

Abstract
Purpose
Inhibition or targeted deletion of histone deacetylase 3 (HDAC3) is neuroprotective in a variety neurodegenerative conditions, including retinal ganglion cells (RGCs) after acute optic nerve damage. Consistent with this, induced HDAC3 expression in cultured cells shows selective toxicity to neurons. Despite an established role for HDAC3 in neuronal pathology, little is known regarding the mechanism of this pathology.
Methods
Induced expression of an HDAC3-mCherry fusion protein in mouse RGCs was accomplished by transduction with AAV2/2-Pgk-HDAC3-mCherry. Increased susceptibility to optic nerve damage in HDAC3-mCherry expressing RGCs was evaluated in transduced mice that received acute optic nerve crush surgery. Expression of HDAC3-FLAG or HDAC3-mCherry was induced by nucleofection or transfection of plasmids into differentiated or undifferentiated 661W tissue culture cells. Immunostaining for cleaved caspase 3, localization of a GFP-BAX fusion protein, and quantitative RT-PCR was used to evaluate HDAC3-induced damage.
Results
Induced expression of exogenous HDAC3 in RGCs by viral-mediated gene transfer resulted in modest levels of cell death but significantly increased the sensitivity of these neurons to axonal damage. Undifferentiated 661W retinal precursor cells were resilient to induced HDAC3 expression, but after differentiation, HDAC3 induced GFP-BAX recruitment to the mitochondria and BAX/BAK dependent activation of caspase 3. This was accompanied by an increase in accumulation of transcripts for the JNK2/3 kinases and the p53-regulated BH3-only gene Bbc3/Puma. Cell cycle arrest of undifferentiated 661W cells did not increase their sensitivity to HDAC3 expression.
Conclusions
Collectively, these results indicate that HDAC3-induced toxicity to neurons is mediated by the intrinsic apoptotic pathway.
Keywords: retinal ganglion cells, glaucoma, 661W cells, histone deacetylase 3 (HDAC3), neurodegeneration, epigenetics, intrinsic apoptosis
Histone deacetylases (HDACs) comprise a large family of proteins spanning three major classes defined by sequence homology, enzyme structure, and the requirement for NAD+ as a co-factor. As the name implies, HDACs play a critical role in balanced acetylation states of histone proteins that regulate chromatin structure, playing a role in modulating gene transcription, DNA synthesis, cell division, and cell death. Additionally, HDACs are the central mediators of post-translational modifications of non-histone targets through acetylation. Growing evidence has linked HDAC activity to age-related changes in health,1 with perhaps the most prominent of these changes involving a variety of neurodegenerative conditions.2
Multiple HDACs have been implicated in neurodegeneration, including the classical class I HDACs, HDAC1, HDAC2, HDAC3 and HDAC6.2 HDAC3, in particular, has been linked to the pathophysiology inherent in ischemic stroke,3,4 Parkinson's disease,2 spinocerebellar ataxias including Friedreich's ataxia,5–7 Huntington's disease,8–10 and Alzheimer's disease.11,12 Additionally, HDAC3 activity has been associated with memory function13,14 and the behavior of cocaine addiction.15 Much of the research centered on HDAC3 function in these conditions has been conducted using selective inhibitors, or HDAC3 knock-down, with a majority indicating that inhibition of HDAC3 provides a protective environment for neurons in animal models of neurodegeneration. Studies on the effects of induced expression of exogenous HDAC3 have been limited to experiments using cultures of immortalized cells derived from a neuronal source, or primary neuron cultures.16,17 These studies show a selective toxicity of neurons to HDAC3 overexpression, ranging from induced cell death to increasing the sensitivity of cells to known damaging stimuli. The mechanism of this selective toxicity is not fully understood and is an ongoing area of investigation.
We have been studying the role of HDAC3 activity in pathologies that cause retinal ganglion cell (RGC) death. RGCs are long projection neurons of the central nervous system and populate the innermost layer of the mammalian retina. Their function is to act as a relay of electrical and chemical activity generated in the retina in response to photon capture by the outermost photoreceptor cells, to visual centers in the brain. Damage to RGC axons in the optic nerve is often deleterious to RGC cell bodies in the retina18 and there are multiple conditions that can lead to such damage and neurodegeneration, some relatively common such as glaucoma19,20 and traumatic brain injury.21,22 Previously, we reported that histone deacetylation was an early event in the pathologic sequence executed by RGCs after both acute and chronic optic nerve damage,23,24 and this phenomenon was associated with the redistribution of HDAC3 from the cytosol to the nucleus in affected cells.23 Conditional deletion of Hdac3 in RGCs in mice, or treatment with the HDAC3-selective inhibitor RGFP966, revealed both a reduction in the level of global deacetylation associated with acute optic nerve damage, and an increase in long-range RGC somata survival.25,26 Because axonal damage leads to the activation of the intrinsic apoptotic pathway in RGCs,27–29 it is possible that HDAC3 activity plays an early and critical role in the molecular sequence of events leading up to the activation of this pathway. Alternatively, the protective effect of either Hdac3 deletion or HDAC3 inhibition, while significantly prolonged (up to 8 weeks) is not comparable to the protective effect of deletion of the intrinsic apoptosis-critical gene, Bax, in RGCs, which blocks RGC death for upwards of 18 months.30 The true contributory role of HDAC3 in neuronal pathology, including RGCs, may be difficult to elucidate in experiments that delete or impair this enzyme because of the “noise” generated by multiple other concurrent molecular events. This study evaluated the role of induced HDAC3 expression with specific emphasis on characterizing its contribution to the activation of the intrinsic apoptotic pathway.
Materials and Methods
Mice
Male and female C57BL/6 mice between the ages of four to six months old were used in these experiments. Mice were housed in microisolator cages and kept on a 12-hour light/dark cycle. They were maintained on a 4% fat diet (8604 M/R; Harland Tekland, Madison, WI, USA). Experiments with mice were approved by the Animal Use Committee at the University of Wisconsin–Madison and conformed to the ARVO statement for use of animals in research. Each treatment group contained a minimum of three mice for all experiments, and each cohort contained an equal proportion of males to females.
Intravitreal Injections and Optic Nerve Crush Procedures
AAV2/2-Pgk-Cre or AAV2/2-Pgk-HDAC3-mCherry were administered intravitreally. In some experiments, each of these viruses was mixed in equal proportion (based on genome copies) with AAV2/2-Pgk-GFP-BAX. The AAV2/2-Pgk-Cre virus, which was used as a non-specific control, was obtained from the University of North Carolina Vector Core (Raleigh, NC). The AAV2/2-Pgk-GFP virus was packaged by the same facility using an entry plasmid developed in our laboratory as previously described.29,31 The AAV2/2-Pgk-HDAC3-mCherry virus was packaged by VectorBuilder/Cyagen Biosciences Inc. (Santa Clara, CA) from a DNA construct (described below) provided to them. Mice were first anesthetized with an intraperitoneal injection of ketamine (6 mg/mL) and xylazine (0.4 mg/mL) and the left eye was numbed with a drop of 0.5% proparacaine. The globe was first punctured near the limbus with the bevel of a 30G needle, and intravitreal injections were conducted using a 10 µL Nanofil syringe with a 35G beveled needle attached (World Precision Instruments, Sarasota, FL) that was positioned into the vitreous chamber through the puncture hole. A volume of 1 µL of virus (equaling approximately 109 genome copies) was injected over a period of 40 seconds, and the needle was held in the eye for another 30 seconds before it was retracted. The puncture site was covered with triple antibiotic ointment and the animals were given an immediate injection (subcutaneous) of buprenorphine and allowed to recover. Retinas were harvested at three, four, five, eight, and 12 weeks after injection for total cell counts and nuclear scoring in the ganglion cell layer (GCL). Retina processing and cell counts were obtained using a modification of the method described in Li et al.32 Briefly, enucleated eyes were immersion fixed in 4% paraformaldehyde in PBS (50 mM phosphate buffer, pH 7.4, 150 mM NaCl) for 60 minutes at 22˚C, followed by 0.4% paraformaldehyde in PBS, where they could be stored at 4˚C. The retinas were then dissected out of the fixed globes and displayed with the ganglion cell layer facing up on Superfrost Plus microscope slides (ThermoFisher Scientific, Waltham, MA, USA). Four relaxing cuts were made in each retina to allow them to splay out flat on the slide in an “iron cross” formation of four lobes. The retinas were then mounted with a drop of Vectashield containing 4ʹ, 6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and coverslipped. Nine digital images, captured with an objective ×40, were taken of each lobe of each whole mount with a Zeiss Axiophot fluorescent microscope (Carl Zeiss Microscopy LLC, White Plains, NY, USA). Neuronal cell numbers were determined from 108 separate 104 µm2 fields for each retina. Likely neurons were defined as cells with round or oval nuclei and prominent nucleoli. Change in cell number for each experimental eye was calculated as a percentage of cell numbers in the corresponding control eye of each mouse.
Optic nerve crush was performed as previously described.32,33 Briefly, at four weeks after bilateral viral injection to allow for maximal expression of the viral transgene, mice were anesthetized as described above, and the optic nerve was exposed after a lateral canthotomy and dissection of the overlying conjunctiva. The exposed nerve was then clamped for three seconds with N7 curved self-closing forceps (Roboz Surgical Instrument Co., Gaithersburg, MD, USA). Mice were then allowed to recover from the procedure as described above. At five days after optic nerve surgery, the animals were euthanized and the extent of damage to the ganglion cell layer was assessed. The time point of five days was chosen because this represents a timepoint when RGCs normally exhibit signs of apoptotic loss induced by the crush surgery such as peak activation of caspase 3.34–36 Damage to RGCs in the ganglion cell layer was assessed by measuring the change in density of TUBB3 positive cells, and by counting the presence or absence of DAPI-stained apoptotic nuclei in TUBB3 positive cells (see immunostaining below). TUBB3 stains for β-III-tubulin, which is highly expressed by RGCs and is often used as an RGC-selective marker.35,37 Damage was also assessed in retinas from eyes that were co-injected with AAV2/2-Pgk-GFP-BAX. In these experiments the percentage of transduced cells exhibiting punctate localization of GFP-BAX, which is a measure of cells executing the intrinsic apoptotic pathway,29,31 was quantified. Last, RGC damage was measured by assessing the number of caspase-3 positive TUBB3 cells in the AAV2/2-Pgk-HDAC3-mCherry and Control AAV2 injected eyes. Image documentation used for quantification was the same as described for DAPI-stained retinas (see above).
Clones and Plasmids
The GFP-Bax construct used in these experiments was described by Semaan et al.,38 the HDAC3-Flag construct was a gift from Eric Verdin (Addgene plasmid no. 13819; Addgene, Cambridge, MA, USA), and Mito-BFP construct was described in Maes et al.39 Generation of the HDAC3-mCherry fusion protein construct was done by first amplifying Hdac3 cDNA from the plasmid pHdac3-tGFP (Origene, Rockville, MD) using the following primers containing Age I sites: FWD-5’-AGGAGATCTACCGGTGCGATCGCCATG-3’ and REV-5ʹ-CATCTCGACCGGTCGCGTACGCGTAATC-3ʹ. The Age I-Hdac3-Age I fragment was then ligated into pmCherry-C1 (Clontech, Mountain View, CA, USA). This plasmid and the empty pmCherry-C1 parent plasmid were used for nucleofection experiments (see below). The HDAC3-mCherry sequence was then excised with Nhe I and Hind III and cloned into a bridge vector created from AAV-Pgk-Cre (a gift from Patrick Aebischer, Addgene plasmid no. 24593) where the Cre coding region was removed and replaced with a restriction enzyme multiple cloning site, to create the AAV2/2-Pgk-HDAC3-mCherry plasmid. This plasmid was digested with Sma I to confirm the presence of inverted terminal repeats, and then sent to VectorBuilder/Cyagen Biosciences Inc. to obtain AAV2/2 virus particles. The pGEM p21 plasmid was a gift from Frederic Mushinski (Addgene plasmid no. 8443).
Immunostaining Mouse Retinal Tissue
Immunostaining of retinal tissue was conducted on whole retinal eyes cups, followed by whole mounting the retina. For immunostaining whole mount retinas, eyecups were prepared from fixed enucleated eyes and the vitreous was removed by wiping the inner surface with a spear made from a Kimwipe (Kimberly-Clark, Irving, TX, USA). Eyecups were equilibrated in 30% sucrose in PBS overnight at 4˚C. A majority of the sucrose was then removed, and each eyecup was subjected to three cycles of a freeze-thaw on dry ice. The entire eyecup was rinsed with PBS and then blocked in blocking buffer (PBS containing 2% to 5% bovine serum albumin [ThermoFisher Scientific] and 0.1% Triton-X 100) overnight at 4˚C, followed by a brief wash in PBS. The eyecup was then submerged in blocking buffer containing one or more of the following primary antibodies: mouse monoclonal against human BRN3A (no. MAB1585, 1:50 dilution) (EMD Millipore Inc., Billerica, MA, USA); mouse monoclonal against mammalian β-III tubulin (TUBB3) (no. ab78078, 1:1000 dilution) (AbCam, Cambridge, MA, USA), rabbit polyclonal antibody to acetylated histone H4 (no. 06-866, 1:100 dilution) (EMD Millipore Inc.), rabbit polyclonal antibody against mCherry (#ab167453, 1:500 dilution) (AbCam), or rabbit polyclonal antibody against activated caspase 3 (no. AF835, 0.5 µg/mL dilution) (R&D Systems, Minneapolis, MN, USA). Incubations with primary antibodies were for four days at 4˚C. Eyecups were then washed in PBS and incubated with blocking buffer containing goat anti-mouse IgG conjugated to FITC or Alexa488 (for BRN3A or TUBB3) or goat anti-rabbit IgG conjugated to Texas Red (for mCherry), Alexa594 (for active caspase 3), or FITC (for acetylated histone H4) (1:1000 dilutions) (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) overnight at 4˚C. Eyecups were then washed in PBS and the retinas were dissected out and whole mounted as described above. Non-confocal imaging and digital capture was conducted using either a Zeiss AxioImager.Z2 epifluorescent microscope or a Zeiss AxioImager.A2 epifluorescent microscope (Carl Zeiss MicroImaging Inc.). Confocal images were taken using a Leica TCS SP8 confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL, USA).
Quantification of Immunostaining for Acetylated Histone H4
Survey images of 4 quadrants of stained retinal whole mounts were captured using a ×40 oil objective lens with constant laser intensity and exposure time (700 msec). Digital images were then imported into ImageJ (imagej.nih.gov) and pixel intensity was measured in a fixed size region of interest of 7 µm in diameter. Ten regions of background intensity were measured and averaged in each image. This value was then subtracted from measurements of intensity taken by moving the region of interest over individually stained nuclei. A minimum of 500 cells were randomly measured per retina and all data from three individual retinas per group was pooled for graphing and analysis.
661W Cell Culture and Differentiation
The 661W cells were cultured in 10% fetal bovine serum supplemented Dulbecco's modified Eagle medium, and cells were incubated at 37°C and 5% CO2 until they reached 70% to 80% confluence before experiments. Cell identity was confirmed as Mus musculus origin by PCR of the mitochondria d-loop40 and their ability to be differentiated.41,42 Differentiation of wild type and BAX-edited 661W cells (line Sg1, described below) was achieved by addition of 316 nM staurosporine (STS; Sigma-Aldrich, St. Louis, MO, USA) dissolved in DMSO and brought up in full media and incubated for 0, 0.5, 1, 6, 12, or 24 hours. Differentiation was carried out 24 hours before transfection for the initial BAX recruitment assay, or differentiation was initiated 24 hours after nucleofection in subsequent experiments. The differentiated state of cells was confirmed by visualization of neurite extensions using differential interference contrast microscopy.
PCR Experiments
For nested reverse transcriptase polymerase chain-reaction (RT-PCR) analysis of the Bak splice variants, frozen pellets of 661W cells or mouse tissues were homogenized and sonicated in Tri-Reagent (Invitrogen, Carlsbad, CA, USA; ThermoFisher Scientific) and total RNA was isolated. First strand cDNA was synthesized from 1 µg of total RNA that had first been pretreated with DNase I to remove genomic DNA contamination, using oligo(dT) as a primer and M-MULV reverse transcriptase (Promega, Madison, WI, USA). Nested RT-PCR was conducted as described previously.43 Briefly, first round PCR was conducted with primers P1 and P2 (Table 1) with 40 cycles of amplification (95°C for 30 seconds, 60°C for 30 seconds, and 72°C for one minute). This reaction was diluted 1:50 with H2O, and 1 µl of this was reamplified using primers P3 and P4 with the same reaction conditions. PCR products were analyzed on 2% agarose gels containing ethidium bromide.
Table 1.
Sequences of Primers Used for PCR Experiments
| Target Gene | Sequence | Comment |
|---|---|---|
| Bak Primer 1 | 5ʹ-atggcatctggacaaggaccaggt-3ʹ | FL gene exon 2 forward |
| Bak Primer 2 | 5ʹ-gtcatgatctgaagaatctgtgtacc-3ʹ | FL gene exon 6 reverse |
| Bak Primer 3 | 5ʹ-cgagatggacaacttgcccctgg-3ʹ | FL gene exon 3 forward |
| Bak Primer 4 | 5ʹ-cagctgatgccactcttaaataggct-3ʹ | FL gene exon 5 reverse |
| Jnk2 Forward | 5ʹ-tgtttggtatgaccccgctg-3ʹ | FL gene exon 9 forward |
| Jnk2 Reverse | 5ʹ-tgcatctgtgctgaaggctg-3ʹ | FL gene exon 11/12 reverse |
| Jnk3 Forward | 5ʹ-gactcaccttccccaagctc-3ʹ | FL gene exon 10 forward |
| Jnk3 Reverse | 5ʹ-cttcgatggtgtgctccctt-3ʹ | FL gene exon 12 reverse |
| S16 Forward | 5ʹ-cactgcaaacggggaaatgg-3ʹ | FL gene exon 2 forward |
| S16 Reverse | 5ʹ-tgagatggactgtcggatgg-3″ | FL gene exon 4 reverse |
For real-time quantitative RT-PCR (qPCR) experiments, total RNA was isolated and first strand cDNA synthesis was performed as described above. For qPCR analysis of Jnk2 and Jnk3, primers were first designed using NCBI primerblast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Table 1). Appropriate targeting and amplification conditions, including sequence confirmation of the PCR product, were determined empirically prior to qPCR analysis. For qPCR, samples were amplified with SyberGreen containing master mix in a QuantStudio 7 Flex real-time PCR machine (Applied Biosystems, Foster City, CA, USA; ThermoFisher Scientific) using the 96 well fast block. For these experiments, S16 ribosomal protein mRNA was used as the reference gene for sample normalization. For expression of BCL2 Homology Domain 3-only (BH3-only) gene products, qPCR was conducted using a custom TaqMan Array card (ThermoFisher Scientific) designed to include optimized primer sets for mouse Bcl2 gene family members.31 Isolation of total RNA was as described above, but first strand cDNA was made using random hexamers. PCR was performed in the QuantStudio 7 Flex machine, using the array card block. For these experiments, 18s rRNA, which is integrated into the array card design, was used as the reference gene for sample normalization. All qPCR data was collected from 3 independent experiments. Data was analyzed using the ΔΔCt method of relative quantification. For Jnk mRNAs, sample comparisons were made between differentiated or undifferentiated cells, expressing HDAC3 relative to the same cells expressing an empty mCherry only vector. For the BH3-only gene comparisons, the ΔCt for each target molecule was first calculated in each of four conditions (undifferentiated 661W cells, expressing either mCherry alone or HDAC3, and differentiated cells, expressing either mCherry alone or HDAC3). The sample with the highest ΔCt value (i.e., with the least abundant amount of target mRNA) was then assigned as the baseline and compared to all the other conditions using the ΔΔCt method.
BAX and BAK Depletion in 661W Cells
CRISPR/Cas9 was used to edit the Bax gene in mouse 661W cells. Here, lentiCRISPRv2 (one vector system), a gift from Feng Zhang (Addgene plasmid #52961) was used because this plasmid contains two expression cassettes, hSpCas9 and the chimeric guide RNA. The plasmid was digested using Bsm BI, and a pair of annealed oligos was ligated into the single guide (Sg) RNA scaffold. Three different oligo pairs for the gRNA were designed based on a 20 bp target site sequence in the first exon of murine Bax and were flanked on the 3ʹ end by a 3 bp NGG PAM sequence (Table 2).
Table 2.
Sequences of Targeting Primers for CRISPR/Cas9 Editing of the Murine Bax Gene (Exon 1)
| Target Sequence | Primers | |
|---|---|---|
| gRNA1 | acgggtccggggagcagcttggg | Bax1F: 5ʹ-caccgACGGGTCCGGGGAGCAGCTT-3ʹ |
| Bax1R: 5ʹ-aaacAAGCTGCTCCCCGGACCCGTc-3ʹ | ||
| gRNA2 | gacgggtccggggagcagcttgg | Bax2F: 5ʹ-caccgGACGGGTCCGGGGAGCAGCT-3ʹ |
| Bax2R: 5ʹ-aaacAGCTGCTCCCCGGACCCGTCc-3ʹ | ||
| gRNA3 | ggcagtgatggacgggtccgggg | Bax3F: 5ʹ-caccgGGCAGTGATGGACGGGTCCG-3ʹ |
| Bax3R: 5ʹ-aaacCGGACCCGTCCATCACTGCCc-3ʹ |
The NGG PAM sequence for each target is shown in bold. Forward (F) and reverse (R) primers for each target are also indicated, with the target sequence shown in upper case letters for each primer.
Briefly, 5 µg of the lentiviral CRISPR plasmid was digested with BsmBI for 30 minutes at 37°C. Then, the digested plasmid was gel purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and eluted in elution buffer. The oligos were annealed, diluted at 1:100, and then ligated into the digested lentiviral CRISPR plasmid. The ligation was transformed into Stbl3 bacteria for propagation. The 661W cells were then transfected with the ligated plasmid and maintained in 1% puromycin to maintain expression of the construct. Control wild type cells were transfected with an empty cassette vector. These cells were used as the wild type controls in all experiments involving CRISPR/Cas9 edited 661W cells. All culturing of 661W cells in these experiments contained puromycin to maintain plasmid expression. Cas9 excision was confirmed using a T7 endonuclease assay (data not shown) and BAX depletion in resulting cell lines was confirmed by Western blotting (see below).
To achieve depletion of pro-apoptotic protein, BAK, ON-TARGETplus mouse Bak1 (12018) siRNA-SMARTpool (no. L-042978-00-0005) and scramble siRNA (no. D-001810-01-05) were obtained from GE Healthcare Dharmacon Inc. (Lafayette, CO, USA). The 661W cells were nucleofected (described below) at a final concentration of 25 nM siRNA/1 × 106 cells, and BAK depletion was confirmed by Western blotting after 48 hours.
Transfection and Nucleofection
Transfection of undifferentiated and differentiated 661W cells was carried out with combinations (as specified) of HDAC3-FLAG (human HDAC3), HDAC3-mCherry (mouse HDAC3), Mito-BFP, pGEMp21, and GFP-BAX plasmids. 661W cells were passaged until they reached 70–80% confluency, then cells were treated with 0.25% trypsin for 5-10 minutes until they became non-adherent. Suspended cells were counted, and cells were plated at 250,000 cells per well of a 6-well plate or 20,000 cells per well of a 4-chamber slide. After 24 hours of incubation, the same 661W cells were differentiated using 316 nM STS. After another 24 hours of incubation, these cells were transfected. To achieve transfection, media was prewarmed and vortexed with 2 µg of each specified plasmid. Then a 2:1 ratio of Transfast reagent (no. E2431) (Promega) was added and vortexed immediately and incubated for 10 to 15 minutes at room temperature. After this incubation step, growth media was removed from cells, and the Transfast reagent/DNA mixture was briefly vortexed before adding to wells containing cells. The cells with the Transfast reagent/DNA mixture was incubated at 37°C for one hour. After one hour of incubation, cells were overlaid with complete medium and returned to the incubator for another 24 hours before assaying. The BAX recruitment assay was used to observe apoptosis in these cells.
In preparation for nucleofection, 661W cells were passaged until they reached 70% to 80% confluency, then cells were treated with 0.25% trypsin for five to 10 minutes until they became nonadherent. Suspended cells were then counted, and 1 × 106 cells were pelleted and resuspended in 100 µL nucleofection solution (Lonza Group, Basel, Switzerland). For all nucleofections, 1 to 5 µg plasmid DNA was added to the cell solution before it was transferred to the nucleofection cuvette and the Neuro-2a Lonza program was run. The 661W cells were then plated at 250,000 cells per well of a 6-well plate or 20,000 cells per well of 4- or 8-chamber cover glass slides (no. 80826 or 80427) (Ibidi USA, Inc., Fitchburg, WI, USA) or 4-or 8-chamber glass slides (no. 354118 or 354114) (Corning, Corning, NY, USA). Nucleofected 661W cells were then incubated for 24 hours at 37°C before differentiating with STS.
For BAK knockdown experiments both wild type and BAX-deficient (Sg1) mouse 661W cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 1% puromycin. Cells were incubated at 37°C and 5% CO2 until they reached 80% confluency. Nucleofections were performed in 12-well strip tubes (Lonza) containing 200,000 cells per tube and 2 to 5 µg of plasmid (2 µg for pmCherry and 5 µg for HDAC3-FLAG) per reaction and 25 nM of siRNA (BAK or scramble). The Neuro-2A program was used on the Amaxa Lonza nucleofector. After nucleofection, 10,000 cells were plated into one well of an 8-well chamber slide and incubated at 37°C and 5% CO2 for 24 hours. Differentiation of cells was achieved by addition of 316 nM of STS. Cells were then incubated for 12 hours. Afterwards, they were fixed for eight minutes using 4% paraformaldehyde in PBS and rinsed in PBS for five minutes. Cells were blocked in 5% BSA in PBS for two hours at room temperature in the dark. Primary antibody: rabbit polyclonal against human activated Caspase-3 (see above) diluted in 3% BSA and 0.1% Triton-X100 was added to cells for 24 hours at 4°C. Cells were washed in PBS three times before adding secondary antibody: goat anti-rabbit IgG conjugated to FITC (1:1000) (Jackson ImmunoResearch Laboratories) in 5% BSA and 0.1% Triton-X100 for two hours at 22°C in the dark. Cells were washed in PBS and counter-stained for 10 minutes with DAPI. After PBS washes, cells were mounted with immunomount and cover slipped. Fluorescent images were obtained using a Zeiss Axioplan 2 Imaging microscope with Axiovision 4.6.3.0 software (Carl Zeiss MicroImaging Inc.) with a ×20 objective. Photos were analyzed using Zen 2.1 lite.
BrdU Immunostaining
Undifferentiated and differentiated 661W cells were incubated in 10 µM 5-bromo-2ʹ-deoxyuridine (BrdU) (ThermoFisher Scientific) for four hours before fixing the cells. After fixation, cells were rinsed in PBS, blocked in 3% H2O2 in PBS for two hours, rinsed again in PBS, and labeled for BrdU using the BrdU in situ detection kit from BD Pharmingen (BD Biosciences, San Jose, CA, USA). Slides were then incubated in hematoxylin for 60 seconds and rinsed in H2O thoroughly before coverslipping and imaging using the Zeiss Imager Axioplan 2 microscope (Carl Zeiss MicroImaging Inc.) and a digital camera attachment. Quantification of BrdU-positive and hematoxylin-positive cells were done by a masked observer from images taken with an objective ×40.
Immunofluorescence of 661W Cells
The 661W cells were grown in 4- or 8-chamber coverslip slides (Ibidi) or 4- or 8-chamber glass slides (Corning), fixed in 4% paraformaldehyde in PBS for 20 minutes and then rinsed in PBS. The cells were then blocked in 5% BSA in PBS for two hours at room temperature and later rinsed in PBS. Primary antibodies included mouse monoclonal against mammalian TUBB3 (see above), mouse monoclonal against FLAG (no. F1804, 1:1000 dilution) (Sigma-Aldrich), and rabbit polyclonal against human activated Caspase-3 (no. ab13847, 1:200 dilution) (AbCam). Cells were incubated in primary antibody for 24 to 48 hours at 4°C and washed in PBS afterward. Secondary antibodies used are described above and were incubated at 22°C in the dark for two hours and washed in PBS. After incubation, cells were counterstained for 10 minutes with 300 ng/mL DAPI (in PBS) and were washed with PBS. Slides were then overlain with Immumount mounting medium (ThermoFisher Scientific) and coverslipped. Fluorescent images were obtained as described above.
Western Blot Analysis
The 661W cell pellets were lysed by sonication in 0.1% phenylmethane sulfonyl fluoride in radioimmunoprecipitation assay buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1.0% IGEPAL, 0.5% Na+ Deoxycholate, 0.1% SDS) and incubated on ice for 30 minutes. Cell lysate was spun in a microcentrifuge (five minutes at 5000 rpm) to separate membranes from lysate, and supernatant was removed into a new tube. Samples were diluted 1:10 in PBS then assayed for protein abundance using the bicinchoninic assay (Pierce, ThermoFisher Scientific) to determine total concentration of protein in each sample. Each sample was then diluted in sample buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue in 125 mM Tris-HCl, pH 6.8) and heated at 42°C for five minutes before loading in triplicate with 20 µg per lane on 12% polyacrylamide gels. After electrophoresis, gels were transblotted onto Immobilon-FL PVDF transfer membrane (Millipore, Inc., Billerica, MA, USA). Blots were blocked in 5% milk in 0.1% Tween-20 in PBS (PBST) for two hours, then rinsed in PBST. Membranes were probed for BAX, BAK, and ACTIN, with primary antibodies diluted in 5% BSA in PBS. For blots probed for BAX, mouse monoclonal antibody to BAX was used at 1:200 (no. sc-23959) (Santa Cruz Biotechnology, Dallas, TX, USA). For blots probed for BAK, a rabbit polyclonal antibody to BAK was used at 1:500 (no. 06-536) (EMD Millipore, Burlington, MA, USA). Both blots were probed for ACTIN using goat polyclonal antibody to ACTIN at 1:200 (no. sc-1616) (Santa Cruz Biotechnology). Blots were incubated in primary antibody overnight, then washed in PBST. For secondary antibody incubations, the blots were first incubated in donkey anti-goat secondary (1:10,000) conjugated to IRDye 800CW (no. 926-32214) (LI-COR Biosciences, Lincoln, NE, USA) in 5% milk in PBST for 2 hours at room temperature to detect the anti-ACTIN antibody, and after washing in PBST, incubated in either goat anti-rabbit secondary (1:10,000) conjugated to IRDye 680RD (no. 926-68071) (LI-COR Biosciences) or goat anti-mouse secondary (1:10,000) conjugated to IRDye 680RD (no. 925-68070) (LI-COR Biosciences) in 5% milk in PBST for two hours at room temperature. Blots were washed in PBST before imaging for labeled bands. Images were scanned and analyzed using the Odyssey Clx (LI-COR Biosciences). Band labeling was quantified using Image Studio software (LI-COR Biosciences), and data were normalized to the ACTIN loading reference on each blot.
Statistical Analysis
Cell count data and Western blot data were collected from independent samples, and the data was presented as the mean ± standard error (bar graphs) or scatter plots with ± standard deviation. All statistical analyses were performed using Welch's two-sample t-test assuming unequal variances. For comparison of multiple groups, we used linear modeling regression analysis (R version 4.0.5). Statistical significance (alpha) set at P ≤ 0.05 for all comparisons.
Results
Characterization of Induced HDAC3-mCherry Expression in Mouse Retinal Ganglion Cells In Vivo
Our previous studies demonstrated that genetic deletion of Hdac3, or selective inhibition using the inhibitor RGFP966, resulted in significant attenuation of RGC death after acute (crush) insult to the optic nerve in mice.25,26 Although these studies demonstrated an important role within the complex molecular changes occurring in damaged RGCs, they did not distinguish between either a primary or a secondary role for HDAC3 activity in the overall activation of the intrinsic apoptotic program being executed by these cells. To evaluate further the hierarchy of the role played by HDAC3 in neuronal cell death, we tested the hypothesis that induced expression of exogenous HDAC3 in otherwise healthy RGCs would lead to activation of RGC pathology and death. Mice were transduced with AAV2/2-Pgk-HDAC3-mCherry in one eye, and aged an additional four to12 weeks. Figure 1A shows a whole mounted retina, 12 weeks after viral transduction, counterstained for mCherry to identify the transgene fusion protein. Immunostaining was used because of the weak signal generated by mCherry in vivo.44 Colocalization with the RGC marker BRN3A indicated that the majority of HDAC3-mCherry expressing cells were RGCs (Fig. 1B). Because deletion or inhibition of HDAC3 in these cells prevents histone deacetylation and chromatin condensation normally associated with optic nerve damage,23–26 we expected induced expression of the protein to lead to wide-spread deacetylation of histones and the formation of heterochromatin in cells of the ganglion cell layer. Immunostaining for pan-acetylated histone H4, however, showed only limited signs of deacetylation in transduced retinas (five weeks shown [Figs. 1C–1E]). Some cells in the transduced retinas appeared to exhibit very little acetylated histone H4, but overall there was only an approximate decrease of 22% of the mean staining intensity in these retinas. At 12 weeks, confocal imaging of whole mounted retinas also showed no evidence of heterochromatin formation in HDAC3-mCherry expressing cells (Figs. 1F–1H). Rather, there appeared to be the opposite effect of an increase in euchromatin formation and shrinkage or dissolution of nucleolar structure in approximately 25% to 30% of HDAC3-mCherry positive cells at this late stage.
Figure 1.

Induced expression of HDAC3-mCherry in retinal ganglion cells fails to stimulate widespread histone deacetylation and heterochromatin formation. Mouse retinas were transduced with AAV2/2-Pgk-HDAC3-mCherry and harvested at times between three to 12 weeks for analysis. (A) A retinal whole mount 12 weeks after transduction and immunostained for mCherry (red) showing widespread expression of the fusion protein in the visible ganglion cell layer. Scale bar: 800 µm. (B) Detail of a retinal whole mount, 12 weeks after transduction, co-immunostained for mCherry and the RGC marker BRN3A (green). In this field, there are seven HDAC3-mCherry expressing cells that do not colocalize with BRN3A (arrows), whereas the majority of cells are positive for both proteins. The lack of BRN3A expression in these cells indicates the possibility of AAV2-mediated transduction of some non-RGCs, or could represent the approximately 15% of RGCs that do not express this protein.124,125 Scale bar: 60 µm. (C) Whole mounted contralateral retina and (D) a retina transduced with AAV2/2-Pgk-HDAC3-mCherry, five weeks after transduction, immunostained for pan-acetyl histone H4 (green). Although both retinas exhibit nuclei with varying levels of fluorescent staining, the transduced retina displays several cells with dramatically reduced staining (arrows in D). Scale bar: 25 µm. (E) A box and whisker plot of fluorescent staining for acetylated histone H4 (1–99 percentiles graphed). The mean staining intensity (defined as arbitrary units per region of interest, which was set to a fixed circle 7 µm in diameter) in the transduced retinas is ∼22% lower than the contralateral retinas (n = 1524 cells, contralateral; n = 2055 cells, HDAC3-transduced; three retinas each condition) (*P < 0.0001). (F–H) Confocal image of a retinal whole mount 12 weeks after transduction, immunostained for mCherry and counterstained with DAPI. RGC nuclei often appear oval or round and display at least one prominent nucleolus. This morphology is evident in a majority of cells expressing HDAC3-mCherry (examples highlighted by arrows). A substantial minority of cells, however, appear to have a marked increase in euchromatin and reduced or absent nucleoli (asterisks). Scale bar: 10 µm.
To assess whether HDAC3-mCherry overexpression induced RGC loss, we first quantified the change in neuronal cell density in the ganglion cell layer over a 12-week interval in whole mounted retinas stained with DAPI. To control for viral transduction, a cohort of age-matched C57BL/6 mice received an injection of an AAV2/2-Pgk-Cre expressing virus. The change in neuronal cell density was quantified relative to the fellow non-viral injected contralateral retina of each mouse. Figure 2 shows the change in density for both the HDAC3-mCherry and control viruses. At four weeks retinas transduced with both viruses showed no change in cell density relative to each other (P = 0.298). Starting at five weeks, one week after the time when maximal transgene expression from an AAV2 vector is normally expected (e.g., four weeks in the mouse retina25), HDAC3-mCherry transduced retinas exhibited a significant decrease in cell density to approximately 90% of the density of cells in nontransduced retinas and transduced retinas at four weeks. This reduced level was maintained at both eight and 12 weeks after transduction. Additionally, eyes injected with the control AAV2/2-Pgk-Cre virus exhibited significantly greater cell density over AAV2/2-Pgk-HDAC3-mCherry transduced retinas at all time points starting at five weeks. Linear regression analysis confirmed these significant changes between the four-week and the five-, eight-, and 12-week time points.
Figure 2.
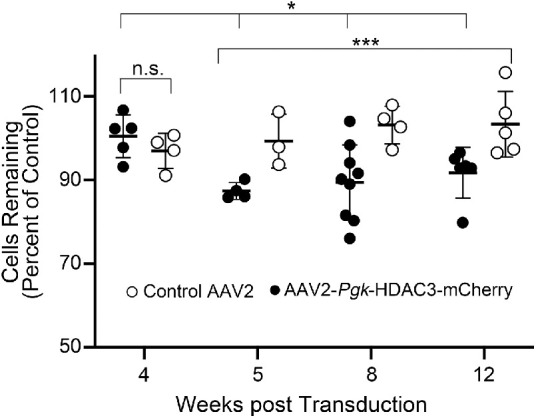
Induced expression of HDAC3-mCherry induces a moderate loss of cells in the ganglion cell layer. Scatter plot of neuron density in the ganglion cell layer of transduced retinas, depicted as a function of the density of contralateral retinas that were not transduced. Each data point represents a single mouse. The control AAV2 was expressing Pgk-driven Cre. Differences in cell density over this time course were assessed using a linear regression model analysis. At four weeks after transduction, there was no change increase in cell density in eyes injected with either virus (n.s., not significant, P = 0.433). At five weeks and later, HDAC3-mCherry transduced retinas exhibit a marked decrease in density compared to transduced retinas at four weeks (*P = 0.018), but there was no significant difference among retinas transduced with HDAC3-mCherry between five, eight, and 12 weeks (P = 0.588). At these later time points, however, HDAC3-mCherry retinas exhibited significantly different levels of cell density relative to control virus (***P = 0.0002).
Although there may be a modest intrinsic ability of HDAC3-mCherry to induce RGC loss, others have shown that induced HDAC3 expression in tissue culture cells also increases the susceptibility of these cells to a damaging stimulus.16 Therefore we examined if RGC death was greater in HDAC3-mCherry transduced eyes when subjected to acute optic nerve crush four weeks after transduction. The time point of four weeks was chosen because this represents a time point when transgene expression reaches peak, and when we do not observe any changes in cell density (Fig. 2). A monoclonal antibody was used to stain TUBB3, which is more highly expressed in RGCs over other retinal neurons and a polyclonal antibody against mCherry was used to identify transgene expression. Optic nerve crush damage to RGCs was examined five days after crush surgery, at a time point when RGC pathology is just beginning to be evident. Quantification of TUBB3 positive cells that exhibited apoptotic nuclei (Supplemental Fig. S1A–S1D) showed a significant increase in apoptotic cells, at five days after crush, relative to control virus transduced cells (P = 0.036) (Supplemental Fig. S1E). Interestingly, many cells with condensed and fragmented nuclei exhibited a marked decrease in mCherry staining. Because DNA degradation and nuclear fragmentation are associated with increased caspase activity,45,46 we hypothesize that the transgene is being degraded by caspases active in these cells. Supporting this, others have described HDACs as substrates for caspases.47,48 HDAC3-mCherry transduced retinas also exhibited a significant decrease in TUBB3-positive cell density over control virus retinas, by five days after optic nerve crush (P = 0.037, Supplemental Fig. S1F). Further evidence of an increase in susceptibility was demonstrated by quantifying the proportion of cells that exhibited translocation of BAX to mitochondria, which is considered a critical step in activation of intrinsic apoptosis in cells.49 RGCs in both retinas were transduced with AAV2/2-Pgk-GFP-BAX virus combined with either AAV2/2-Pgk-Cre or AAV2/2-Pgk-HDAC3-mCherry. After four weeks, one eye of each mouse was subjected to optic nerve crush and both retinas were analyzed for GFP-BAX localization after 5 days (Figs. 3A–3D). Crush induced a significant increase in cells with punctate distribution of BAX, indicative of mitochondrial translocation,50 regardless of which virus they were transduced with. Crushed retinas transduced with HDAC3-mCherry, however, exhibited significantly more punctate GFP-BAX cells than retinas transduced with the control virus (Fig. 3D). A caveat to this experimental paradigm is that increased cell loss in HDAC3-mCherry expressing RGCs may be a cumulative effect of intrinsic RGC susceptibility to the HDAC and additional cells responding to crush. To overcome the intrinsic effect, we transduced both eyes of mice with either the control or HDAC3 expressing virus and waited 6 weeks until independent HDAC3-mediated loss had stabilized (see Fig. 2). Eyes were then subjected to crush surgery and retinas were harvested and stained for activated caspase-3 and TUBB3, 5 days later. Contralateral retinas exhibited virtually no active caspase 3 staining in TUBB3+ cells (one cell in five retinas for control virus and 15 cells in six retinas for HDAC3-mCherry). After optic nerve crush, however, HDAC3-mCherry transduced retinas exhibited significantly more TUBB3+ cells that were positive for active caspase-3 than the crushed retinas expressing the control virus (P = 0.0061) (Fig. 3G). Collectively, these results suggest that HDAC3 may function as a primary activator of downstream apoptotic events in a subset of cells, while further sensitizing additional RGCs to a damaging stimulus such as optic nerve crush.
Figure 3.

Induced expression of HDAC3-mCherry sensitizes retinal ganglion cells to optic nerve damage. To validate that transduced cells were executing the intrinsic apoptotic pathway, retinas were co-transduced with AAV2/2-Pgk-GFP-BAX and either AAV2/2-Pgk-Cre (control virus) or AAV2/2-Pgk-HDAC3-mCherry. At four weeks after transduction eyes were subjected to crush surgery and assessed five days later. (A–C) Detail of co-transduced cells in the ganglion cell layer after crush. Two co-transduced cells exhibit punctate localization of GFP-BAX (arrows) indicating recruitment of BAX to the mitochondria. A third co-transduced cell exhibits cytosolic localization of GFP-BAX (asterisk). Scale bar: 15 µm. (D) Scatter plot showing percentage of transduced cells exhibiting punctate GFP-BAX five days after optic nerve crush (ONC). Retinas transduced with either virus exhibited a significant increase in punctate BAX cells after crush compared to contralateral retinas (**P < 0.001), but HDAC3-mCherry co-transduced cells showed significantly greater percentages of punctate BAX than cells co-transduced with the control virus (*P = 0.0034). Each point represents a single microscopic field (minimum of four fields/mouse from three separate mice). To further confirm that transduced cells were dying, retinas were transduced with AAV2/2-Pgk-Cre (control virus) or AAV2/2-Pgk-HDAC3-mCherry. At six weeks after transduction, eyes were subjected to optic nerve crush surgery and assessed five days later. Images of whole mounts labeled for caspase-3 (red) and DAPI (blue) (TUBB3 staining not shown) indicate more visible caspase-3–labeled cells in the HDAC3 overexpressing retina (E, F). Scale bar: 60 µm. Cell count data indicate a significantly higher number of TUBB3 positive/caspase-3 positive cells in HDAC3-mCherry transduced retinas than control retinas (**P = 0.0061). Each point represents a single retina.
Induced HDAC3 Expression Selectively Stimulates Recruitment of BAX to Mitochondria in Neuronally-Differentiated 661W Retinal Precursor Cells
To better evaluate the pathologic molecular events associated with HDAC3 expression, we initiated studies in tissue culture cells. Since it was previously shown that induced HDAC3 expression was selectively toxic to primary neurons and neuroblastoma cell lines, whereas non-neuronal cells were resistant,16 we reasoned that we could duplicate this effect in a single neuronally-derived cell line that can be converted from an undifferentiated to a differentiated state. For these experiments we chose 661W cells, which were derived from retinal tumors of transgenic mice expressing the SV40 virus large T-antigen under the control of the promoter for human interphotoreceptor retinol-binding protein (IRBP).51 These cells can be converted from an immortalized phenotype (undifferentiated) into a differentiated phenotype, where they stop dividing and extend neurite-like processes, by incubating them for 24 hours in 316 nM STS.42 To examine the selective toxicity in these cells in their differentiated and undifferentiated states, we first transfected cells in both conditions with plasmids that enabled expression of mito-blue fluorescent protein (BFP) to label mitochondria and the GFP-BAX fusion protein (Figs. 4A–4C). Some cells were also cotransfected with a plasmid expressing a human HDAC3-FLAG fusion protein (Fig. 4D–F). After 24 hours, cells were fixed and examined for translocation of GFP-BAX to mitochondria, resulting in a punctate pattern of localization. Differentiated cells not expressing HDAC3-FLAG typically showed diffuse, cytosolic localization of GFP-BAX (Fig. 4C), whereas differentiated cells that were transfected with HDAC3-FLAG were more likely to show punctate GFP-BAX labeling that colocalized with mito-BFP–labeled mitochondria (Fig. 4F). Quantification of both undifferentiated and differentiated cells showed that only differentiated cells transfected with HDAC3-FLAG exhibited significantly more cells with punctate GFP-BAX (P ≤ 0.005) (Supplemental Fig. S2A). In a separate experiment, we also stained transfected cells with an antibody against the FLAG epitope and scored them for apoptotic nuclei. As with GFP-BAX localization, only differentiated cells showed a significant increase in the presence of apoptotic nuclei (P ≤ 0.005) (Supplemental Fig. S2B).
Figure 4.

Differentiated 661W cells are selectively sensitive to HDAC3-induced BAX recruitment and caspase 3 activation. The activation of the BAX proapoptotic protein is a hallmark of the mitochondrial-dependent apoptotic pathway and is often considered the irreversible step in the death process.49 The recruitment of BAX was visualized in cells expressing a GFP-BAX fusion protein. (A–C) A 661W cell, differentiated with 316 nM STS, transfected with plasmids expressing GFP-BAX and mito-BFP to label mitochondria. In cells lacking induced HDAC3 expression, GFP-BAX occupies a cytosolic distribution indicative of inactive latent protein. (D–F) A differentiated cell transfected with GFP-BAX, mito-BFP, and a plasmid expressing HDAC3-FLAG. Induced HDAC3 expression induces recruitment of GFP-BAX to the mitochondria, which is evident by the punctate labeled aggregates of GFP-BAX colocalized with the mito-BFP, starting between 11 to 14 hours after transfection. Scale bar: 5 µm. (G–N) 661W cells nucleofected with HDAC3-mCherry and GFP-BAX before incubation without or with STS to induce differentiation. Undifferentiated cells retain diffuse localization of both fusion proteins (G–J), whereas differentiated cells exhibit some concentration of HDAC3-mCherry in the nucleus and punctate/aggregations of GFP-BAX (K–N). DAPI counterstain. Scale bar: 5 µm. (O) Quantification of cells with GFP-BAX localization at different time points after STS addition. Differentiated cells show significant punctate GFP-BAX localization after 24 hours (*P ≤ 0.032, relative to the other groups at 24 hours).
A drawback with our transfection protocol was the transfection efficiency, which was less than 5% of the cells after differentiation. Nucleofection can increase the efficiency of introducing DNA into cells. We found that nucleofection was able to introduce plasmids into 73.68% ± 10.42% of undifferentiated cells, but this decreased to 17.93% ± 3.66% of cells if they were differentiated. Because undifferentiated cells appeared to be resistant to induced HDAC3 expression, we tested whether nucleofecting undifferentiated cells with HDAC3-mCherry followed by differentiation was able to replicate the selective sensitivity to this enzyme demonstrated in the transfection experiments. Cells were nucleofected with GFP-BAX and either mouse HDAC3-mCherry or mCherry containing plasmids. After 24 hours, cells were treated with 316 nM STS or left undifferentiated, and the localization of GFP-BAX was scored over the next 24 hours (Figs. 4G–4O). The process of differentiation induced the translocation of GFP-BAX to the mitochondrial membrane in cells with induced expression of HDAC3-mCherry to significant levels over the next 24 hours, relative to undifferentiated cells or cells expressing mCherry alone (P ≤ 0.032) (Fig. 4O).
Induced Expression of HDAC3 Induces Intrinsic Apoptosis in Differentiated 661W Cells
The ability of induced HDAC3 expression to induce GFP-BAX translocation implies that these cells are activating the intrinsic apoptotic pathway. However, BAX activation is often a feature of other cell death pathways, which will recruit the mitochondrial dependent intrinsic pathway by secondary activation of specific BH3-only proteins.52,53 Alternatively, intrinsic apoptosis is exclusively dependent on the function of proapoptotic proteins such as BAX and BAK. To assess the dependence of HDAC3-induced toxicity on BAX function, we selectively expressed a CRISPR/Cas9 cassette with different gRNAs targeting regions of exon 1 of the Bax gene in wild type 661W cells. CRISPR editing successfully reduced or eliminated BAX protein levels (Fig. 5A), with the greatest reduction using the Sg1 gRNA (Fig. 5B). As a consequence, all further experiments were conducted with this cell line, which was termed Sg1. Sg1 cells treated with 316 nM STS exhibited expected signs of differentiation, including neurite extension with βIII-tubulin expression (Figs. 5C–5J and Supplemental Figs. S3A–S3H) and cell cycle arrest (Figs. 5K–5O). Undifferentiated wild type and Sg1 cells were then nucleofected with HDAC3-mCherry, differentiated, and assessed for cell death by immunostaining for activated caspase 3 (Figs. 6A–6P). Editing of the Bax gene in Sg1 cells conferred significant, but only partial, attenuation of HDAC3-mediated cell death (P = 0.0006 relative to wild type cells expressing HDAC3-mCherry) (Fig. 6Q).
Figure 5.

Characterization of 661W cells after CRISPR/Cas9 editing of the Bax gene. The Bax gene was edited using lentiCRISPRv2 transfected into undifferentiated 661W cells. Plasmid expression was maintained by growing the cells in puromycin, which was subsequently added to the media for all experiments using these cells. Additionally, WT cells were transfected with the lentiCRISPRv2 plasmid, but without a guide RNA. (A) LICOR-generated image of a representative western blot showing endogenous BAX and ACTIN levels in WT and 3 lines edited with different gRNAs against the first exon of the Bax gene. (B) Quantification of three independent Western blots showing BAX protein levels normalized to the ACTIN reference. All three lines showed significant depletion of BAX (P < 0.0001). Sg1 cells were used for further experiments. (C–J) WT and edited Sg1 cells show typical features of differentiation including the extension of neurite-like processes and staining for βIII-tubulin (green—see also Supplemental Fig. S3). Panels show different times after addition of 316 nM STS to induce differentiation. Scale bar: 10 µm. DAPI counterstain. (K–N) Differentiation causes cell cycle arrest in both WT and Sg1 cells. Undifferentiated cells exhibit incorporation of BrdU (brown immunoreaction product) (Scale bar: 15 µm), which is reduced to negligible levels by 24 hours in culture with STS (O). (*P < 0.001).
Figure 6.

Differentiated Bax-edited 661W cells (Sg1) show reduced apoptotic death in response to induced HDAC3 expression. Wild type (WT) and Sg1 cells were nucleofected with HDAC3-mCherry and then differentiated with 316 nM staurosporine or left undifferentiated. After 24 hours, cells were fixed and immunostained for activated caspase 3. (A–D) Undifferentiated cells show diffuse HDAC3-mCherry and no active caspase 3, but differentiated cells expressing the HDAC3 plasmid are positive for this active protease and exhibit concentration of HDAC3-mCherry in the nucleus (E–H). (I–L) Undifferentiated Sg1 cells also exhibit diffuse HDAC3-mCherry distribution. (M–P) Some differentiated Sg1 cells showed a concentration of HDAC3-mCherry to the nucleus and perinuclear region of the cell and activation of caspase 3. Scale bar: 10 µm. (Q) Quantification of HDAC3-mCherry expressing cells that were positive for activated caspase 3. Differentiated WT and Sg1 both exhibited higher levels of activated caspase 3 relative to undifferentiated cells (*P < 1.0E-07), but Sg1 cells exhibited a reduction of 52% of nucleofected cells showing activation of caspase 3 at this time point (**P = 0.0006).
Partial attenuation of cell death by Bax deletion is a feature of cells that express both BAX and BAK.54 Downregulation of both genes is required to achieve full protection. Most neurons, however, execute a splice variant of the Bak gene transcript, which introduces a weak 20 bp exon that creates a premature stop codon, resulting in a transcript called N(neuronal)-Bak.43 This transcript also reportedly exhibits impaired translation in neurons.55,56 As a consequence, Bax-deletion in mice effectively results in a Bax/Bak double knock-out phenotype and confers a complete block of intrinsic apoptosis of a variety of neuronal populations,57–60 including RGCs.27,28,30,61 N-Bak in 661W cells was first examined by nested RT-PCR, which revealed the absence of the neuronal splice variant and the presence of the full-length variant in these cells (Fig. 7A). The expression of full-length BAK protein was also confirmed by western blotting extracts from both wild type and Sg1 661W cells (Fig. 7B). To reduce BAK levels, we nucleofected both wild type and Sg1 cells with an siRNA directed against mouse Bak mRNA along with a plasmid expressing HDAC3-FLAG. The siRNA significantly reduced the levels of BAK protein in both cell lines relative to cells treated with a scrambled siRNA (Figs. 7C and 7D) but did not completely eliminate it. These cells were then differentiated and examined for activated caspase 3 after 24 hours. Wild type cells with BAK knockdown exhibited a 43.5% reduction in labeling for activated caspase 3 relative to scramble siRNA treated cells (P < 0.0005). Conversely, Sg1 cells exposed to the scrambled siRNA exhibited a 65.4% decrease (P = 0.0003, relative to wild type cells treated with Bak siRNA). Sg1 cells treated with the Bak siRNA exhibited an 82% reduction of caspase 3 activation (P = 0.005, relative to Sg1 cells treated with scrambled siRNA) (Fig. 7E). These results indicate that HDAC3-induced toxicity results in activation of the intrinsic apoptotic program.
Figure 7.

Characterization of BAK expression and knock-down in WT and Sg1 661W cells. (A) Nested PCR yields a 200 bp product for transcripts from the full length Bak splice variant (FL-Bak), which is present in the liver, brain, and both wild-type (WT) and Sg1 661W cells. The brain sample also shows the neuronal Bak (N-Bak) splice variant containing a weak 20 bp exon. N-Bak is not detected in 661W cells. Extraneous lanes were removed for presentation of this figure. (B) Western blot showing full length BAK in spleen and both undifferentiated (Un) and differentiated (D) 661W cells. Full length BAK is virtually undetectable in normal mouse retina. (C) Western blot showing levels of BAK protein in differentiated WT and Sg1 661W cells treated with a scrambled siRNA or an siRNA directed against Bak. Extraneous lanes were removed for presentation of this figure. (D) Bak siRNA results in depletion of BAK protein. Mean (± SD) of 4 independent western blots shown. (*P ≤ 0.033). (E) Graph showing the level of caspase 3 activation in cells expressing HDAC3-FLAG. All data were collected 24 hours after induction of differentiation. WT cells show a significant increase in activated caspase 3 staining after differentiation, which is not affected by scrambled siRNA. Nucleofection with Bak siRNA; however, results in a significant reduction in cells with activated caspase 3. Sg1 cells also show a significant increase in activated caspase 3 staining after differentiation, but this increase is reduced relative to WT cells (no siRNA treatment, P < 0.0001, not indicated on graph) consistent with the results in Figure 6. Nucleofection of scrambled siRNA does not affect caspase 3 activation in 661W cells, but Bak siRNA induces a further reduction in caspase 3 activation over Sg1 cells alone, and over WT cells nucleofected with the Bak siRNA. Values shown are the mean ± (SEM) of three independent experiments. At least 120 cells were scored for each treatment group in each experiment. (*P = 0.0052; **P ≤ 0.0005; ***P < 0.0001).
Induced Expression of HDAC3 Stimulates Jnk2/3 and BH3-Only Bbc3(Puma) Gene Expression in Differentiated Cells
In neurons, including RGCs, damage stimulates a cascade of kinase activation that originates with the activation of dual leucine zipper kinase and ultimately the activation of JUN N-terminal kinases (JNKs) (Fig. 8A). With respect to BAX activation, this pathway leads to the transcription of BH3-only genes, which function to antagonize anti-apoptotic proteins such as BCL-X and facilitate a conformational change in latent BAX to allow it to interact and permeabilize the mitochondrial outer membrane. Two transcription factors downstream of JNK activation are cJUN and p53, which are activated or stabilized, respectively. Phosphorylated cJUN activates Bim and Hrk gene transcription, whereas p53 activates Bbc3(Puma) and Noxa gene transcription. First, we conducted a qPCR analysis of Jnk2 and Jnk3 transcript abundance, comparing either undifferentiated or differentiated cells with or without induced HDAC3 expression, since these molecules represent a pivotal point in this activation cascade (Fig. 8B). Undifferentiated cells showed less than a onefold increase in transcript abundance for these genes in response to induced HDAC3 expression. However, once the cells were differentiated, they showed a significantly greater induction of Jnk2 and Jnk3 gene expression in response to HDAC3 overexpression. We then examined the change in expression for 4 different BH3-only gene transcripts (Fig. 8C). We observed no significant change in the abundance of mRNA for the cJUN-dependent BH3-only gene Bim, while Hrk mRNA levels were below the level of detection. Alternatively, HDAC3 induced significant upregulation of the p53-dependent gene Bbc3(Puma) in differentiated cells (P ≤ 0.002). Noxa mRNA levels were elevated by induced HDAC3 expression in undifferentiated cells, and in differentiated cells not expressing exogenous HDAC3. In differentiated cells, induced HDAC3 expression produced no further increase in Noxa expression. Based on these results, we predict that HDAC3 induces JNK2/3 signaling in differentiated cells, which results in p53 activation.
Figure 8.

Increased transcript abundance of Jnk2, Jnk3, and Bbc3(Puma) by induced HDAC3 expression in differentiated 661W cells. (A) Schematic of the dual leucine zipper kinase–JNK activation pathway. Previous studies indicate that this pathway is prevalent in damaged neurons including retinal ganglion cells after optic nerve damage. The result of JNK activation is the phosphorylation and activation of two regulatory transcription factors.94,126 Activation of cJUN leads to the expression of the BH3-only proteins BIM127 and HRK,128 whereas p53 directs the transcription of the BH3-only proteins BBC395 and NOXA.129 BH3-only proteins, in turn, lead to the activation of BAX. (B) Real time qPCR analysis of transcript abundance of Jnk2 and Jnk3. The data shown represents the fold change in abundance of either undifferentiated, or differentiated, 661W cells expressing HDAC3-mCherry for 24 hours, relative to cells expressing mCherry only (*P < 0.02). (C) Real time qPCR of three BH3-only gene products representative of p53-directed (Bbc3 and Noxa) or JUN-directed (Bim) transcription. Induced HDAC3-mCherry expression in undifferentiated cells induces a significant increase in Noxa expression. Differentiation of 661W cells also induces significant expression of this BH3-only gene that is not increased further by HDAC3-mCherry. Conversely, HDAC3-mCherry expression in differentiated 661W cells induces a significant increase in Bbc3 transcript abundance. (*P ≤ 0.016; **P = 0.002; ***P = 3.28E-05). The mean (± SD) of three independent experiments is shown for all qPCR experiments.
HDAC3-Mediated Death of 661W Cells is Not Dependent on Cell-Cycle Arrest
Differentiation of 661W cells causes cell cycle arrest (Fig. 5). Since one of the functions of HDAC activity is to promote transition of cells from G1 to S phase,62 it is possible that the differential response to increased HDAC3 activity between undifferentiated dividing 661W cells, and differentiated non-dividing 661W cells, is a consequence of induced entry into the cell cycle. Forcing terminally differentiated cells, such as neurons, into S phase frequently induces cell death.63 Additionally, previous studies showing the selective sensitivity of neurons to HDAC3 expression did not explicitly examine the effect of cell cycle arrest on non-neuronal cell lines.16 To examine the possible contribution of cell cycle arrest in HDAC3 sensitivity, we nucleofected undifferentiated 661W cells with a plasmid expressing high levels of p21WAF-1/Cip-1, which will create a G1-S phase block and push cells into a G0-like state of quiescence.64 Cells were nucleofected with (or without) p21WAF-1/Cip-1, GFP-BAX, and HDAC3-mCherry. Cell cycle arrest was validated in some cultures of p21WAF-1/Cip-1-expressing 661W cells by monitoring the incorporation of BrdU (Figs. 9A and 9B), which was reduced by 24 hours and completely eliminated by 48 hours. After 24 and 36 hours nucleofected cells were scored for the formation of GFP-BAX puncta (Figs. 9C and 9D). As a control, we nucleofected the same plasmids into HeLa cells to assess if cell cycle arrest would also sensitize a cell line not derived from a neuronal lineage. The results showed that p21WAF-1/Cip-1–induced cell cycle arrest had no effect in sensitizing either cell line to induced HDAC3-mCherry expression at these time points. The time point of 36 hours was chosen in anticipation of observing cells that were transitioning into cell cycle arrest, however, in a separate experiment, we also examined if HDAC3 induced GFP-BAX recruitment in undifferentiated 661W cells at 48 after p21WAF-1/Cip-1 nucleofection. This experiment also showed no evidence of an increase in sensitivity to HDAC3 (-p21, 30.0 ± 15.3% cells with punctate GFP-BAX; +p21, 23.3% ± 9.7% cells with punctate GFP-BAX, P = 0.718). The implication of these results is that other elements, specific to neuronal differentiation, are required to make cells susceptible to HDAC3 toxicity.
Figure 9.

Cell cycle arrest mediated by p21WAF-1/Cip-1 does not sensitize undifferentiated cells to induced HDAC3 expression. Immunohistochemical labeling showing BrdU incorporation into (A) undifferentiated 661W cells or (B) undifferentiated cells 36 hours after exogenous expression of p21WAF-1/Cip-1. Scale bar: 10 µm. (C) Quantification of BrdU-positive staining cells showing reduced incorporation after 24 hours, and significantly reduced incorporation after 48 hours (*P = 1.10E-07). (D, E) Quantification of GFP-BAX recruitment in cell cycle-arrested undifferentiated 661W cells with or without induced HDAC3 expression. In this experiment, p21WAF-1/Cip-1 -arrested HeLa cells were used as a non-neuronal cell line for comparison. (D) At 24 hours, and (E) 36 hours, induced HDAC3 expression in both p21WAF-1/Cip-1 nucleofected HeLa and 661W cells failed to induce any increase in GFP-BAX recruitment relative to normal dividing cells. The time points represent the time from nucleofection with plasmids for GFP-BAX, HDAC3-mCherry, and a p21WAF-1/Cip-1 expressing plasmid. Data from three independent experiments are plotted.
Discussion
Previous studies with Hdac3 conditional knock-out mice25 or with an HDAC3 selective inhibitor26 revealed a considerable role for this enzyme in the process of RGC death after acute optic nerve injury. These studies did not, however, reveal the temporal importance of HDAC3 activity in the activation of other downstream apoptotic events. Induced expression of HDAC3 in mouse RGCs showed a modest intrinsic effect stimulating cell loss around five weeks after transduction. This timing likely reflects the beginning of peak transgene expression, which takes up to four weeks to reach optimal levels in AAV2 transduced RGCs.25 After five weeks, no further loss of cells was detected by eight and 12 weeks after transduction (Fig. 1). Although HDAC3 overexpression was able to directly induce loss of some cells, it had a greater effect on sensitizing the RGCs to axonal damage. This lowering of the tolerance threshold of cells was also reported in neuronal cell cultures transfected with HDAC3.16
The limited amount of cell loss induced by direct HDAC3 transduction could be explained by differential expression of the transgene, such that cells exhibiting the highest levels of HADC3 expression were more likely to spontaneously die. It is difficult to control the level of expression in cells using the paradigm of viral-mediated transduction and it is clear that there is a heterogeneous population of cells exhibiting different levels of HDAC3-mCherry staining after transduction (Fig. 1B). Additionally, cells with robust transgene expression clearly survived as late as 12 weeks after transduction. It is possible that the cells that spontaneously died represented a class of RGCs (or transduced non-RGCs) that were selectively more sensitive to HDAC3 expression. A similar phenomenon was described by Tran and colleagues65 in the mouse retina after optic nerve crush damage. Using single cell RNA-Seq, this group was able to classify 46 different sub-types of RGCs on the basis of their transcriptome and show that select classes of these cells were more or less resilient to the optic nerve injury. Future experiments should be directed at determining if a similar selectivity to HDAC3 expression exists within different RGC subtypes. It is also important to note that AAV2, while having a high trophism for RGCs, can also transduce other retinal cell types in the naïve retina, principally Müller cells, but also some amacrine cells and rod bipolar cells. Because we used the nonselective Pgk promoter to ensure maintenance of transgene expression after injury, our experiments cannot rule out that possibility that increased RGC sensitivity was influenced by the expression of the transgene in these other cell types as well.
To better evaluate the mechanisms by which HDAC3 induces cellular toxicity, intrinsically, we examined the effects of induced expression of this enzyme in a cell culture system using the neuronally-derived 661W cells. Transfection of these cells in both the un- and differentiated states showed that induced expression of exogenous HDAC3 was selectively toxic to differentiated cells. Our initial assay for toxicity was monitoring the recruitment of a GFP-BAX fusion protein that was introduced into the cells along with the plasmid expressing HDAC3. BAX (and its homologous protein BAK) recruitment to the mitochondrial outer membrane is often considered the point of no return in the intrinsic apoptotic pathway49 and precedes irreparable events of the apoptotic pathway such as the release of cytochrome c, mitochondrial fragmentation, and caspase activation.39,49,66–69 Although BAX activation is a distinct feature of intrinsic apoptosis, this event can also occur during execution of the extrinsic pathway by caspase 8-mediated cleavage of the BH3-only protein BID.52 To evaluate the critical nature of BAX (and BAK) function in HDAC3-mediated cell death, the Bax gene was edited in 661W cells, whereas BAK protein levels were reduced using siRNA. Combined depletion of both of these full-length pro-apoptotic proteins significantly abrogated HDAC3-dependent cell death in differentiated 661W cells, strongly implicating the activation of the intrinsic apoptotic program by induced expression of this HDAC. Consistent with this, induced HDAC3 expression also stimulated an increase in gene elements associated with JNK signaling pathway and resulted in an increase in Bbc3(Puma) expression and activated caspase 3.
HDAC3 and Chromatin Changes Associated With Apoptosis
Conditional Hdac3 ablation,25 or treatment with HDAC3-selective inhibitors,26 results in the prevention of global histone deacetylation normally observed during the early stages of RGC death induced by acute optic nerve damage. Additionally, the kinetics of histone H4 deacetylation after optic nerve damage is consistent with the translocation of HDAC3 from a predominantly cytosolic localization to the nuclei of RGCs.23,24 From these results, we have attributed the process of death-associated histone deacetylation to HDAC3. Therefore it was unexpected to observe that induced HDAC3 expression only resulted in moderate levels of histone H4 deacetylation in mouse RGCs. Additionally, we did not observe an increase in nuclei with a heterochromatic appearance. In fact, we observed the opposite, where the nuclei of a large proportion of transduced cells appeared to become more euchromatic (Fig. 1). It is probable that a subpopulation of the HDAC3mCherry expressing cells were amacrine or ipRGCs, since BRN3A does not label these cell types. This could explain why some cells did not undergo apoptosis, and euchromatic nuclei were observed. This phenomenon requires further study.
There are a variety of explanations as to why induced HDAC3 expression did not lead to a major increase in histone H4 deacetylation and heterochromatin formation. HDAC3 is the core epigenetic subunit of the nuclear repressor co-receptor (NCoR)/silencing mediator of retinoid and thyroid hormone receptor (SMRT) co-repressor complex. It is this complex that mediates the interaction of HDAC3 with targeted areas of chromatin structure in the nucleus, which leads to transcriptional repression.70 Simply upregulating HDAC3 expression, as we have done in our experiments, may not necessarily stimulate an increase in NCoR/SMRT repressor complex activation. Similarly, activity of the NCoR/SMRT corepressor can be influenced by interacting with other molecules such as c-MYC71 and peroxisome proliferator-activated receptor γ.72,73 Up- or down-regulation of additional factors that control the repression complex may be a consequence of early signaling steps in the RGC cell death program. HDAC3 itself may require cell death-induced post-translational modifications such as deubiquitinization74 or phosphorylation,75,76 which lead to stabilization and increased activity, respectively. It is telling that while induced expression in RGCs yielded moderate cell loss, these cells became sensitized to optic nerve crush, suggesting that axon-induced damaged was required to activate one or more secondary factors that were then able to catalyze and accelerate RGC pathology in the presence of high levels of HDAC3. Increased sensitivity to a damaging stimulus was also observed in neuronal tissue culture cells expressing HDAC3.16
Curiously, however, differentiated 661W cells exhibited almost complete activation of apoptosis in response to induced HDAC3 expression. It is possible that factors required to mediate HDAC3-induced pathology were already present in these cells, perhaps as a result of the differentiation process by sub-lethal levels of STS. Additionally, tissue culture cells do not benefit from a more stabilized environment with supporting glial cells that RGCs likely enjoy. Alternatively, 661W cells differ from RGCs in that they express both BAX and BAK (see below), which may differentially sensitize them to the effects of induced HDAC3 expression. While these pro-apoptotic proteins are known to function similarly to induce mitochondrial outer membrane permeabilization, their activation is not uniform to the spectrum of damaging stimuli. In mouse embryonic fibroblasts, for example, BAK is preferentially activated in response to DNA damaging radiation, whereas BAX is preferentially activated by trophic factor withdrawal.54
661W Cells as a Model System
We reasoned that a better understanding of why neurons are selectively sensitized to HDAC3 expression may require a simplified experimental platform for study, rather than conducting experiments in vivo. Such a platform could be enhanced if we could show that cells from the same line acquired sensitivity, thereby eliminating confounding factors associated with lineage specific genetic and epigenetic differences encountered when conducting comparative studies between divergent cell lines. We chose 661W cells because of the relative ease at which they could be differentiated and because of their origin from the mouse retina. These cells were originally derived from the tumors of transgenic mice expressing the SV40 large T-antigen under the control of the promoter for IRBP.51 Initial studies with these cells showed that they could be induced to express cone photoreceptor-specific antigens,51 and subsequently they have often been considered to be a cell culture model of cones. More recently, however, Sayyad and colleagues77 reported that 661W cells expressed both cone-specific and RGC-specific markers, suggesting that T-antigen transformation may have occurred in neuroblasts that were transitioning near the specification points of RGCs and cones. Non-supporters of this idea might argue that IRBP expression is restricted to photoreceptors in the adult retina,78 but IRBP expression has also been reported early in mouse retinal development (embryonic day 11- E11) and during this period is colocalized with BrdU, indicating its expression in dividing neuroblasts.79 E11 marks the time point where both RGCs80 and cone photoreceptors81 begin to form from daughter cells of neuroblasts. As a consequence, 661W cells may represent a retinal precursor cell that is transitioning between these two cell types, and it may be erroneous to categorize them as strictly cone-lineage cells.
Further evidence that 661W cells represent a retinal precursor cell is the fact that they still express the full-length Bak transcript. Differentiated neurons exhibit a neuron-specific splice variant of this transcript that incorporates a weak 20 bp exon embedded in the fourth intron of the Bak gene.43,82 Insertion of this exon creates a premature STOP codon in the coding sequence, resulting in a truncated peptide that contains only the BH3-domain of the full-length BAK protein (N-BAK),82,83 although there is some evidence that the spliced transcript is also inefficiently translated.55,56 When neuroblasts switch to the splice variant is not known, but full-length BAK is expressed by glia in the CNS43,82,84 (note the presence of both the N-Bak and full-length transcripts in mouse brain in Fig. 7) and preliminary studies done by our group indicate that full length Bak transcripts are present in Müller cells (data not shown). Because Müller cells are derived late from the same neuroblast lineage that retinal neurons arise from, it stands to reason that full length Bak expression is conserved throughout the life of dividing retinoblast cells. This is consistent with studies showing full length BAK protein expression in other neuronal precursor cells85,86 and the appearance of the N-Bak splice variant coinciding with the ending phase of neurogenesis in the mouse cortex.87
Regardless of the origin of 661W cells, our experiments documenting the toxicity of HDAC3 expression in this line may be equally relevant to both RGC and cone photoreceptor biology in disease. Broad spectrum HDAC inhibitors protect both rod and cone photoreceptors in models of outer retinal degeneration88,89 and HDAC activity is increased in both classes of photoreceptors by day PN30 in rd1 mice and PN59 in rd10 mice.90
The Relationship Between p53 and HDAC Activity
Our results indicate that induced HDAC3 expression in differentiated 661W cells leads to the upregulation of Jnk2 and Jnk3, consistent with these molecules being activated in dying neurons.91 In cultured cerebellar granular neurons, HDAC3 overexpression precipitates an increase in cJUN protein levels,17 an expected outcome for the activation of cJUN in this pathway. We observed no change in BH3-only gene expression attributed to activated phospho-cJUN (Bim and Hrk),92,93 however. Instead, we observed that HDAC3 expression selectively induced an increase in the p53-regulated gene Noxa in undifferentiated cells and Bbc3(Puma)94,95 in differentiated 661W cells. NOXA is a weak BH3-only protein and has a minimal impact on cell viability alone, but appears to amplify apoptosis when expressed in conjunction with other BH3-only proteins.96 Conversely, BBC3(PUMA) is a potent killer,96,97 which may account for the increased activation of BAX recruitment by HDAC3 in differentiated cells. There is extensive literature suggesting that p53 mediates the neuronal toxicity of HDAC expression. The activity and selectivity of promoter binding of p53 is influenced by both its level of phosphorylation and acetylation.98 Acetylation of p53 at specific sites prevents its activity,99,100 while HDAC inhibition or conditional deletion of Hdacs in neurons, such as RGCs, reduces p53-dependent gene expression and p53-dependent death.101,102 Interestingly, the opposite appears to be true for dividing cells, where inhibition of HDAC activity can stimulate p53-mediated apoptosis,103–105 including undifferentiated 661W cells.106 Alternatively, one of the reasons why differentiated 661W cells were selectively sensitive to HDAC3 was because the process of differentiation removed them from the cell cycle. However, undifferentiated 661W cells that were placed in cell cycle arrest by the expression of p21WAF-1/Cip-1 showed no increase in sensitivity to induced HDAC3 expression.
The Mechanism of Neuronal Susceptibility
It is clear that HDAC3 plays an important role in neuronal pathology. This is evidenced by the growing number of studies showing that blocking the activity of this HDAC has a protective effect in a variety of different neurodegenerative conditions (reviewed by D'Mello107). The fact that induced HDAC3 expression in RGCs does not trigger widespread death of these neurons, however, would seem to suggest that the action of this enzyme is not a master regulator of downstream apoptotic reactions. A caveat to this conclusion is that the function of HDAC3 in the death process may require additional co-factors that are independently generated as an early step in the apoptotic pathway. For example, this may take the form of additional components to allow for the formation of active NCoR/SMRT complexes.
Alternatively, pathologic HDAC3 activity in RGCs may require secondary modifications such as phosphorylation by glycogen synthetase kinase 3β (GSK3β).16 Regulation of GSK3β is mediated in many cells by the activity of the AKT/PI3 Kinase complex.108,109 AKT phosphorylates GSK3β rendering it inactive, but inhibition (or loss of activity) of AKT results in accumulation of dephosphorylated GSK3β and its activation. The loss of AKT activity is considered one of the primary events associated with loss of neurotrophin support when axons of RGCs are damaged. Supporting this, depletion of GSK3β by siRNA knock-down has a protective effect in RGCs after optic nerve damage.110 It is reasonable to speculate that the delayed toxicity of induced HDAC3 expression is mediated by a requirement to activate GSK3β through optic nerve damage. Interestingly, this may not be the only event leading to HDAC3 toxicity, since GSK3β commonly requires an initial primary modification of its substrates before it will phosphorylate them.108 If this is the case for RGCs, then HDAC3 requires an additional post-translational modification in these cells by a mechanism that is not yet identified.
How HDAC3 selectively kills neurons is still unresolved. Our studies suggest that activation of p53, leading to the activation of intrinsic apoptosis, is a principal mechanism. The selectivity for neurons may be a function of the genes that p53 targets for transcriptional control, which can change depending on the post-translational modification of p53 and the chromatin structure of target genes with either strong or weak p53-binding sites.98,111–115 Alternatively, a recent proteomics study of rat cerebellar granule neurons expressing HDAC3 showed the upregulation of several proteins that are linked to neurotoxicity, including NPTX1 (neuronal pentraxin), HDAC9, TEX10, TFG, TSC1, and NFL.17 Of these, we were able to amplify a transcript for Nptx1 in 661W cells, but abundance of this mRNA was not changed by induced HDAC3 expression (data not shown). Pentraxins are intriguing, however, because they regulate the complement pathway116 and studies suggest this is an important regulator of pathology in most neurodegenerative conditions.117 Deletion of the complement activation pathway, for example, is protective in the DBA/2J mouse model of glaucoma,118–120 where Nptx1 is up-regulated in the optic nerve head early in the progression of the disease.118
Last, the pathologic effects of HDAC3 may not be directly related to its role in histone modification/transcriptional regulation. Regulation of the activity of non-nuclear targets, such as kinases,121 metabolic enzymes,122 and structural elements of the cytoskeleton123 by HDAC3-mediated deacetylation have also been described. At this point, it is not possible to rule out that HDAC3 may be acting on one of these “nonconventional” substrates to mediate its effect on neurons.
Supplementary Material
Acknowledgments
The authors thank Joel Dietz for maintaining the mice used in this study, Satoshi Kinoshita and the Translational Research Initiative in Pathology Laboratory at the University of Wisconsin-Madison for cutting retinal sections analyzed in this study, and Mark Banghart for statistical review of the data analysis.
Supported by National Eye Institute Grants R01 EY012223 (RWN), R01 EY030123 (RWN), R01 EY029809 (LWG), R01 EY029809 (LWG) and a Vision Research CORE grant P30 EY016665, NRSA grant T32 GM081061, by an unrestricted research grant from Research to Prevent Blindness, Inc., and by a University of Wisconsin-Madison Vilas Life Cycle award and the Frederick A. Davis Research Chair (RWN).
Disclosure: H.M. Schmitt, None; R.L. Fehrman, None; M.E. Maes, None; H. Yang, None; L.-W. Guo, None; C.L. Schlamp, None; H.R. Pelzel, None; R.W. Nickells, None
References
- 1.McIntyre RL, Daniels EG, Molenaars M, Houtkooper RH, Janssens GE.. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol Med. 2019; 11: e9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas EA, D'Mello SR. Complex neuroprotective and neurotoxic effects of histone deacetylases. J Neurochem. 2018; 145: 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Wu Q, Zhang L, Feng L.. Inhibition of histone deacetylase 3 (HDAC3) mediates ischemic preconditioning and protects cortical neurons against ischemia in rats. Front Mol Neurosci. 2016; 9: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao H, Li G, Zhang S, et al.. Inhibition of histone deacetylase 3 by MiR-494 alleviates neuronal loss and improves neurological recovery in experimental stroke. J Cereb Blood Flow Met. 2019; 39: 2392–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu C, Soragni E, Chou CJ, et al.. Chemical probes identify a role for histone deacetylase 3 in Friedreich's ataxia gene silencing. Chem Biol. 2009; 16: 980–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soragni E, Chou CJ, Rusche JR, Gottesfeld JM.. Mechanism of action of action of 2-aminobenzamide HDAC inhibitors in reversing gene silencing in Friedreich's ataxia. Front Neurology. 2015; 6: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandi C, Pinto RM, Al-Mahdawi S, et al.. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol Dis. 2011; 42: 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia H, Pallos J, Jacques V, et al.. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease. Neurobiol Dis. 2012; 46: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bardai FH, Verma P, Smith C, et al.. Disassociation of histone deacetylase-3 from normal Huntingtin underlies mutant Huntingtin neurotoxicity. J Neurosci. 2013; 33: 11833–11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia H, Wang Y, Morris CD, et al.. The effects of pharmacological inhibition of histone deacetylase 3 (HDAC3) in Huntington's disease mice. PLoS One. 2016; 11: e0152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janczura KJ, Volmar CH, Sartor GC, et al.. Inhibition of HDAC3 reverses Alzheimer's disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc Natl Acad Sci USA. 2018; 115: E11148–E11157. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Yu L, Liu Y, Jin Y, et al.. Lentivirus-mediated HDAC3 inhibition attenuates oxidative stress in APPswe/PS1dE9 mice. J Alzheimers Dis. 2018; 61: 1411–1424. [DOI] [PubMed] [Google Scholar]
- 13.McQuown SC, Barrett RM, Matheos DP, et al.. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011; 31: 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bieszczad K, Bechay K, Rusche JR, et al.. Histone deacetylase inhibition via RGFP966 releases the brakes on sensory cortical plasticity and the specificity of memory formation. J Neurosci. 2015; 35: 13124–13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malvaez M, McQuown SC, Rogge GA, et al.. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci USA. 2013; 110: 2647–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bardai FH, D'Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3b. J Neurosci. 2011; 31: 1746–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu Z, D'Mello SR. Proteomic analysis identifies NPTX1 and HIP1R as potential targets of histone deacetylase-3-mediated neurodegeneration. Exp Biol Med. 2018; 243: 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galindo-Romero C, Aviles-Trigueros M, Jimenez-Lopez M, et al.. Axotomy-induced retinal ganglion cell death in adult mice: quantitative and topographic time course analyses. Exp Eye Res. 2011; 92: 377–387. [DOI] [PubMed] [Google Scholar]
- 19.Quigley HA, Broman AT.. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006; 90: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tham YC, Li X, Wong TY, et al.. Global prevalence of glaucoma and projections of glaucoma burden through 2040. Ophthalmology. 2014; 121: 2081–2090. [DOI] [PubMed] [Google Scholar]
- 21.Steinsapir KD, Goldberg RA.. Traumatic optic neuropathy. Surv Ophthalmol. 1994; 38: 487–518. [DOI] [PubMed] [Google Scholar]
- 22.Chen YJ, Liang CM, Tai MC, et al.. Longitudinal relationship between traumatic brain injury and the risk of incident optic neuropathy: a 10-year follow-up nationally representative Taiwan survey. Oncotarget. 2017; 8: 86924–86933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pelzel HR, Schlamp CL, Nickells RW.. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010; 11: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelzel HR, Schlamp CL, Waclawski M, Shaw MK, Nickells RW.. Silencing of Fem1cR3 gene expression in the DBA/2J mouse precedes retinal ganglion cell death and is associated with Histone Deacetylase activity. Invest Ophthalmol Vis Sci. 2012; 53: 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt HM, Pelzel HR, Schlamp CL, Nickells RW.. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol Neurodegen. 2014; 9: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt HM, Schlamp CL, Nickells RW.. Targeting HDAC3 activity with RGFP966 protects against retinal ganglion cell nuclear atrophy and apoptosis after optic nerve injury. J Ocul Pharmacol Ther. 2018; 34: 260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Schlamp CL, Poulsen KP, Nickells RW.. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000; 71: 209–213. [DOI] [PubMed] [Google Scholar]
- 28.Libby RT, Li Y, Savinova OV, et al.. Susceptibility to neurodegeneration in glaucoma is modified by Bax gene dosage. PLoS Genet. 2005; 1: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maes ME, Schlamp CL, Nickells RW.. BAX to basics: how the BCL2 gene family controls the death of retinal ganglion cells. Prog Retin Eye Res. 2017; 57: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semaan SJ, Li Y, Nickells RW.. A single nucleotide polymorphism in the Bax gene promoter affects transcription and influences retinal ganglion cell death. ASN Neuro. 2010; 2: e00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donahue RJ, Maes ME, Grosser JA, Nickells RW.. BAX depleted retinal ganglion cells survive and become quiescent following optic nerve damage. Mol Neurobiol. 2020; 57: 1070–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Semaan SJ, Schlamp CL, Nickells RW.. Dominant inheritance of retinal ganglion cell resistance to optic nerve crush in mice. BMC Neurosci. 2007; 8: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Schlamp CL, Nickells RW.. Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 1999; 40: 1004–1008. [PubMed] [Google Scholar]
- 34.Harder JM, Libby RT.. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegen. 2011; 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandes KA, Harder JM, Fornarola LB, et al.. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012; 46: 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sánchez-Migallón MC, Valiente-Soriano FJ, Nadal-Nicolas FM, Vidal-Sanz M, Agudo-Barriuso M.. Apoptotic retinal ganglion cell death after optic nerve transection or crush in mice: delayed RGC loss with BDNF or a caspase 3 inhibitor. Invest Ophthalmol Vis Sci. 2016; 57: 81–93. [DOI] [PubMed] [Google Scholar]
- 37.Cui Q, Yip HK, Zhao RCH, So KF, Harvey AR.. Intraocular elevation of cyclic AMP potentiates ciliary neurotrophic factor-induced regeneration of adult rat retinal ganglion cell axons. Mol Cell Neurosci. 2003; 22: 49–61. [DOI] [PubMed] [Google Scholar]
- 38.Semaan SJ, Nickells RW.. The apoptotic response in HCT116BAX-/− cancer cells becomes rapidly saturated with increasing expression of a GFP-BAX fusion protein. BMC Cancer. 2010; 10: 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maes ME, Schlamp CL, Nickells RW.. Live-cell imaging to measure BAX recruitment kinetics to mitochondria during apoptosis. PLoS One. 2017; 12: e0184434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Bergen NJ, Wood JPM, Chidlow G, et al.. Recharacterization of the RGC-5 retinal ganglion cell line. Invest Ophthalmol Vis Sci. 2009; 50: 4267–4272. [DOI] [PubMed] [Google Scholar]
- 41.Lieven CJ, Millet LE, NHoegger MJ, Levin LA.. Induction of axon and dendrite formation during RGC-5 cell differentiation. Exp Eye Res. 2007; 85: 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson AF, Crowe ME, Lieven CJ, Levin LA.. Induction of neuronal morphology in the 661W cone photoreceptor cell line with staurosporine. PLoS One. 2015; 10: e0145270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uo T, Kinoshita Y, Morrison RS.. Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J Biol Chem. 2005; 280: 9065–9073. [DOI] [PubMed] [Google Scholar]
- 44.Heppert JK, Dickinson DJ, Pani AM, et al.. Comparative assessment of fluorescent proteins for in vivo imaging in an animal model system. Mol Biol Cell. 2016; 27: 3385–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Enari M, Sakahira H, Yokoyama H, et al.. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998; 391: 43–50. [DOI] [PubMed] [Google Scholar]
- 46.Sahara S, Aoto M, Eguchi Y, et al.. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 1999; 401: 168–173. [DOI] [PubMed] [Google Scholar]
- 47.Escaffit F, Vaute O, Chevillard-Briet M, et al.. Cleavage and cytoplasmic relocalization of histone deacetylase 3 are important for apoptosis progression. Mol Cell Biol. 2007; 27: 554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paroni G, Mizzau M, Henderson C, et al.. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell. 2004; 15: 2804–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang LK, Putcha GV, Deshmukh M, EM Johnson Jr. Mitochondrial involvement in the point of no return in neuronal apoptosis. Biochimie. 2002; 84: 223–231. [DOI] [PubMed] [Google Scholar]
- 50.Wolter KG, Hsu YT, Smith CL, et al.. Movement of bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997; 139: 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan E, Ding XQ, Saadi A, et al.. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Invest Ophthalmol Vis Sci. 2004; 45: 764–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Zhu H, Xu CJ, Yuan J.. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998; 94: 491–501. [DOI] [PubMed] [Google Scholar]
- 53.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X.. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998; 94: 481–490. [DOI] [PubMed] [Google Scholar]
- 54.Wei MC, Zong WX, Cheng EHY, et al.. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001; 292: 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jakobson M, Jakobson M, Llano O, Palgi J, Arumae U.. Multiple mechanisms repress N-Bak mRNA translation in the healthy and apoptotic neurons. Cell Death Disease. 2013; 4: e777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jakobson M, Lintulahti A, Arumae U.. mRNA for N-Bak, a neuron-specific BH3-only splice isoform of Bak, escapes nonsense-mediated decay and is translationally repressed in neurons. Cell Death Disease. 2012; 3: e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deckwerth TL, Elliot JL, Knudson CM, et al.. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996; 17: 401–411. [DOI] [PubMed] [Google Scholar]
- 58.White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD.. Widespread elimination of naturally occurring neuronal death in bax-deficient mice. J Neurosci. 1998; 18: 1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deckwerth TL, Easton RM, Knudson CM, Korsmeyer SJ, Johnson EM Jr. Placement of the BCL2family member BAX in the death pathway of sympathetic neurons activated by trophic factor deprivation. Exp Neurol. 1998; 152: 150–162. [DOI] [PubMed] [Google Scholar]
- 60.Sun W, Oppenheim RW.. Response of motoneurons to neonatal sciatic nerve axotomy in Bax-knockout mice. Mol Cell Neurosci. 2003; 24: 875–886. [DOI] [PubMed] [Google Scholar]
- 61.Ogilvie JM, Deckwerth TL, White FA, et al.. Bax knockout mice have an increased survival of retinal neurons. Invest Ophthalmol Vis Sci. 1997; 38: S33. [Google Scholar]
- 62.Yamaguchi T, Cubizolles F, Zhang Y, et al.. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010; 24: 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma R, Kumar D, Kumar Jha N, et al.. Re-expression of cell cycle markers in aged neurons and muscles: whether cells should divide or die? Biochim Biophys Acta. 2017; 1863: 324–336. [DOI] [PubMed] [Google Scholar]
- 64.Spencer SL, Cappell SD, Tsai FC, et al.. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013; 155: 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tran NM, Shekhar K, Whitney IE, et al.. Single-cell profiles of retinal ganglion cells differing in resilience to injury reveal neuroprotective genes. Neuron. 2019; 104: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jurgensmeier JM, Xie ZH, Deveraux Q, et al.. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998; 95: 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC.. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. 2000; 345: 271–278. [PMC free article] [PubMed] [Google Scholar]
- 68.Karbowski M, Lee YJ, Gaume B, et al.. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002; 159: 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Slee EA, Harte MT, Kluck RM, et al.. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999; 144: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karagianni P, Wong J.. HDAC3: taking the SMRT-N-CoRrect road to expression. Oncogene. 2007; 26: 5439–5449. [DOI] [PubMed] [Google Scholar]
- 71.Zhuang Q, Li W, Benda C, et al.. NCoR/SMRT co-repressors cooperate with c-MYC to create an epigenetic barrier to somatic cell reprogramming. Nat Cell Biol. 2018; 20: 400–412. [DOI] [PubMed] [Google Scholar]
- 72.Villapol S.Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol Neurobiol. 2018; 38: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vallée A, Lecarpentier Y.. Alzheimer Disease: Crosstalk between the canonical Wnt/Beta-Catenin pathway and PPARs alpha and gamma. Front Neurosci. 2016; 10: 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng Q, Miao Y, Ge J, et al.. ATXN3 positively regulates type I IFN antiviral response by deubiquitinating and stabilizing HDAC3. J Immunol. 2018; 201: 675–687. [DOI] [PubMed] [Google Scholar]
- 75.Zhang X, Ozawa Y, Lee H, et al.. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005; 19: 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanigan TW, Aboukhatwa SM, Taha TY, Frasor J, Petukhov PA.. Divergent JNK phosphorylation of HDAC3 in triple-negative breast cancer cells determines HDAC inhibitor binding and selectivity. Cell Chem Biol. 2017; 24: 1356–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sayyad Z, Sirohi K, Radha V, Swarup G.. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci Reports. 2017; 7: 16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yokoyama T, Liou GI, Caldwell RB, Overbeek PA.. Photoreceptor-specific activity of the human interphotoreceptor retinoid-binding protein promoter in transgenic mice. Exp Eye Res. 1992; 55: 225–233. [DOI] [PubMed] [Google Scholar]
- 79.Liou GI, Wang M, Matragoon S.. Timing of interphotoreceptor retinoid-binding protein (IRBP) gene expression and hypomethylation in developing mouse retina. Dev Biol. 1994; 161: 345–356. [DOI] [PubMed] [Google Scholar]
- 80.Wang SW, Kim BS, Ding K, et al.. Requirement for math5 in the development of retinal ganglion cells. Genes Dev. 2001; 15: 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brzezinski JA, Reh TA.. Photoreceptor cell fate specification in vertebrates. Development. 2015; 142: 3263–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun YF, Yu LY, Saarma M, Timmusk T, Arumae U.. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001; 276: 16240–16247. [DOI] [PubMed] [Google Scholar]
- 83.Sun YF, Yu LY, Saarma M, Arumae U.. Mutational analysis of N-Bak reveals different structural requirements for antiapoptotic activity in neurons and proapoptotic activity in nonneuronal cells. Mol Cell Neurosci. 2003; 23: 134–143. [DOI] [PubMed] [Google Scholar]
- 84.Kitamura Y, Ota T, Matsuoka Y, et al.. Hydrogen peroxide-induced apoptosis mediated by p53 protein in glial cells. Glia. 1999; 25: 154–164. [PubMed] [Google Scholar]
- 85.Lindsten T, Golden JA, Zong WX, et al.. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci. 2003; 23: 11112–11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Milosevic J, Storch A, Schwarz J.. Spontaneous apoptosis in murine free-floating neurospheres. Exp Cell Res. 2004; 294: 9–17. [DOI] [PubMed] [Google Scholar]
- 87.Lin L, Zhang M, Stoilov P, Chen L, Zheng S.. Developmental attenuation of neuronal apoptosis by neural-specific splicing of Bak1 microexon. Neuron. 2020; 107: 1180–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mitton KP, Guzman AE, Deshpande M, et al.. Different effects of valproic acid on photoreceptor loss in Rd1 and Rd10 retinal degeneration mice. Mol Vis. 2014; 20: 1527–1544. [PMC free article] [PubMed] [Google Scholar]
- 89.Trifunovic D, Arango-Gonzalez B, Comitato A, et al.. HDAC inhibition in the cpfl1 mouse protects degenerating cone photoreceptors in vivo. Hum Mol Genet. 2016; 25: 4462–4472. [DOI] [PubMed] [Google Scholar]
- 90.Samardzija M, Corna A, Gomez-Sintes R, et al.. HDAC inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death Differ. 2021; 28: 1317–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hollville E, Romero SE, Deshmukh M.. Apoptotic cell death regulation in neurons. FEBS J. 2019; 286: 3276–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma C, Ying C, Yuan Z, et al.. dp5/HRK is a c-Jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J Biol Chem. 2007; 282: 30901–30909. [DOI] [PubMed] [Google Scholar]
- 93.Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J.. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001; 29: 629–643. [DOI] [PubMed] [Google Scholar]
- 94.Wong HK, Fricker M, Wyttenbach A, et al.. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol Cell Biol. 2005; 25: 8732–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nakano K, Vousden KH.. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001; 7: 683–694. [DOI] [PubMed] [Google Scholar]
- 96.Chen L, Willis SN, Wei A, et al.. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005; 17: 393–403. [DOI] [PubMed] [Google Scholar]
- 97.Adams JM, Cory S.. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007; 26: 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knights CD, Catania J, Di Giovanni S, et al.. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006; 173: 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chao C, Wu Z, Mazur SJ, et al.. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol. 2006; 26: 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brochier C, Dennis G, Rivieccio MA, et al.. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J Neurosci. 2013; 33: 8621–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Uo T, Veenstra TD, Morrison RS.. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009; 29: 2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lebrun-Julien F, Suter U.. Combined HDAC1 and HDAC2 depletion promotes retinal ganglion cell survival after injury through reduction of p53 target gene expression. ASN Neuro. 2015; 7: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mrakovcic M, Bohner L, Hanisch M, Fröhlich LF.. Epigenetic targeting of autophagy via HDAC inhibition in tumor cells: Role of p53. Int J Mol Sci. 2018; 19: 3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yun T, Yu K, Yang SS, et al.. Acetylation of p53 protein at lysine 120 up-regulates Apaf-1 protein and sensitizes the mitochondrial apoptotic pathway. J Biol Chem. 2106; 291: 7386–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wagner T, Brand P, Heinzel T, Krämer OH.. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta. 2014; 1846: 524–538. [DOI] [PubMed] [Google Scholar]
- 106.Wallace DM, Cotter TG.. Histone deacetylase activity in conjunction with E2F-1 and p53 regulates Apaf-1 expression in 661W cells and the retina. J Neurosci Res. 2009; 87: 887–905. [DOI] [PubMed] [Google Scholar]
- 107.D'Mello SR. Histone deacetylase-3: friend and foe of the brain. Exp Biol Med. 2020; 245: 1130–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beurel E, Grieco SF, Jope RS.. Glygone synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol Ther. 2015; 148: 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grabinski T, Kanaan NM.. Novel non-phosphorylated serine 9/21 GSK3. Front Mol Neurosci. 2016; 9: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ahmed Z, Morgan-Warren PJ, Berry M, Scott RAH, Logan A.. Effects of siRNA-mediated knockdown of GSK3. Cells. 2019; 8: 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen J.The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016; 6: a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tanikawa C, Espinosa M, Suzuki A, et al.. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat Commun. 2012; 3: 676. [DOI] [PubMed] [Google Scholar]
- 113.Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, et al.. DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2010; 38: 356–368. [DOI] [PubMed] [Google Scholar]
- 114.Millau JF, Bandele OJ, Perron J, et al.. Formation of stress-specific p53 binding patterns is influenced by chromatin but not by modulation of p53 binding affinity to response elements. Nucleic Acids Res. 2011; 39: 3053–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Horvath MM, Wang X, Resnick MA, Bell DA.. Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS Genet. 2007; 3: e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haapasalo K, Meri S.. Regulation of the complement system by pentraxins. Front Immunol. 2019; 10: 1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Donahue RJ, Moller-Trane R, Nickells RW.. Meta-analysis of transcriptomic changes in optic nerve injury and neurodegenerative models reveals a fundamental response to injury throughout the central nervous system. Mol Vis. 2017; 23: 987–1005. [PMC free article] [PubMed] [Google Scholar]
- 118.Howell GR, Macalinao DG, Sousa GL, et al.. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011; 121: 1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Williams PA, Tribble JR, Pepper KW, et al.. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegen. 2016; 11: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harder JM, Braine CE, Williams PA, et al.. Early immune responses are independent of RGC dysfunction in glaucoma with complement component C3 being protective. Proc Natl Acad Sci USA. 2017; 114: E3839–E3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Long J, Fang WY, Chang L, et al.. Targeting HDAC3, a new partner protein of AKT in the reveral of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia. 2017; 31: 2761–2770. [DOI] [PubMed] [Google Scholar]
- 122.Wang S, Jiang B, Zhang T, et al.. Insulin and mTOR pathway regulate HDAC3-mediated deacetylation and activation of PGK1. PLoS Biol. 2015; 13: e1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li X, Liu X, Gao M, et al.. HDAC3 promotes meiotic apparatus assembly in mouse oocytes by modulating tubulin acetylation. Development. 2017; 144: 3789–3797. [DOI] [PubMed] [Google Scholar]
- 124.Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, et al.. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009; 50: 3860–3868. [DOI] [PubMed] [Google Scholar]
- 125.Nadal-Nicolas FM, Jimenez-Lopez M, Salinas-Navarro M, et al.. Whole number, distribution and co-expression of Brn3 transcription factors in retinal ganglion cells of adult albino and pigmented rats. PLoS One. 2012; 7: e49830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fuchs SY, Adler V, Pincus MR, Ronai Z.. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci USA. 1998; 95: 10541–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wakabayshi T, Kosaka J, Oshika T.. JNK inhibitory kinase is up-regulated in retinal ganglion cells after axotomy and enhances BimEL expression levels in neuronal cells. J Neurochem. 2005; 95: 526–536. [DOI] [PubMed] [Google Scholar]
- 128.Fernandes KA, Harder JM, Kim J, Libby RT.. JUN regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp Eye Res. 2013; 112: 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kiryu-Seo S, Hirayama T, Kato R, Kiyama H.. Noxa is a critical mediator of p53-dependent motor neuron death after nerve injury in adult mouse. J Neurosci. 2005; 25: 1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.