

Abstract
HIV-1 infection and nicotine addiction are global public health crises. In the central nervous system, HIV-1 causes a devastating neurodegenerative disease. It is well recognized that microglial cells play a pivotal role in the neuropathogenesis of HIV-1 and that drugs of abuse not only contribute to the spread of this agent but may facilitate viral expression in these brain macrophages. Nicotine has been shown to stimulate the production of HIV-1 by in vitro-infected alveolar macrophages, and the HIV-1 protein gp120 binds to nicotinic receptors. In this study, we demonstrated the constitutive expression of nicotinic acetylcholine receptor mRNA in primary human microglial cells and showed that the pretreatment of microglia with nicotine increased HIV-1 expression in a concentration-dependent manner, as measured by p24 antigen levels in culture supernatants. We also found that nicotine robustly altered the gene expression profile of HIV-1-infected microglia and that the transforming growth factor-β1 is involved in the enhanced expression of HIV-1 by nicotine.
Keywords: nicotine, HIV-1, microglia, neuropathogenesis, TGF-β1
Nicotine is one of over 3,800 components of cigarette smoke and is the addictive element of tobacco. Unique among these components, nicotine has been identified as the major immunosuppressive agent of tobacco (Sopori and Kozak 1998). The manner in which nicotine expresses its immunosuppressive properties is by promoting T cell anergy, inhibiting the Th1 response, increasing leukocyte interleukin (IL)-4 production, and inhibiting macrophage production of the proinflammatory cytokines IL-1, IL-6, and tumor necrosis factor (TNF) (Sopori and Kozak 1998). In addition, nicotine has been shown to be chemotactic for neutrophils (Totti et al. 1984), enhance the production of the chemokine CCL3 by rat alveolar macrophages (Chong et al. 2002), inhibit lipopolysaccharide (LPS)-induced production of proinflammatory cytokines (IL-1, IL-8) and prostaglandin E2 production by a macrophage cell line (Sugano et al. 1998), and suppress dendritic cell function (Nouri-Shirazi and Guinet 2003). In one model, the α−7 subunit of the nicotinic acetylcholine receptor (nAChR) has been implicated as the essential regulator of inflammation (Wang et al. 2003).
The effect of nicotine on the resident macrophages of the brain, i.e., microglial cells, has received limited research attention. nAChRs are found on murine microglia (Shytle et al. 2004). The effects of nicotine on the functional activities of microglial cells may have implications regarding the neuropathogenesis of infections of the central nervous system (CNS) and may be an important confounder in the study of drugs of abuse on such infections. Reports of studies on the effects of nicotine on HIV-1 pathogenesis are sparse. In one study, the HIV-1 protein gp120 was shown to bind to nAChRs (Bracci et al. 1992). In addition, nicotine has been reported to enhance the production of HIV-1 by in vitro-infected alveolar macrophages from otherwise healthy cigarette smokers (Abbud et al. 1995). Finally, the limited examination of the neuropathogenic effects of nicotine on CNS-related HIV-1 infection has shown that galantamine and nicotine have a synergistic effect on the inhibition of microglial cell activation induced by HIV-1 gp120 (Giunta et al. 2004).
Because nicotine possesses immunomodulatory properties and has been shown to affect HIV-1 interaction with macrophages, we set out in this study to test the hypothesis that nicotine would enhance HIV-1 expression in human microglial cells and if so, to explore the mechanism underlying this property of nicotine.
Materials and methods
Reagents
Nicotine: nicotine hydrogen tartrate salt; nAChR antagonists: D-tubocurarine (D-TC) and α-bungarotoxin (α-BGTX); Dulbecco’s modified Eagle’s medium (DMEM), Hank’s balanced salt solution (HBSS), penicillin, streptomycin, trypsin (Sigma Chemical, St. Louis, MO, USA); transforming growth factor (TGF)-β1 specific polyclonal chicken IgY antibodies (R&D Systems, Minneapolis, MN, USA); HIV-1SF162: obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health (Bethesda, MD, USA).
Microglial cell cultures and HIV-1 infection
Highly enriched cultures of human microglia were isolated from 16- to 20-week-old fetal brain tissue specimens using a protocol approved by the Human Subject Research Committee at our institution as previously described (Peterson et al. 2004). Briefly, brain tissues were dissociated after trypsinization (0.25%) and plated into 75 cm2 Falcon culture flasks in DMEM containing 6% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml). The medium was replenished 4 days later. Microglia were harvested after 10–14 days with purity of >99% (stained by anti-CD68 antibody, a macrophage marker).
HIV-1SF162, a monocytotropic (R5) variant, was grown in peripheral blood mononuclear cell (PBMC) cultures that had been prepared with cells obtained by a standard Ficoll separation technique yielding cells that were <98% PBMC. PBMC cultures were stimulated with 4 mg/ml of phytohemagglutinin (PHA) and 5 U/ml interleukin (IL)-2 for 3 days before infection. After 21 days of incubation with media changes every 3 days and fresh PBMCs every week, supernatants were harvested, centrifuged at 3,000 rpm for 20 min to eliminate cell debris, and stored at −80°C. The MOI of HIV-1 in this study was based on the tissue culture infectious dose (TCID) 50, which was determined by exposing peripheral blood mononuclear cells to HIV-1. HIV-1SF162 was added to microglia at a multiplicity of infection (MOI) of 0.02. After 24 h of absorption with HIV-1 at 37°C, nicotine-pretreated and untreated control cells were washed three times and resuspended in culture medium (DMEM, 6% FBS) containing the indicated concentrations of drugs or drug combinations.
Assessment of HIV-1 expression
Supernatants from microglial cell cultures were collected at 7 days post infection and assayed for HIV-1 p24 Ag production by ELISA (Beckman Coulter, Fullerton, CA, USA) as previously described (Peterson et al. 2004).
RT-PCR assay for nACh receptor expression
Total RNA isolated with Qiagen RNeasy Mini kit (Qiagen, Valencia, CA, USA) from microglia was treated with DNase (Ambion, Austin, TX, USA) followed by reverse transcription for cDNA synthesis with SuperScript™ II reverse transcriptase (RT), 5x cDNA synthesis buffer, 0.1 M dithiothreitol (DTT) (Invitrogen, Carlsbad, CA, USA), dNTP, oligo (dT)12–15, and random primer (GE HealthCare, Piscataway, NJ, USA) to a final volume of 20 μl. The same amounts of RNA were used in no RT reaction as controls to monitor DNA contamination. The cDNA was amplified by PCR for 40 cycles with the Stratagene RoboCycler (Stratagene, La Jolla, CA, USA) with primers specific for nAChRs and the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The amplified PCR products were electrophoresed for size verification. The primer sequences were: sense 5′-ATTGGAGAGTACCTCCTGTTCACCAT-3′ and antisense 5′-GCTTCTCGTGAGGTTAGCACTGAAAT-3′ for α3 (357 bp); sense 5′-AGCTGACTATGAGAATGTCACCTCCATC-3′ and antisense 5′-ACAGCACTCGTACTTCCTGGTGTTGTAG-3′ for α4 (373 bp); sense 5′-GGTAACCGTCTTCGCTATCA ACATTC-3′ and antisense 5′-CAATCTTCAACAACC TCACGGACATC-3′ for α5 (273 bp); sense 5′-AGAGGAGTGAAAGGTTCTATGAGTGCTG-3′ and antisense 5′-TTC AGAAGGATGACTCTGGTCCACTT-3′ for α7 (400 bp); sense 5′-GAGAAGATGACGTTGTGCATCTCAGT-3′ and antisense 5′-GTACTTCCAGTCCTCACTCACGCTCT-3′ for β2 (570 bp); sense 5′-CGGTCTTGGTTTCTCTGA CAGTTTTC-3′and antisense 5′-AGGACTTTGCCTTT CACTACTGGTTG-3′ for β3 (310 bp); sense 5′-CTCTCATCGGCAAGTACCTCATGTT-3′ and antisense 5′-TACTTCCAGTCCTCAACGACACTCTG-3′ for β4 (481 bp); sense 5′-CCACCCATGGCAAATTCCATGGCA-3′ and antisense 5′-TCTAGACGGCAGGTCAGGTCCACC-3′ for GAPDH (600 bp).
Oligonucelotide microarray
Array analysis was performed using Oligo GEArray® Human HIV Infection and Host Response arrays (SuperArray, Frederick, MD, USA), which contains 120 distinct HIV-1-related genes, and the hybridization procedures were performed following the manufacturer’s instructions. Human microglial cells were pretreated with 300 μM nicotine (vs medium alone [control]) for 24 h and then infected with HIV-1SF162 (vs uninfected control cells) and total RNA was extracted 24 h and 7 days post infection using the RNeasy Mini Kit (Qiagen). The stated gene expression values are from several replicate determinations in a single assay. Chemiluminescent detection steps were performed, and positive spots on the arrays were scanned using a Kodak Image Station and were quantified using the GEArray Analysis Suite software (SuperArray). Normalization of values and group comparisons were also determined by the GEArray Analysis Suite software (SuperArray). The method by which genes were selected as significant was twofold or greater value.
Treatment with TGF-β1 antibodies
Microglial cells were pretreated with nicotine for 24 h at 300 μM in the presence or absence of anti-TGF-β1 antibodies 30 min before at the indicated concentrations, followed by infection with HIV-1SF162.
Statistical analysis
For the analysis of the effect of nicotine on viral expression, p24 Ag levels in supernatants of cells treated with nicotine and nACh receptor antagonists were expressed as the percentage of inhibition relative to the p24 Ag levels in supernatants of untreated (control) cells and analyzed by Student’s t test. Similar analysis was performed for the evaluation of the effect of anti-TGF-β1 antibodies.
Results
Human microglial cells constitutively express nAChRs
Examination of the various subunits of the nAChR was conducted using RT-PCR of primary human microglial cells, which revealed the constitutive expression of nAChR mRNA in samples of microglia tested. Specifically, α3, α5, α7, and β4 subunits were present, whereas α4, β2, and β3 subunits were absent (Fig. 1a). These findings not only establish a potential receptor by which nicotine may exert its effects on microglial cells, but demonstrate the presence of the α7 subunit, which has been shown to be critical in expressing the immunomodulatory effects of nicotine (Wang et al. 2003).
Fig. 1.
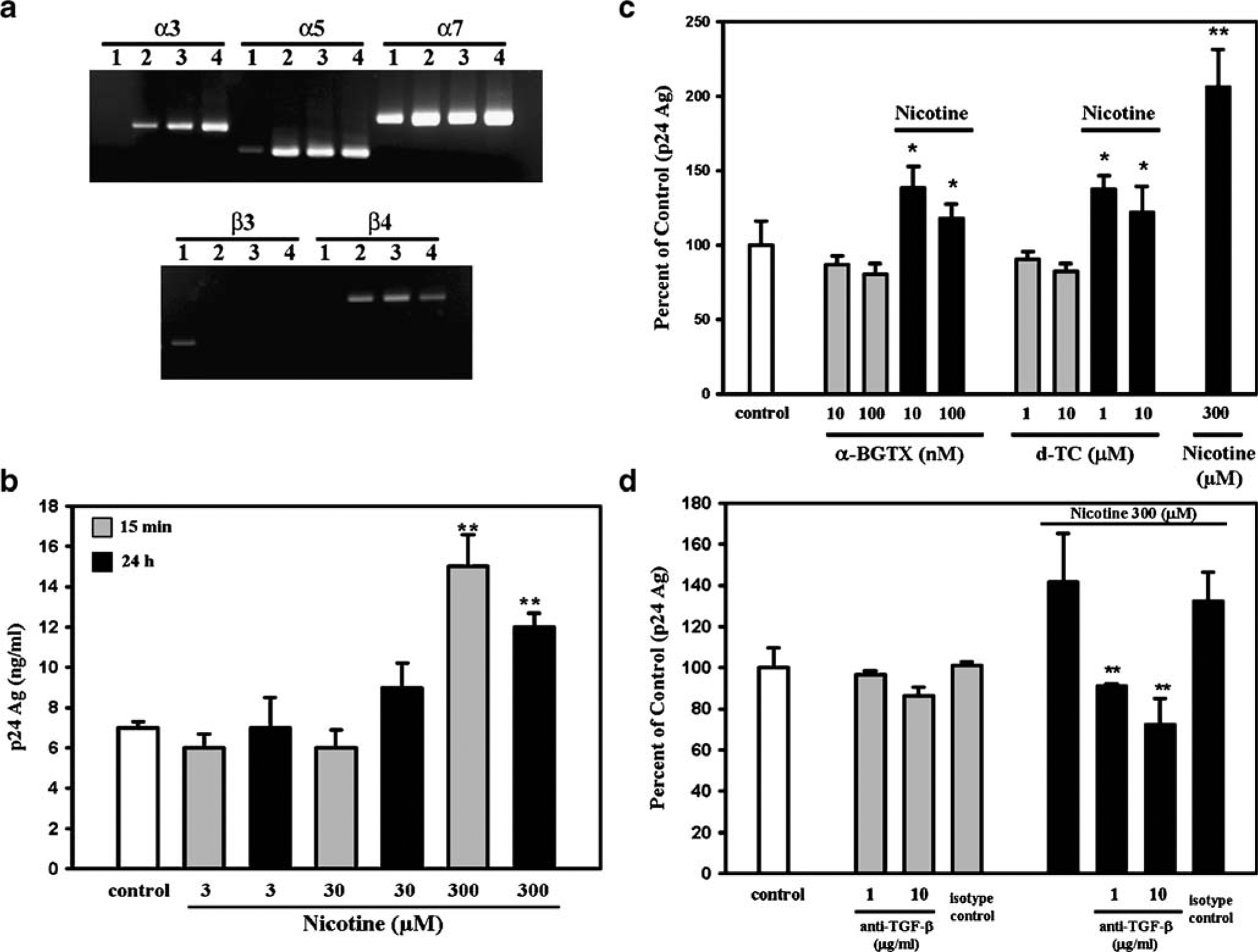
Nicotinic receptors and effects of nicotine on HIV-1 expression in microglia. a Constitutive expression of nAChRs in human microglial cells. A representative gel shows the presence of mRNA of α3, α5, α7, and β4 subunits. mRNA for the β3 subunit was absent. mRNA for subunits α4 and β2 were also absent (data not shown). Lanes 1 to 4 microglial cell cultures derived from four different brain tissue specimens. No PCR products were found in RT control of DNase-treated RNA (data not shown). b Effects of nicotine pretreatment on viral expression. Microglial cells were pretreated with nicotine for 15 min or 24 h at the indicated concentrations, followed by infection with HIV-1. After 7 days of infection, supernatants were then collected for measurement of p24 antigen (Ag) levels. Data are presented as the mean±SEM of three separate experiments using microglia from different brain specimens. **p<0.01 vs untreated (control) cells by Student’s t test. c Effects of α-BGTX and D-TC on nicotine pretreatment of HIV-1-infected microglia. Microglial cells were pretreated with nicotine for 24 h at 300 μM in the presence or absence of either α-BGTX or D-TC 30 min before at the indicated concentrations, followed by infection with HIV-1SF162. After 7 days of infection, supernatants were then collected for measurement of p24 Ag levels. Data are presented as the mean±SEM of five separate experiments using microglia from different brain specimens. *p<0.05 vs nicotine alone by Student’s t test; **p<0.01 vs untreated (control) cells by Student’s t test. d Role of TGF-β1 on nicotine effects on HIV-1-infected microglia. Microglial cells were pretreated with nicotine for 24 h at 300 μM in the presence or absence (isotype control) of anti-TGF-β1 antibodies 30 min before at the indicated concentrations, followed by infection with HIV-1SF162. After 7 days of infection, supernatants were then collected for measurement of p24 Ag levels. Data are presented as the mean±SEM of two separate experiments using microglia from different brain specimens. **p<0.01 vs nicotine alone by Student’s t test
Nicotine pretreatment promotes HIV-1 expression
As stated previously, nicotine has demonstrable effects on factors that relate to the pathogenesis of HIV-1, including binding of gp120 to nicotinic receptors (Bracci et al. 1992) and enhanced production of HIV-1 by in vitro-infected alveolar macrophages (Abbud et al. 1995). Based on these reports, we investigated nicotine’s effect on HIV-1-infected human microglial cells, the primary target of this virus within the CNS. Microglial cells were pretreated with nicotine and infected with HIV-1SF162. As hypothesized, pretreatment with nicotine increased HIV-1 expression in a concentration-dependent manner, as measured by p24 antigen levels in microglial cell culture supernatants (Fig. 1b). Also, the nAChR antagonists α-BGTX and D-TC abrogated nicotine’s effect on HIV-1 expression in microglia (Fig. 1c).
Nicotine alters microglial cell gene expression of HIV-related cellular pathways
To look for potential mediators involved in nicotine’s enhancement of viral expression, we performed a gene array analysis which focused explicitly on cellular processes that are associated with HIV-1 pathogenesis. Using Oligo GEArray® Human HIV Infection and Host Response arrays, we demonstrated that nicotine robustly altered the gene profile of HIV-1-infected microglia (Table 1). Among the genes upregulated at day 7 were TGF-β1, IL-4, CX3CL1, CCR2, and CXCR6 (Table 1). Among the genes that were downregulated at day 7 were chemokines/cytokines IL-8, IL-10, TNF, CCL2, and the chemokine receptor CXCR4 (Table 1). When added to uninfected microglia or mock control, nicotine had no effect on this same set of HIV-1-associated genes (data not shown).
Table 1.
Effect of nicotine on HIV-1-infected microglial cell gene expression of specific HIV-related cellular pathways
| Symbol | Gene description | Fold change | |
|---|---|---|---|
| 24 h p.i. | 7 days p.i. | ||
| Genes upregulated in HIV-infected human microglial cells (pretreated vs untreated) with 300 μM nicotine at 24 h and 7 days post infection | |||
| CCL4 | Chemokine (C-C motif) ligand 4 | 2.52 | - |
| CCR7 | Chemokine (C-C motif) receptor 7 | 2.92 | - |
| XPO1 | Exportin 1 | 2.58 | 1.24 |
| STAT3 | Transcription factor STAT3 | 2.47 | 0.11 |
| NFKB1A | NF-κB 1A | 2.35 | 0.01 |
| APOBEC3F | Alipoprotein B mRNA editing enzyme 3F | - | 368.83 |
| ABCE1 | ATP-binding cassette, subfamily E member 1 | - | 208.07 |
| KIR3DL1 | Killer cell immunoglobulin-like receptor | - | 184.41 |
| CD209 | CD209 antigen | 0.69 | 68.22 |
| SERPINA1 | Serpin peptidase inhibitor member 1 | 0.72 | 66.77 |
| KLRD1 | Killer cell lectin-like receptor subfamily D 1 | - | 61.91 |
| BANF1 | Barrier to autointegration factor 1 | - | 59.56 |
| HTATSF1 | HIV-1 Tat specific factor 1 | - | 59.05 |
| CX3CL1 | Chemokine (C-X3-C motif) ligand 1 | 0.91 | 41.97 |
| TGF-β1 | Transforming growth factor-β1 | - | 41.87 |
| CASP3 | Caspase-3 | - | 22.66 |
| FCAR | Receptor for Fc fragment of IgA | - | 20.56 |
| CCR2 | Chemokine (C-C motif) receptor 2 | - | 16.47 |
| TNFSF10 | TNF ligand superfamily member 10 | - | 15.90 |
| CXCR6 | Chemokine (C-X-C motif) receptor 6 | - | 14.29 |
| APEX1 | APEX nuclease 1 | 3.53 | 11.70 |
| CX3CL12 | Chemokine (C-X3-C motif) ligand 12 | 1.77 | 11.43 |
| CREBBP | CREB binding protein | 1.06 | 8.95 |
| IL16 | Interleukin 16 | - | 8.05 |
| PIK3C2B | Phosphoinositide-3-kinase, class 2, β-peptide | - | 4.38 |
| IL4 | Interleukin 4 | - | 4.16 |
| HLA-E | Major histocompatibility complex, class I, E | 0.48 | 3.85 |
| RBL2 | Retinoblastoma-like 2 | 1.67 | 2.52 |
| SELL | Selectin L | 1.01 | 2.10 |
| Genes downregulated in HIV-infected human microglial cells (pretreated vs untreated) with 300 μM nicotine at 24 h and 7 days post infection | |||
| GLI2 | GLI-Kruppel family member GLI2 | 5.47 | 0.03 |
| CD69 | CD69 antigen | 2.31 | 0.63 |
| CD3Z | CD3Z antigen | 2.09 | 0.81 |
| HLA-E | Major histocompatibility complex, class I, E | 2.06 | 0.25 |
| CD47 | CD47 antigen | 1.81 | - |
| HLA-A | Major histocompatibility complex, class I, A | 1.11 | 150.76 |
| IRF1 | Interferon regulatory factor 1 | 0.79 | 148.46 |
| HLA-C | Major histocompatibility complex, class I, C | 1.11 | 137.05 |
| TSG101 | Tumor susceptibility gene 101 | 0.55 | 130.01 |
| IL8 | Interleukin-8 | 0.60 | 111.20 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 1.05 | 96.77 |
| TNFRSF1B | TNF receptor superfamily member 1B | 1.11 | 91.29 |
| NFKB1A | NF-κB 1A | 0.42 | 77.04 |
| LTBR | Lymphotoxin β receptor | 0.65 | 65.67 |
| CXCR4 | Chemokine (C-X-C motif) receptor 4 | 0.52 | 58.80 |
| CD4 | CD4 antigen | 0.52 | 57.36 |
| CIITA | Class II, major histocompatibility complex | 0.67 | 45.27 |
| PPIA | Peptidylprolyl isomerase A (cyclophilin A) | 1.11 | 25.58 |
| ZBTB7A | Zinc finger and BTB domain containing 7A | 0.58 | 22.74 |
| COPS6 | COP9 photomorphogenic homolog subunit 6 | 0.69 | 22.72 |
| CEBPB | CCAAT/enhancer binding protein (C/EBP), beta | 0.58 | 20.75 |
| JUN | V-jun sarcoma virus 17 oncogene homolog | 0.77 | 20.15 |
| GABBR1 | GABA B receptor, 1 | 1.39 | 19.78 |
| Genes downregulated in HIV-infected human microglial cells (pretreated vs untreated) with 300 μM nicotine at 24 h and 7 days post infection | |||
| STAT1 | Transcription factor STAT1 | 0.91 | 16.21 |
| HTATIP2 | HIV-1 Tat interactive protein 2 | 0.68 | 15.83 |
| IL10 | Interleukin-10 | 0.37 | 12.67 |
| STAT3 | Transcription factor STAT3 | 0.40 | 8.38 |
| TNF | Tumor necrosis factor | 1.46 | 7.34 |
| CDK9 | Cyclin-dependent kinase 9 | 1.32 | 7.24 |
| NFIB | Nuclear factor I/B | - | 5.95 |
| CBX5 | Chromobox homolog | 0.69 | 5.85 |
| SLPI | Secretory leukocyte peptidase inhibitor | 2.51 | 4.85 |
| CCL2 | Chemokine (C-C motif) ligand 2 | 0.51 | 4.45 |
| HCK | Hemopoietic cell kinase | 3.78 | 3.81 |
| NFKB2 | NF-κB 2 | 1.02 | 3.72 |
| CD44 | CD44 antigen | 0.52 | 2.76 |
| YY1 | YY1 transcription factor | 0.55 | 2.58 |
Results are expressed in fold change
TGF-β1 is involved in the enhanced expression of HIV by nicotine
The finding that nicotine upregulated TGF-β1 expression in HIV-1-infected microglia suggested that this cytokine could be involved in the potentiating effect of nicotine, as we had previously found that cocaine-mediated enhancement of HIV-1 expression in microglia involved TGF-β1 (Gekker et al. 2006). To test this hypothesis, microglial cells were pretreated with anti-TGF-β1 antibodies and then treated with nicotine and infected with HIV-1SF162. Treatment with anti-TGF-β1 antibodies was found to completely block nicotine’s stimulatory effect on HIV-1 expression (Fig. 1d), supporting the involvement of this cytokine in the mechanism underlying nicotine’s enhancement of viral expression.
Discussion
The results of this study demonstrate that human microglia possess nAChR and support our hypothesis that nicotine enhances HIV-1 expression by these brain macrophages. That nicotine’s potentiating effect on HIV-1 expression is at least in part mediated by nAChR was demonstrated by the reduced expression of HIV-1 in the presence of nAChR’s antagonists α-BGTX and D-TC.
Whereas it is well recognized that microglia play an important role in the neuropathogenesis of HIV-1 and that drugs of abuse appear to foster the development of HIV-associated dementia (HAD), before this study, essentially nothing was known about the effects of nicotine on HIV-1-infected human microglial cells and whether this addictive substance could impact the neuropathogenic mechanisms of HIV-1. Other drugs of abuse, such as opiates and cocaine, have been shown to enhance HIV-1 replication in mononuclear phagocytes (Peterson et al. 1991; Ho et al. 2003; Roth et al. 2005) and mounting evidence suggests that these drugs foster the neuropathogenesis of HIV-1 (Tomlinson et al. 1999; Nath et al. 2002; Bell 2004). Because of the widespread dependence on nicotine in opiate- and cocaine-addicted individuals, it is possible that nicotine could confound some of the effects attributed to opiates and cocaine on HIV-1 neuropathogenesis.
Whereas the findings in this study suggest a novel role for nicotine in HIV-1 infection in the CNS, this hypothesis needs to be tested in vivo before any firm conclusions are made. Our findings in an acute in vitro model may differ from an in vivo chronic exposure model, which would more closely model real-world smoking exposure or treatment with nAChR ligands. Finally, whereas the nicotine concentrations used in this study encompassed physiologically achievable levels (Ghosheh et al. 2001; Shytle et al. 2004; Suzuki et al. 2006), the concentrations that were found to be most effective were achieved at a level most would consider to be very high.
In this study, we also found, by microarray analysis of genes known to play a role in HIV-1 expression, that having nicotine in the environment of HIV-1-infected microglia markedly affected the expression of a number of genes. The consequences of the effects of nicotine on the expression of these genes are unknown, but it is conceivable that the alterations of cytokine and chemokine mRNA expression could be relevant to the development of HAD as the products of these genes are known to be involved in HIV-1 neuropathogenesis. The results of this analysis should lay the framework for further investigation. Of note, we found that TGF-β1 mRNA was robustly upregulated in HIV-1-infected microglial cells pretreated with nicotine. Because TGF-β1 has been previously shown to be involved in enhanced HIV-1 expression in microglial cells treated with cocaine (Gekker et al. 2006) and enhanced HIV-1 expression in monocyte-derived macrophages and peripheral blood mononuclear cells treated with this drug of abuse (Peterson et al. 1991), and based on the data from our microarray analysis, we hypothesized that TGF-β1 may be involved in the effects of nicotine on HIV-1 expression in microglial cells. The finding that anti-TGF-β1 antibodies completely abrogated the potentiating effect of nicotine on viral expression supports the involvement of TGF-β1 in the enhancing effect of nicotine.
Acknowledgments
This work was supported by grants from National Institute on Drug Abuse, DA-020398, and was also supported in part by the U.S. Public Health Service grant DA020398.
Footnotes
Presented in part at the 13th Annual Conference of the Society on NeuroImmune Pharmacology, Salt Lake City, Utah; April 10–14, 2007; #T-33.
Human fetal brain tissue was obtained under the protocol approved by the Human Subjects Research Committee at our institution.
None of the authors has a commercial or other association that might pose a conflict of interest with the current study.
References
- Abbud RA, Finegan CK, Guay LA, Rich EA (1995) Enhanced production of human immunodeficiency virus type 1 by in vitro-infected alveolar macrophages from otherwise healthy cigarette smokers. J Infect Dis 172:859–863 [DOI] [PubMed] [Google Scholar]
- Bell JE (2004) An update on the neuropathology of HIV in the HAART era. Histopathology 45:549–559 [DOI] [PubMed] [Google Scholar]
- Bracci L, Lozzi L, Rustici M, Neri P (1992) Binding of HIV-1 gp120 to the nicotinic receptor. FEBS Lett 311:115–118 [DOI] [PubMed] [Google Scholar]
- Chong IW, Lin SR, Hwang JJ, Huang MS, Wang TH, Hung JY, Paulauskis JD (2002) Expression and regulation of the macrophage inflammatory protein-1 alpha gene by nicotine in rat alveolar macrophages. Eur Cytokine Netw 13:242–249 [PubMed] [Google Scholar]
- Gekker G, Hu S, Sheng WS, Rock RB, Lokensgard JR, Peterson PK (2006) Cocaine-induced HIV-1 expression in microglia involves sigma-1 receptors and transforming growth factor-beta1. Int Immunopharmacol 6:1029–1033 [DOI] [PubMed] [Google Scholar]
- Ghosheh OA, Dwoskin LP, Miller DK, Crooks PA (2001) Accumulation of nicotine and its metabolites in rat brain after intermittent or continuous peripheral administration of [2′-(14)C]nicotine. Drug Metab Dispos 29:645–651 [PubMed] [Google Scholar]
- Giunta B, Ehrhart J, Townsend K, Sun N, Vendrame M, Shytle D, Tan J, Fernandez F (2004) Galantamine and nicotine have a synergistic effect on inhibition of microglial activation induced by HIV-1 gp120. Brain Res Bull 64:165–170 [DOI] [PubMed] [Google Scholar]
- Ho WZ, Guo CJ, Yuan CS, Douglas SD, Moss J (2003) Methylnaltrexone antagonizes opioid-mediated enhancement of HIV infection of human blood mononuclear phagocytes. J Pharmacol Exp Ther 307:1158–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT (2002) Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr 31(Suppl 2):S62–S69 [DOI] [PubMed] [Google Scholar]
- Nouri-Shirazi M, Guinet E (2003) Evidence for the immunosuppressive role of nicotine on human dendritic cell functions. Immunology 109:365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Chao CC, Schut R, Molitor TW, Balfour HH Jr (1991) Cocaine potentiates HIV-1 replication in human peripheral blood mononuclear cell cocultures. Involvement of transforming growth factor-beta. J Immunol 146:81–84 [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Hu S, Cabral G, Lokensgard JR (2004) Cannabinoids and morphine differentially affect HIV-1 expression in CD4(+) lymphocyte and microglial cell cultures. J Neuroimmunol 147:123–126 [DOI] [PubMed] [Google Scholar]
- Roth MD, Whittaker KM, Choi R, Tashkin DP, Baldwin GC (2005) Cocaine and sigma-1 receptors modulate HIV infection, chemokine receptors, and the HPA axis in the huPBL-SCID model. J Leukoc Biol 78:1198–1203 [DOI] [PubMed] [Google Scholar]
- Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 89:337–343 [DOI] [PubMed] [Google Scholar]
- Sopori ML, Kozak W (1998) Immunomodulatory effects of cigarette smoke. J Neuroimmunol 83:148–156 [DOI] [PubMed] [Google Scholar]
- Sugano N, Shimada K, Ito K, Murai S (1998) Nicotine inhibits the production of inflammatory mediators in U937 cells through modulation of nuclear factor-kappaB activation. Biochem Biophys Res Commun 252:25–28 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, Andra M, Matsubayashi H, Sakai N, Kohsaka S, Inoue K, Nakata Y (2006) Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 83:1461–1470 [DOI] [PubMed] [Google Scholar]
- Tomlinson GS, Simmonds P, Busuttil A, Chiswick A, Bell JE (1999) Upregulation of microglia in drug users with and without pre-symptomatic HIV infection. Neuropathol Appl Neurobiol 25: 369–379 [DOI] [PubMed] [Google Scholar]
- Totti N 3rd, McCusker KT, Campbell EJ, Griffin GL, Senior RM (1984) Nicotine is chemotactic for neutrophils and enhances neutrophil responsiveness to chemotactic peptides. Science 223:169–171 [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388 [DOI] [PubMed] [Google Scholar]