atypicals) and a Novel Mechanism Antidepressant (Glutamate Antagonist).
| Mean Sx Reduction with Placebo |
Mean Sx Reduction with Antidepressant |
Mean Drug-Placebo Difference in Sx Reduction |
Mean Effect Size |
|||||
| Tricyclics [19 trials, 19 tx arms] |
29.0% | 41.2% | 12.1% | ---a | ||||
| SSRIs [38 trials, 52 tx arms] |
31.6% | 42.8% | 10.9% | 0.31 | ||||
| SNRIs [29 trials, 41 tx arms] |
34.3% | 46.6% | 12.2% | 0.31 | ||||
| Atypicals [19 trials, 22 tx arms] |
33.8% | 42.1% | 7.7% | 0.23 | ||||
| Glutamate (NMDA receptor) antagonist [1 trial, 1 tx arm] |
42.4% | 53.5% | 11.1% | 0.29 | ||||
| ANOVA | ---b | p = 0.07c | p = 0.18c | p = 0.281d |
aeffect size could not be calculated for these data due to lack of sufficient details such as p-values or standard error
bANOVA test could not be run to compare the values above due to duplication of placebo values between trials.
ctest evaluates differences between data from Tricyclics, SSRIs, SNRIs, and Atypicals
dtest evaluates differences between data from SSRIs, SNRIs, and Atypicals.
Regulatory Trial Attributes that may Contribute to the Flattening of Effects
Effect sizes and symptom reduction seen with modern antidepressants in regulatory clinical trials have remained remarkably stable over the years. As can be seen in Table 5, there is very little variation from an effect size of 0.3 across all four classes of antidepressants. Furthermore, drug-placebo difference in symptom reduction hovers closely around 11 percent. Interestingly older generation tricyclics, which were assumed to be more potent based on pre-regulatory trial research, show very similar percent symptom reduction as modern antidepressants when evaluated in FDA regulated trials, converging with evidence from published studies.27
Even drugs with novel mechanisms of action such as the esketamine nasal spray show the same effect size and look nearly identical to other antidepressants when evaluated in the regulatory context (42% symptom reduction with placebo, 54% with drug, effect size 0.29). However, it must be taken under consideration that this trial was unique from the others in that it was an adjunctive study of esketamine nasal spray in treatment resistant patients. It is worth noting that two short-term trials conducted for regulatory approval of esketamine but not included in the label did not reach statistical significance (P = 0.058 and P = 0.088).28 Independent analysis of these esketamine trial data submitted to the FDA show that despite expectations from small-scale preliminary studies, esketamine performs modestly in patients with treatment resistant depression in the context of large, regulatory trials.29 These authors also raised concerns about the potential lack of specificity of drug effects and the risk of side effects demonstrated in these trials.
As of now, this phenomenon of equivalent effect sizes and percent symptom reduction across antidepressants has not been adequately investigated. Given the stubborn persistence of such results from antidepressant regulatory trials specifically, we theorize that aspects of regulatory involvement in clinical trials has had a minimizing and equalizing effect. Namely, the Kefauver-Harris Act invited the entry of non-clinical scientists employed by the US Federal Government into the design, analysis, and interpretation of clinical trials—the product of this involvement has been a mechanization and desensitization of modern antidepressant trials. Following are the specific changes that may have contributed to the significant narrowing of antidepressant-placebo differences and reduction of sensitivity and clinical relevance of modern regulatory antidepressant trials.
Changes in Depression Diagnosis and Measurement
First of these changes relates to the indisputable fact that depression is a heterogenous disorder. Intrinsically, the depressive syndrome consists of multiple variations of symptoms and signs. Some depression patients tend to have more subjective psychological symptoms: sadness, anhedonia, hopelessness, helplessness, being overwhelmed or fearful, and suicidality. On the other hand, some depressed patients tend to experience more somatic symptoms such as sleep disturbances (insomnia or hypersomnia), appetite disturbance (increased or decreased), weight changes (gain or loss), diminished concentration, lethargy, sexual dysfunction, and hypochondriacal preoccupations.
As elaborated in the work by pioneering Dutch psychologists Fried and Nesse,30 these various symptoms and signs can lead to over 52 various specific complaints and the combinations of these subjective and objective symptoms are innumerable. Not only are the presenting combinations at the time of diagnosis innumerable, but individual symptoms can fluctuate from day to day and from week to week. This varied clinical presentation is further complicated by the fact that patients who enter trials must meet DSM diagnostic criteria whereas the dependent outcome measures used in the trials, such as the previously used HAM-D and the current standard MADRS scale, use an entirely different set of symptoms.
Furthermore, rating scales highlight and describe symptoms very differently from one another. The widespread discord among depression rating scales is clearly illustrated in the diagram by author Fried,31 which we have included as Figure 2 below. Given the fact that none of the current dependent measures incorporate all 52 signs and symptoms described in this figure, we assert that methods to assess depression are currently lacking in adequacy. Furthermore, naturally occurring fluctuations are not measured at all, with the assumption that randomization will effectively control for such oscillations. Not surprisingly, this type of assumption can lead to minimizing any significant changes.
Figure 2.
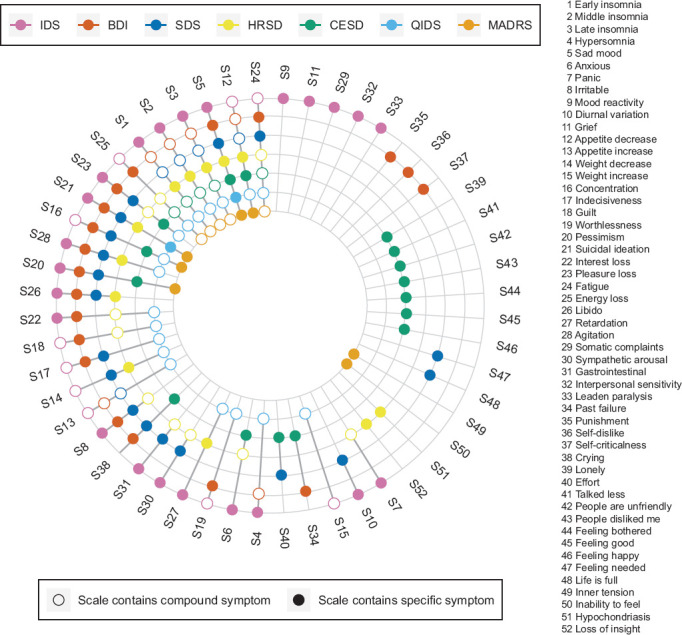
Co-Occurrence of 52 Depression Symptoms Across 7 Depression Rating Scales Colored Circles for a Symptom Indicate that a Scale Directly Assesses that Symptom, While Empty Circles Indicate that a Scale Only Measures a Symptom Indirectly. for Instance, the IDS Assesses Item 4 Hypersomnia Directly; the BDI Measures Item 4 Indirectly Via a General Question On Sleep Problems; and the SDS Does Not Capture Item 4 at All. Note that the 9 QIDS Items Analyzed Correspond Exactly to the DSM-5 Criterion Symptoms for MDD
Reprinted from Journal of Affective Disorders, vol. 208, Eiko I Fried, The 52 symptoms of major depression: Lack of content overlap among seven common depression scales, 191–197. Copyright (2017), with permission from Elsevier.
Of greater concern is the fact that the complex phenomenon of depression has been reduced to a single, composite total score. This is typically selected as the primary outcome measure by the US FDA, meaning that the success or failure of a putative new antidepressant rests heavily on this measure. Such a score adds together all the assigned numerical values given to each symptom, which are disjointed as is, and this value is intended to represent “severity” of the depressive syndrome.
The use and overuse of composite scores in efficacy analysis by the US FDA promotes fallacious, misleading, and inaccurate assumptions. For example, a score of 30 on MADRS may have an entirely different cluster of symptoms and signs for each patient and these may or may not be the most relevant or impactful symptoms for that individual patient.32 Consider the comparison of a 25-year-old teacher who is depressed, anxious, and fearful and a 55-year-old unemployed construction worker who just lays in bed most of the time. While depression presents in a vastly different manner between these two cases, their composite total scores may be identical. Moreover, the way in which antidepressants interact with a patient’s unique cluster of symptoms cannot be captured in these scales and antidepressants may have differential effects for different symptoms.33
A depersonalized composite score starkly contrasts against the vivid clinical vignettes used to illustrate the effects of depression treatments in the years of early antidepressant development. Furthermore, it has become customary to accept this composite score as a substitute for an overall clinical impression—however, it is important to note that the validity of this substitution has not been substantiated with any data and is generally questioned by practitioners. While it is impossible to measure the amount of information that is “lost in translation” when patients are deduced to a numeric value, one can safely assume that the clinical relevance of such information is certainly diminished. The assumption that a person’s total score on a depression scale can serve as a stand-in for the individual themselves is an inbuilt error in the dependent measures used in regulatory trials.34
Not surprisingly, repeat use of such erroneous composite scores (such as weekly batteries of MADRS at trial visits) leads to diminishing sensitivity of the dependent measure. By asking repeated identical questions in order to increase reliability, patients learn the rote of evaluation and the response tends to become less thoughtful, less detailed, and more contrived. This phenomenon is likely a significant underlying factor contributing to a gradual decrease in the severity of reported symptoms across all subject scores. Due to this normalizing effect, we suggest that there may be a dilution of treatment effects as a result of repetitive use of scales that combine vastly divergent symptom profiles to create total composite scores. In addition, treatment effects may be further dampened by their reliance on patient recall and self-report.
Unfortunately, as it stands the only regulatory-approved scales are the MADRS and HAM-D, which have shown to have no apparent difference from one another in detecting treatment effects in regulatory antidepressant trials.35 Eliminating repeat use of scales and creating a dependent measure more focused on clinically relevant and treatment-sensitive symptoms would be one avenue to pursue. Additionally, we suggest a measure that incorporates clinical judgment and interpretation in the overall assessment of treatment effects.
Changes in Statistical Procedures
There are vast differences in the procedures for statistical assessment in modern regulatory trials compared to trials conducted prior to 1978. The pre-regulatory antidepressant trials consisted of a simple model involving categorical endpoints—initially, physicians observed depressed patients over time and declared how many were better after treatment with an investigational compound. Not surprisingly, this method appears to have favored the proportion of responders. With the need to quantify further what qualified as a therapeutic response, a simple seven-point scale called the Clinical Global Impression Scale (CGI) was developed.36 However, the CGI was still felt to be inadequate and hence, in the 1960s a scale that quantified signs and symptoms was developed. The HAM-D was introduced as a tool intended to measure the entire syndrome of depression with the assumption that this continuous variable would more specifically characterize the symptoms involved in therapeutic response (e.g. differential effects of antidepressants on insomnia or agitation). It was also intended to provide better estimates of the magnitude of drug effects.
Despite the increased specificity of the HAM-D, clinicians were more inclined to determine overall response rates for antidepressants which provided simple statistics to report to patients in their practice. Therefore HAM-D scores on individual symptoms were totaled, essentially becoming a composite score of global symptom improvement. Furthermore, the investigators moved towards dichotomizing into responders vs. non-responders based on a simple and arbitrary 50% cutoff for reduction in symptoms from baseline. Raters were fully aware of the baseline scores of patients and how to differentiate a therapeutic response. In effect, the bias of categorical endpoints favoring greater drug-placebo differences was replicated with a continuous measure by imposing dichotomization. This bias favoring greater drug-placebo differences can be seen in Table 3 from Rickels et al16 in the introduction.
Usually in these pre-regulatory trials, depressed patients who could not tolerate the medication or failed to show any signs of early response were excused and not counted as trial participants. Because data would be excluded from analysis unless the patient made it to the end of the trial, this technique was termed “completer analysis”. Although analyses at multiple points during the AD trial were performed, these week by week results did not serve as primary outcome measures. As described earlier, the scientists and statisticians at regulatory agencies considered these types of data handling as a free pass to putative investigational antidepressants and strongly disfavored such data analysis techniques.
With regulatory involvement, depression trials transitioned from completer analysis to an alternative method of accounting for dropouts throughout the trial, known as Last Observation Carried Forward (LOCF). In this technique, it is assumed that any patient who leaves the trial early will maintain the same severity of symptoms as measured by the last recorded composite score. The LOCF method may reduce any antidepressant-placebo difference for two reasons. First, patients who cannot tolerate the antidepressant are counted in the efficacy analysis. Also, differences between the rate of drop out and the reasons for drop out in the different treatment assignments may serve to decrease the estimated drug response and increase placebo.37 LOCF as a statistical method has been widely disputed and has been shown to both inflate and deflate drug effects in different circumstances.38,39
Alternatives to LOCF are being developed. Mixed-effects model repeated measures (MMRM) have been used in some cases and appear to afford greater power in the statistical handling of regulatory trial data, which results in greater success rates but unaffected effect sizes. Multiple imputation (MI) involves creating multiple complete datasets based on the margin of variability in the distribution of observed data and essentially averaging the results from each dataset. Studies have demonstrated that there may be some advantages of this technique, in that it tends to be less biased than others.40 and has in certain studies been shown to nearly replicate the results obtained from an actual complete dataset in a clinical trial setting.41 For these reasons, MI may provide a better model for handling missing data in regulatory trials of antidepressants although further studies would be needed to draw conclusions about the effect of this method on effect sizes.
Another factor that has changed in the analysis and interpretation of AD trials relates to the inclusion of all subjects in the efficacy analysis who took a single dose of medication (termed intent to treat). In earlier trials, the analysis only included subjects who had had an adequate duration of exposure to the antidepressant: between 2 and 4 weeks. Currently the FDA analysis of efficacy data includes all participants regardless of circumstances, potentially leading to diminishing antidepressant-placebo differences.
Regulatory involvement also saw the end of categorical analyses of responder rates as primary endpoints. The selected endpoint used in efficacy analysis in regulatory trials of antidepressants is almost uniformly the average change in total HAM-D scores from baseline. Assessing these change scores over the course of the trial, and in some cases week-by-week, required much more complicated statistical procedures such as ANOVA for repeated measures and mixed models. It is likely that the increasingly complex statistical handling of these efficacy data has brought more rigor to regulatory statistical analysis but at the cost of decreased sensitivity to therapeutic effects. The goal of decreasing the statistical favorability toward drug effects seen early pre-regulatory trials has likely been achieved with these changes in statistical analysis procedures employed by regulatory agencies.
Take Home Points
Historically it was thought that antidepressant trials would result in two-thirds of depressed patients responding to antidepressants and one-third to placebo. This is no longer applicable for the antidepressant trials conducted in the past 40 years. Changes in study design, conduct, and data analysis from the introduction of regulatory oversight have systematically altered the landscape for antidepressant development. In other words, we can now reliably expect that the contemporary antidepressant clinical trials will show an effect size of 0.3 regardless of the class of antidepressant. Most importantly, without major reform in the way that trials are conducted, modest antidepressant effects will likely continue to dominate regulatory trial research.
However, given the extent of regulatory involvement in depression trials in the last 40 years, it stands to reason that these regulatory trial data may not represent the true effectiveness of antidepressants. In this context, it notable that modern antidepressants are extensively prescribed (in England, nearly 71 million prescriptions for antidepressants were given out in 2018)42 and antidepressants are generally felt to be useful among prescribers. In addition, many reviews indicate that clinical use is more nuanced than in clinical trials.16,43–45 It is especially important to consider that antidepressants in practice are applied to patients who vastly differ from clinical trial participants.4
Of practical importance, because of the current state of modest effects in antidepressant trials it is essential to include a large enough sample size to power for an effect size of 0.3 or smaller. Small trials can easily create both over and under-estimation errors in both placebo and drug groups. False negatives are well-known risks of small sized studies. However, it is equally important to note that if we do not enroll adequate sample sizes we will continue run the serious risk of getting an inflated false positive resulting in an overestimate of treatment effects that is not replicable (as was the case with many of the earlier regulatory trials, which tended to have small sample sizes).25 This is especially pertinent for early pilot studies of investigational antidepressants (phase I and II trials), which are not always subject to the same regulatory statutes of later stage trials. This phenomenon is illustrated by the dramatic decline of treatment effect sizes seen with esketamine over the course of development (from small pilot studies to large regulatory trials). Although regulatory agencies allow for more lenient methods for exploratory purposes, this method may yield misleading conclusions because these small trials are invariably under-powered. Specifically, these exploratory trials may end up with an erroneously low placebo response and thus a falsely inflated estimate of effect size.46 This possibility is under appreciated by many investigators but should be strongly considered given the persistence of modest effect sizes in regulatory trials of antidepressants.
It is also important to keep in mind that this randomized, double-blind, placebo-controlled model is limited to chemical treatments— we cannot apply this model to other interventions such as ECT, acupuncture, and behavioral therapy and compare across treatment modalities. These regulatory trials are highly restrictive in format and do not allow or encourage innovation in depression treatment.
Finally, we must appreciate that the regulatory placebo-controlled, double blind model is not applicable to the real world. It is simply a hurdle to clear; a sterilized and insular creation for a narrow purpose. These are not representative patients and the practices of a clinical trial are not reflective of how depression medications and treatments are managed in the real world. We must not stretch the results of these trials beyond their actual capacity to inform clinical practice. They are, after all, effectively artifacts of reactive regulatory decisions. FDA trials are solely designed to establish if the drug is more effective than placebo and to determine basic safety. While we recognize that clinical trials serve the important role of establishing a general superiority of antidepressants over placebo, the extent of treatment benefits that antidepressants provide for patients is best measured in clinical practice—where the drugs are discerningly applied, where the symptoms most relevant to individual patients are weighted, and where practitioners can observe the effects of treatment on symptoms as they present in the context of a unique patient’s life.
Acknowledgments
The authors of this work do not wish to make any special acknowledgments.
References
- 1.Khan A, Faucett J, Lichtenberg P, Kirsch I, Brown WA. A systematic review of comparative efficacy of treatments and controls for depression. PLoS ONE. 2012;7(7):e41778. doi: 10.1371/journal.pone.0041778. DOI: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirsch I, Huedo-Medina TB, Pigott HE, Johnson BT. Do outcomes of clinical trials resemble those of “real world” patients? A reanalysis of the STAR*D antidepressant data set. Psychology of Consciousness. Theory, Research, and Practice. 2018;5(4):339–345. [Google Scholar]
- 3.Winerman L. By the numbers: antidepressant use on the rise. APA Monitor on Psychology. 2017;48(10):120. [Google Scholar]
- 4.Zimmerman M, Mattia JI, Posternak MA. Are subjects in pharmacological treatment trials of depression representative of patients in routine clinical practice. Am J Psychiatry. 2002;159(3):469–73. doi: 10.1176/appi.ajp.159.3.469. [DOI] [PubMed] [Google Scholar]
- 5.Kramer PD. New York, NY: 2016. Ordinarily Well: The Case for Antidepressants. Farrar, Straus, and Giroux. [Google Scholar]
- 6.Ramachandraih CT, Subramanyam N, Jurgen Bar K, Baker G, Yeragani VK. Antidepressants: from MAOIs to SSRIs and more. Indian J Psychiatry. 2011;53(2):180–182. doi: 10.4103/0019-5545.82567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kline NS. Clinical experience with iproniazid (marsilid) J Clinical and Experimental Psychopathology. 1958;19(2):72–79. [PubMed] [Google Scholar]
- 8.Loomer HP, Saunders JC, Kline NS. Drugs and affects: a clinical and pharmacodynamic evaluation of iproniazid as a psychic energizer. Psychiatric Research Reports. 1957:129–141. [PubMed] [Google Scholar]
- 9.Furst W. Therapeutic re-orientation in some depressive states: Clinical evaluation of a new mono amine oxidase inhibitor (W-1544-A) (phenelzine (Nardil)) Am J Psychiatry. 1959;116:429–434. doi: 10.1176/ajp.116.5.429. [DOI] [PubMed] [Google Scholar]
- 10.Arnow LE. Phenelzine: A therapeutic agent for mental depression. Clinical Medicine. 1959;6:1573–1577. [Google Scholar]
- 11.Sawyer-Foner GJ, Koranyi EK, Meszaros A, Grauer H. Depressive states and drugs II: The study of phenelzine dihydrogen sulfate (Nardil) in open psychiatric settings. Can Med Assoc J. 1959;81:991–996. [PMC free article] [PubMed] [Google Scholar]
- 12.Brown WA, Rosdolsky M. The clinical discovery of imipramine. Am J Psychiatry. 2015;172:426–429. doi: 10.1176/appi.ajp.2015.14101336. [DOI] [PubMed] [Google Scholar]
- 13.Klerman GL, Cole JO. Clinical pharmacology of imipramine and related antidepressant compounds. Pharmacol Rev. 1965;17(2):101–141. [PubMed] [Google Scholar]
- 14.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rickels K, Feighner JP, Smith WT. Alprazolam, amitriptyline, doxepin, and placebo in the treatment of depression. Arch Gen Psychiatry. 1985;142:134–141. doi: 10.1001/archpsyc.1985.01790250028004. [DOI] [PubMed] [Google Scholar]
- 16.Rickels K, Chung HR, Csanalosi IB, Hurowitz AM, London J, Wiseman K, Kaplan M, Amsterdam JD. Alprazolam, diazepam, imipramine, and placebo in outpatients with major depression. Arch Gen Psychiatry. 1987;44:862–866. doi: 10.1001/archpsyc.1987.01800220024005. [DOI] [PubMed] [Google Scholar]
- 17.Green JA, Podolsky SH. Reform, regulation, and pharmaceuticals — the Kefauver–Harris Amendments at 50. NEJM. 2012;367(16):1481–1483. doi: 10.1056/NEJMp1210007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darrow JJ, Avorn J, Kesselheim AS. FDA approval and regulation of pharmaceuticals, 1983–2018. JAMA. 2020;323(2):164–176. doi: 10.1001/jama.2019.20288. doi: [DOI] [PubMed] [Google Scholar]
- 19.BMRC: British Medical Research Council. Clinical trial of the treatment of depressive illness . BMJ. 1965;1:881–886. doi: 10.1136/bmj.1.5439.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalmers I. Why the 1948 MRC trial of streptomycin used treatment allocation based on random numbers. J R Soc Med. 2011;104(9):383–386. doi: 10.1258/jrsm.2011.11k023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan A, Brodhead AE, Kolts RL. Relative sensitivity of the Montgomery-Asberg depression rating scale, the Hamilton depression rating scale and the Clinical Global Impressions rating scale in antidepressant clinical trials: a replication analysis. Int Clin Psychopharmacol. 2004;19:1–4. doi: 10.1097/00004850-200405000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Khan A, Warner HA, Brown WA. Symptom reduction and suicide risk in patients treated with placebo in antidepressant clinical trials. Arch Gen Psychiatry. 2000;57:311–317. doi: 10.1001/archpsyc.57.4.311. [DOI] [PubMed] [Google Scholar]
- 23.Khan A, Fahl Mar K, Faucett J, Khan Schilling S, Brown WA. Has the rising placebo response impacted antidepressant clinical trial outcome? Data from the US Food and Drug Administration 1987–2013. World Psychiatry. 2017;16:181–192. doi: 10.1002/wps.20421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, Leucht S, Ruhe HG, Turner EH, Higgins JPT, Egger M, Takeshima N, Hayasaka Y, Imai H, Shinohara K, Tajika A, Ioannidis JPA, Geddes JR. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391(10128):1357–1366. doi: 10.1016/S0140-6736(17)32802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chevance A, Naudet F, Gaillard R, Ravaud P, Porcher R. Power behind the throne: a clinical trial simulation study evaluating the impact of controllable design factors on the power of antidepressant trials. Int J Methods Psychiatr Res. 2019:e1779. doi: 10.1002/mpr.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munkholm K, Paludan-Muller AS, Boesen K. Considering the methodological limitations in the evidence base of antidepressants for depression: a reanalysis of a network meta-analysis. BMJ Open. 2019;9:e024886. doi: 10.1136/bmjopen-2018-024886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Undurraga J, Baldessarini RJ. Comparison of tricyclic and serotonin-reuptake inhibitor antidepressants in randomized head-to-head trials in acute major depression: systematic review and meta-analysis. J Psychopharmcology. 2017;31(9):1184–1189. doi: 10.1177/0269881117711709. [DOI] [PubMed] [Google Scholar]
- 28.Wilkinson ST, Howard DH, Busch SH. Psychiatric practice patterns and barriers to the adoption of esketamine. JAMA. 2019;322(11):1023–1116. doi: 10.1001/jama.2019.10728. [DOI] [PubMed] [Google Scholar]
- 29.Gastaldon C, Papola D, Ostuzzi G,, Barbui C. Esketamine for treatment resistant depression: a trick of smoke and mirrors. Epidemiology and Scientific Sciences. 2019;29(e79):1–4. doi: 10.1017/S2045796019000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fried EI, Nesse RM. Depression sum-scores don’t add up: why analyzing specific depression symptoms is essential. BMC Med. 2015;13:72. doi: 10.1186/s12916-015-0325-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fried E. The 52 symptoms of major depression: Lack of content overlap among seven common depression scales. J Affect Disord. 2017;208:191–197. doi: 10.1016/j.jad.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 32.Fried E. Problematic assumptions have slowed down depression research: why symptoms, not syndromes are the way forward. Front Psychol. 2015;6:309. doi: 10.3389/fpsyg.2015.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballard ED, Yarrington JS, Farmer CA, Lener MS, Kadriu B, Lally N, Williams D, Machado-Vieira R, Niciu MJ,, Park L, Zarate CA., Jr Parsing the heterogeneity of depression: an exploratory factor analysis across commonly used depression rating scales. J Affect Disord. 2018 doi: 10.1016/j.jad.2018.01.027. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan A, Fahl Mar K, Brown WA. The conundrum of depression clinical trials: one size does not fit all. Int Clin Psychopharmacol. 2018;33:239–248. doi: 10.1097/YIC.0000000000000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan A, Khan SR, Shankles EB, Polissar NL. Relative sensitive of the Montgomery-Asberg Depression Rating Scale, the Hamilton Depression Rating Scale and the Clinical Global Impressions rating scale in antidepressant clinical trials. Int Clin Psychopharmacol. 2002;17:281–285. doi: 10.1097/00004850-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Guy W. Rockville, MD: US Department of Health, Education, and Welfare Public Health Service Alcohol, Drug Abuse, and Mental Health Administration; 1976. Clinical Global Impressions. In: ECDEU Assessment Manual for Psychopharmacology. [Google Scholar]
- 37.Schalkwijk S, Undurraga J, Tondo L, Baldessarini RJ. Declining efficacy in controlled trials of antidepressants: effects of placebo dropout. Int J Neuropsychopharmacology. 2014;17:1343–1352. doi: 10.1017/S1461145714000224. [DOI] [PubMed] [Google Scholar]
- 38.Hamer RM, Simpson PM. Last observation carried forward versus mixed models in the analysis of psychiatric clinical trials. Am J Psychiatry. 2009;166:6. doi: 10.1176/appi.ajp.2009.09040458. [DOI] [PubMed] [Google Scholar]
- 39.Lachin JM. Fallacies of last observation carried forward. Clin Trials. 2016;13(2):161–168. doi: 10.1177/1740774515602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sterne JAC, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, Wood AM, Carpenter JR. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;338:b2393. doi: 10.1136/bmj.b2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen AW, Lundstrom LH, Wetterslev J, Astrup A, Gotsche PC. Comparison of results from different imputation techniques for missing data from an anti-obesity drug trial. PLOS One. 2014;9(11):e111964. doi: 10.1371/journal.pone.0111964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iacobucci G. NHS prescribed record number of antidepressants last year. BMJ. 2019;364:l1508. doi: 10.1136/bmj.l1508. [DOI] [PubMed] [Google Scholar]
- 43.Moller HJ. Isn’t the efficacy of antidepressants clinically relevant? A critical comment on the results of the meta-analysis by Kirsch et al. 2008. Eur Arch Psychiatry Clin Neurosci. 2008;258:451–455. doi: 10.1007/s00406-008-0836-5. [DOI] [PubMed] [Google Scholar]
- 44.Hegerl U, Mergl R. The clinical significance of antidepressant treatment effects cannot be derived from placebo-verum response differences. J Psychopharmacol. 2009;24:445–448. doi: 10.1177/0269881109106930. [DOI] [PubMed] [Google Scholar]
- 45.Baghai TC, Blier P, Baldwin DS, Bauer M, Goodwin GM, Fountoulakis KN, Kasper S, Leonard BE, Malt UF, Stein D, Versiani M, Moller H., WPA General and comparative efficacy and effectiveness of antidepressants in the acute treatment of depressive disorders: a report by the WPA section of pharmacopsychiatry. Eur Arch Psychiatry Clin Neurosci. 2011;261(Suppl 3):S207–S245. doi: 10.1007/s00406-011-0259-6. [DOI] [PubMed] [Google Scholar]
- 46.Reinhart A. San Francisco, CA: No Starch Press, Inc; 2015. Statistics done wrong: A woefully complete guide. [Google Scholar]