

Abstract
The faithful propagation of cellular signals in most organisms relies on the coordinated functions of a large family of protein kinases that share a conserved catalytic domain. The catalytic domain is a dynamic scaffold that undergoes large conformational changes upon activation. Most of these conformational changes, such as movement of the regulatory αC-helix from an “out” to “in” conformation, hinge on a conserved, but understudied, loop termed the αC-β4 loop, which mediates conserved interactions to tether flexible structural elements to the kinase core. We previously showed that the αC-β4 loop is a unique feature of eukaryotic protein kinases. Here, we review the emerging roles of this loop in kinase structure, function, regulation, and diseases. Through a kinome-wide analysis, we define the boundaries of the loop for the first time and show that sequence and structural variation in the loop correlate with conformational and regulatory variation. Many recurrent disease mutations map to the αC-β4 loop and contribute to drug resistance and abnormal kinase activation by relieving key auto-inhibitory interactions associated with αC-helix and inter-lobe movement. The αC-β4 loop is a hotspot for post-translational modifications, protein–protein interaction, and Hsp90 mediated folding. Our kinome-wide analysis provides insights for hypothesis-driven characterization of understudied kinases and the development of allosteric protein kinase inhibitors.
Keywords: cancer mutation, conformational regulation, disease mutation, drug resistance, Hsp-90, molecular brake, post-translational modifications, protein kinase
1 |. INTRODUCTION
Protein kinases are one of the largest gene families in the human genome and regulate virtually all cellular processes. Dysregulation of protein kinase activity can lead to a variety of disease phenotypes such as cancer,1 diabetes,2 neurodegeneration,3 and cardiovascular disease.4 Consequently, there is a need to understand the diverse regulatory mechanisms of protein kinases as a foundation for developing protein kinase inhibitors. To this end, comparative studies on protein kinase sequence and structure have provided important insights into protein kinase activation, regulation, evolution, and inhibition.5–7
Drug discovery efforts on protein kinases have traditionally focused on the conserved catalytic domain, which adopts a bi-lobal fold. The N-terminal ATP binding lobe consists of five strands and a helix, while the larger C-terminal substrate binding lobe is primarily composed of helices. Extensive structural studies on the catalytic domain and comparisons of active and inactive conformations have highlighted the role of key flexible elements in kinase conformational regulation. The activation segment8,9 and αC-helix10,11 are two such flexible elements that undergo dramatic conformational changes upon activation of most protein kinases.12 Another critical example of a flexible element is the dynamic assembly of the regulatory spine (RS),11,13 a spatially connected network of hydrophobic interactions spanning the ATP and substrate binding lobes. RS assembly is correlated with kinase activation and conformational strain in the catalytic loop.14
At the advent of the post-genomic era, the new-found wealth of sequencing information allowed large-scale comparisons across diverse protein kinases. In particular, quantitative comparisons of the evolutionary constraints acting on eukaryotic and distantly related eukaryotic-like kinases in prokaryotes revealed that conformational flexibility and allosteric regulation evolved progressively in protein kinases through addition of key flexible elements such as the activation loop.7 In addition to the activation loop and the substrate binding lobe, sequence motifs in the αC-β4 loop were also identified as unique to eukaryotic protein kinases.7 Structurally, the αC-β4 loop immediately follows the αC-helix and connects to the β8 strand, which immediately precedes the activation loop DFG motif. The αC-β4 loop also serves as a hinge point for inter-lobe movement.
The αC-β4 loop resides at the intersection of many essential regulatory mechanisms for protein kinase function. Disease-related mutations in this region are capable of altering kinase activity and drug response.15–17 While our knowledge of the kinase activation loop is quite extensive, relatively little is known about the role of the αC-β4 loop in kinase function. In this review, we aim to provide a comprehensive review on the αC-β4 loop region and centralize the knowledge to facilitate comparisons across the protein kinome.
2 |. CONSERVATION AND VARIATION IN THE αC-β4 LOOP OF PROTEIN KINASES
2.1 |. Structure and sequence conservation of αC-β4 loop
The αC-β4 loop is located on the N-lobe of the kinase domain and connects the αC-helix to the β4 strand (Figure 1a). To provide an unbiased overview on the kinase αC-β4 loop, we mined the Protein Data Bank (PDB) for kinase structures solved by X-ray crystallography (4,900 structures, 8,122 chains). We generated a non-redundant dataset of kinase structures by only including kinase chains with unique Uniprot IDs. During this filtering procedure, priority was given to structures with high resolution and fully resolved αC-β4 loops. The final filtered dataset contained 426 kinase chains and was used for all subsequent structural analyses.
FIGURE 1.
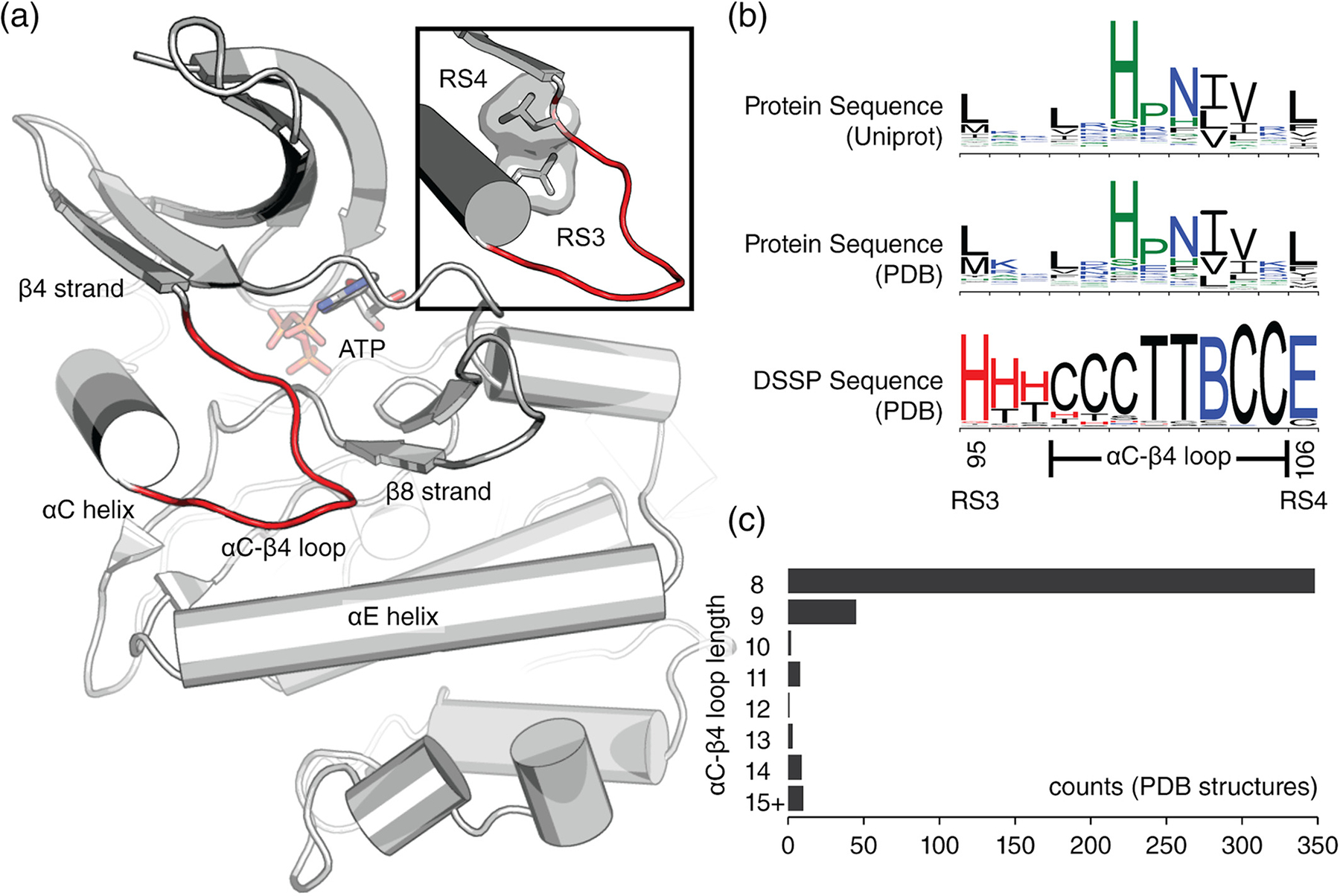
Definition of the αC-β4 loop. (a) The αC-β4 loop (red) of protein kinase A (PDB ID: 1ATP).18 Structural regions near the αC-β4 loop are labeled for reference. At the top-right corner, a close-up shows the αC-β4 loop flanked by the RS3 and RS4 residues. (b) Sequence logo plots spanning from RS3 to RS4 are shown. This span of residues was used on all logo plots throughout the review. Sequence logo plots include amino acid sequences from Uniprot proteomes (top), amino acid sequences from PDB (middle), and secondary structure sequences from PDB (bottom). Secondary structure sequences were defined by DSSP where red = helix, blue = strand, black = coil. DSSP classifications: G = 310 helix, H = α-helix, I = π-helix, B = isolated β-bridge, E = extended β-strand, T = hydrogen bonded turn, S = non-hydrogen bonded bend, C = coil. (c) Histogram showing the distribution of αC-β4 loop lengths calculated from unique protein kinase structures in the PDB. The 15+ category includes lengths greater than or equal to 15. DSSP, Define Secondary Structure of Proteins; PDB, Protein Data Bank
To analyze the sequence conservation of the αC-β4 loop, we identified and aligned 600,734 protein kinase sequences from Uniprot proteomes.19 We also converted our filtered dataset of 426 kinase chains into amino acid sequences. Amino acid sequence logos from Uniprot (Figure 1b, top) and PDB (Figure 1b, middle) are similar, suggesting that our filtered dataset of kinase structures is representative.
To define the boundaries of the αC-β4 loop, we assigned a secondary structure sequence to each kinase structure using the Define Secondary Structure of Proteins (DSSP) algorithm20 (Figure 1b). Based on secondary structure propensity, we defined the αC-β4 as an 8-residue segment starting from RS3 + 3 (PKA position 98) and ending at RS4–1 (PKA position 105). While the αC-β4 loop is typically 8 residues in length (Figure 1c), we note exceptions in multiple kinase structures (Table 1).
TABLE 1.
List of kinases containing an extended αC-β4 loop
| PDB chain | αC-β4 length | Group | Name |
|---|---|---|---|
| 4jr7_A21 | 47 | CMGC | scCK2α (Saccharomyces cerevisiae) |
| 5oat_A22 | 23 | Other | PINK1 (Tribolium castaneum) |
| 6bru_A | 21 | CK1 | VRK1 (Homo sapiens) |
| 2v62_A23 | 21 | CK1 | VRK2 (Homo sapiens) |
| 2jii_A23 | 21 | CK1 | VRK3 (Homo sapiens) |
| 1q8y_A24 | 19 | CMGC | SKY1 (Saccharomyces cerevisiae) |
| 4qtc_A25 | 17 | Other | HASPIN (Homo sapiens) |
| 5my8_A26 | 16 | CMGC | SRPK1 (Homo sapiens) |
| 2×7g_A | 16 | CMGC | SRPK2 (Homo sapiens) |
| 5yk0_A27 | 15 | pknB | Rv3197 (Mycobacterium tuberculosis) |
| 6s14_A | 14 | CMGC | DYRK1A (Homo sapiens) |
| 6fyv_A28 | 14 | CMGC | CLK4 (Homo sapiens) |
| 6fyr_A28 | 14 | CMGC | CLK3 (Homo sapiens) |
| 6fyl_A28 | 14 | CMGC | CLK2 (Homo sapiens) |
| 6ft8_A29 | 14 | CMGC | CLK1 (Homo sapiens) |
| 5y86_A30 | 14 | CMGC | DYRK3 (Homo sapiens) |
| 5lxc_A31 | 14 | CMGC | DYRK2 (Homo sapiens) |
| 4iir_A32 | 14 | CMGC | PRPF4B (Homo sapiens) |
| 3llt_A | 13 | CMGC | PF3D7_1445400 (Plasmodium falciparum) |
| 6p5s_A33 | 13 | CMGC | HIPK2 (Homo sapiens) |
| 4nt4_A34 | 13 | CK1 | Gilgamesh (Drosophila melanogaster) |
| 5wtk_A35 | 12 | Other | cas13a (Leptotrichia shahii) |
| 4×7q_B36 | 12 | CAMK | PIM2 (Homo sapiens) |
Note: Examples were retrieved from the aforementioned dataset of 426 representative kinase chains which was filtered by unique Uniprot ID’s with priority given to high resolution structures and fully resolved αC-β4 loops. Abbreviation: PDB, Protein Data Bank.
2.2 |. The HxN motif
The αC-β4 loop and the associated HxN motif are uniquely conserved in eukaryotic protein kinases including pseudokinases,37 but is absent/divergent in distantly related atypical protein kinases and eukaryotic-like small molecule kinases.7 To investigate sequence variations in the αC-β4 loop, we analyzed sequence conservation within each kinase group using the aforementioned Uniprot dataset of 600,734 protein kinase sequences (Figure 2a). This analysis revealed a consensus sequence for the αC-β4 loop: Φ-X-H-X-N-Φ-Φ-X (Figure 2a, top-left), where Φ represents a hydrophobic residue and X represents any amino acid (wildcard). The HxN motif facilitates a β-turn connecting the αC-helix and β4 strand (Figure 2b, left). The HxN-Asn hydrogen bonds the backbone of the β8 strand via an isolated β-bridge and the carboxamide side chain (Figure 3a). These interactions tether the αC-β4 loop to the hinge region of the protein kinase domain. The HxN wildcard residue is usually a proline and shows varying levels of conservation across different kinase groups.
FIGURE 2.
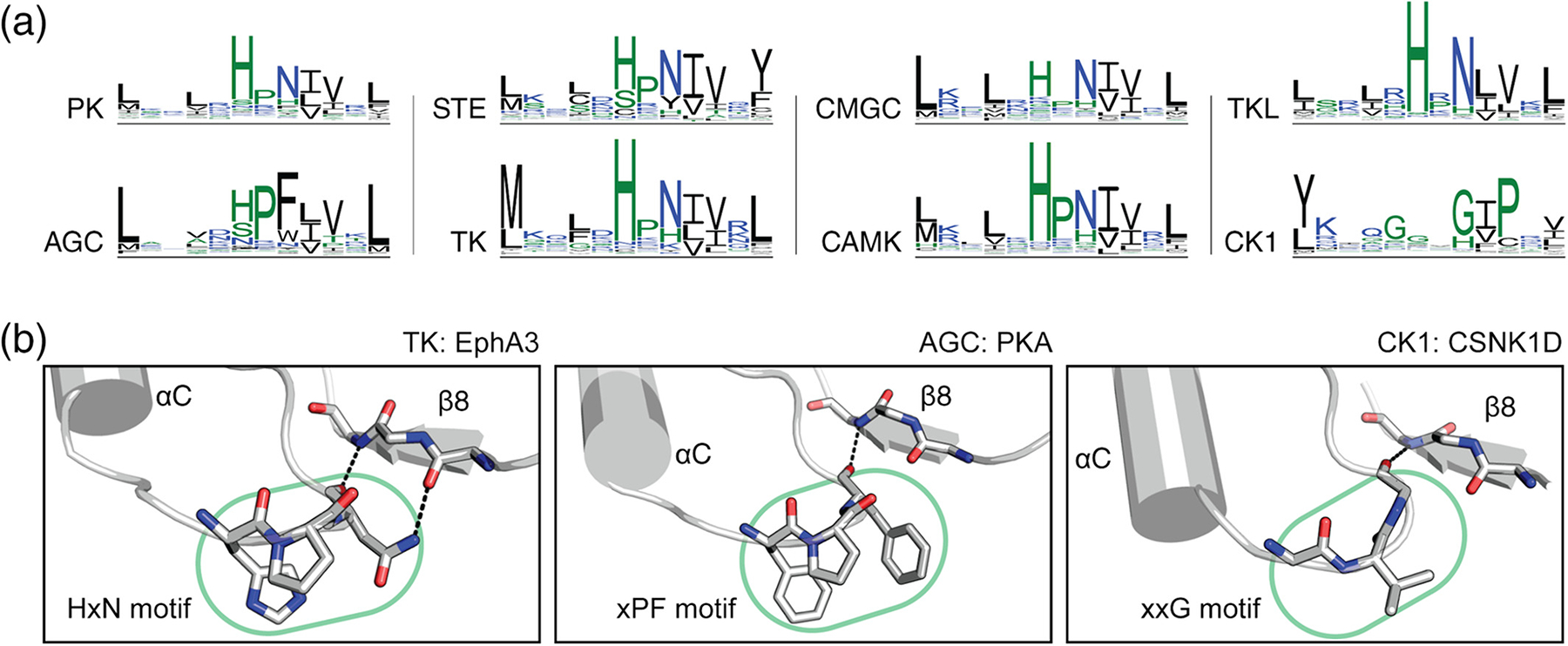
Sequence conservation of the αC-β4 loop. (a) Sequence logo plots spanning from RS3 to RS4 are shown for different protein kinase groups. (b) The canonical HxN motif is shown in tyrosine kinase EphA3 (PDB ID: 3dzq) (left). The AGC-specific variant is shown in PKA (PDB ID: 1atp)18 (middle). The CK1- specific xxG motif is shown in CSNK1D (PDB ID: 4twc)38 (right). Residue numbers (not shown) are provided: 679–681 for the HxN motif in EphA3, 100–102 for xPF motif in PKA, and 62–64 for the xxG motif in CSNK1D. Side chains are not shown for the β8 strand. PDB, Protein Data Bank
FIGURE 3.
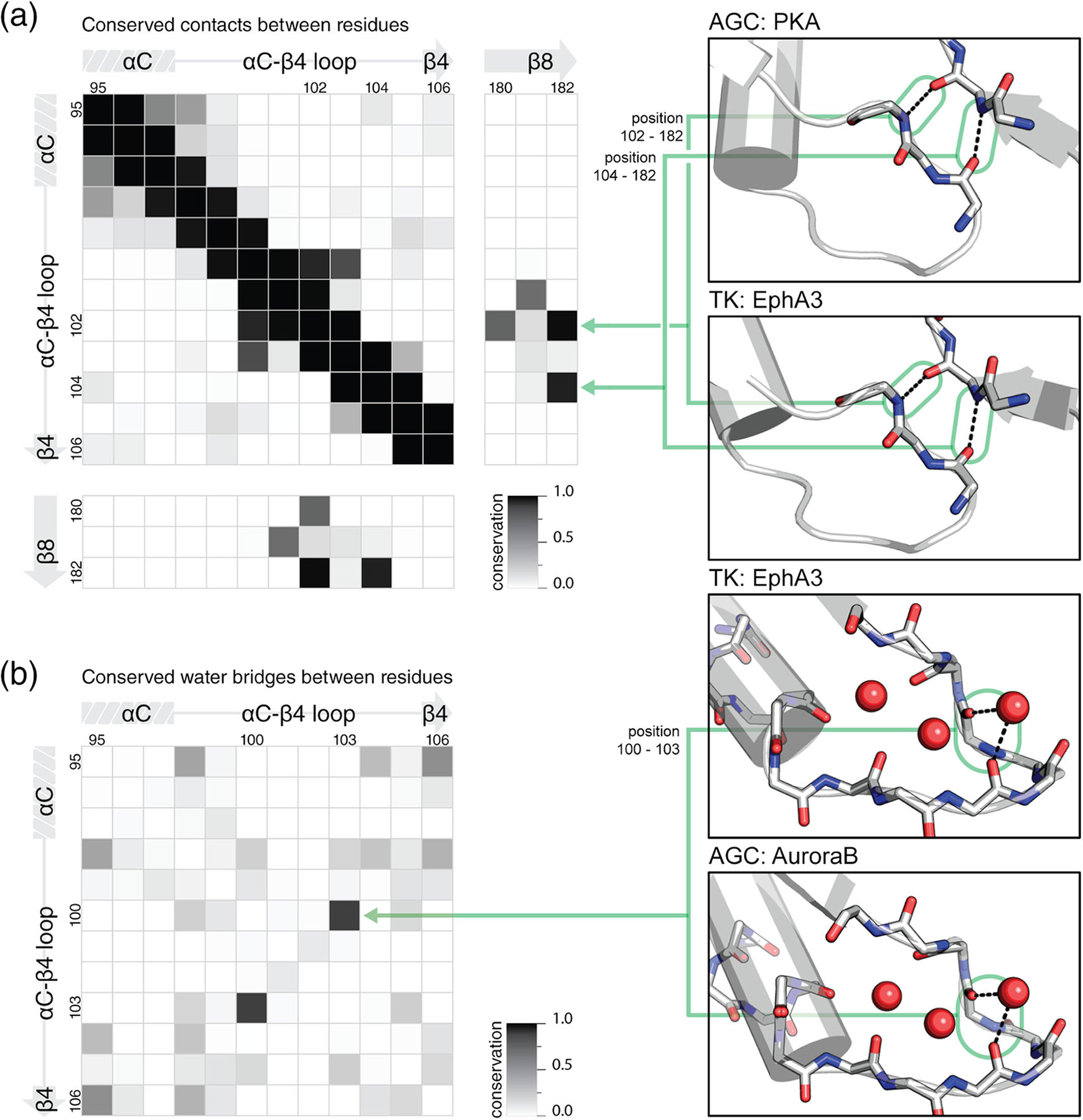
Conserved interactions within the αC-β4 loop. Conservation was defined by the fraction of structures containing the interaction. All heatmaps use PKA numbering for residue positions. (a) Conserved contacts found in kinase structures are shown in an all-versus-all comparison of residues in the αC-β4 loop and β8 strand. Two examples of the two most highly conserved long-range contacts in the αC-β4 loop and β8 strand are shown in PKA (PDB ID: 1atp)18 and EphA3 (PDB ID: 3dzq). (b) Conserved water bridges found in kinase structures are shown in an all-versus-all comparison of residues in the αC-β4 loop. Two examples of a highly conserved water bridge is shown in EphA3 (PDB ID: 3dzq) and AuroraB (PDB ID: 2vrx).39 PDB, Protein Data Bank
AGC and CK1 kinases display group-specific variations within the HxN motif to accommodate unique regulatory functions (Figure 2a). The AGC-specific xPF motif (Figure 2b, middle) facilitates cis-interactions with the C-terminal tail and is hypothesized to modulate ATP binding and inter-lobe movement.40,41 However, the CK1-specific xxG motif (Figure 2b, right) is not well understood. Similar to the HxN-Asn, the xPF-Phe and xxF-Gly form isolated β-bridges with the β8 strand (Figure 2b).
2.3 |. Conserved interactions involving the αC-β4 loop
The αC-β4 loop mediates many conserved structural interactions.40,41 We independently quantified these interactions using the aforementioned dataset of 426 representative kinase chains with unique Uniprot IDs. To identify conserved contacts, we performed an all-versus-all residue comparison for residue pairs within 3.2 Å (heavy atom distance) (Figure 3a, left). This cutoff was chosen to be greater than hydrogen bond distance and less than the van der Waals contact distance.42 This should account for uncertainty in electron density mapping while excluding hydrophobic packing interactions. Residue pairs within this cutoff are expected to either be covalently linked or hydrogen bonded.
Our analysis identified conserved hydrogen bonds involving the αC-β4 loop (Figure 3a, left). When considering interactions between all possible residue pairs within the αC-β4 loop, we identified a single conserved β-turn between position 100 (HxN-His) and 103 (HxN + 1) (Figure 3a, left). By extending our analysis to the entire kinase domain, we further identified conserved contacts between the αC-β4 loop and the β8 strand. On the β8 strand, 182 forms two highly conserved isolated β-bridges with 102 and 104 on the αC-β4 loop (Figure 3a, right). The isolated β-bridge with 102 (HxN-Asn) was described in the previous section (Figure 2b). These isolated β-bridge are observed in CK1 and AGC kinases, despite the absence of the HxN motif. Furthermore, many kinases with extended αC-β4 loops also maintain an isolated β-bridge with the β8 strand. A lesser conserved contact is detected between 102 and 180. This is only conserved amongst HxN containing kinases and reflects hydrogen bonds allowed by the carboxamide side chain of the HxN-Asn (Figure 2b, left).
Our analysis also identifies conserved water bridges in the αC-β4 loop (Figure 3b, left). Water bridges were defined as two residues residing within 3.2 Å (heavy atom distance) of a shared water molecule. To identify conserved water bridges, we perform an all-versus-all residue comparison for residue pairs within the αC-β4 residues. Crystal structures that lack water densities were excluded. We identified a single highly conserved water bridge connecting position 100 (HxN-His) and 103 (HxN + 1) (Figure 3b, right). This water bridge helps stabilize the conserved β-turn.43 While our analysis did not cover water–water interactions, we note a conserved network of water molecules in some high resolution structures.
The αC-β4 loop contains three conserved hydrophobic positions (HxN-2, HxN + 1, and HxN + 2) which are buried within the kinase core, forming hydrophobic packing interactions with the RS. Furthermore, the HxN + 2 residue takes part in a hydrophobic ensemble critical for RS assembly and thus catalytic activation.44 In addition, recent studies suggest that conservative substitution of these hydrophobic residues can modify the shape of the active site cleft.45
2.4 |. Extended αC-β4 loop conformations
Although the length of the αC-β4 is typically conserved across the protein kinase superfamily, we observed extended conformations in multiple kinase crystal structures. Extended αC-β4 loops are most commonly found in the CK1 and CMGC groups (Figure 4a) usually in the form of a short helical insert (Figure 4b). In many cases, extended αC-β4 loops seem to be linked to constitutive enzyme activity.21,23,49,50 Table 1 shows a list of kinases containing an extended αC-β4 loop.
FIGURE 4.
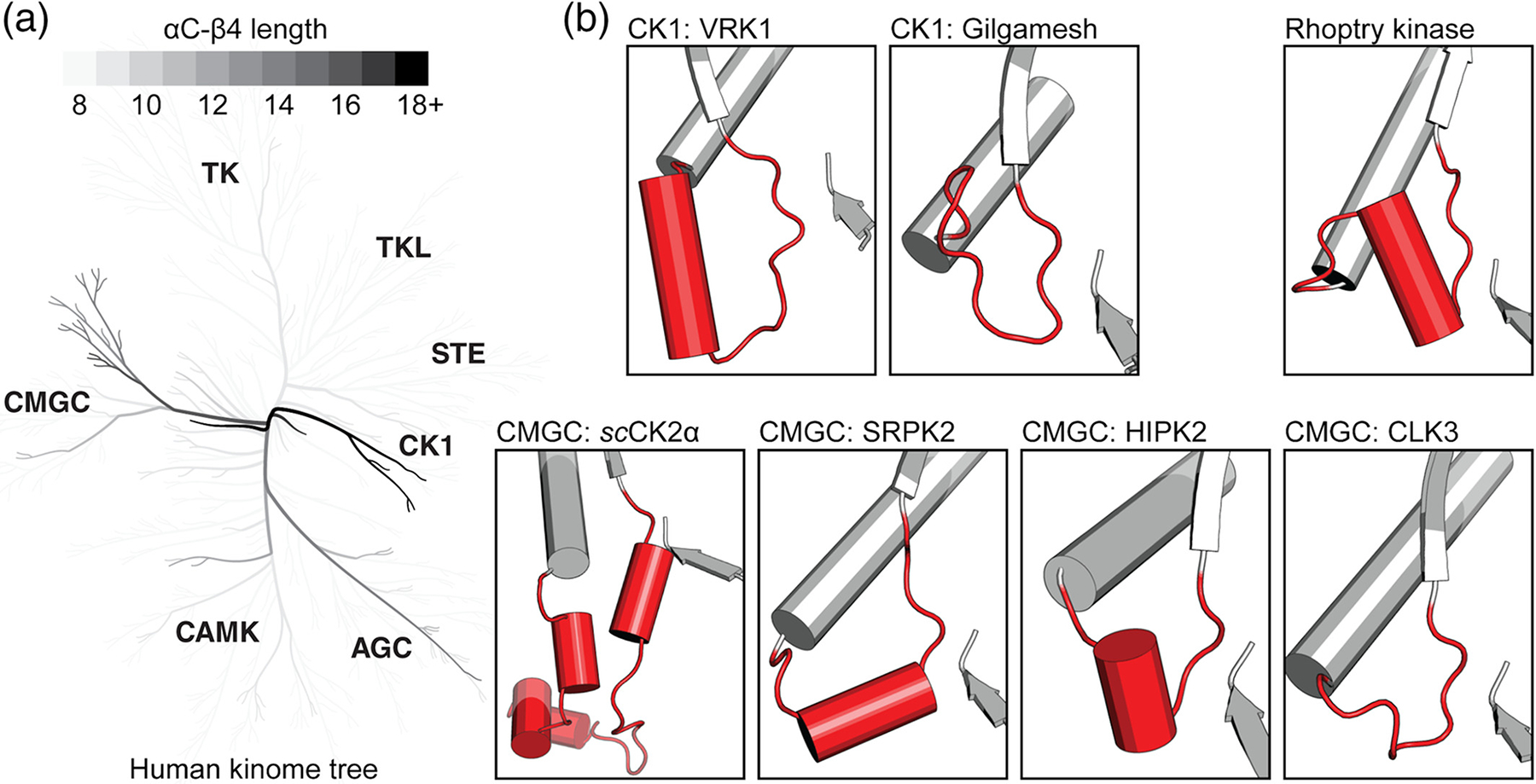
Extended conformations of the αC-β4 loop in the protein kinome. (a) A phylogenetic tree shows αC-β4 loop lengths of human protein kinases.46,47 Branches containing longer αC-β4 loops are colored darker. The 18+ color category includes lengths greater than or equal to 18. (b) Structural examples of extended αC-β4 loops are shown in red. The β8 strand is shown for reference. Protein names are provided alongside its kinase group. Locations for αC-β4 loops are provided: 93–135 in scCKA1 (PDB ID: 4jr7),21 147–160 in SRPK2 (PDB ID: 2×7g), 253–262 in HIPK2 (PDB ID: 6p5s),33 212–222 in CLK3 (PDB ID: 6fyr),28 92–112 in VRK1 (PDB ID: 6bru), 95–106 in Gilgamesh (PDB ID: 4 nt4),34 and 322–340 in Rhoptry kinase (PDB ID: 3byv).48 More information about these structures can be found in Table 1. PDB, Protein Data Bank
In the CMGC group, Saccharomyces cerevisiae CK2α (scCK2α) contains the longest resolved αC-β4 loop (47 residues).21 scCK2α is a homologue of human CK2α1, a member of one of the most phylogenetically ancient CMGC kinases families. We note that the human homologue only has a 9-residue αC-β4 loop. Experimentally, scCK2α has broad substrate specificity and is constitutively active.21 A crystal structure of scCK2α reveals that the extended αC-β4 loop is tethered to the surface of the kinase C-lobe and interacts with both the N and C-terminal tails flanking the kinase domain. Deletion of the elongated segment negatively impacts ATP binding and results in a six-fold increase of Km.51 CMGC kinases SRPK249 and HIPK233 contain a short helical insertion in their extended αC-β4 loop (Figure 4b). SRPK1 has been shown to maintain constitutive activity in vitro despite extensive mutation at the activation loop.49 This short helical segment is not found in structures of CLK52 and DYRK1A. However, DYRK1A maintains a similar resilience against inactivation.53,54
In the CK1 group, the VRK family also contains an elongated αC-β4 loop with a helical insert.23 The αC-helix is tightly linked with the αE helix of the kinase domain through aromatic packing interactions, presumably rigidifying the αC-helix into an active conformation.23,55 Consequently, VRK1 and VRK2 are constitutively active, while VRK3 is a pseudokinase lacking ATP binding and phosphoryl-transfer activity.23 A homologue of human CK1-γ, Gilgamesh kinase from Drosophila melanogaster also carries an extended αC-β4 loop.34 This insertion is not observed in the sequence of the human homologue, and its function is yet to be determined.
Rhoptry kinases also contain an elongated αC-β4 loop with a helical insert.48 The rhoptry kinases are specific to the protozoan parasite Toxoplasma gondii and have also been shown to be important virulence factors secreted by coccidian parasites.56 Comparative sequence analyses identified the helical insert to be one of the most distinguishing features of rhoptry kinases56 (Figure 3). However, the biological role of the conserved helical insert is not well understood.
3 |. DISEASE VARIANTS IN THE αC-β4 LOOP
Many mutations in protein kinases play a direct role in cancer progression. To explore cancer-related variants in the αC-β4 loop, we retrieved missense mutations deposited in the Catalogue of Somatic Mutations in Cancer (COSMIC) v9057 (Figure 5a,b). To evenly sample all protein kinases, we only considered mutations from genome-wide screens. Furthermore, we removed redundancy caused by alternative transcripts by only including mutations with a unique tumor sample and genomic location. The resulting dataset contains a diverse combination of cancer driver mutations, passenger mutations, and drug resistance mutations. The filtered list of disease mutations that map to the αC-β4 loop is provided in Table S1.
FIGURE 5.
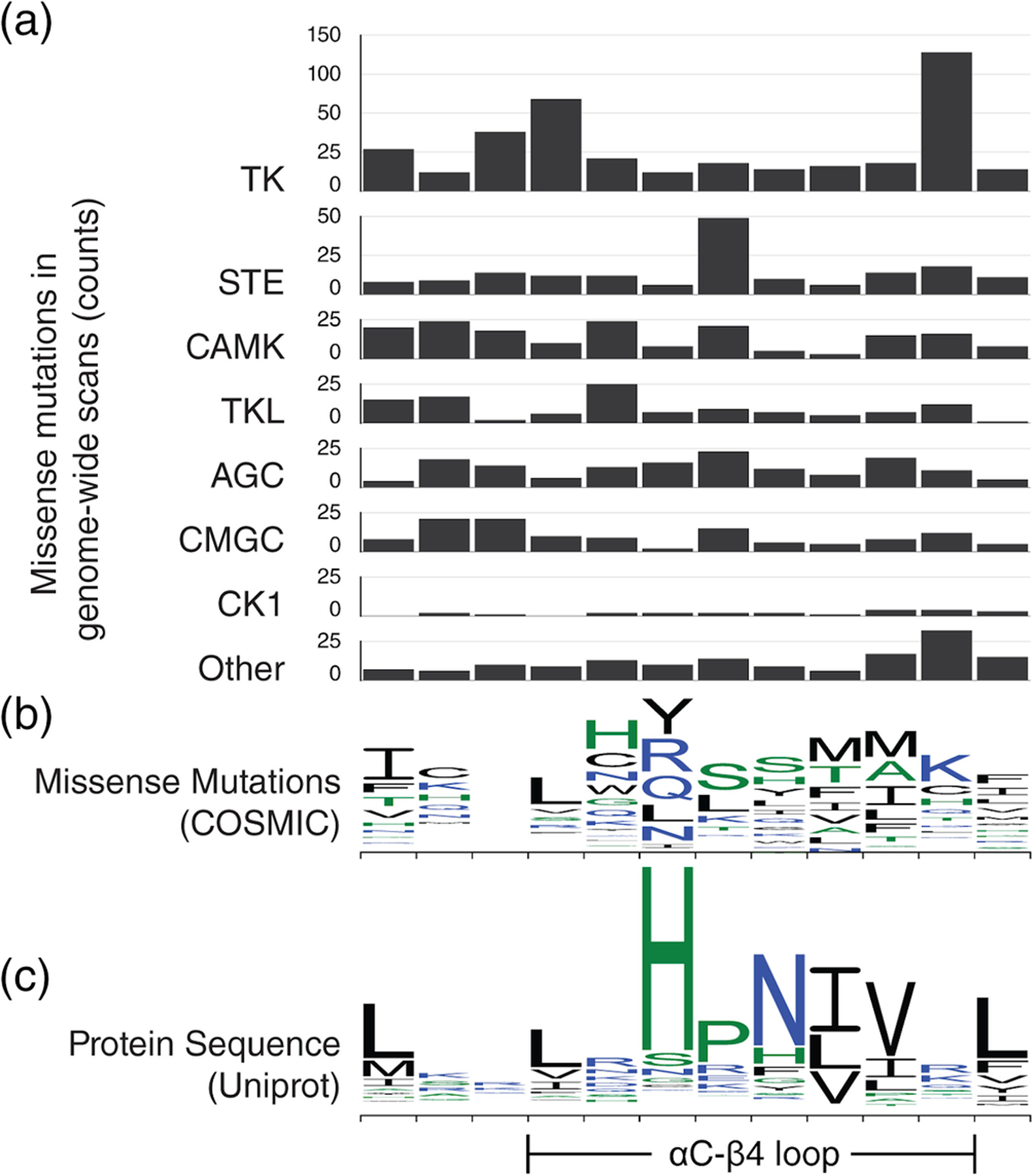
Missense mutations in the αC-β4 loop. For easy comparison, residue position on the x-axis is kept consistent throughout all graphs. (a) Bar graphs showing the number of missense mutations in the αC-β4 loop from 7 different protein kinase groups. The y-axis scale is consistent across all bar graphs to allow cross comparison. (b) The missense mutations are shown using a sequence logo plot. Similar to a sequence logo, each column shows the relative frequency of all substitutions occurring at that position. (c) A sequence logo for wildtype αC-β4 sequences from Uniprot is provided as reference
3.1 |. The molecular brake is a mutational hotspot in the αC-β4 loop
The most prevalent αC-β4 loop mutations occur at the HxN + 3 position in tyrosine kinases (Figure 5a). The HxN + 3 position has been known to take part in the “molecular brake,” a regulatory mechanism conserved in receptor tyrosine kinases (RTKs).58 The molecular brake is a hydrogen bonding network mediated by three polar residues located at the kinase hinge region, including HxN + 3. This regulatory mechanism was first identified in fibroblast growth factor receptor 2 (FGFR2) and extended to include several other RTKs through sequence comparison. In FGFR2, the molecular brake triad (N549, E565, and K641) locks the kinase in an inactive conformation. Mutations at the HxN + 3, FGFR2N549H/T, disengage the brake and activate the kinase.58
Within the COSMIC genome-wide screens dataset, the majority of HxN + 3 missense mutations substitute the RTK-conserved arginine (Figure 2a) for lysine (Figure 5b). For example, cancer-related HxN + 3 mutations have been found in three FGFR family members, including FGFR1N546K, FGFR2N549H, and FGFR3N540K/S. These mutations have all been experimentally determined to be gain-of-function.58–62 In other RTKs, PDGFRAN659K/S relieves the molecular brake, triggers constitutive STAT5 phosphorylation, and results in growth factor-independent cell proliferation.63,64 Similarly, FLT3N676K (HxN + 3 position) increases autophosphorylation and downstream AKT/MAPK phosphorylation.65,66 EGFRR776H also increases autophosphorylation and preferentially adopts the acceptor position in the EGFR asymmetric dimer.67 These examples suggest that HxN + 3 position mutations are a common mechanism for tyrosine kinase activation in cancer cells.
3.2 |. Molecular brake mutations alter drug response
Comparative sequence studies have hypothesized that the αC-β4 loop is coupled with protein kinase activation by regulating inter-lobe movement and αC dynamics.50,67 Supporting this notion, biophysical and biochemical studies suggest that the αC-β4 loop maintains auto-inhibitory interactions to prevent inadvertent kinase activation.50,58,67 For example, nuclear magnetic resonance and hydrogen–deuterium exchange mass spectrometry (MS) experiments on FGFR1 suggest that the molecular brake mechanism is coupled to activation loop conformation and active–inactive transition.7,40
By taking advantage of this coupling, it is possible that molecular brake mutations in the αC-β4 loop (HxN + 3) may confer drug resistance by altering the conformational equilibrium of a kinase, as opposed to directly altering the active site cleft.68 The molecular brake stabilizes the auto-inhibited conformation of the kinase. HxN + 3 mutations typically disrupt this inhibitory interaction and push the equilibrium toward the active conformation. Mutations that favor the active conformation (activating mutations) are generally resistant against Type II inhibitors, which target the inactive conformation.69 At the HxN + 3 position, examples of activating mutations that resist Type II inhibitors include KitN655K against imatinib and sunitinib70,71 and FLT3-ITDN676K (FLT3N676K with internal tandem duplication) resistance against quizartinib (AC220).65,66,72,73 Conversely, activating mutations can also result in sensitivity toward Type I inhibitors, which target the active conformation. For instance, FLT3N676K is sensitive to Type I inhibitor crenolanib.66,74
3.3 |. Gatekeeper-proximal mutations in the αC-β4 loop associated with drug resistance
In the αC-β4 loop, the HxN + 1 and HxN + 2 positions are spatially proximal to the gatekeeper position: a well-studied hotspot for secondary drug resistance mutations.75,76 Mutations at HxN + 1 and HxN + 2 positions have been associated with drug resistance in several tyrosine kinases. Both HxN + 1 and HxN + 2 take part in hydrophobic packing interactions that help form the kinase active site cleft in the N-lobe. Drawing parallels to resistance mutations at the gatekeeper position, mutations in HxN + 1 and HxN + 2 may alter the shape and packing of the active site cleft, which could sterically block or disfavor drug binding.69
AblL298V (HxN + 1) and AblV299L (HxN + 2) have been shown to confer secondary drug resistance in leukemia patients.15,17,77 Computational docking and molecular dynamics studies have predicted that these secondary mutations raise the free energy barrier of drug binding.45 In another RTK, c-KitV654A (HxN + 2) has been documented in imatinib-resistant gastrointestinal stromal tumors.78 Although c-KitV654A does not result in constitutive kinase activity by itself, it can occur in conjunction with cooccurring mutations such as c-KitV560G, resulting in elevated kinase activity and factor-independent growth.78
To the best of our knowledge, experimental characterization of HxN + 1 and HxN + 2 mutants are currently limited to the tyrosine kinases. However, HxN + 2 seems to be a mutational hotspot across all seven eukaryotic protein kinase groups (Figure 5a). The position is highly conserved as a valine in all major protein kinase groups except CK1 (Figure 2a). Although examples are currently limited to the tyrosine kinases, HxN + 1 and HxN + 2 mutations may be capable of conferring drug resistance in all protein kinases by modifying the shape/biochemical environment of the active site cleft.
3.4 |. Insertion mutations in the αC-β4 loop
Drug resistance mutations have been reported for EGFR and HER2 at exon 20.79–81 Exon 20 overlaps the αC-β4 loop and is a hotspot for insertion mutations. Historically, EGFR exon 20 insertion mutations are associated with resistance to first and second generation TK inhibitors. However, recent studies demonstrate differential responses to irreversible covalent inhibitors.16 These differential responses depend on the sequence and location of the insertion. Further detailed characterization of these αC-β4 insertion mutations is crucial for understanding drug resistant mechanisms and, ultimately, for the development of effective protein kinase inhibitors.16,50
3.5 |. Cis domain interactions affected by αC-β4 loop mutations
Many disease mutations target cis-interactions and interfere with normal kinase regulation. In TGF β-receptor I family, the regulatory GS domain interacts with a family-conserved arginine at the HxN-1 position.82–84 Phosphorylation of the GS domain results in a conformational change that activates the kinase.83 This conserved arginine interacts with the GS domain and shields it from phosphorylation. Disease mutations such as ACVR1R258G/S destabilize this interaction and result in constitutive kinase activity.84 Constitutive activation of ACVR1 leads to fibrodysplasia ossificans progressiva, a rare disorder in extraskeletal bone formation.85 In this example, the αC-β4 loop is capable of controlling kinase activity through interactions with regulatory domains.
Many kinases are regulated by long disordered regions flanking the kinase domain, also referred to as “tails.” EGFR is negatively regulated by its C-terminal tail, which makes electrostatic interactions with the αC-β4 loop and the hinge region of the kinase domain.86 The auto-inhibitory interaction at the αC-β4 loop is compromised by oncogenic mutations at the HxN + 3 position, EGFRR776H/C.87 Molecular dynamics and cell-based assays suggest that EGFRR776H weakens the inhibitory interaction and results in constitutive autophosphorylation.67 Equivalent mutations are observed in HER2 and HER4, suggesting that disrupting this inhibitory mechanism is a common strategy for cancer cells to activate members of the EGFR family. In addition to the EGFR family, we note more examples of C-terminal tail interactions mediated by the αC-β4 loop in MAPK family88,89 and IGF-1R.90
Mutations that alter cis-interactions can also confer drug resistance. MEK1P124L/S was discovered to be resistant against selumetinib (AZD6244) in a random mutagenesis study.91 MEK1P124 is the HxN wildcard residue and packs against the MEK1 A-helix, a negative regulatory element located N-terminal to the kinase domain. Being a highly specific inhibitor, it is possible that selumetinib targets the inactive conformation which is disfavored in the absence of the A-helix interaction.
Another example can be found in ALK, an RTK. ALK is inhibited by its N-terminal juxtamembrane (JM) segment which makes hydrophobic contacts with the αC-helix and αC-β4 loop.92 Phosphorylation of JM tyrosines results in ALK activation by disengaging the JM segment. ALKF1174L is a recurring oncogenic mutation that disrupts pi-stacking interactions between the αC-β4 loop and the JM domain. Furthermore, ALKF1174L results in constitutive kinase activity and confers resistance to crizotinib.93
3.6 |. Protein–protein interactions affected by αC-β4 loop mutations
Mutations in the αC-β4 loop can also interfere with protein–protein interaction interfaces. In ERK2, the αC-β4 loop takes part in the D-recruitment site which helps the kinase bind to effector proteins.94,95 A patient derived mutation on the HxN wildcard residue, ERK2E81K, activates the kinase.94 Furthermore, ERK2E81K may disrupt negative regulation by DUSP6 phosphatase.94
4 |. POST-TRANSLATIONAL MODIFICATIONS IN THE αC-β4 LOOP
The catalytic function of many protein kinases is regulated by post-translational modifications (PTMs). For example, phosphorylation of the activation loop segment is required for the activation of many kinases.8 The αC-β4 loop is also targeted by a variety of PTMs. To explore the landscape of PTMs within the αC-β4 loop, we retrieved a variety of mammalian PTMs from the PhosphoSitePlus database96 (Figure 6a). The majority of PTM sites were identified by MS and filtered by a statistical cutoff for assignment (p < .05). Within the database, available PTM assignments included ubiquitination, phosphorylation, acetylation, sumoylation, methylation, O-GlcNAc, and O-GalNAc. O-GalNAc was the only PTM without assignments to the αC-β4 loop. A table of PTMs that map to the αC-β4 loop is provided in Table S2.
FIGURE 6.
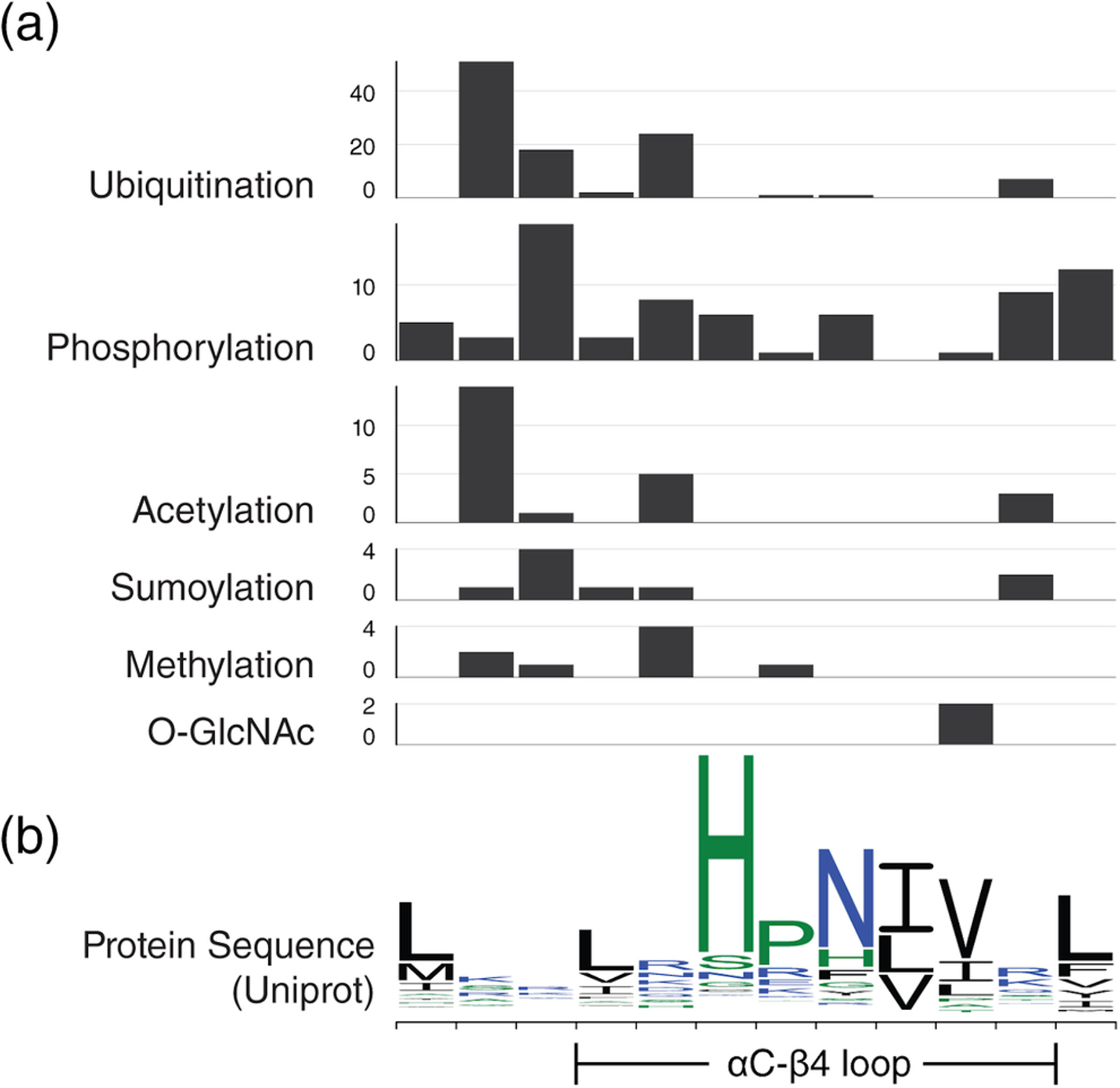
Post-translational modifications in the αC-β4 loop. For easy comparison, residue position on the x-axis is kept consistent throughout all graphs. (a) Bar graphs showing the number of PTMs found at each αC-β4 position separated by PTM. Please note that the y-axis scale is not consistent across bar graphs. (b) A sequence logo for wildtype αC-β4 sequences from Uniprot is provided as reference
Phosphorylation, one of the most abundant PTMs, plays a major role in modulating conformation and protein–protein interfaces. In the MST family, the HxN motif is replaced by a phosphorylatable SPx motif at the equivalent position. JNK phosphorylates MST1S82 which is the SPx-Ser position. Phosphorylation of MST1S82 enhances MST1 activity and promotes apoptosis.97 The SPx wildcard residue is sometimes phosphorylatable. For instance, c-Abl phosphorylates MST2Y81 at the SPx wildcard position. Phosphorylation of MST2Y81 prevents MST2 from interacting with Raf-1 and promotes MST2 homodimerization.98
O-linked β-N-acetylglucosamine (O-GlcNAc) is another important PTM that varies in response to many factors such as extracellular stress, cell cycle, and development.99 Western blot and MS assignments have revealed many O-GlcNAcylation sites on PKC family in rats.100 Within the PKC family, O-GlcNAcylation sites were assigned to the αC-β4 loop of rat PKCA and PKCB. Some of these glycosylation sites intersect with phosphorylation sites suggesting that these modifications may modulate each other.101 Although not in the αC-β4 loop, examples of kinase regulations via cross-talk between glycosylation and phosphorylation have been described in CaMKIV.101
S-nitrosylation is also an important cysteine PTM that provides a mechanism for redox-based regulation.102 In human InsR kinase, a modifiable cysteine replaces HxNHis. A study on cultured skeletal muscle cells demonstrates that S-nitrosylation of InsRC1083 results in the inhibition of kinase activity.103
5 |. THE αC-β4 LOOP MEDIATES PROTEIN–PROTEIN INTERACTIONS
5.1 |. The αC-β4 loop is involved in kinase dimer interfaces
Many protein kinases are regulated by the formation of dimeric complexes. RAF kinases are activated by a side-to-side dimer interface involving the αC-β4 loop.104,105 A mutation at the HxN-1 position, BRAFR509H, impairs dimer formation and results in the kinase-dead phenotype.106 Pseudokinase KSR is also capable of dimerizing with BRAF. Consequently, the equivalent mutation KSRR665H also results in the loss of BRAF activity.107
The αC-β4 loop can also take part in a symmetric back-to-back dimer interface. This conformation exposes the active site cleft and is usually associated with catalytically active kinase. One of the first examples was discovered in PKR, where the active back-to-back dimer was solved in two different crystallographic environments.108 The IRE1 back-to-back dimer is also associated with an active kinase and high RNase activity.109 Similarly, the Nek7-Nek9 back-to-back heterodimer is associated with rapid autophosphorylation of Nek7. Autophosphorylation assays showed that Nek7N90R (HxN-1) resulted in reduced kinase activity.110 The proposed mechanism for PknB activation suggests that a back-to-back active dimer is induced by ligand binding to the PknB extracellular sensor domain.111,112 PknE has also been crystallized in the back-to-back conformation.113
5.2 |. The αC-β4 loop plays an important role in Hsp90-mediated kinase folding
One of the most important roles of the αC-β4 loop is the recognition of molecular chaperone Hsp90 and co-chaperone cdc37. Hsp90 promotes proper folding in many proteins including 60% of the human kinome.114 This discovery started from an observation that human EGFR neither requires nor associates with Hsp90, a stark contrast to its paralog, HER2.115,116 However, mutation of the HER2 αC-β4 loop to the EGFR sequence abolished Hsp90 association in HER2.116,117 Furthermore, FerY616, an αC-β4 loop residue, is essential for Hsp90 association and kinase activity.118 Cryo-EM experiments have shown that co-chaperone cdc37 mimics the conformation of the αC-β4 loop and uses the HxN motif to form hinge interactions with the client kinase.119 Although the full mechanism for Hsp90-recognition remains a mystery, results have shown that the HxN motif plays a role in cdc37-mediated folding for more than half of the human kinome.114
6 |. CONCLUDING REMARKS AND PREDICTIONS ON UNDERSTUDIED DARK KINASES
In this article, we have highlighted the αC-β4 loop as a central hub for many essential regulatory mechanisms for protein kinase function. To investigate the diverse functions of this region, we have compiled a list of disease-related mutations and PTMs that localize to the αC-β4 loop. We provide many examples of disease-related mutations linked to aberrant signaling and drug resistance. Experimental characterization shows that these mutations can alter both conserved and family-specific regulatory mechanisms. We believe that the αC-β4 loop is a conserved, yet understudied hotspot for regulatory interactions within the eukaryotic protein kinome.
Our kinome-wide analysis provides a useful resource for investigating understudied kinases. Recently, the NIH common fund program initiated a large-scale effort to identify and characterize new druggable proteins within the human genome. To guide research efforts, this initiative has maintained a list of understudied kinases (last updated on June 2019), collectively referred to as the dark kinome.
Interestingly, we find that nearly all kinases with extended αC-β4 loop segments have been classified as dark kinases (Figure 4a). In the CMGC group, most members of the DYRK, HIPK, CLK, and SRPK families are classified as dark kinases. As opposed to the typical 8-residue loop, these families form a large clade whose members have a conserved ~14-residue αC-β4 loop. Members such as HIPK2, DYRK1A, and SRPK2 are some of the few characterized members of this clade. Using existing knowledge, we noticed clade-specific trends such as constitutive activity and helical inserts. These observations can guide hypothesis-driven research in the clade’s understudied members. This approach can also be applied to the VRK family of the CK1 group. This family of dark kinases has a single well-characterized member, VRK1, and contains a conserved 21-residue αC-β4 loop. From a drug discovery perspective, these extended loop conformations may also provide a targetable interface for high-specificity protein kinase inhibitors.
The αC-β4 loop remains a regulatory hotspot within the complex web of interactions that modulate protein kinase activity. As such, mutations within this region can trigger a variety of human diseases. An in-depth understanding of the structure, function, and evolution of the αC-β4 loop will provide new insights on kinase regulation and enhance the discovery of novel protein kinase inhibitors.
Supplementary Material
ACKNOWLEDGMENTS
Funding for N. K. from the National Institutes of Health (5R01GM114409 and 1U01CA239106) is acknowledged. Z. R. is the recipient of 2017 Innovative and Interdisciplinary Research Grant for Doctoral Students (IIRG). We thank the members of the N. K. Lab for providing helpful comments and suggestions.
Abbreviations:
- Abl
Abelson tyrosine-protein kinase
- ACVR1
activin receptor type 1
- AGC
protein kinase A, G, and C group
- ALK
anaplastic lymphoma kinase
- CAMK
Ca/Calmodulin kinases group
- CK1
casein kinase I group
- CK2α
casein kinase 2 alpha
- CLK3
CDC-like kinase 3
- CMGC
CDK, MAPK, GSK, and close relatives group
- CSNK1D
casein kinase 1 delta
- DSSP
define secondary structure of proteins
- DUSP6
dual specificity protein phosphatase 6
- DYRK1A
dual specificity tyrosine-phosphorylation-regulated kinase 1A
- EGFR
epidermal growth factor receptor
- EphA3
ephrin receptor A3
- ERK2
extracellular signal-regulated kinase 2
- FGFR2
fibroblast growth factor receptor 2
- FLT3
Fms-like tyrosine kinase 3
- HIPK2
homeodomain-interacting protein kinase 2
- Hsp90
heat shock protein 90
- IGF-1R
insulin-like growth factor 1 receptor
- JNK
c-Jun N-terminal kinase
- JM
juxtamembrane
- MEK1
MAPK/ERK kinase 1
- MST2
mammalian STE20-like protein kinase 2
- KSR
kinase suppressor of Ras
- PDGRFA
platelet-derived growth factor receptor
- PKA
protein kinase A
- PKR
protein kinase R
- PTM
post translational modification
- RS
regulatory spine
- RTK
receptor tyrosine kinases
- SRPK2
serine/arginine-rich splicing factor kinase 2
- STAT5
signal transducer and activator of transcription 5
- TGF β-receptor I
transforming growth factor β-receptor I
- TK
tyrosine kinase group
- TKL
tyrosine kinase-like group
- VRK1
vaccinia related kinase 1
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015; 125:1780–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fountas A, Diamantopoulos L-N, Tsatsoulis A. Tyrosine kinase inhibitors and diabetes: A novel treatment paradigm? Trends Endocrinol. Metabolism. 2015;26:643–656. [DOI] [PubMed] [Google Scholar]
- 3.Li J-Q, Tan L, Yu J-T. The role of the LRRK2 gene in parkinsonism. Mol Neurodegener. 2014;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar R, Singh VP, Baker KM. Kinase inhibitors for cardiovascular disease. J Mol Cell Cardiol. 2007;42:1–11. [DOI] [PubMed] [Google Scholar]
- 5.Ubersax JA, Ferrell JE. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. [DOI] [PubMed] [Google Scholar]
- 6.Scheeff ED, Bourne PE. Structural evolution of the protein kinase-like superfamily. PLoS Comput Biol. 2005;1:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kannan N, Neuwald AF. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J Mol Biol. 2005;351:956–972. [DOI] [PubMed] [Google Scholar]
- 8.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. [DOI] [PubMed] [Google Scholar]
- 9.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: Structural basis for regulation. Cell. 1996;85:149–158. [DOI] [PubMed] [Google Scholar]
- 10.Palmieri L, Rastelli G. αC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov Today. 2013;18:407–414. [DOI] [PubMed] [Google Scholar]
- 11.Taylor SS, Shaw AS, Kannan N, Kornev AP. Integration of signaling in the kinome: Architecture and regulation of the αC helix. Biochim Biophys Acta. 2015;1854:1567–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. [DOI] [PubMed] [Google Scholar]
- 13.Taylor SS, Kornev AP. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oruganty K, Talathi NS, Wood ZA, Kannan N. Identification of a hidden strain switch provides clues to an ancient structural mechanism in protein kinases. Proc Natl Acad Sci U S A. 2013;110:924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones D, Chen SS, Jabbour E, Rios MB, Kantarjian H, Cortes J. Uncommon BCR-ABL kinase domain mutations in kinase inhibitor-resistant chronic myelogenous leukemia and Ph+ acute lymphoblastic leukemia show high rates of regression, suggesting weak selective effects. Blood. 2010;115:5428–5429. [DOI] [PubMed] [Google Scholar]
- 16.Kosaka T, Tanizaki J, Paranal RM, et al. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 2017;77:2712–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolini FE, Corm S, Lê QH, et al. Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: A retrospective analysis from the French intergroup of CML (Fi (phi)-LMC GROUP). Leukemia. 2006;20:1061–1066. [DOI] [PubMed] [Google Scholar]
- 18.Zheng J, Trafny EA, Knighton DR, et al. 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D Biol Crystallogr. 1993;49:362–365. [DOI] [PubMed] [Google Scholar]
- 19.Neuwald AF. Rapid detection, classification and accurate alignment of up to a million or more related protein sequences. Bioinformatics. 2009;25:1869–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Wang H, Teng M, Li X. The multiple nucleotide-divalent cation binding modes of Saccharomyces cerevisiae CK2α indicate a possible co-substrate hydrolysis product (ADP/GDP) release pathway. Acta Crystallogr D Biol Crystallogr. 2014;70:501–513. [DOI] [PubMed] [Google Scholar]
- 22.Kumar A, Tamjar J, Waddell AD, et al. Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife. 2017;6:e29985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheeff ED, Eswaran J, Bunkoczi G, Knapp S, Manning G. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure. 2009;17:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nolen B, Ngo J, Chakrabarti S, Vu D, Adams JA, Ghosh G. Nucleotide-induced conformational changes in the Saccharomyces cerevisiae SR protein kinase, Sky 1p, revealed by X-ray crystallography. Biochemistry. 2003;42:9575–9585. [DOI] [PubMed] [Google Scholar]
- 25.Chaikuad A, Tacconi EMC, Zimmer J, et al. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol. 2014;10:853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batson J, Toop HD, Redondo C, et al. Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem Biol. 2017;12:825–832. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Q, Liu H, Liu X, et al. Discovery of the first macrolide antibiotic binding protein in mycobacterium tuberculosis: A new antibiotic resistance drug target. Protein Cell. 2018;9: 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kallen J, Bergsdorf C, Arnaud B, et al. X-ray structures and feasibility assessment of CLK2 inhibitors for phelan-McDermid syndrome. Chem Med Chem. 2018;13:1997–2007. [DOI] [PubMed] [Google Scholar]
- 29.Walter A, Chaikuad A, Helmer R, et al. Molecular structures of cdc 2-like kinases in complex with a new inhibitor chemotype. PLoS One. 2018;13:e0196761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim K, Cha JS, Cho Y-S, et al. Crystal structure of human dual-specificity tyrosine-regulated kinase 3 reveals new structural features and insights into its auto-phosphorylation. J Mol Biol. 2018;430:1521–1530. [DOI] [PubMed] [Google Scholar]
- 31.Chaikuad A, Diharce J, Schröder M, et al. An unusual binding model of the methyl 9-anilinothiazolo [5, 4-f] quinazoline-2-carbimidates (EHT 1610 and EHT 5372) confers high selectivity for dual-specificity tyrosine phosphorylation-regulated kinases. J Med Chem. 2016;59:10315–10321. [DOI] [PubMed] [Google Scholar]
- 32.Gao Q, Mechin I, Kothari N, et al. Evaluation of cancer dependence and druggability of PRP4 kinase using cellular, biochemical, and structural approaches. J Biol Chem. 2013; 288:30125–30138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agnew C, Liu L, Liu S, et al. The crystal structure of the protein kinase HIPK2 reveals a unique architecture of its CMGC-insert region. J Biol Chem. 2019;294:13545–13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han N, Chen C, Shi Z, Cheng D. Structure of the kinase domain of Gilgamesh from Drosophila melanogaster. Acta Crystallogr F Struct Biol Commun. 2014;70:438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L, Li X, Wang J, et al. Two distant catalytic sites are responsible for c2c2 rnase activities. Cell. 2017;168: 121–134.e12. [DOI] [PubMed] [Google Scholar]
- 36.Gu S, Sushko O, Deery E, Warren MJ, Pickersgill RW. Crystal structure of cob K reveals strand-swapping between Rossmann-fold domains and molecular basis of the reduced precorrin product trap. Sci Rep. 2015;5:16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon A, Scott S, Taujale R, et al. Tracing the origin and evolution of pseudokinases across the tree of life. Sci Signal. 2019; 12:eaav3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bischof J, Leban J, Zaja M, et al. 2-Benzamido-N-(1H-benzo [d]imidazol-2-yl)thiazole-4-carboxamide derivatives as potent inhibitors of CK1δ/ε. Amino Acids. 2012;43:1577–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girdler F, Sessa F, Patercoli S, Villa F, Musacchio A, Taylor S. Molecular basis of drug resistance in aurora kinases. Chem Biol. 2008;15:552–562. [DOI] [PubMed] [Google Scholar]
- 40.Kannan N, Haste N, Taylor SS, Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A. 2007;104: 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannan N, Neuwald AF, Taylor SS. Analogous regulatory sites within the alpha C-beta 4 loop regions of ZAP-70 tyrosine kinase and AGC kinases. Biochim Biophys Acta. 2008; 1784:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bondi A van der Waals Volumes and Radii. J Phys Chem. 1964;68:441–451. [Google Scholar]
- 43.Thanki N, Umrania Y, Thornton JM, Goodfellow JM. Analysis of protein main-chain solvation as a function of secondary structure. J Mol Biol. 1991;221:669–691. [DOI] [PubMed] [Google Scholar]
- 44.Meharena HS, Chang P, Keshwani MM, et al. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013;11:e1001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibbons DL, Pricl S, Posocco P, et al. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc Natl Acad Sci U S A. 2014;111:3550–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metz KS, Deoudes EM, Berginski ME, et al. Coral: Clear and customizable visualization of human kinome data. Cell Syst. 2018;7:347–350.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 48.Qiu W, Wernimont A, Tang K, et al. Novel structural and regulatory features of rhoptry secretory kinases in toxoplasma gondii. EMBO J. 2009;28:969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ngo JCK, Gullingsrud J, Giang K, et al. SR protein kinase 1 is resilient to inactivation. Structure. 2007;15:123–133. [DOI] [PubMed] [Google Scholar]
- 50.Ruan Z, Kannan N. Altered conformational landscape and dimerization dependency underpins the activation of EGFR by αC-β4 loop insertion mutations. Proc Natl Acad Sci USA. 2018;115:E8162–E 8171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sajnaga E, Kubiński K, Szyszka R. Catalytic activity of mutants of yeast protein kinase CK2alpha. Acta Biochim Pol. 2008;55:767–776. [PubMed] [Google Scholar]
- 52.Bullock AN, Das S, Debreczeni JE, et al. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure. 2009;17:352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soundararajan M, Roos AK, Savitsky P, et al. Structures of down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure. 2013; 21:986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adayev T, Chen-Hwang M-C, Murakami N, Lee E, Bolton DC, Hwang Y-W. Dual-specificity tyrosine phosphorylation-regulated kinase 1A does not require tyrosine phosphorylation for activity in vitro. Biochemistry. 2007; 46:7614–7624. [DOI] [PubMed] [Google Scholar]
- 55.Couñago RM, Allerston CK, Savitsky P, et al. Structural characterization of human Vaccinia-related kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations. Sci Rep. 2017;7:7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Talevich E, Kannan N. Structural and evolutionary adaptation of rhoptry kinases and pseudokinases, a family of coccidian virulence factors. BMC Evol Biol. 2013;13:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tate JG, Bamford S, Jubb HC, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47: D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen H, Ma J, Li W, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patani H, Bunney TD, Thiyagarajan N, et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget. 2016; 7:24252–24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lew ED, Furdui CM, Anderson KS, Schlessinger J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci Signal. 2009;2:ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of fibroblast growth factor receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015;26:425–449. [DOI] [PubMed] [Google Scholar]
- 62.Rand V, Huang J, Stockwell T, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A. 2005;102:14344–14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23: 5357–5364. [DOI] [PubMed] [Google Scholar]
- 64.Elling C, Erben P, Walz C, et al. Novel imatinib-sensitive PDGFRA-activating point mutations in hypereosinophilic syndrome induce growth factor independence and leukemia-like disease. Blood. 2011;117:2935–2943. [DOI] [PubMed] [Google Scholar]
- 65.Opatz S, Polzer H, Herold T, et al. Exome sequencing identifies recurring FLT3 N676K mutations in core-binding factor leukemia. Blood. 2013;122:1761–1769. [DOI] [PubMed] [Google Scholar]
- 66.Huang K, Yang M, Pan Z, et al. Leukemogenic potency of the novel FLT3-N676K mutant. Ann Hematol. 2016;95: 783–791. [DOI] [PubMed] [Google Scholar]
- 67.Ruan Z, Kannan N. Mechanistic insights into R776H mediated activation of epidermal growth factor receptor kinase. Biochemistry. 2015;54:4216–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003;4:75–85. [DOI] [PubMed] [Google Scholar]
- 69.Roskoski R Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48. [DOI] [PubMed] [Google Scholar]
- 70.Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106:1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20: 5745–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zorn JA, Wang Q, Fujimura E, Barros T, Kuriyan J. Crystal structure of the FLT3 kinase domain bound to the inhibitor Quizartinib (AC220). PLoS One. 2015;10:e0121177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith CC, Zhang C, Lin KC, et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “gatekeeper” F691L mutation with PLX 3397. Cancer Discov. 2015;5:668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galanis A, Rajkhowa T, Muralidhara C, Ramachandran A, Levis MJ. Crenolanib is a highly potent, selective, FLT3 TKI with activity against D835 mutations. Blood. 2012;120: 1341–1341. [Google Scholar]
- 75.Yun C-H, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105: 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threo-nine. Nat Struct Mol Biol. 2008;15:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jabbour E, Morris V, Kantarjian H, Yin CC, Burton E, Cortes J. Characteristics and outcomes of patients with V299L BCR-ABL kinase domain mutation after therapy with tyrosine kinase inhibitors. Blood. 2012;120:3382–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roberts KG, Odell AF, Byrnes EM, et al. Resistance to c-KIT kinase inhibitors conferred by V654A mutation. Mol Cancer Ther. 2007;6:1159–1166. [DOI] [PubMed] [Google Scholar]
- 79.Yasuda H, Park E, Yun C-H, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5:216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012;13:e23–e31. [DOI] [PubMed] [Google Scholar]
- 81.Hirotsu Y, Nakagomi H, Amemiya K, et al. Intrinsic HER2 V777L mutation mediates resistance to trastuzumab in a breast cancer patient. Med Oncol. 2017;34:3. [DOI] [PubMed] [Google Scholar]
- 82.Huse M, Chen YG, Massagué J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–436. [DOI] [PubMed] [Google Scholar]
- 83.Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massagué J. The TGF beta receptor activation process: An inhibitor- to substrate-binding switch. Mol Cell. 2001;8:671–682. [DOI] [PubMed] [Google Scholar]
- 84.Chaikuad A, Alfano I, Kerr G, et al. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem. 2012;287: 36990–36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaplan FS, Kobori JA, Orellana C, et al. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (ACVR1 c.772G>A; R258G): A report of two patients. Am J Med Genet A. 2015;167A:2265–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kovacs E, Das R, Wang Q, et al. Analysis of the role of the C-terminal tail in the regulation of the epidermal growth factor receptor. Mol Cell Biol. 2015;35:3083–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McSkimming DI, Dastgheib S, Talevich E, et al. Pro kin O: A unified resource for mining the cancer kinome. Hum Mutat. 2015;36:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Salvador JM, Mittelstadt PR, Guszczynski T, et al. Alternative p 38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol. 2005;6:390–395. [DOI] [PubMed] [Google Scholar]
- 89.Diskin R, Lebendiker M, Engelberg D, Livnah O. Structures of p38alpha active mutants reveal conformational changes in L16 loop that induce autophosphorylation and activation. J Mol Biol. 2007;365:66–76. [DOI] [PubMed] [Google Scholar]
- 90.Kelly GM, Buckley DA, Kiely PA, Adams DR, O’Connor R. Serine phosphorylation of the insulin-like growth factor I (IGF-1) receptor C-terminal tail restrains kinase activity and cell growth. J Biol Chem. 2012;287: 28180–28194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang Q, Johnson TW, Bailey S, et al. Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib. J Med Chem. 2014; 57:1170–1187. [DOI] [PubMed] [Google Scholar]
- 93.Berry T, Luther W, Bhatnagar N, et al. The ALK (F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brenan L, Andreev A, Cohen O, et al. Phenotypic characterization of a comprehensive set of MAPK1/ERK2 missense mutants. Cell Rep. 2016;17:1171–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alexa A, Gógl G, Glatz G, et al. Structural assembly of the signaling competent ERK2-RSK1 heterodimeric protein kinase complex. Proc Natl Acad Sci U S A. 2015;112:2711–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. Phospho site plus, 2014: Mutations. PTMs and recalibrations Nucleic Acids Res. 2015;43: D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bi W, Xiao L, Jia Y, et al. C-Jun N-terminal kinase enhances MST1-mediated pro-apoptotic signaling through phosphorylation at serine 82. J Biol Chem. 2010;285:6259–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu W, Wu J, Xiao L, et al. Regulation of neuronal cell death by c-Abl-Hippo/MST2 signaling pathway. PLoS One. 2012;7: e36562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wells L, Whelan SA, Hart GW. O-Glc NAc: A regulatory post-translational modification. Biochem Biophys Res Commun. 2003;302:435–441. [DOI] [PubMed] [Google Scholar]
- 100.Robles-Flores M, Meléndez L, García W, et al. Posttranslational modifications on protein kinase c isozymes. Effects of epinephrine and phorbol esters. Biochim Biophys Acta. 2008; 1783:695–712. [DOI] [PubMed] [Google Scholar]
- 101.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-Glc NAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-nitrosylation: Purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. [DOI] [PubMed] [Google Scholar]
- 103.Schmid E, Hotz-Wagenblatt A, Dröge W. Inhibition of the insulin receptor kinase phosphorylation by nitric oxide: Functional and structural aspects. Antioxid Redox Signal. 1999;1: 45–53. [DOI] [PubMed] [Google Scholar]
- 104.Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. [DOI] [PubMed] [Google Scholar]
- 105.Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. [DOI] [PubMed] [Google Scholar]
- 106.Hu J, Stites EC, Yu H, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154: 1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lavoie H, Sahmi M, Maisonneuve P, et al. MEK drives BRAF activation through allosteric control of KSR proteins. Nature. 2018;554:549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. [DOI] [PubMed] [Google Scholar]
- 109.Joshi A, Newbatt Y, McAndrew PC, et al. Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget. 2015;6:13019–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Haq T, Richards MW, Burgess SG, et al. Mechanistic basis of Nek 7 activation through Nek 9 binding and induced dimerization. Nat Commun. 2015;6:8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Young TA, Delagoutte B, Endrizzi JA, Falick AM, Alber T. Structure of mycobacterium tuberculosis Pkn B supports a universal activation mechanism for Ser/Thr protein kinases. Nat Struct Biol. 2003;10:168–174. [DOI] [PubMed] [Google Scholar]
- 112.Lombana TN, Echols N, Good MC, et al. Allosteric activation mechanism of the mycobacterium tuberculosis receptor Ser/Thr protein kinase, Pkn B. Structure. 2010;18:1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gay LM, Ng H-L, Alber T. A conserved dimer and global conformational changes in the structure of apo-Pkn E Ser/Thr protein kinase from mycobacterium tuberculosis. J Mol Biol. 2006;360:409–420. [DOI] [PubMed] [Google Scholar]
- 114.Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu W, Soga S, Beebe K, et al. Sensitivity of epidermal growth factor receptor and Erb B2 exon 20 insertion mutants to Hsp 90 inhibition. Br J Cancer. 2007;97:741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. Surface charge and hydrophobicity determine Erb B2 binding to the Hsp 90 chaperone complex. Nat Struct Mol Biol. 2005; 12:120–126. [DOI] [PubMed] [Google Scholar]
- 117.Citri A, Harari D, Shohat G, et al. Hsp 90 recognizes a common surface on client kinases. J Biol Chem. 2006;281:14361–14369. [DOI] [PubMed] [Google Scholar]
- 118.Hikri E, Shpungin S, Nir U. Hsp 90 and a tyrosine embedded in the Hsp 90 recognition loop are required for the Fer tyrosine kinase activity. Cell Signal. 2009;21:588–596. [DOI] [PubMed] [Google Scholar]
- 119.Verba KA, Agard DA. How hsp 90 and cdc 37 lubricate kinase molecular switches. Trends Biochem Sci. 2017;42:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.