

Abstract
Purpose:
The study goal was to determine safety, antitumor activity, and pharmacodynamic profile of mogamulizumab, an anti-CCR4 monoclonal antibody (mAb) targeting effector regulatory T cells (eTregs), in combination with mAb checkpoint inhibitors durvalumab or tremelimumab.
Patients and Methods:
This was a multicenter, phase I, dose-escalation study, followed by disease-specific cohort expansion (NCT02301130). Mogamulizumab dose escalation proceeded with concurrent dose escalation of durvalumab or tremelimumab in patients with advanced solid tumors. Cohort expansion occurred with mogamulizumab 1 mg/kg plus durvalumab 10 mg/kg or tremelimumab 10 mg/kg in patients with advanced pancreatic cancer.
Results:
Forty patients were enrolled during dose escalation, followed by 24 patients during dose expansion. No dose-limiting toxicities occurred during dose escalation. No new or unexpected toxicities were seen. Tolerability, the primary endpoint, was acceptable utilizing mogamulizumab 1 mg/kg plus durvalumab or tremelimumab 10 mg/kg in the combined dose-escalation and dose-expansion cohorts (each n = 19). At these doses, the objective response rate was 5.3% (95% CI: 0.1%, 26.0%) [1 partial response] with each combination treatment. At all doses, mogamulizumab treatment led to almost complete depletion of peripheral eTregs as well as reduction of intratumoral Tregs in the majority of patients. There was no clear correlation of clinical response with peripheral or intratumoral reduction in CCR4+ eTregs or with baseline degree of CCR4+ expression.
Conclusions:
Mogamulizumab in combination with durvalumab or tremelimumab did not result in potent antitumor efficacy in patients with advanced solid tumors. Tolerability of mogamulizumab 1 mg/kg combined with durvalumab or tremelimumab 10 mg/kg was acceptable.
Introduction
Monoclonal antibodies (mAbs) targeting cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death 1/programmed cell death ligand 1 (PD-1/PD-L1) immune checkpoints have demonstrated clinical benefit in several cancer types, although responses have generally been low, limited to a minority of patients, and are frequently not durable. The combination of checkpoint inhibitors with immunomodulatory mAbs that act via different mechanisms may offer an approach to improve therapeutic outcome (1, 2). One potential combination partner for checkpoint inhibitors might be afforded by an agent that is able to deplete regulatory T cells (Tregs), given that Tregs play a pivotal role in maintaining immunological tolerance that can inhibit antitumor immune responses and may mediate resistance to immunomodulatory therapy targeting CTLA-4 or PD-1/PD-L1 (3–5).
C-C chemokine receptor 4 (CCR4) is a lymphocyte receptor recognizing two chemokines: CC ligand 17 (CCL17) [also known as thymus and activation-regulated chemokine (TARC)] and CCL22 [also known as macrophage-derived chemokine (MDC)] (6). CCR4 is expressed on Th2 cells, various T-cell malignancies, and a unique effector subset of normal human Tregs (eTregs) (6, 7). CCL17 and CCL22 chemokine production by tumor cells attracts CCR4+ Treg cells into the tumor microenvironment where they favor tumor escape by suppression of the host antitumor immune response (8). CCR4 has therefore been suggested as a therapeutic target. High CCR4+ Treg levels have been detected in a wide range of murine and human solid tumors, including breast, colorectal, oral squamous, prostate, lung, renal, hepatic, and ovarian cancer and melanoma and/or have been associated with tumor progression or metastasis (9–19). Mogamulizumab (KW-0761), a first-in-class defucosylated humanized anti-CCR4 mAb, was recently FDA-approved for adult patients with relapsed or refractory mycosis fungoides or Sézary syndrome, which are both subtypes of cutaneous T-cell lymphoma (20). Mogamulizumab has been shown to deplete Tregs from peripheral blood in patients with solid tumors (21). The combination of mogamulizumab with checkpoint inhibitors might therefore improve clinical outcomes of patients with advanced malignancies (21).
Durvalumab (MEDI4736) is a human immunoglobulin G1 kappa (IgGκ) mAb that blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1) on immune cells (22, 23). It is FDA-approved for the treatment of patients with locally advanced or metastatic urothelial carcinoma and patients with unresectable, Stage III non-small cell lung cancer (NSCLC) whose disease has not progressed following concurrent platinum-based chemotherapy and radiation therapy.
Tremelimumab (CP-675,206) is a human IgG2 mAb directed against CTLA-4 cluster of differentiation (CD152), a cell surface receptor that is expressed primarily on activated T cells and acts to inhibit their activation, that is undergoing clinical investigation. Tremelimumab blocks the interaction of CTLA-4 with CD80 and CD86, resulting in increased release of cytokines from human T cells (24). This blockade markedly enhances T-cell activation and antitumor activity in animal models, including killing of established murine solid tumors and induction of protective antitumor immunity (24). Tremelimumab has demonstrated activity in clinical trials of patients with hepatocellular carcinoma as monotherapy (25) and in combination with durvalumab in malignant mesothelioma (26).
The aim of the present clinical study was to evaluate whether CCR4+ Treg depletion by mogamulizumab enhances antitumor response in combination with the checkpoint inhibitors durvalumab or tremelimumab in patients with advanced solid tumors.
Materials and Methods
Patients
Eligible patients included adult patients (≥18 years) with measurable, histologically or cytologically confirmed locally advanced or metastatic solid tumors that had been previously treated and for which no additional standard or approved therapy options were available. They had to have adequate organ and bone marrow function, Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1, and a life expectancy >3 months. Detailed inclusion/exclusion criteria are provided as Supplementary Information. Advice on standard restrictions to avoid pregnancy was given to female patients of child-bearing potential and to male patients with a partner of child-bearing potential.
Study design and objectives
This was a two-part, multicenter, Phase I open-label, dose-escalation, cohort-expansion study of mogamulizumab + durvalumab (Arm A) and mogamulizumab + tremelimumab (Arm B) in adults with locally advanced or metastatic solid tumors. Part 1 had a parallel 3+3 design to identify the maximum tolerated dose (MTD) or the highest protocol-defined dose in the absence of MTD for each combination. That dose level was used for cohort expansion in Part 2.
The dose-escalation period followed the standard 3+3 design. Patients who did not receive all infusions in Cycle 1 at the assigned doses or did not complete safety follow-up (until 1 week after end of Cycle 1) were replaced. The doses administered with the two treatments are summarized in Table 1. In the initial study protocol, three dose levels were planned for each treatment (Cohorts 1A–3A and 1B–3B) to establish the recommended dose level for cohort expansion, which in actuality occurred at the highest dose level for each combination treatment (Cohorts 5A and 5B). Following protocol amendment, an additional, higher dose-escalation cohort was added for each treatment (Cohorts 4A and 4B) after cohort expansion was completed in Cohorts 5A and 5B. Expansion of Cohorts 4A and 4B was only planned for ≥2 responses in either cohort, which would have provided an 80% chance of a true objective response rate (ORR) of the treatment >14%.
Table 1.
Doses administered in the dose-escalation and dose-expansion cohorts and number of patients enrolled
| Mogamulizumab (Moga) + Durvalumab (Durva) | ||||
|---|---|---|---|---|
| Cohort 1A (n = 4) [dose-escalation] |
Cohort 2A (n = 3) [dose-escalation] |
Cohort 3A (n = 7) [dose-escalation] |
Cohort 4A (n = 7) [dose-escalation] |
Cohort 5A (n = 12) [dose-expansion] |
| Moga 0.3 mg/kg + Durva 3 mg/kg |
Moga 1 mg/kg + Durva 3 mg/kg |
Moga 1 mg/kg + Durva 10 mg/kg |
Moga 3 mg/kg + Durva 10 mg/kg |
Moga 1 mg/kg + Durva 10 mg/kg |
| Mogamulizumab (Moga) + Tremelimumab (Treme) | ||||
|
| ||||
| Cohort 1B (n = 3) [dose-escalation] |
Cohort 2B (n = 3) [dose-escalation] |
Cohort 3B (n = 7) [dose-escalation] |
Cohort 4B (n = 6) [dose-escalation] |
Cohort 5B (n = 12) [dose-escalation] |
| Moga 0.3 mg/kg + Treme 3 mg/kg |
Moga 1 mg/kg + Treme 3 mg/kg |
Moga 1 mg/kg + Treme 10 mg/kg |
Moga 3 mg/kg + Treme 10 mg/kg |
Moga 1 mg/kg + Treme 10 mg/kg |
During cohort expansion in the initial protocol, it was intended to recruit patients with pancreatic cancer, NSCLC, or head and neck cancer as consecutive disease strata (12 patients per tumor type). Following the dose escalation, a decision was made to first recruit patients with pancreatic cancer to the expansion Cohorts 5A and 5B, after which the trial was stopped. Study termination was considered after ORR was evaluated for the first 12 patients in each expansion cohort. The study used an efficacy cut-off for which there was an 80% chance that the true ORR was >20% for the mogamulizumab + durvalumab combination and >15% for the mogamulizumab + tremelimumab combination. A stop to cohort expansion would also have been considered if the proportion of patients with dose-limiting toxicity (DLT) became statistically significant (>16.7%) with either treatment combination.
The primary objective was to characterize the safety and tolerability, and to determine the MTD of the combinations of mogamulizumab + durvalumab and mogamulizumab + tremelimumab in patients with advanced solid tumors. Secondary objectives were to evaluate the clinical activity of the combinations and to evaluate the pharmacokinetics and immunogenicity of mogamulizumab, durvalumab, and tremelimumab. An exploratory objective was to determine the pharmacodynamic profile of the combinations and whether any biomarkers were correlated with safety or activity.
Study drug administration
The drug combinations used in this study had not been previously administered in humans. Lower-than-approved doses were therefore used to start dose escalation. The rationale for dose selection is elaborated in more detail as Supplementary Information.
Treatment cycles were 28 days. Mogamulizumab (0.3, 1, and 3 mg/kg) was administered by intravenous (IV) infusion over ≥1 hour on Days 1, 8, 15, and 22 of the Cycle 1 and on Days 1 and 15 of each subsequent cycle. Mogamulizumab 3 mg/kg was administered over ≥3 hours for the first infusion. If well-tolerated, subsequent infusions could be administered over 1.5 hours. A minimum 1-hour post-dose observation period was required after each infusion. After the 1-hour mogamulizumab observation period, durvalumab (3 and 10 mg/kg) was administered by IV infusion over ≥1 hour on Days 1 and 15 of each cycle. After the 1-hour mogamulizumab observation period, tremelimumab (3 and 10 mg/kg) was administered by IV infusion over ≥1 hour on Day 1 of each cycle for the first six cycles and then every 12 weeks. A minimum 4-hour post-dose observation period was required after the first two infusions of durvalumab or tremelimumab, which was reduced to a minimum of 1 hour after subsequent infusions. All infusions were made using an infusion pump with a 0.22- or 0.2-µm inline filter. Mogamulizumab + durvalumab was administered for up to 12 months and mogamulizumab + tremelimumab for up to 24 months.
Patients were premedicated with oral acetaminophen and IV diphenhydramine 50 mg (or equivalent) prior to the first mogamulizumab infusion. If a patient experienced an infusion-related reaction, pre-medication was recommended prior to subsequent infusions.
Concurrent treatments that were prohibited during the study are detailed as Supplementary Information.
Definition of dose-limiting toxicity
DLT was determined from the first dose until 1 week after the last dose of Cycle 1. DLTs were defined as: any adverse event (AE) Grade ≥4 (except for Grade 4 neutropenia not associated with fever or systemic infection that improved by ≥1 grade within 3 days or Grade 4 lymphopenia); Grade ≥3 non-infectious pneumonitis, colitis, or febrile neutropenia; any Grade 3 immune-mediated AE that did not downgrade to Grade 2 within 3 days despite optimal management or did not downgrade to Grade ≤1 or baseline within 14 days; liver transaminase elevation >8 times the upper limit of normal (ULN) or total bilirubin >5 times ULN; Grade 2 pneumonitis that did not resolve to Grade ≤1 within 3 days of starting maximal supportive care; and Stevens-Johnson syndrome or toxic epidermal necrolysis. This excluded any AEs clearly and directly related to the primary disease or to another etiology. More detailed DLT definitions are provided as Supplementary Information.
Assessments
Demographics and medical/cancer history were recorded at screening (up to 4 weeks prior to first dose). Vital signs were recorded at every visit. Physical examination was undertaken at screening and at end of treatment. Hematology profile was determined at screening, on Days 1, 8, 15, 22, and 22 of Cycle 1, on Days 1 and 15, of Cycles ≥2, and at end of treatment. Serum chemistry profile was determined at screening, on Days 1, 8, 15, 22, and 22 of Cycle 1, on Day 1 of Cycles ≥2, and at end of treatment. Urinalysis was undertaken at screening, on Days 1 and 15 of Cycle 1, on Day 1 of Cycles ≥2, and at end of treatment. Coagulation profile and 12-lead ECG were performed at screening, on Day 1 of Cycles ≥1, and at end of treatment. Thyroid function testing was undertaken at screening, on Day 1 of Cycles ≥2, and at end of treatment. ECOG performance status was determined at screening, on Day 1 of Cycle 1, and end of treatment. Virus testing was performed at screening. Serum pregnancy testing was undertaken at screening and urinary pregnancy testing was performed on Day 1 of Cycles ≥1, and at end of treatment in women of child-bearing potential.
Tumor response and safety
Tumor assessment was performed at screening and every 2 cycles in the first year and every 3 cycles in the second year using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 criteria (27). Evaluation included serum tumor markers applicable to a patient’s tumor type. ORR was determined for the percentage of patients with either complete response (CR) or partial response (PR) confirmed ≥4 weeks later. Patients who did not meet CR/PR were classified as stable disease (SD) if assessed as SD (or better) ≥6 weeks after first dose of investigational medicinal product (IMP).
Adverse events (AEs) were recorded following observation by the investigator or in response to non-leading questioning during clinic visits, after spontaneous reporting by the patient, or on the basis of clinical or laboratory tests. AEs were graded by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) v4.03 and classified by the investigator with respect to relationship to treatment (definitely, probably, possibly, or unrelated). Treatment-related AEs included those considered definitely, probably, or possibly related to treatment. Serious AEs (SAEs) were reported in an expedited manner. Safety was analyzed in the safety analysis set that included all patients who received at least one dose (even a partial dose) of IMP.
Antidrug antibody (ADA) testing was undertaken for mogamulizumab in each combination arm using blood samples pre-dose at the start of Cycles 1–5 and 90 days after the end of treatment. Samples were also taken for ADA testing for durvalumab and tremelimumab but were not analyzed as the study was terminated early.
Biomarker assessments
Blood samples were taken for biomarker and pharmacodynamic assessment pre-dose on Days 1 and 15 of Cycle 1, pre-dose at the start of Cycles 2–5 and the last cycle, and 90 days after the end of treatment. The main biomarker parameters to be measured included circulating CCR4+ Tregs, activated T-cell populations, and other immune cell populations by flow cytometry and immunohistochemtistry. For flow cytometry, the following antibody probes were used: CD3 V510; clone 510 (BioLegend, San Diego, CA), CD4 PECy7; clone SK3 (BD), CCR6 APC; clone G034E3 (BioLegend), CD183 PerCPCy5.5; clone G025H7 (BioLegend), CCR10 PE; clone 6588–5 (BioLegend), CCR5 FITC; clone J418F1 (BioLegend), CTLA-4 APC; clone BNI3 (BD), CD8 V421; clone SK1 (BioLegend), PD-1 PerCPCy5.5; clone EH12.1 (BD), Ki67 PE; clone Ki-67 (BioLegend), CD45 FITC, CD69 APC, ICOS PE, CD38 PerCPCy5.5, CD45 FITC; clone HI30 (BioLegend), CCR6 APC; clone 53103 (R&D Systems), CCR5 FITC: clone 2D7 (BD), CCR10 PE: clone 1B5 (BD), CCR4 V421; clone 1G1 (BD), and CD183 PerCPCy5.5; clone G025H7 (BioLegend). Briefly, for surface staining, whole blood was added to fluorochrome-conjugated monoclonal antibodies and incubated at room temperature. Samples were lysed and washed once with 1× Dulbecco’s phosphate-buffered saline (DPBS). Samples were resuspended in 1× DPBS for acquisition on the flow cytometer. For flow cytometric intracellular staining, whole blood was added to fluorochrome-conjugated monoclonal surface antibodies and incubated at room temperature. Samples were lysed and washed once with 1× DPBS. Samples were washed again with 1× permeabilization buffer (perm buffer) and then resuspended in 1× perm buffer with intracellular antibody(s). Samples were incubated at room temperature and washed twice with 1× perm buffer. Samples were resuspended in 1× DPBS for acquisition on the flow cytometer. For CCR4/FoxP3 staining, whole blood was washed in Stain Buffer (BSA) and then added to fluorochrome-conjugated monoclonal surface antibodies for 1 hour at 2–8°C. Samples were lysed and washed once with Stain Buffer. Samples were fixed and permeabilized using the FoxP3 Buffer Kit (BD) according to the manufacturer’s instructions. Samples were then resuspended in Stain Buffer along with FoxP3 or isotype control. Samples were incubated at room temperature and then washed twice with Stain Buffer. Samples were resuspended in Stain Buffer for acquisition on the flow cytometer.
Fluorescence immunohistochemistry (F-IHC) staining for CCR4 and other cell markers was undertaken for tumor biopsy samples taken at baseline and at the end of Cycle 2 or Cycle 4. The following antibodies were used: polyclonal anti-CCR4 (Sigma Life Science), CD25; clone SP176 (Novus), anti-FOXP3; clone D2W8E (Cell Signaling Technologies, anti-CD16; clone 0.N.82 (Abcam), CD56 and anti-PD1; clone NAT105 (Biocare Medical), and anti-CD4; clone 4B12, anti-CD8; clone C8/144B, and polyclonal rabbit anti-cytokeratin, wide spectrum screening (DAKO). F-IHC was performed as previously described by (28). Briefly, slide-mounted, formalin-fixed, paraffin-embedded tumor biopsy slices were dewaxed and used for staining using an automated Vectra 2 Intelligent Slide Analysis System (Perkin Elmer, Waltham, MA) (29) to examine tissue regions of interest. Tumor regions of interest were identified by the presence of cytokeratin or S100 depending on the tissue types. Imaging analysis was performed by fully automated AQUA Technology (Perkin-Elmer).
Statistical analysis
No formal sample size calculation was performed for this study. Part 1 of the study for each combination utilized a standard 3+3 design in four cohorts. In Part 2, an additional 36 patients were intended to be recruited in the dose-expansion cohort of each combination treatment.
Demographic, baseline characteristics, and efficacy and safety endpoints were summarized descriptively. Frequency and percentages were used for categorical variables and summary statistics (number, mean, standard deviation [SD], median, minimum, and maximum) were calculated for continuous variables.
Efficacy endpoints were analyzed using the efficacy analysis set which included all patients who had measurable disease and completed the first cycle of combination therapy and who had baseline and at least one post-baseline on-study assessment for response. ORR, progression-free survival (PFS), and overall survival (OS) were reported in patients in the dose-escalation cohort plus the dose-expansion cohort who were treated with the same dose regimen of mogamulizumab 1 mg/kg + durvalumab 10 mg/kg (Cohorts 3A and 5A) and mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg (Cohorts 3B and 5B). Two-sided 95% exact confidence interval (CI) for the ORR was derived using the Clopper-Pearson exact binomial method (30). Duration of response, PFS, and OS were defined conventionally, and median values, along with two-sided 95% CI (31), were estimated using the Kaplan-Meier method.
Ethics
The study was conducted in accordance with the Declaration of Helsinki and International Conference for Harmonization of Good Clinical Practice Guidelines. The protocol and its amendments were approved the local institutional review boards at the participating centers. All patients provided written informed consent prior to study registration. The study was registered in ClinicalTrials.gov (NCT02301130).
Results
Patient characteristics
The study was conducted between 26 November 2014 and 5 March 2018 at 7 US centers (MD Anderson Cancer Center, Houston, TX; The Angeles Clinic and Research Institute, Los Angeles, CA; Smilow Cancer Hospital, New Haven, CT; Memorial Sloan Kettering Cancer Center, New York, NY; UCLA Hematology and Oncology Clinic, Los Angeles, CA; Georgia Cancer Center, Augusta, GA). A total of 64 patients were enrolled and treated: Part 1 (n = 40) and Part 2 (n = 24). Baseline clinical and demographic characteristics of the patients are summarized in Table 2. Patients recruited to the dose-expansion cohorts had pancreatic cancer exclusively.
Table 2.
Baseline patient demographics and clinical characteristics
| Variable | Dose-escalation cohorts |
Dose-expansion cohort |
||
|---|---|---|---|---|
| Mogamulizumab + durvalumab (n = 21) |
Mogamulizumab + tremelimumab (n = 19) |
Mogamulizumab + durvalumab (n = 12) |
Mogamulizumab + tremelimumab (n = 12) |
|
| Age (years), median (range) | 63.0 (23–80) | 57.0 (33–76) | 68.0 (29–76) | 64.5 (46–80) |
| ≥65 years, n (%) | 8 (38.1) | 4 (21.1) | 9 (75.0) | 6 (50.0) |
|
| ||||
| Gender, n (%) | ||||
| Male | 10 (47.6) | 11 (57.9) | 6 (50.0) | 7 (58.3) |
| Female | 11 (52.4) | 8 (42.1) | 6 (50.0) | 5 (41.7) |
|
| ||||
| Race, n (%) | ||||
| White | 19 (90.5) | 12 (63.2) | 12 (100.0) | 9 (75.0) |
| Asian | 0 | 3 (15.8) | 0 | 1 (8.3) |
| Black or African American | 2 (9.5) | 2 (10.5) | 0 | 1 (8.3) |
| Not reported | 0 | 2 (10.5) | 0 | 1 (8.3) |
|
| ||||
| ECOG performance status, n (%) | ||||
| 0 | 7 (33.3) | 9 (47.4) | 5 (41.7) | 3 (25.0) |
| 1 | 14 (66.7) | 10 (52.6) | 7 (58.3) | 9 (75.0) |
|
| ||||
| Time from diagnosis (months), median (range) | 43.9 (8.2–403.3) | 42.1 (12.5–230.5) | 25.3 (7.9–59.0) | 20.5 (8.4–50.6) |
|
| ||||
| Primary tumor type, n (%) | ||||
| Pancreatic | 1 (4.8) | 2 (10.5) | 12 (100.0)a | 12 (100.0)a |
| Colorectal | 5 (23.8) | 5 (26.3) | – | – |
| Sarcoma | 5 (23.8) | 0 | – | – |
| Head and neck | 1 (4.8) | 3 (15.8) | – | – |
| Renal cell | 1 (4.8) | 3 (15.8) | – | – |
| Ovarian | 2 (9.5) | 1 (5.3) | – | – |
| Prostate | 1 (4.8) | 1 (5.3) | – | – |
| NSCLC, non-squamous | 1 (4.8) | 1 (5.3) | – | – |
| NSCLC, squamous | 1 (4.8) | 0 | – | – |
| Anal | 0 | 1 (5.3) | – | – |
| Breast | 1 (4.8) | 0 | – | – |
| Other | 2 (9.5) | 2 (10.5) | – | – |
|
| ||||
| No. of prior cancer regimens, n (%) | ||||
| 0 | 1 (4.8) | 0 | 0 | 0 |
| 1 | 1 (4.8) | 3 (15.8) | 0 | 2 (16.7) |
| 2 | 2 (9.5) | 2 (10.5) | 3 (25.0) | 2 (16.7) |
| 3 | 5 (23.8) | 2 (10.5) | 2 (16.7) | 4 (33.3) |
| 4 | 6 (28.6) | 2 (10.5) | 3 (25.0) | 4 (33.3) |
| ≥5 | 6 (28.6) | 10 (52.6) | 4 (33.3) | 0 |
Only patients with pancreatic cancer enrolled in dose-expansion cohort. ECOG, Eastern Cooperative Oncology Group; NSCLC, non-small cell lung cancer.
All patients were included in the safety analysis set, which included all patients who received treatment. One patient receiving mogamulizumab + durvalumab was not evaluated for efficacy because of lack of post-baseline assessment. Patient disposition during the study is detailed as Supplementary Information (Fig. S1). Reasons for discontinuation from the study were progressive disease (n = 36), adverse events (n = 9), consent withdrawal (n = 5), and lost to follow-up (n = 2); three patients completed treatment. Drug exposure for the different cohorts is detailed as Supplementary Information (Tables S1–S4). Median relative dose intensity for mogamulizumab, durvalumab, and tremelimumab was essentially 100% across cohorts (99.3%–100.6%; range, 95.6%–102.7%).
Dose-limiting toxicity and safety
No DLTs occurred in any patients in the dose-escalation cohorts with either combination treatment. Cohort expansion therefore occurred with mogamulizumab 1 mg/kg + durvalumab 10 mg/kg and mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg, i.e. the highest intended dose in the original protocol based on maximum pharmacodynamic effect (defined by CCR4+ Treg depletion) achieved with mogamulizumab 1 mg/kg. The higher dose cohorts 4A and 4B (with mogamulizumab 3 mg/kg in each combination) were added by protocol amendment after cohort expansion had started in Cohorts 5A and 5B.
The most common and all Grade ≥3 treatment-related AEs in the same-dose cohorts (dose escalation + cohort expansion) for each combination treatment are shown in Table 3. The most common and AE Grade ≥3 treatment-related AEs were maculopapular rash for both treatment arms: mogamulizumab 1 mg/kg + durvalumab 10 mg/kg (36.8% and 21.1%, respectively) and mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg (26.3% and 10.5%, respectively). Over the whole study (dose-escalation and dose-expansion phases), five patients died during the study in the mogamulizumab + durvalumab arm and two in the mogamulizumab + tremelimumab arm, none of which were related to the study treatments. In the same-dose cohorts (dose escalation + dose-expansion), SAEs occurred in 16 of 19 (84.2%) of patients in the mogamulizumab 1 mg/kg + durvalumab 10 mg/kg arm and in 10 of 19 (52.6%) in the mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg arm, of which six (31.6%) and two (10.5%), respectively, were considered related to treatment. Discontinuation of any IMP due to treatment-emergent AEs occurred in six (31.6%) patients in the mogamulizumab 1 mg/kg + durvalumab 10 mg/kg arm and in two (10.5%) patients in the mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg arm. Details for AEs of special interest are detailed as Supplementary Information. There were no unanticipated laboratory safety signals or any consistent or clinically meaningful differences between the groups in vital signs, physical examinations, or ECG parameters with either combination treatment. One patient developed anti-mogamulizumab antibodies (not neutralizing) while receiving mogamulizumab + tremelimumab, which became negative at 90 days after the end of treatment.
Table 3.
Treatment-related adverse events reported in ≥3 patients or Grade ≥3 in the same dose cohorts (dose escalation + cohort expansion) for each combination treatment.
| Adverse event | No. of patients (%) |
|||
|---|---|---|---|---|
| Mogamulizumab 1 mg/kg + durvalumab 10 mg/kg (n = 19) |
Mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg (n = 19) |
|||
| Any Grade | Grade ≥3 | Any Grade | Grade ≥3 | |
| Rash maculopapular | 7 (36.8) | 4 (21.1) | 5 (26.3) | 2 (10.5) |
| Fatigue | 6 (31.6) | 1 (5.3) | 3 (15.8) | 0 |
| Pruritis | 5 (26.3) | 0 | 3 (15.8) | 1 (5.5) |
| Infusion-related reactions | 4 (21.1) | 0 | 7 (36.8) | 0 |
| Diarrhea | 4 (21.1) | 0 | 4 (21.1) | 0 |
| Hypothyroidism | 3 (15.8) | 0 | 0 | 0 |
| Stomatitis | 1 (5.3) | 1 (5.3) | 4 (21.1) | 0 |
| Rash | 1 (5.3) | 0 | 3 (15.8) | 1 (5.3) |
| Colitis | 1 (5.3) | 1 (5.3) | 3 (15.8) | 1 (5.3) |
| Decreased lymphocytes | 1 (5.3) | 0 | 1 (5.3) | 1 (5.3) |
| Transaminases increased | 1 (5.3) | 0 | 1 (5.3) | 1 (5.3) |
| Autoimmune hepatitis | 1 (5.3) | 0 | 1 (5.3) | 1 (5.3) |
| Gastritis | 1 (5.3) | 1 (5.3) | 0 | 0 |
| Blood CPK increased | 1 (5.3) | 1 (5.3) | 0 | 0 |
| Hyperglycemia | 1 (5.3) | 1 (5.3) | 0 | 0 |
| Vomiting | 0 | 0 | 1 (5.3) | 1 (5.3) |
| Abnormal liver function test | 0 | 0 | 1 (5.3) | 1 (5.3) |
| Hypertension | 0 | 0 | 1 (5.3) | 1 (5.3) |
CPK, creatine phosphokinase.
Antitumor activity
Efficacy (as ORR and OS) were evaluated in patients in the dose-escalation cohort plus the dose-expansion cohort who were treated with the same dose regimen of mogamulizumab 1 mg/kg + durvalumab 10 mg/kg (Cohorts 3A and 5A) and mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg (Cohorts 3B and 5B) [Table 5]. ORR was the same in both treatment groups, 5.3% (95% CI: 0.1, 26). There was only one patient with PR in each of these two treatment groups. Each of the patients with PR occurred in the dose-escalation cohorts. The duration of response was 10.6 months for a patient with alveolar soft part sarcoma in the mogamulizumab 1 mg/kg + durvalumab 10 mg/kg arm and 3.7 months for a patient with prostate cancer in the mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg arm. Five (26.3%) patients in the mogamulizumab 1 mg/kg + durvalumab 10 mg/kg arm and 7 (36.8%) patients in the mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg arm had SD. The median OS of patients in the mogamulizumab 1 mg/kg + durvalumab 10 mg/kg arm was 8.9 months (95% CI: 4.3, 18.4) and that for patients in the mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg arm was 4.4 months (95% CI: 2.5, 13.4). The OS curves are presented in Supplementary Information (Fig. S2). Both treatments had similar response survival function. However, the survival estimate in the mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg arm drops rapidly after 3 months, while that for mogamulizumab 1 mg/kg + durvalumab 10 mg/kg arm drops after a longer time period of about 9 months. No responses occurred with either combination treatment during dose-expansion in the patients with pancreatic cancer. The stop criterion for further expansion was therefore reached. Additionally, there was a strategic decision to not expand into head and neck or lung tumor types; PD-1/checkpoint blockade had been generally adopted as a standard of care for these tumor types during the course of this trial, making accrual of patients with those tumor types problematic. The changes in tumor burden over time for each treatment combination are shown as spider plots in Supplementary Information (Figs S3 and S4).
Besides the two patients with a PR response observed in the efficacy data set with the same dose regimen of mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg and mogamulizumab 1 mg/kg + durvalumab 10 mg/kg, one additional patient with renal cell carcinoma in the dose-escalation cohort of mogamulizumab 3 mg/kg + durvalumab 10 mg/kg had a PR. None of the patients with a PR response had prior treatment with an immune checkpoint inhibitor. Five patients had received prior PD-1/PD-L1 blockade and were enrolled in the mogamulizumab + tremelimumab cohorts. Two of the patients (both with renal cell carcinoma) demonstrated SD as best response, while the remaining patients (1 with NSCLC and 2 with colorectal cancer) had PD.
Pharmacodynamics
Biomarker assessment on the study included quantification of the Tregs and other immune cell populations in peripheral blood and tumors. Various Treg populations in peripheral blood were defined as previously (7, 32). There was no apparent therapeutic relationship between clinical response of patients in both combination treatment arms and the degree of baseline CCR4+ expression on various T-cell subsets, including CD4, CD8, naive Tregs (CCR4+CD45RA+FoxP3lo), eTregs (CCR4+CD45RA–FoxP3hi), and non-suppressor Tregs (CD4+CD45RA–FoxP3lo) (Fig. 1A). Therapy with mogamulizumab resulted in reduction of peripheral blood CCR4+ eTregs in both combination treatment arms at all dose levels, with maintenance of depletion throughout treatment (Fig. 1B). Assessment of other cell populations revealed concomitant general depletion of CCR4+CD4 (Fig. 1C) and CCR4+CD8 lymphocytes (Fig. 1D). Reductions in natural killer, Th2, Th17, and Th22 cells were also seen (Supplementary Information, Table S5). There did not appear to be correlation between clinical response and the depletion of CCR4+ eTreg, CCR4+CD4, or CCR4+CD8 populations (Fig. 1B–D). Analysis of CD8 T-cell activation markers in peripheral blood, such as CD38, CD69, CD134, and HLA-DR demonstrated evidence of T-cell activation in response to therapy in some patients; however, this did not correlate with response (Supplementary Information, Fig. S5).
Figure 1.
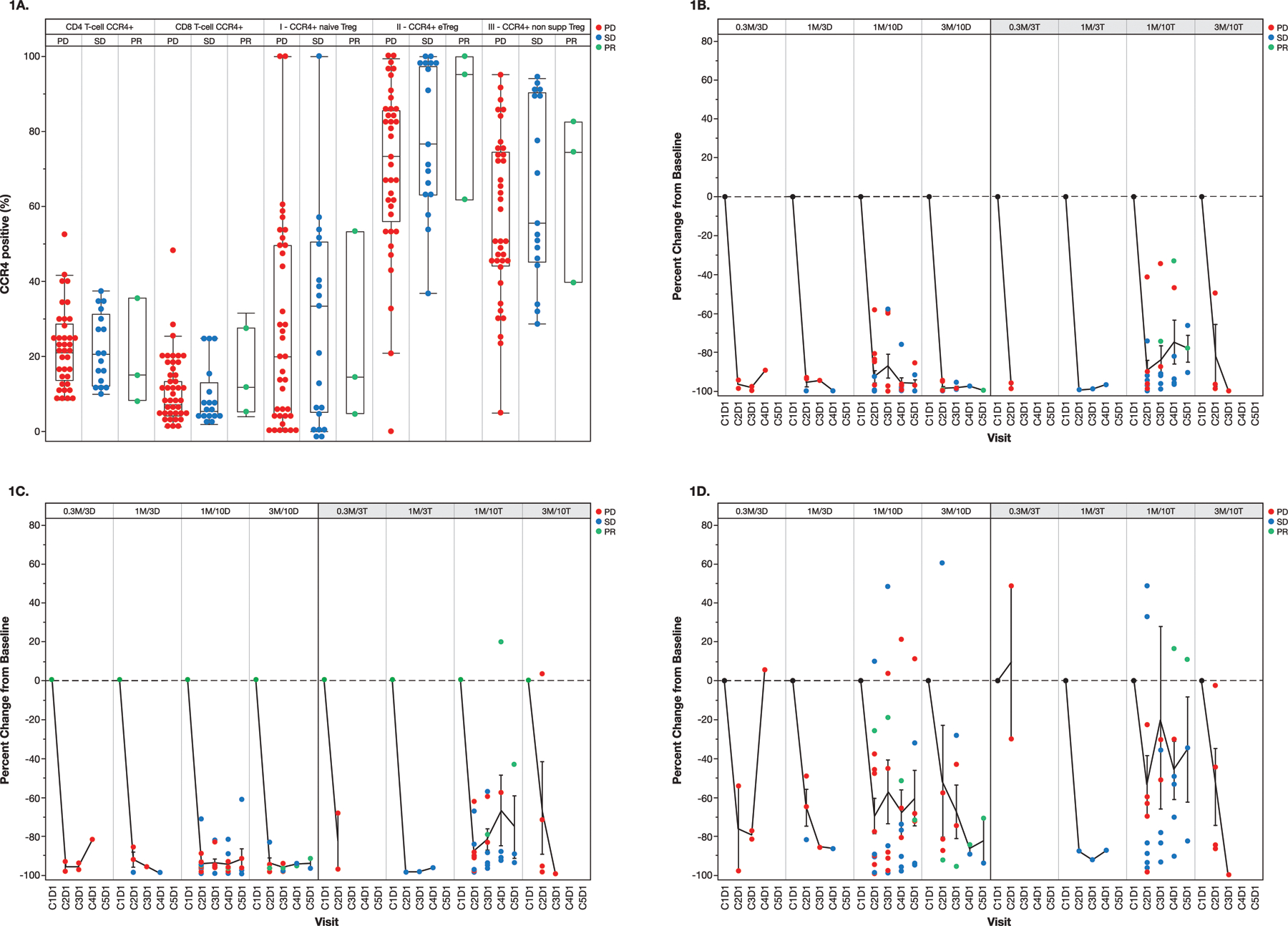
(A) Relationship between clinical response and baseline expression of CCR4 by various T-cell subsets in peripheral blood. Box and whisker plots (median and interquartile ranges) are shown. T-cell subsets were defined by flow cytometry as follows: naive Treg (regulatory T cell) = CD4+CD45RA+FoxP3lo; eTreg (effector Treg) = CD4+CD45RA–FoxP3hi; and non-supp Treg (non-suppressor) Treg = CD4+CD45RA–FoxP3lo. (B-D) Changes in peripheral blood eTregs and other CCR4+ T-cell populations in response to therapy. Mean (± standard deviation) change from baseline in (B) CCR4+ effector regulatory T cells (eTregs), (C) CCR4+ CD4 T cells, and (D) CCR4+ CD8 T cells. X-axis delineates treatment Cycle (C) and Day (D). See Table 1 for patient numbers involved. D, durvalumab; M, mogamulizumab; PD, progressive disease; PR, partial response; SD, stable disease; T, tremelimumab.
In patients in whom pre- and on-treatment biopsies could be performed and yielded sufficient tissue, quantification of Tregs and other immune cell subsets in tumor microenvironment was performed using F-IHC. In the majority of the analyzed cases, there was a relative reduction of tumor-infiltrating Tregs such as CCR4+FoxP3+ and CD25+FoxP3+ in response to therapy to a varying degree (Fig. 2A–B). There were no consistent trends in other cell populations, including overall CD4+ lymphocytes, CD8+ lymphocytes, or NK cells (CD16+CD56+). Similar to peripheral blood findings, changes in the intratumoral immune cell populations, including the degree of Treg depletion, did not appear to correlate with clinical response. There was not sufficient tissue remaining to perform PD-L1 testing or genetic analyses such as tumor mutational burden.
Figure 2.

Mean percent change from baseline of cell populations in individual patient tumor biopsies. (A) Mogamulizumab + Durvalumab; BR*=0.3M/3D; BR**=M/10D; BR***=3M/10D. (B) Mogamulizumab + Tremelimumab; BR**=1M/10T; BR***=3M/10T. All biopsies were performed during Cycle 2, at the same time as scans. BR, best response; M, mogamulizumab; PD, progressive disease; PR, partial response; SCLC, small cell lung cancer; SD, stable disease; T, tremelimumab.
Discussion
Regulatory T cells play a major immunosuppressive role in the tumor microenvironment and may represent a major obstacle to the efficacy of cancer immunotherapies (33). Across cancer types, Treg infiltration is associated with poor outcomes (34). Studies in preclinical models have indicated that depletion of Tregs may afford therapeutic benefit, particularly when used in combination with other interventions, such as radiation or immune checkpoint blockade (35, 36). Therapeutic depletion of Tregs, however, has been limited by a lack of unique surface marker that is not expressed on effector T-cell populations. For example, the initial enthusiasm for targeting of Tregs with the anti-CD25 antibody daclizumab was dampened due to its concomitant depletion of activated effector T cells (37–39). Recent studies, however, have demonstrated that modification of the Fc portion of the anti-CD25 antibody to bind the inhibitory Fc gamma receptor in tumors can enhance its anti-tumor activity (40).
CCR4 is highly and predominantly expressed on the population of regulatory T cells that have been shown to possess the most potent immunosuppressive activity. Prior studies have demonstrated that mogamulizumab could potently deplete peripheral blood CCR4+ Tregs (7, 21). Mogamulizumab has been engineered with a defucosylated Fc region with enhanced ability for ADCC due to more efficient Fc gamma receptor binding (41), thus making it an attractive candidate for intratumoral T-cell depletion.
Despite the reasonable therapeutic rationale for combinatorial therapy of mogamulizumab with immune checkpoint blockade, the current clinical trial suggests that although Treg population depletion by mogamulizumab in combination with checkpoint inhibitors might be useful in inducing antitumor immunity, it does not appear to be the only factor sufficient to induce potent antitumor efficacy as ORR was 5.3% (95% CI; 0.1%, 26.0%) in each treatment arm (mogamulizumab 1 mg/kg in combination with durvalumab 10 mg/kg or tremelimumab 10 mg/kg). In addition, no further expansion was pursued in the cohorts containing mogamulizumab 3 mg/kg given the lack of any objective responses in these cohorts. Prior studies with mogamulizumab in solid tumors noted evidence of single-agent clinical activity of this drug (21). While several patients on our study developed durable responses, it is not possible to conclude whether the clinical activity could be attributed to mogamulizumab or its immune checkpoint inhibitor partner. Results of a Phase I study of mogamulizumab in combination with the anti-PD-1 antibody, nivolumab, in 90 patients with advanced or metastatic solid tumors have recently been published [42]. The safety profile was acceptable and similar to that in the present study. ORR across all tumor types was 12%. ORR was 7% among the 15 patients with pancreatic adenocarcinoma, which is similar to the present study. Higher ORR was reported in 15 patients with non-small cell lung cancer (20%) and in 15 patients with hepatocellular carcinoma (27%). Interestingly, in the current study, a response was observed in prostate cancer, which is considered an immunologically “cold” tumor. While CCR4+ regulatory T cells have been shown to be associated with poor prognosis in prostate cancer (43), there is evidence of response of prostate cancer to single-agent CTLA-4 blockade (44, 45). Thus, it is unclear whether our responding patient could have benefitted from tremelimumab alone, or whether addition of mogamulizumab was beneficial.
With respect to safety, the primary endpoint, the combination of mogamulizumab with either durvalumab or tremelimumab proved tolerable in the treatment of patients with solid tumors. AEs were manageable and generally mild to moderate in intensity. No DLTs occurred in the dose-escalation cohorts and no MTD was established with the tested doses of mogamulizumab up to 3 mg/kg in combination with durvalumab or tremelimumab 10 mg/kg. The most common treatment-related AEs included maculopapular rash, fatigue, pruritus, infusion-related reactions, and diarrhea. The most common treatment-related AEs grade ≥3 were maculopapular rash, pruritus, and colitis. The overall AE profiles including AEs of special interest during the combination treatments are similar to those previously reported with mogamulizumab (20, 46), durvalumab (47, 48), or tremelimumab (49–51) monotherapy. No new safety concerns were identified with either combination treatment.
The study was stopped early since it had already accumulated sufficient safety data (primary objective); therefore, there was no need to continue to collect more once it was apparent that there was no apparent efficacy (secondary objective). Due to this study stop, it was decided to undertake no analysis of pharmacokinetic data, which had been another secondary endpoint of the study. In addition, analyses of pharmacokinetic/pharmacodynamic interactions and exposure/response relationship were not undertaken. Exploratory pharmacodynamic analyses are still warranted as potentially meaningful to undertake.
Mogamulizumab proof-of-pharmacologic activity was demonstrated by a reduction in the number of CCR4+ eTregs, as seen in other studies of mogamulizumab monotherapy in patients with solid tumors (7). Depletion of CCR4+ eTregs or differentiated CD4 T cells appeared relatively constant when increasing mogamulizumab dose over the range from 0.3 to 3 mg/kg. Similarly, reduction of intratumoral Tregs to a varying degree was observed in the majority of the patients from whom pre- and on-treatment tissue was available; however, there was no apparent correlation between Treg reduction and clinical response. Similarly, a correlation between baseline CCR4+ expression and clinical response could not be established due to small numbers of patients with response.
We suspect that there are several reasons for why marked depletion of CCR4+ Tregs was achieved in peripheral blood, but not in tumors and why therapeutic enhancement of combination therapies was not seen. First of all, it is unclear how much antibody was actually able to penetrate the tumors. Second, depletion of Tregs by mogamulizumab is likely dependent on presence of cells expressing activating Fc receptors, such as NK cells (via antibody-dependent cell-mediated cytotoxicity) and possibly phagocytes. Absence of these additional cell populations from the tumor microenvironment could potentially limit the efficacy of mogamulizumab, even when the drug is present in sufficient quantities. Third, in peripheral blood, we observed significant depletion of non-eTreg T-cell populations, both in the CD4+ and CD8+ T-cell compartments. It is thus possible that concomitant depletion of effector T-cell populations could negatively offset the therapeutic effect afforded by Treg depletion. Fourth, it is unknown whether Tregs play a key immunosuppressive role in the cancer types evaluated in the study. Lastly, the majority of the patients in the study had advanced solid tumors such as pancreatic cancer that typically do not respond to immune checkpoint blockade and it is likely that targeting of additional mechanisms of immunosuppression would be necessary to achieve therapeutic efficacy in these patients.
In conclusion, the efficacy of durvalumab or tremelimumab was not enhanced by the addition of mogamulizumab in patients with advanced solid tumors despite achieving significant depletion of eTregs in both peripheral blood and tumor. It is likely that peripheral eTreg depletion alone is not sufficient to reverse the immunosuppressive effect driving therapeutic resistance to immune checkpoint blockade. Further research into the strategies to enhance depletion of Tregs using combinations of mogamulizumab with other strategies may be warranted.
Supplementary Material
Table 4.
Efficacy results in the dose-escalation and dose-expansion cohorts receiving the same dose combined.
| Mogamulizumab 1 mg/kg + durvalumab 10 mg/kg (n = 19) |
Mogamulizumab 1 mg/kg + tremelimumab 10 mg/kg (n = 19) |
|
|---|---|---|
| ORR, n (%) [95% CI] | 1 (5.3) [0.1, 26.0] | 1 (5.3) [0.1, 26.0] |
| CR, n (%) | 0 | 0 |
| PR, n (%) | 1a (5.3) | 1b (5.3) |
| SD, n (%) | 5 (26.3) | 7 (36.8) |
| PD, n (%) | 12 (63.2) | 9 (47.4) |
| NE, n (%) | 1 (5.3) | 2 (10.5) |
| Median OS, months (95% CI) | 8.9 (4.3, 18.4) | 4.4 (2.5, 13.4) |
| Median PFS, months (95% CI) | 1.9 (1.7, 4.4) | 1.9 (1.4, 3.7) |
Duration of response was 10.6 months and time to response was 3.68 months in a patient with alveolar soft part sarcoma.
Duration of response was 3.7 months and time to response was 1.84 months in a patient with prostate cancer.
CI, confidence interval; CR, complete response; NE, not evaluable; PD, progressive disease; PFS, progression-free survival; PR, partial response; OS, overall survival; SD, stable disease.
Translational Relevance.
Mogamulizumab is a monoclonal antibody targeting CCR4, which is highly expressed by eTregs. This phase I study in advanced solid tumors evaluated whether depletion of eTregs with mogamulizumab was safe and improved the efficacy of immune checkpoint inhibitors durvalumab or tremelimumab, targeting PD-L1 or CTLA-4, respectively. No dose-limiting toxicity occurred with either combination. No additional efficacy was observed with addition of mogamulizumab compared to that expected with durvalumab or tremelimumab monotherapy. Mogamulizumab proof-of-pharmacologic activity was demonstrated by a reduction in the number of peripheral blood CCR4+ eTregs and intratumoral Tregs; however, there was no clear correlation of clinical response with reduction in peripheral blood CCR4+ eTregs or with baseline degree of CCR4+ expression. These observations suggest that although Treg population depletion by mogamulizumab in combination with checkpoint inhibitors might be useful in inducing antitumor immunity, it does not appear to be the only factor sufficient to induce potent antitumor efficacy.
Acknowledgments
This study was sponsored by Kyowa Kirin Pharmaceutical Development, Inc. D. Zamarin is funded in part by the MSK Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (P30CA008748). Medical writing and editorial support was provided by P.A. Todd of Tajut Ltd. (Kaiapoi, New Zealand), and S.E. Johnson of S. E. Johnson Consulting, LLC (New Hope, PA), which was funded by Kyowa Kirin Pharmaceutical Development, Inc. (Princeton, NJ).
Footnotes
Disclosure of Potential Conflicts of Interest
D. Zamarin reports consulting fees from Merck, Synlogic Therapeutics, Western Oncolytics, Tizona Therapeutics, and Tesaro.
S. Sahebjam reports research support from Merck, Bristol Myers Squibb, and Brooklyn ImmunoTherapeutics.
M. Sznol reports personal or advisory fees from Intensity Therapeutics, Adaptimmune, AstraZeneca/MedImmune, Baxalta/Shire, Biodesix, BristolMyers Squibb, Genentech/Roche, Inovio Pharmaceuticals, Nektar, Lilly, Merck Sharp & Dohme, Modulate, Molecular Partners, Newlink Genetics, Novartis, Omniox, Pfizer, Pierre Fabre, Seattle Genetics, Theravance, AcademicCME, DAVAOncology, Haymarket Media, Physician Education Resource Research to Practice, Symphogen, Nextcure, Verastem, Innate, Incyte, Iovance, Genmab, Celldex, Abbvie, Immunocore, Almac, Hinge, Anaeropharma, Array, Biontech, Pieris, Torque, and Gritstone; and stock options from Actym, Adaptive Biotechnologies, Amphivena, Nextcure, and Torque.
F.E. Fox is an employee of Kyowa Kirin Pharmaceutical Development, Inc.
A. Collaku and M.A. Marshall are former employees of Kyowa Kirin Pharmaceutical Development, Inc.
D.S. Hong reports research/grant funding from AbbVie, Adaptimmune, Aldi-Norte, Amgen, Astra-Zeneca, Bayer, BristolMyers Squibb, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, Ignyta, Infinity, Kite, Kyowa, Lilly, LOXO, Merck, MedImmune, Mirati, miRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda, and Turning Point Therapeutics; travel, accommodation, and expenses from LOXO, miRNA, Genmab, AACR, ASCO, and SITC; consulting or advisory roles for Alpha Insights, Amgen, Axiom, Adaptimmune, Baxter, Bayer, Genentech, GLG, Group H, Guidepoint, Infinity, Janssen, Merrimack, Medscape, Numab, Pfizer, Prime Oncology, Seattle Genetics, Takeda, Trieza Therapeutics, and WebMD; and ownership interests in Molecular Match (Advisor), OncoResponse (Founder), and Presagia Inc. (Advisor).
O. Hamid and A. Nayak-Kapoor report no conflicts of interest.
References
- 1.Cohen J, Sznol M. Therapeutic combinations of immune-modulating antibodies in melanoma and beyond. Semin Oncol 2015;42:488–94. [DOI] [PubMed] [Google Scholar]
- 2.Pennock GK, Chow LQ. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist 2015;20:812–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol 2016;28:401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Franco F, Ho PC. Metabolic regulation of Tregs in cancer: opportunities for immunotherapy. Trends Cancer 2017;3:583–92. [DOI] [PubMed] [Google Scholar]
- 5.Yan S, Zhang Y, Sun B. The function and potential drug targets of tumour-associated Tregs for cancer immunotherapy. Sci China Life Sci 2019;62:179–86. [DOI] [PubMed] [Google Scholar]
- 6.Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol 2015;27:11–20. [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA 2013;110:17945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci 2006;97:1139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10:942–9. [DOI] [PubMed] [Google Scholar]
- 10.Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 2009;69:2000–9. [DOI] [PubMed] [Google Scholar]
- 11.Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res 2009;69:5996–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe Y, Katou F, Ohtani H, Nakayama T, Yoshie O, Hashimoto K. Tumor-infiltrating lymphocytes, particularly the balance between CD8+ T cells and CCR4+ regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;109:744–52. [DOI] [PubMed] [Google Scholar]
- 13.Svensson H, Olofsson V, Lundin S, Yakkala C, Björck S, Börjesson L, et al. Accumulation of CCR4⁺CTLA-4hiFOXP3⁺CD25hi regulatory T cells in colon adenocarcinomas correlate to reduced activation of conventional T cells. PLoS One 2012;7:e30695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med 2013;5:200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berlato C, Khan MN, Schioppa T, Thompson R, Maniati E, Montfort A, et al. A CCR4 antagonist reverses the tumor-promoting microenvironment of renal cancer. J Clin Invest 2017;127:801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng X, Wu H, Jin ZJ, Ma D, Yuen S, Jing XQ, et al. Up-regulation of chemokine receptor CCR4 is associated with human hepatocellular carcinoma malignant behavior. Sci Rep 2017;7:12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein A, Sagi-Assif O, Meshel T, Telerman A, Izraely S, Ben-Menachem S, et al. CCR4 is a determinant of melanoma brain metastasis. Oncotarget 2017;8:31079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maolake A, Izumi K, Shigehara K, Natsagdorj A, Iwamoto H, Kadomoto S, et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017;8:9739–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karasaki T, Qiang G, Anraku M, Sun Y, Shinozaki-Ushiku A, Sato E, et al. High CCR4 expression in the tumor microenvironment is a poor prognostic indicator in lung adenocarcinoma. J Thorac Dis 2018;10:4741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, et al. ; MAVORIC Investigators. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol 2018;19:1192–204. [DOI] [PubMed] [Google Scholar]
- 21.Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res 2015;21:4327–36. [DOI] [PubMed] [Google Scholar]
- 22.Creelan B Update on immune checkpoint inhibitors in lung cancer. Cancer Control 2014;21:80–9. [DOI] [PubMed] [Google Scholar]
- 23.Wills S, Deshmukh R. Durvalumab: a newly approved checkpoint inhibitor for the treatment of urothelial carcinoma. Curr Probl Cancer 2019;43:181–94. [DOI] [PubMed] [Google Scholar]
- 24.Suarez N, Alfaro C, Dubrot J, Palazon A, Bolaños E, Erro L, et al. Synergistic effects of CTLA-4 blockade with tremelimumab and elimination of regulatory T lymphocytes in vitro and in vivo. Int J Cancer 2011;129:374–86. [DOI] [PubMed] [Google Scholar]
- 25.Sangro B, Gomez-Martin C, de la Mata M, Iñarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81–8. [DOI] [PubMed] [Google Scholar]
- 26.Calabrò L, Morra A, Giannarelli D, Amato G, D’Incecco A, Covre A, et al. Tremelimumab combined with durvalumab in patients with mesothelioma (NIBIT-MESO-1): an open-label, non-randomised, phase 2 study. Lancet Respir Med 2018;6:451–60. [DOI] [PubMed] [Google Scholar]
- 27.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumors: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 28.Johnson DB, Bordeaux J, Kim JY, Vaupel C, Rimm DL, Ho TH, et al. Quantitative spatial profiling of PD-1/PD-L1 interaction and HLA-DR/IDO-1 predicts improved outcomes of anti-PD-1 therapies in metastatic melanoma. Clin Cancer Res 2018;24:5250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014;70:46–58. [DOI] [PubMed] [Google Scholar]
- 30.Hollander M, Wolfe DA. Nonparametric statistical methods John Wiley & Sons, Inc; 1973. [Google Scholar]
- 31.Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics 1982;38:29–41. [Google Scholar]
- 32.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009;30:899–911. [DOI] [PubMed] [Google Scholar]
- 33.Han S, Toker A, Liu ZQ, Ohashi PS. Turning the tide against regulatory T cells. Front Oncol 2019;9:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 2015;5:15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med 2008;205:2125–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 2013;210:2435–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res (1999)59:3128–33. [PubMed] [Google Scholar]
- 38.Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP Brouwer HM, Scharenborg NM, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res 2010;16:5067–78. [DOI] [PubMed] [Google Scholar]
- 39.Rech AJ, Mick R, Martin S, Recio A, Aqui N., Powell DJ Jr., et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med 2012;4:134ra162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 2017;46:577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereira NA, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs 2018;10:693–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res 2019;25:6614–22. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe M, Kanao K, Suzuki S, Muramatsu H, Morinaga S, Kajikawa K, et al. Increased infiltration of CCR4-positive regulatory T cells in prostate cancer tissue is associated with a poor prognosis. Prostate 2019;79:1658–65. [DOI] [PubMed] [Google Scholar]
- 44.Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase iii trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40–7. [DOI] [PubMed] [Google Scholar]
- 45.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 2014;15:700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips AA, Fields PA, Hermine O, Ramos JC, Beltran BE, Pereira J, et al. ; 0761–009 Study Group. Mogamulizumab versus investigator choice of chemotherapy regimen in relapsed/refractory adult T-cell leukemia/lymphoma. Haematologica 2019;104:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. ; PACIFIC Investigators. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med 2017;377:1919–29. [DOI] [PubMed] [Google Scholar]
- 48.Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol 2017;3:e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirkwood JM, Lorigan P, Hersey P, Hauschild A, Robert C, McDermott D, et al. Phase II trial tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin Cancer Res 2010:16:1042–8. [DOI] [PubMed] [Google Scholar]
- 50.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol 2013:31:616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maio M, Scherpereel A, Calabrò L, Aerts J, Cedres Perez S, Bearz A, et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): a multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol 2017;18:1261–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.