

Abstract
Activatable contrast agents are of ongoing research interest because they offer low background and high specificity to the imaging target. Engineered sensitivity to protease activity is particularly desirable because proteases are critical biomarkers in cancer, infectious disease, inflammatory disorders, and so forth. Herein, we developed and characterized a set of peptide-linked cyanine conjugates for dual-modal detection of protease activity via photoacoustic (PA) and fluorescence imaging. The peptide–dye conjugates were designed to undergo contact quenching via intramolecular dimerization and contained n dyes (n = 2, 3, or 4) with n – 1 cleavable peptide substrates. The absorption peaks of the conjugates were blue-shifted 50 nm relative to the free dye and had quenched fluorescence. This effect was sensitive to solvent polarity and could be reversed by solvent switching from water to dimethyl sulfoxide. Employing trypsin as a model protease, we observed a 2.5-fold recovery of the peak absorbance, 330–4600-fold fluorescent enhancement, and picomolar detection limits following proteolysis. The dimer probe was further characterized for PA activation. Proteolysis released single dye–peptide fragments that produced a 5-fold PA enhancement through the increased absorption at 680 nm with nanomolar sensitivity to trypsin. The peptide substrate could also be tuned for protease selectivity; as a proof-of-concept, we detected the main protease (Mpro) associated with the viral replication in SARS-CoV-2 infection. Last, the activated probe was imaged subcutaneously in mice and signal was linearly correlated to the cleaved probe. Overall, these results demonstrate a tunable scaffold for the PA molecular imaging of protease activity with potential value in areas such as disease monitoring, tumor imaging, intraoperative imaging, in vitro diagnostics, and point-of-care sensing.
Keywords: photoacoustic, cyanine, protease, activity, dimer, H-aggregate, trypsin, Mpro
Graphical Abstract
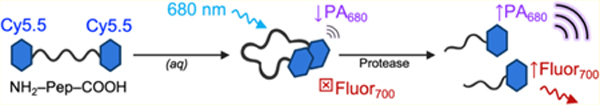
Photoacoustic-ultrasound (PA-US) imaging is a fast-growing imaging modality that combines near-infrared (NIR) optical excitation with ultrasonic detection.1,2 This technique relies on the PA effect—the generation of ultrasound waves by an absorbing molecule following optical absorption and thermoelastic expansion.3 While conventional ultrasound can image anatomical structures in real time, it can only generate contrast from tissue interfaces that differ in their acoustic impedance (i.e., attenuation of the speed of sound). This constraint has motivated the development of ultrasound contrast agents4,5 and also highlighted a benefit of PA-US: the enhancement of contrast through the dependence of PA signal on optical absorption.6–8 In vivo, PA contrast is determined by endogenous molecules with high absorption coefficients in the NIR such as melanin, oxy- and deoxyhemoglobin, and lipids.9–11
Endogenous PA contrast enables a variety of clinical applications that are under active development,12–14 but the range of imaging applications can be further extended with activatable contrast agents that modulate their optical absorbance.15–17 The targets of molecular imaging agents can include cells, nucleic acids, proteins, and other biological molecules; among these, enzymes are particularly attractive due to their critical role in many diseases.18 Proteases often possess highly specific activity and are implicated in cancer, Alzheimer’s disease, cardiovascular diseases, infections, and inflammatory diseases.19,20 Matrix metalloproteases, for example, have been widely studied in this area and motivated the cell-penetrating peptide strategy developed by Tsien and co-workers for fluorescence imaging.21 While fluorescence molecular imaging has proved to be tremendously valuable, particularly for image-guided surgery/resection and in vitro studies, its poor penetration depth restricts it to surface-weighted applications. In contrast, PA imaging can achieve deep tissue imaging, though it commonly sacrifices sensitivity relative to fluorescence.
The primary challenge for PA detection of activity is the efficient generation of an enzyme-specific PA signal.22 These probes can take many forms—one general approach that has been explored involves the self-assembly or aggregation of absorbers following enzymatic cleavage.23 These approaches leverage the macrocyclization of dye molecules or the aggregation of nanomaterials.24–27 Another strategy is the use of dual-absorber conjugates, for example, nanoparticle-dye or dye-quencher systems.28–30 In these designs, two spectrally unique absorbers are linked by a cleavable peptide substrate. These have primarily relied upon one-half of the probe retaining at the target site (e.g., intratumoral or intracellular) after proteolysis, while the other half diffuses away. Though powerful, this technique is limited to applications in which the cleaved fragment can quickly and efficiently diffuse out of the imaging plane. One motivation for this work was to develop a simple probe scaffold with activatable PA signal that does not rely upon in vivo clearance of one of the probe components. Some initial efforts in this area validated the difference in PA lifetime contrast between methylene blue monomers/dimers and proposed an enzyme-activatable mechanism.31,32
Our work uses cyanine dyes because of their utility in chemical detection.33–37 These dyes offer high absorption coefficients, tunable fluorescence quantum yields, and long excitation and emission wavelengths. In biological settings, they have exhibited excellent biocompatibility and low toxicity—indocyanine green, a cyanine derivative, has been in clinical use for over 60 years.38 These favorable properties have driven their integration into a wide variety of chemosensors that can undergo colorimetric and fluorescence changes for the measurement of inorganic ions, pH, small molecules, and biological macromolecules.34,39–41
A fundamental property of cyanine dyes is their propensity to aggregate under concentration-dependent conditions in solution.41 This underpins the well-known formation of H-aggregates and J-aggregates.42 These molecular assemblies differ according to the relative orientations of the transition dipole moments of their constituent molecules whereby H-aggregates maintain a “side-by-side” orientation and J-aggregates maintain a “head-to-tail” orientation.43 Spectroscopically, H-aggregates exhibit a hypsochromic shift in their peak absorbance (blue shift) relative to the monomeric dye, while J-aggregates exhibit a bathochromic shift (red shift).44 In addition to the blue shift, H-aggregate dimers undergo complete fluorescent quenching. The quenching is due to the rapid internal conversion from the upper to lower exciton singlet state, followed by the intersystem crossing to the triplet state (radiative transitions from the lower exciton state are formally forbidden).45 This characteristic previously informed designs on scaffolds that enabled fluorescent monitoring of protein binding or enzyme activity.46,47 However, the PA signal is a function of absorbance, and thus, our interest here was the modulation of the absorbance spectrum following proteolysis. Recently, this property was leveraged in a DNA nanosensor for the PA detection of interferon gamma via J-aggregate forming phthalocyanine dyes.48 In this work, we investigated a set of pentamethine cyanine dye–peptide scaffolds as a platform for PA-based protease activity measurement with tandem fluorescence via the proteolytic conversion of H-aggregate dimers to monomers.
METHODS
Reagents.
Cy5.5-NHS ester was purchased from Lumiprobe Inc. (Maryland, USA). Peptides (ArgArgLys, ArgArgLysArgArgLys, ArgArgLysArgArgLysArgArgLys, GlyHisLys, and GlyThrSerAlaValLeuGlnSerGlyPheArgLys) were purchased from GenScript Inc. (New Jersey, USA). Pooled human saliva was purchased from Lee Biosolutions (Maryland Heights, MO). Dimethyl sulfoxide (DMSO), triethylamine (TEA), trypsin from porcine pancreas, leupeptin, phosphate-buffered saline, sodium chloride, and ammonium bicarbonate were purchased from Sigma-Aldrich (Missouri, USA).
Expression and Purification of Mpro.
The SARS-CoV-2 Mpro plasmid was provided by Rolf Hilgenfeld, University of Lübeck, Germany49 and transformed into Escherichia coli strain BL21-Gold (DE3). The expression and purification of the protein has been described in detail previously.50 Briefly, cells were lysed by homogenization in 20 mM Tris–HCl, pH 7.8, 150 mM NaCl, 0.25 mM DTT, and 5% glycerol, and the lysate was cleared by centrifugation, followed by filtration through a 0.45 μM membrane. Soluble Mpro was purified by Ni2+ chelation chromatography (HisTrap FF, GE Healthcare Life Sciences), and the eluted protein was processed by PreScission protease (GenScript) to remove the C-terminal His-tag. Mpro was separated from the PreScission protease and His-tag using GSTrap FF and HisTrap FF column (GE Healthcare Life Sciences). Active fractions from the flow-through were pooled and stored at −80 °C in 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM DTT, and 5% glycerol.
Synthesis of Dye–Peptide Conjugates.
Dye–peptide conjugates were synthesized via the amide linkage of succinimidyl ester (NHS)-activated dyes with the primary amines of peptides at an equimolar ratio of dye/amine (Scheme S1). For a representative reaction, RRK (0.4 mg, 0.87 μmol) was mixed with Cy5.5-NHS (1.3 mg, 1.74 μmol) in anhydrous DMSO (200 μL) with a 1.5-fold molar excess of TEA (1.30 μmol). Reactions were stirred in the dark at 300 rpm for 12 h at 30 °C. The reaction mixture was dried under vacuum centrifugation using a Vacufuge Plus (Eppendorf, Hamburg) at 60 °C for 2–3 h.
Purification.
Crude reaction pellets were resuspended in MeCN/H2O (50:50 v/v) and purified via analytical reversed phase-high-performance liquid chromatography (RP-HPLC) with a Shimadzu LC-40 on a Shim-pack GIS C18 column (5 μm). Crude reaction mixtures were separated with a 50–95% B gradient [A: H2O 0.05% trifluoroacetyl (TFA) and B: MeCN (0.05% TFA)] over 45 min. Fractions were collected and diluted to 90% (v/v) MeOH before analysis with electrospray ionization mass spectrometry (ESI-MS) in positive mode (centroid scan) on a Micromass Quattro Ultima Triple Quadrupole mass spectrometer. Products were concentrated via vacuum centrifugation or lyophilization and stored in the dark.
Absorbance and Fluorescence Spectroscopy.
Absorbance and fluorescence spectra were measured with a BioTek Synergy H1 plate reader. Samples were measured at the 100 μL scale in 96-well plates. Unless otherwise noted, absorbance scans were collected from 500 to 850 nm and fluorescence emission scans were collected with an excitation wavelength of 600 nm and an emission wavelength range of 660–860 nm in 2 nm increments.
Nuclear Magnetic Resonance Spectroscopy.
The dry compound was dissolved in a deuterated solvent, and 1H NMR spectra were collected in mixtures of D2O/DMSO-d6 with a 300 MHz Bruker spectrometer. Solvent mixtures were varied by drying the compound between scans and then resuspending with different D2O/DMSO-d6 ratios.
PA Imaging.
PA images of in vitro samples were acquired with a Vevo 2100 LAZR (VisualSonics) using a 21 MHz transducer (LZ-250). Samples (20 μL) were loaded into individual 0.86 mm polyethylene tubes and fixed in parallel within a 3D-printed holder 1 cm below the transducer in a vessel filled with water.51 Single-wavelength scans were operated at 680 nm with a repetition rate of 20 Hz (pulse width = 4–6 ns, ∼45 mJ/pulse at source).52 To generate 3D images, the transducer was scanned with a stepper motor (step size = 0.054 mm) along the axial dimension of the tubes and B-mode frames were registered via maximum intensity projection. PA spectra were measured from 680 to 900 nm (2 nm step size). Whole human blood was collected in citrate tubes from a healthy male donor according to institutional guidelines. All animal experiments were performed in accordance with NIH guidelines approved by the Institutional Animal Care and Use Committee (IACUC) under protocol S15050 at the University of California, San Diego. Nude mice (NU/J) were anesthetized with isoflurane and injected subcutaneously with 100 μL of the probe or buffer in 50% (v/v) Matrigel. Mice were imaged with a 40 MHz PA/ultrasound transducer (LZ-550).
RESULTS AND DISCUSSION
Synthesis of Dye–Peptide Conjugates.
The goal of this study was to synthesize and optically characterize a set of peptide–dye conjugates for their utility in PA detection of proteolysis (Figure 1A). The ideal molecular scaffold could be generalizable and repurposed to a variety of target proteases by tuning its cleavable sequence. Here, we studied the utility of carbocyanine dimers formed via forced proximity with peptide linkers and assayed using the representative protease trypsin. Peptide–dye conjugates were synthesized via the reaction of succinimidyl ester activated dyes with the N-terminal primary amines and C-terminal lysine side chains of the peptides reported herein (Scheme S1 and Figure S1). The primary scaffold we studied was [Cy5.5]2[RRK]1 (Figure S1A); Cy5.5 was chosen for its absorbance in the NIR and arginine residues were included for recognition and cleavage by trypsin, a readily available and well-understood protease.
Figure 1.
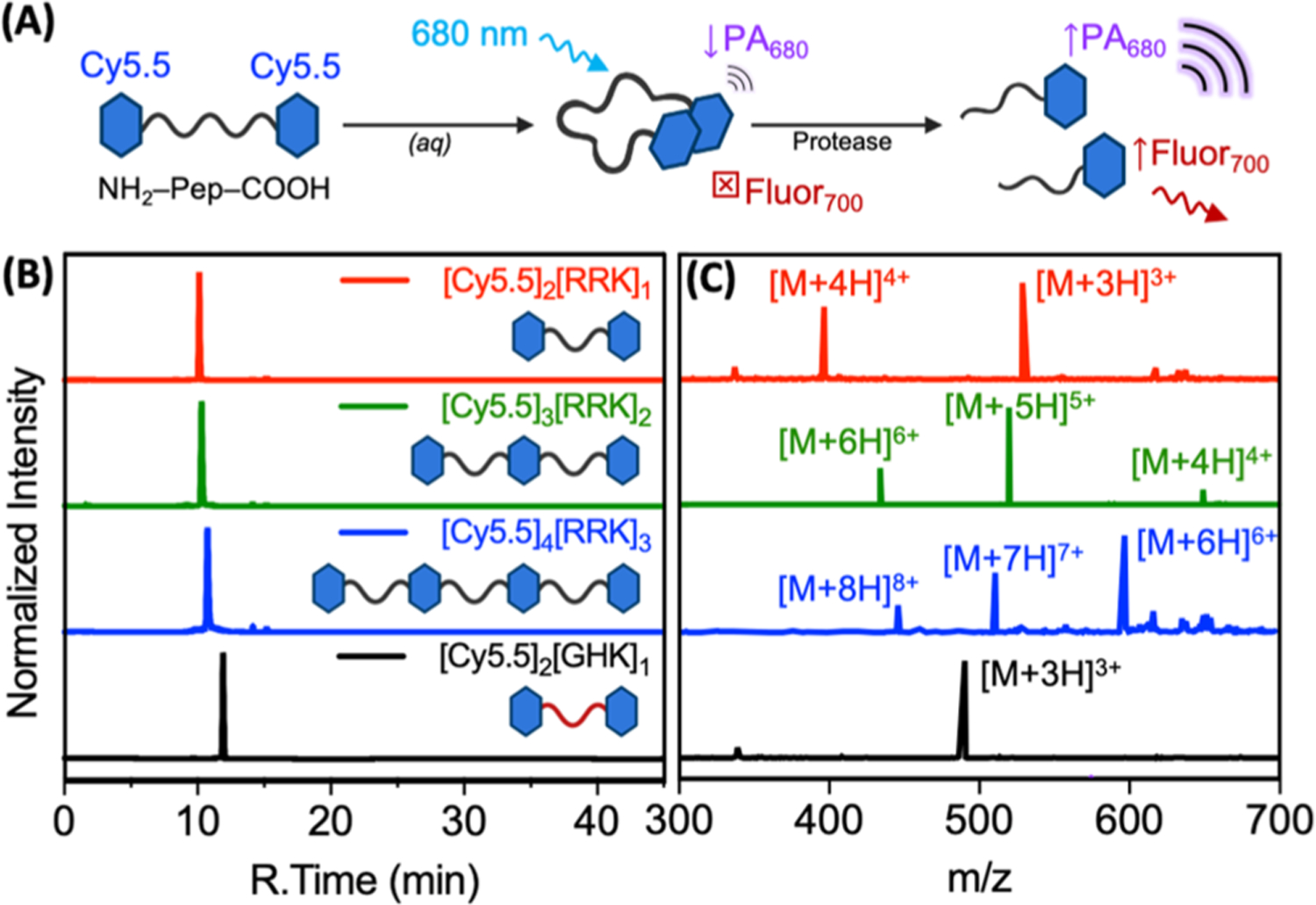
RP-HPLC and ESI-MS of protease-responsive cyanine–peptide conjugates. (A) Schematic of the probe design. (B) Liquid chromatograms of purified [Cy5.5]2[RRK]1 (96.7%), [Cy5.5]3[RRK]2 (91.5%), [Cy5.5]4[RRK]3 (96.5%), and [Cy5.5]2[GHK]1 (97.2%) monitored via HPLC. The retention times were 10.1, 10.3, 10.8, 11.9, and 12.7 min, respectively. Samples were dissolved in 25% acetonitrile and eluted at 1 mL/min with a 25 min gradient from 25 to 95% B [A: water (0.05% TFA) and B: acetonitrile (0.05% TFA)]. The gradient was held at 95% B for 10 min and reduced to 25% B over the next 10 min (total time: 45 min). (C) ESI-MS of the respective conjugates. [Cy5.5]2[RRK]1 expected [M + 4H]4+: 398.0, detected: 398.1, expected [M + 3H]3+: 530.3, and detected: 530.5 (Figure S2). [Cy5.5]3[RRK]2 expected [M + 6H]6+: 433.1, detected: 433.3, expected [M + 5H]5+: 519.5, detected: 519.8, expected [M + 4H]4+: 649.1, and detected: 649.5 (Figure S3). [Cy5.5]4[RRK]3 expected [M + 8H]8+: 450.8, detected: 450.9, expected [M + 7H]7+: 515.0, detected: 515.2, expected [M + 6H]6+: 600.7, and detected: 600.9 (Figure S4). [Cy5.5]2[GHK]1 expected [M + 3H]3+: 490.9 and detected: 490.9 (Figure S5).
Our utilization of a tripeptide linker stemmed from two design goals: (1) maximizing the likelihood of contact quenching between the dyes and (2) minimizing the likelihood of a nonspecific cleavage by off-target proteases. Though RRK can be cleaved at its first or second peptide bond, both cleavage sites were included to mitigate against the possibility of steric hindrance by the dyes. These conjugates were purified with RP-HPLC (Figure 1B) and validated with ESI-MS (Figure 1C). In order to explore the effect of multiple cleavage sites and dye molecules, we also synthesized conjugates with multiple dye–peptide subunits, that is, a dye trimer, [Cy5.5]3[RRK]2 (Figure S1B), and a dye tetramer, [Cy5.5]4[RRK]3 (Figure S1C). The column retention times of these conjugates increased with the addition of peptide–dye subunits: 10.1, 10.3, and 10.8 min for [Cy5.5]2[RRK]1, [Cy5.5]3[RRK]2, and [Cy5.5]4[RRK]3, respectively. We also synthesized [Cy5.5]2[GHK]1 as a negative-control probe for trypsin proteolysis (Figure S1D).
Solvatochromic Properties of Conjugates.
It is well known that cyanine dyes in an aqueous solution exhibit intermolecular interactions observable as shifts in their peak absorbance wavelength (λmax). Here, we used peptide linkers to facilitate these dimerizing interactions intramolecularly, that is, between two or more dye structures within a single conjugate (e.g., the covalent dimer, [Cy5.5]2[RRK]1). We also synthesized [Cy5.5]3[RRK]2 and [Cy5.5]4[RRK]3 conjugates, which are referred throughout as trimers and tetramers, respectively.
In order to assess optical differences in these molecules, we first suspended them in DMSO to solubilize the dyes, favoring the monomeric state, and titrated their concentrations to an equivalent λmax. One might expect solubilization of these molecules in DMSO to fully neutralize the dye–dye interactions. However, the absorbance spectra of these molecules revealed a minor increase in the ratio of OD640/OD686 relative to the free dye (Figure 2A), and this ratio increased with respect to the number of dye moieties. This trend corresponded to the increased fluorescence quenching (Figure 2B). However, the absorbance shift and fluorescence quenching were significantly more prominent when the conjugates were suspended in water (1% DMSO, Figure 2C,D). The λmax shifted from 676 nm for free dye to 624 nm for [Cy5.5]2[RRK]1, 616 nm for [Cy5.5]3[RRK]2, and 612 nm for [Cy5.5]4[RRK]3 (Figure 2C). These shifts were accompanied by intensity reductions of the hypsochromic peak and peak broadening with increasing dye units; the total area under the curve (AUC) remained relatively constant (<1% RSD). Significant changes in the absorption at the monomeric peak (676 nm) were not observed between the three conjugates. Fluorescence quenching efficiencies (QEs) of the molecules in water relative to DMSO were greater than 98% for [Cy5.5]2[RRK]1, [Cy5.5]3[RRK]2, and [Cy5.5]4[RRK]3 (Figure 2D). When compared to the free dye QE (69.5%), which is due to the aggregation-associated intermolecular quenching, these values reveal the significant effect of intramolecular quenching. We confirmed the role of π–π stacking and hydrophobic effects in the absorbance peak shift and fluorescence quenching for [Cy5.5]2[RRK]1 by monitoring the upfield shift of its aromatic hydrogens after water addition via 1H NMR (Figure S6).53,54
Figure 2.
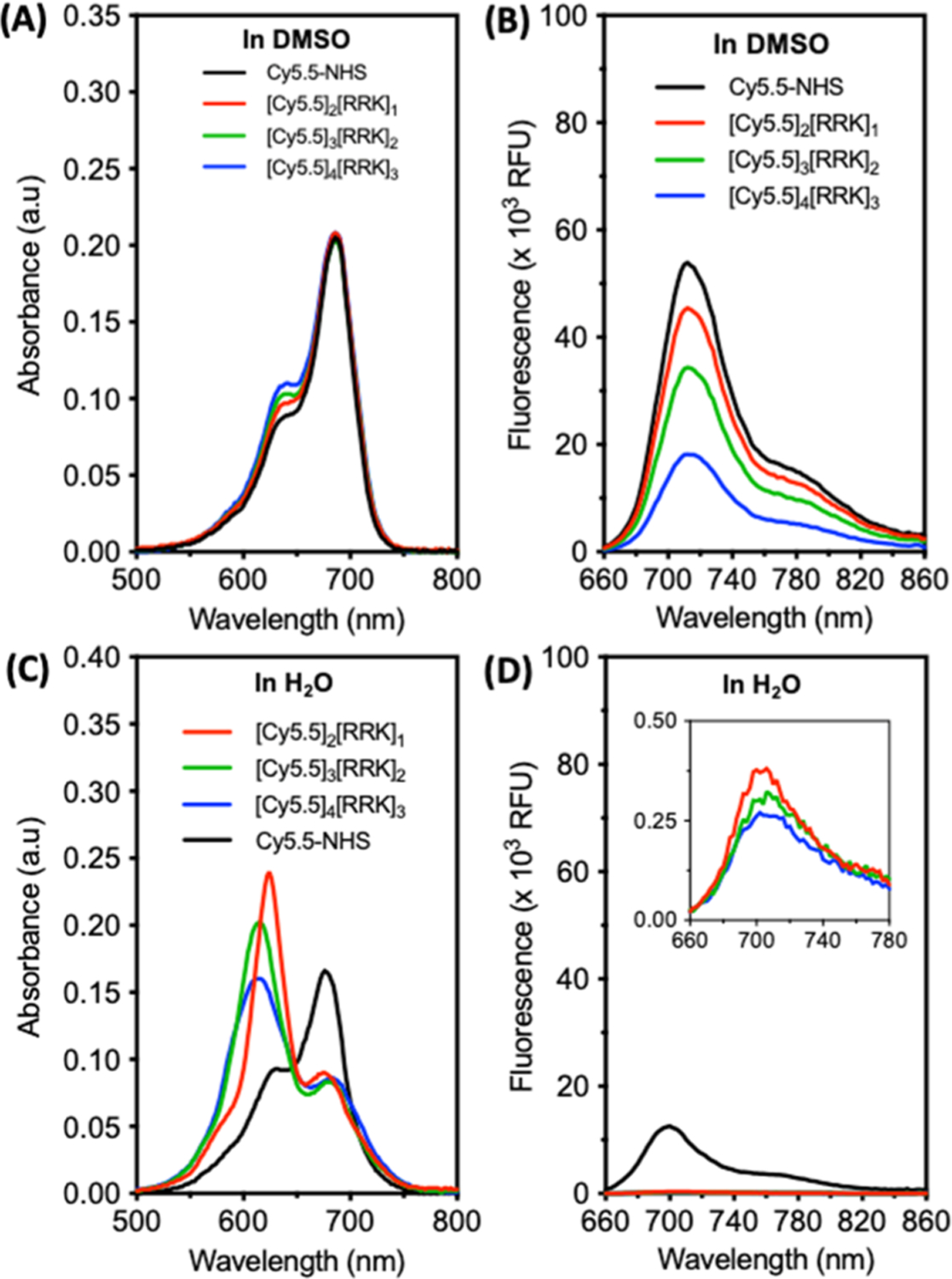
Absorbance and fluorescence spectra of cyanine–peptide conjugates are modulated by the self-association of their dye moieties. (A) Absorbance spectra of free dye, dimer, trimer, and tetramer conjugates after suspension in DMSO at an equimolar concentration of dye moieties. This was done by titrating each solution to an equivalent λmax corresponding to the absorbance maximum of Cy5.5-NHS (OD686 = 0.21). (B) The emission spectra in DMSO revealed a decrease in fluorescence from dimer to trimer to tetramer, indicating more persistent π–π interactions in the higher-order species (ex: 600 nm and em: 660–860 nm). (C) Absorbance spectra of the conjugates diluted to the same concentrations in water (1% v/v, DMSO/H2O) reveal significant blue shifts in each case and peak broadening for the trimers and tetramers. (D) The emission spectra in water show >98% fluorescence quenching for all conjugates (inset: magnification reveals minor relative differences in QEs).
Absorbance- and Fluorescence-Based Activity Measurements.
The absorbance spectra of probes at equimolar concentrations were monitored before and after the addition of trypsin in the ammonium bicarbonate buffer. In the buffer, charge screening induced by the higher ionic strength favors dye aggregation and reduces the intensity of absorption peaks observed in Figure 3A. Nevertheless, the AUC of the probes increased from dimer to trimer to tetramer, indicative of the increasing dye content across the three conjugates. The ability for trypsin to cleave these probes was first confirmed for [Cy5.5]2[RRK]1 using HPLC and ESI-MS (Figure S7). The intact probe eluted at 9 min; after incubation with trypsin, new peaks were observed at 6.5, 6.9, 8.2, and 8.3 min. The latter two peaks were collected as a single fraction, and it contained Cy5.5-R measured via ESI-MS. The 6.9 min fraction contained RK-Cy5.5. A noncleavable sequence was used to synthesize the control probe, [Cy5.5]2[GHK]1, which did not exhibit trypsin-catalyzed activation (Figure S8). This confirmed the specificity of the synthesized probes.
Figure 3.
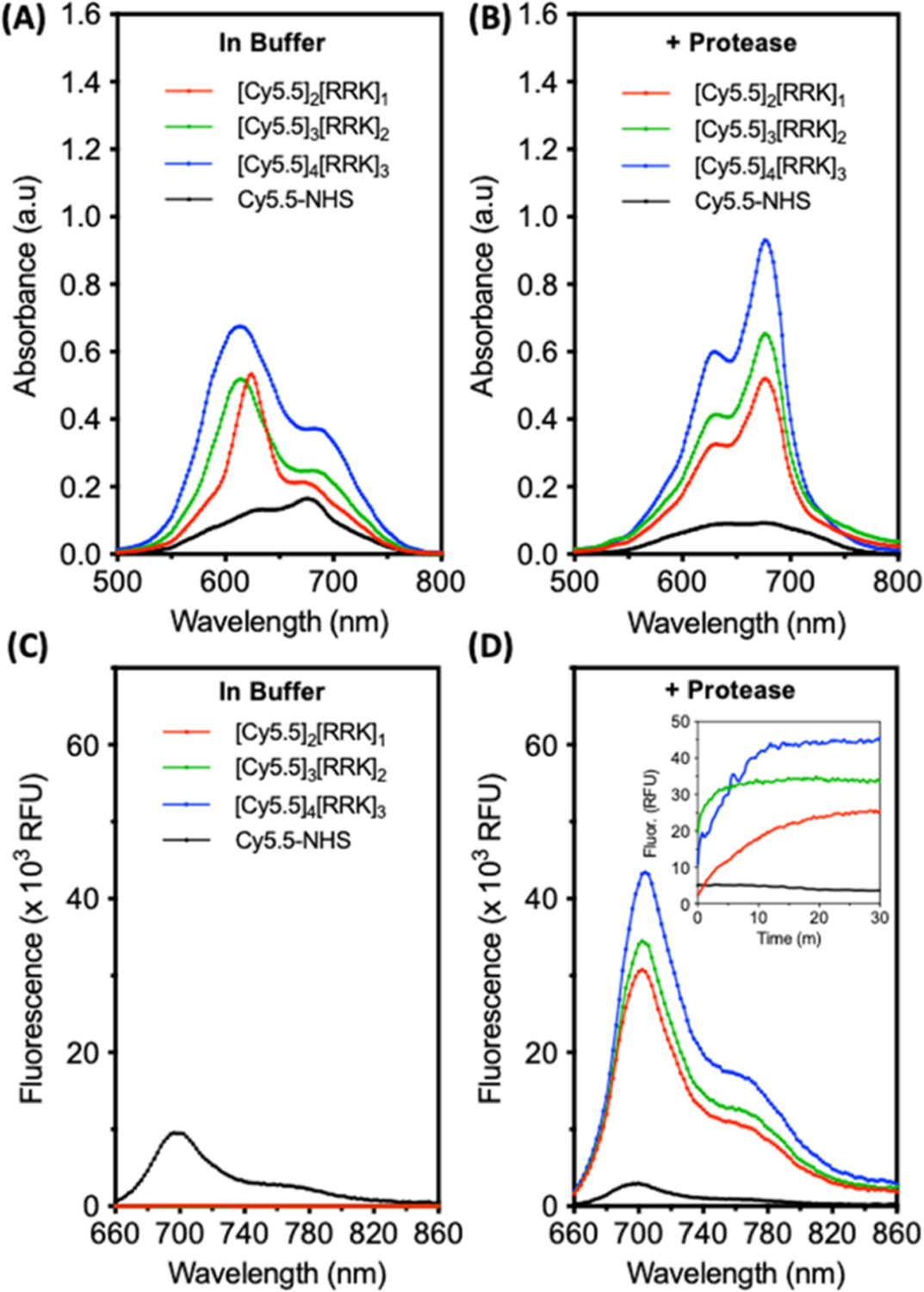
Proteolysis of cyanine–peptide conjugates in the buffer induces a red shift and fluorescent activation. (A) Absorbance spectra of free dye, dimer, trimer, and tetramer conjugates at 7 μM in buffer (10 mM NH4HCO3, 1% DMSO, and pH 8.0). (B) Absorbance spectra of the molecules after incubation with 5 μM trypsin (30 min, 37 °C). (C) Fluorescence spectra of the molecules in the buffer (ex: 600 nm); inset: kinetic monitoring of fluorescence after the addition of 5 μM trypsin. (D) Fluorescence spectra after 30 min of incubation with trypsin. Inset: fluorescence activation from 0 to 30 min.
After proteolysis, the monomeric spectroscopic peak shape of the probes was regained with relatively similar ΔOD676 values between pre- and postcleaved conjugates of 2.46, 2.64, and 2.51 for the dimer, trimer, and tetramer, respectively (Figure 3B). Prior to proteolysis, fluorescence intensities at 700 nm were 92, 14, and 9 RFU for the dimer, trimer, and tetramer, respectively (Figure 3C). These increased 332-fold, 2436-fold, and 4652-fold, respectively, after 30 min of incubation in 5 μM trypsin (Figure 3D). It is interesting to note that the fluorescence contrast increased with a higher dye content, but ΔOD676 remained relatively constant between probes. For spectral comparison, the absorbance and fluorescence of free dye (Cy5.5-NHS) were also measured. It is clear that the free dye does not enhance absorbance or fluorescence after incubation with trypsin. Instead, we observed some broadening of the absorbance and reduction in fluorescence over the measurement period due to the hydrolysis of the activated dye, contributing to the self-association of the dye monomers and gradual precipitation (leading to contact quenching). There is also a possibility that some activated dye bound to exposed lysine residues on trypsin. Nevertheless, we conclude that the observed optical enhancements of the probes (red-shifting and turn-on fluorescence) were not caused by the nonspecific interaction of the dyes with trypsin. This is further supported by the lack of red-shifting and fluorescence for the noncleavable [Cy5.5]2[GHK]1 control probe upon incubation with trypsin (Figure S8).
Fluorescence Sensitivity and Kinetics.
According to fluorescence kinetics, [Cy5.5]2[RRK]1 exhibited maximum activity at approximately 7 μM (Figure 4A). The initial rate decreased at higher concentrations, likely due to the decreasing solubility at these values. The concentrations for peak activity were slightly lower for the trimer and tetramer, though the rate appeared to increase proportionally to the number of peptide–dye subunits (Figure 4A). The increase in signal magnitude was logical since the trimer and tetramer have 1.5 times and 2 times more dye content than the dimer.
Figure 4.
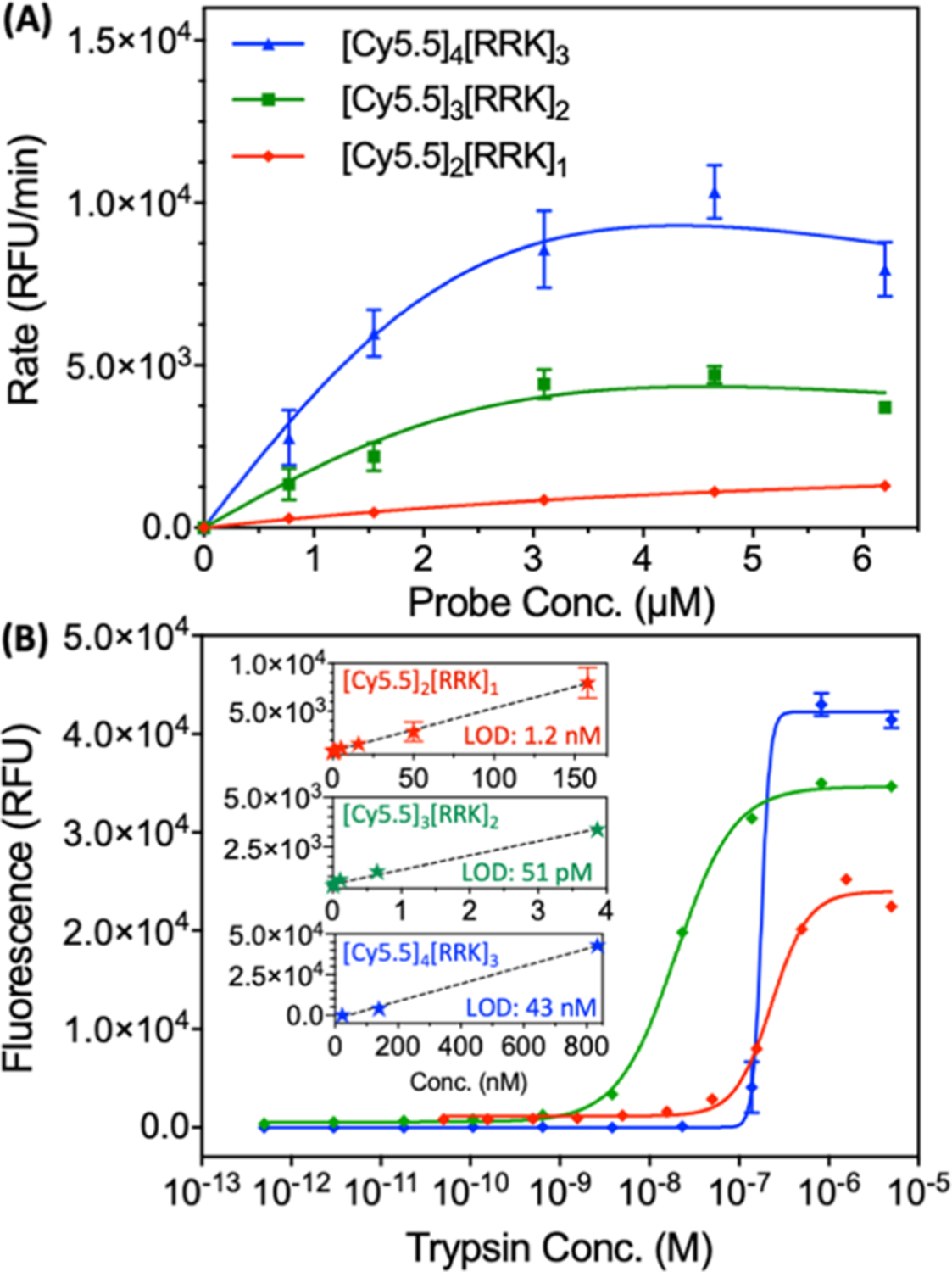
Kinetic measurements and fluorescent LODs for [Cy5.5]2[RRK]1, [Cy5.5]3[RRK]2, and [Cy5.5]4[RRK]3. (A) Probe concentration vs initial rate for [Cy5.5]2[RRK]1, [Cy5.5]3[RRK]2, and [Cy5.5]4[RRK]3 with 5 μM trypsin (ex: 600 and em: 700). Higher concentrations were not included due to the low aqueous solubility of the [Cy5.5]3[RRK]2 and [Cy5.5]4[RRK]3 probes at these values. Measurements were performed in triplicate (error bars = standard error of the mean). (B) Fluorescent LODs for all probes at 7 μM incubated with a trypsin gradient in 10 mM NH4HCO3 and 1% DMSO for 1 h at 37° C (ex: 600 nm). LODs were calculated to be 1.2 nM for [Cy5.5]2[RRK]1, 51 pM for [Cy5.5]3[RRK]2, and 43 nM for [Cy5.5]4[RRK]3. Insets show the linear regions used to calculate LODs. Measurements were performed in triplicate (error bars = standard deviation).
We also calculated and compared the fluorescent limits of detection (LODs) for trypsin of the three conjugates at an equimolar probe concentration (Figure 4B). Representative spectral titrations are included for [Cy5.5]2[RRK]1 (Figure S9A), [Cy5.5]3[RRK]2 (Figure S9B), and [Cy5.5]4[RRK]3 (Figure S9C). The LODs were calculated based on the linear ranges from 0.05 to 158 nM (R2 = 0.9297) for [Cy5.5]2[RRK]1, 0.0005–3.9 nM (R2 = 0.9210) for [Cy5.5]3[RRK]2, and 23–834 nM (R2 = 0.9929) for [Cy5.5]4[RRK]3 according to the method described by Armbruster et al.55 The LODs were 1.2 nM, 51 pM, and 43 nM, respectively. By comparison, the best fluorescent LODs reported for trypsin probes are in the low picomolar range.56
One possible explanation for the differences in sensitivity is the fact that the addition of [RRK][Cy5.5] subunits increases the total charge (z) of the molecule, increasing its affinity for the negatively charged catalytic pocket. However, it also decreases the solubility as evidenced by the longer LC retention times (Figure 1B). The increased affinity hypothesis is supported by comparison of the initial rates of activation for each probe (Figure 4A). Indeed, the average rate of product formation increased from dimer (z = +4) to trimer (z = +7) to tetramer (z = +10). Due to the higher rate of ES formation (kf), it would be reasonable for [Cy5.5]3[RRK]2 to have a lower LOD than [Cy5.5]2[RRK]1. However, increasing the charge further (z = +10 for [Cy5.5]4[RRK]3) proved to be detrimental to the sensitivity of the probe to low trypsin concentrations.
For this bulky and highly charged probe, unfavorable steric or electrostatic interactions with enzyme residues adjacent to the active site could potentially increase kr > kcat. Another possibility is that the lower solubility of the tetramer led to gradual aggregation and reduction of the number of accessible cleavage sites as the probe incubated, causing poor sensitivity to low concentrations of trypsin. Nevertheless, at protease concentrations well above the LOD, the rate of probe activation was proportional to the number of peptide–dye subunits. Kinetic constants for the probes were not extracted due to low aqueous solubilities at higher concentrations.
PA Characterization of Conjugates.
A range of probe concentrations were incubated with equimolar trypsin to determine the minimum probe concentration required for imaging, that is, that would maximize the PA signal-to-background ratio (SBR) (Figures S10 and 5). Concentrations between 1 and 4.5 μM were insufficient for strong contrast between the cleaved and uncleaved probe because noise-free PA images could not be collected (Figure S10A). The cleaved probe at 1.12 and 2.25 μM showed no significant difference in the PA intensity and 4.50 μM had low contrast (Figure S10B). However, concentrations between 6.75 and 11.25 μM had a significant contrast and were therefore suitable for PA imaging (Figure 5E,F). Of note, the fold-differences quantitated in Figure 5F are underestimated because white pixels in Figure 5E are saturated pixels.
Figure 5.
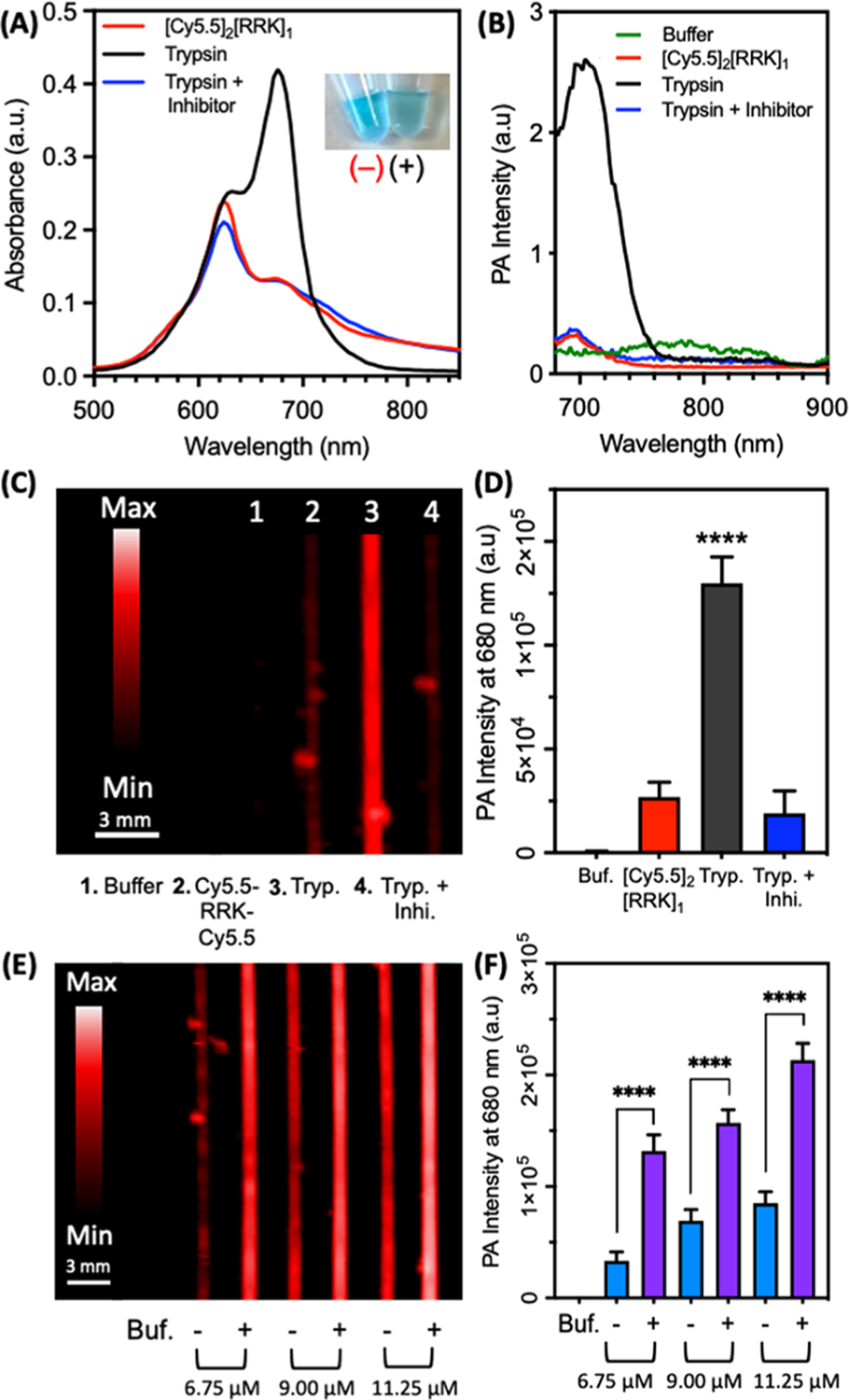
PA imaging of the protease-dependent [Cy5.5]2[RRK]1 activation. (A) Absorbance spectra of [Cy5.5]2[RRK]1 (7 μM) with trypsin (5 μM) and trypsin/inhibitor (50 μM leupeptin) after 1 h of incubation in 100 mM NH5CO3 (1% DMSO) at 37° C. Photographic inset shows probe before (−) and after (+) cleavage by trypsin. The color dims due to the increased contribution of longer wavelength absorption. (B) PA spectra of the same samples, showing signal activation only in the probe + trypsin sample. Units of PA intensity are arbitrary. (C) PA image of the samples at 680 nm. (D) Quantitation of (C) via integrated pixel density. Asterisks denote the significant difference between [Cy5.5]2[RRK]1 and trypsin and trypsin with inhibitor (unpaired t-test, p-value < 0.0001, n = 8 regions of interest, error bars = SD). (E) PA image of [Cy5.5]2[RRK]1 at three concentrations before (−) and after (+) incubation with trypsin. (F) Quantitation of (E) via integrated pixel intensity. Asterisks denote significant difference (unpaired t-test, p-value < 0.0001, n = 8 regions of interest, error bars = SD).
The specificity of the probe was verified by further examining the absorbance and fluorescence in parallel to PAs with trypsin alone and trypsin inhibited by the small molecule, leupeptin (Figure 5).57 Following the cleavage by trypsin, the probe increased the absorbance at 680 nm, leading to a noticeable color change (Figure 5A) and activation of fluorescence (Figure S11). The PA spectrum followed a similar trend as the absorbance (Figure 5B); this correlation is generally predicted from the principle that the PA signal intensity of a molecule is determined by the amount of light it absorbs. Indeed, validation of this hypothesis for a peptide-linked cyanine dye pair was a primary goal of this investigation. Single-wavelength PA scans at 680 nm showed a significant contrast between the inactivated and cleaved probe (Figure 5C). The signal enhancement between these two species was about 5-fold (Figure 5D). Additionally, mixtures of probe, trypsin, and inhibitor did not lead to PA activation. The PA sensitivity of [Cy5.5]2[RRK]1 to enzyme was lower than its fluorescence- and absorbance-based sensitivity (Figure S12). The absorbance spectra for the probe with 0–5 μM trypsin revealed an increase in the ratio of OD680/OD624 at increasing protease concentrations (Figure S12A), with distinct intensities. Fluorescent activation at 700 nm showed a similar trend (Figure S12B). PA imaging at 680 nm was unable to resolve the difference between 0 and 40 nM enzyme after 1 h; however, the higher tested concentrations were readily detectable (Figure S12C,D). Overall, this difference in sensitivity was not surprising, as PA imaging commonly involves a trade-off between sensitivity and the ability to perform real-time imaging with high penetration depths.
To validate the tunability of the probe scaffold for other protease targets, we synthesized a variation of the homodimer by exchanging the RRK peptide with GTSAVLQSGFRK (Figure S13). The peptide sequence AVLQSGFR is a previously reported substrate for the cleavage at Q/S by the main protease (Mpro or 3CLpro) involved in the replication of SARS-CoV-2 during viral infection.49,58 This has motivated the development of both inhibitors and other sensors targeting Mpro.59 After the synthesis and purification of the probe [Cy5.5]2[GTSAVLQSGFRK]1, we used HPLC and ESI-MS to validate the cleavage of the probe with recombinant Mpro and observed the expected fragments (Cy5.5-GTSAVLQ and SGFRK-Cy5.5) after incubation of the probe with 100 nM Mpro for 1 h (Figure S13A,B). We measured an approximate Km of 3.5 μM (Figure 6A) via fluorescence and used this probe concentration for titration with Mpro (0–200 nM). Similar enhancements of absorbance, fluorescence, and PA intensity were observed (Figure 6B–F) as with [Cy5.5]2[RRK]1 and trypsin. We detected a lower limit of 50 nM Mpro in buffer with photoacoustics. Aside from the difference in the amino acids used, the major difference was the length of the peptide linker (12 vs 3 residues). We attribute the retained activity to the flexibility of the peptide backbone enabling intramolecular dimerization to occur even with a longer linker. To the best of our knowledge, this is the first demonstration of a PA sensor for a SARS-CoV-2 biomarker. Nevertheless, more work is required to demonstrate its value in preclinical models of disease or clinical samples.
Figure 6.
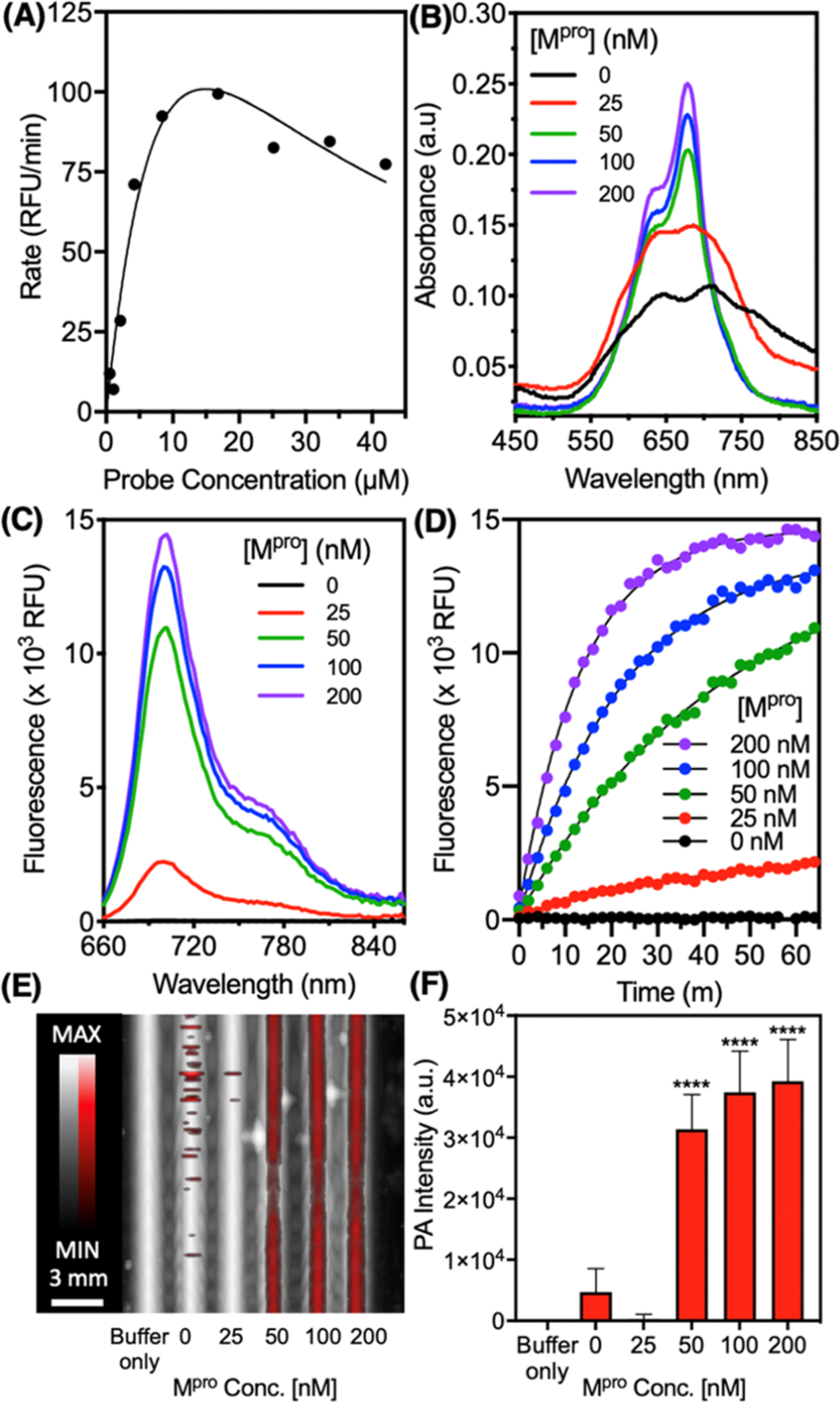
Optical and PA properties of the Mpro-responsive probe [Cy5.5]2[GTSAVLQSGFRK]1. (A) Concentration-dependent probe activity. The probe was incubated at different concentrations with 50 nM Mpro (20 mM Tris, 150 mM NaCl, and pH 8.0) at 37 °C and the initial rate of fluorescent activation was recorded. (B) The probe (3.5 μM) was incubated with increasing concentrations of Mpro and the absorbance spectra were recorded after 1 h, showing enhancement at 680 nm. (C) Fluorescence spectra after 1 h and (D) fluorescence kinetics were recorded (ex: 600 nm and em: 700 nm). (E) Dual PA-US image of the samples excited at 680 nm. The PA signal (red) is overlaid on ultrasound (white). (F) Quantitation of Panel E via the raw integrated density of the PA signal (n = 8 regions of interest, error bars = SD), and asterisks denote the significant difference from probe-only (unpaired t-test, p < 0.05).
One challenge for the SBR of the probe is the presence of exogenous absorbers such as hemoglobin. While we do not envision this probe design for applications involving intra-venous delivery, we measured the PA intensity of the molecule (30 μM) in 25 and 50% whole blood and found that the signal was enhanced relative to blood alone (Figure S14A-C). We also observed that blood improves the photostability of the probes, which normally have a PA half-life of only a few minutes due to the photobleaching of the carbocyanine dyes (Figure S14D). Unfortunately, cleaved probe was not readily distinguished from the uncleaved in solution with 25 and 50% blood. We attribute this difficulty to the significant absorption of hemoglobin, which has a concentration of ∼15 g/dL in whole blood.60 In human tissue, the average concentration of hemoglobin is actually much lower, ranging from ∼0.18 g/dL in healthy tissue to ∼0.58 g/dL in cancerous tissue.61 Alternatively, there may be background activation in blood for the “uncleaved” samples. Follow-up work will explore the efficacy of the probe design in such conditions.
Activatable imaging was achieved in saliva (Figure S15)—a biofluid that contains numerous proteases associated with oral disease. For example, gingipains are a class of trypsin-like protease expressed by the periodontal pathogen Porphyromonas gingivalis and have been found in the gingival sulcus at micromolar concentrations.62 After spiking 50% saliva with trypsin, we observed a 2-fold increase in the PA intensity between the cleaved and uncleaved probe and inhibition of the signal in the presence of an inhibitor (leupeptin). Activatable probes may be of use for diagnostic imaging in future PA-based dental applications.7,63,64
Last, we characterized the in vivo PA intensity and LOD for activated [Cy5.5]2[RRK]1 via subcutaneous injection in nude mice (Figure 7). First, the probe was cleaved in solution via incubation with trypsin and titrated across a range of concentrations (Figure S16). Absorbance red shifts, fluorescence enhancement, and PA enhancement were observed for each of the tested concentrations. While first demonstrating the efficacy of the probe across a range of concentrations, this data also confirmed that intramolecular interactions were not surpassed by intermolecular interactions even at higher concentrations. After the subcutaneous injection, the PA intensity was quantified via integrated density (Figure S17) and these values correlated with the probe concentration for the tested range between 0 and 30 μM (Figure 7A–D). The minimum amount of detectable probe was determined to be 0.3 μM (Figure 7E); of course, higher concentrations (e.g., 10–30 μM) would provide a higher SBR in tissue for further applications of this scaffold. Nevertheless, for surface-weighted imaging applications, we found that the injected probe could be spectrally distinguished from the background signal in the surrounding dermis and tissue—this spectral acoustic imaging is a key strength of the PA imaging modality (Figure 7F).
Figure 7.
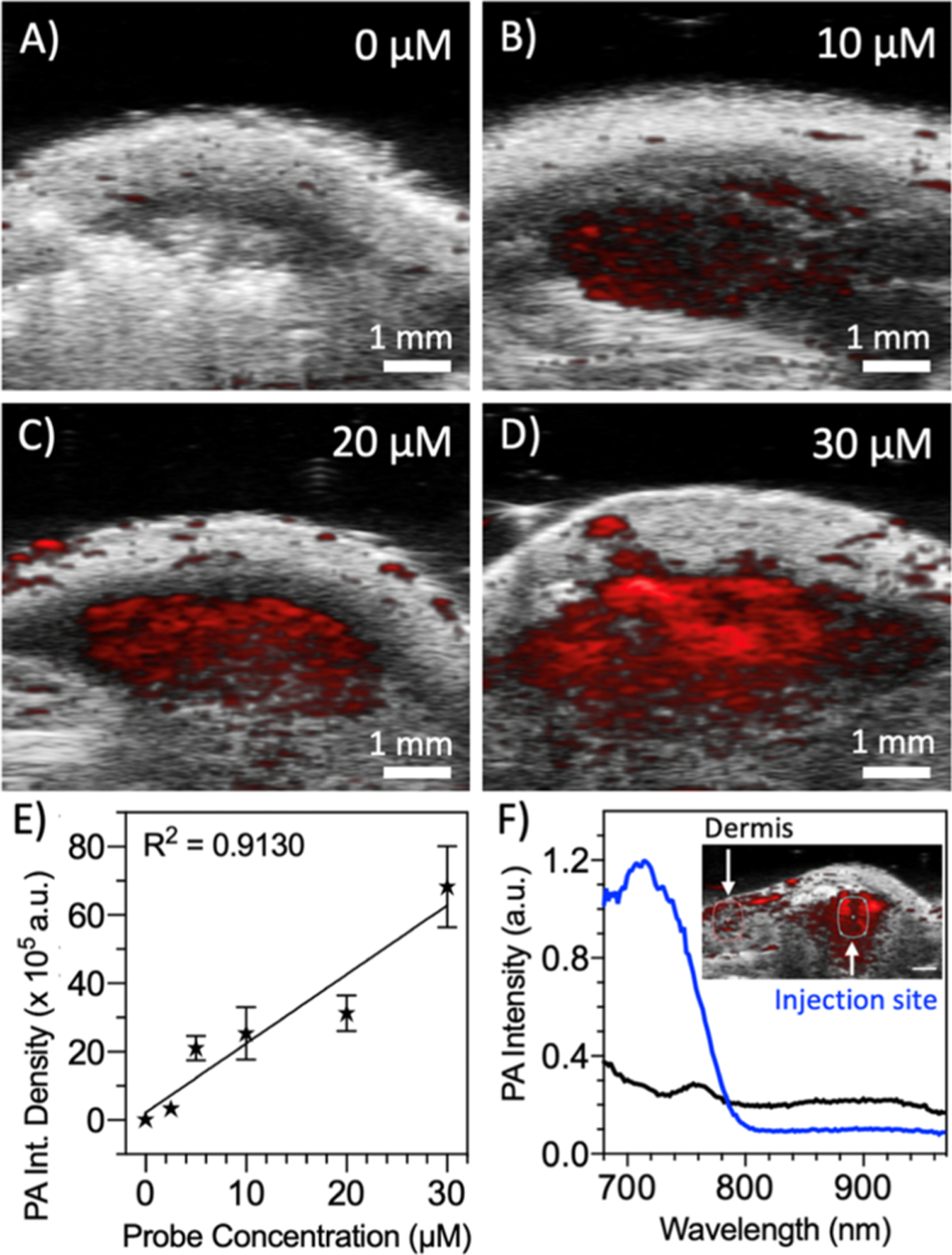
PA imaging of activated [Cy5.5]2[RRK]1 in vivo via subcutaneous injection. A stock solution of [Cy5.5]2[RRK]1 (40 μM) was incubated with 5 μM trypsin for 1 h in a NH5CO3 buffer and diluted to five lower concentrations for the subcutaneous injection in nude mice (n = 3) after mixing with 50% v/v Matrigel: probe. Representative B-mode PA/US images are shown for (A) vehicle only, (B) 10, (C) 20, and (D) 30 μM probes. (E) Regions of subcutaneous PA intensity were quantitated via the integrated pixel density and plotted vs the probe concentration to determine the LOD of the cleaved probe (y = 20,206 × x + 218,48, R2 = 0.9130, LOD = 0.3 μM, n = 3, error bars = SD). (F) PA spectra of the injection site for the 20 μM probe showed a peak corresponding to [Cy5.5]2[RRK]1, distinguishing it from the relatively flat signal from the dermis (inset: PA-US image with ROIs for spectral analysis).
Quantitative protease imaging in vivo is a challenge—many factors can influence the measured PA intensity at any given time. These may include differences in the optical fluence due to tissue variation, laser fluctuations, inflammation contributing to changes in blood perfusion (and total hemoglobin), photostability, and changes in the local concentration of the probe. One approach to improve the quantitative ability of the probes in vivo is to engineer a ratiometric signal. Future efforts will investigate the coupling of more red-shifted dyes to potentially leverage ratiometric PA imaging in the excitable range of existing PA scanners (e.g., 680–970 nm).
CONCLUSIONS
We investigated the performance of a peptide-linked cyanine dimer, [Cy5.5]2[RRK]1, for the simultaneous PA imaging and fluorescent monitoring of the enzyme activity. The dyes were attached to the N-terminus and C-terminal lysine of a trypsin-cleavable peptide [RRK] via amide linkage. This conjugate exploited the intramolecular association of cyanine dyes to blue-shift its peak absorbance by >50 nm and undergo contact quenching, achieving an off state. Following the cleavage of the peptide substrate, the dyes were separated, and PA and fluorescent signals were activated. We compared the cleavable dimer to a trimer and tetramer, such that n – 1 peptide spacers [RRK] were linked between n cyanine dyes. A 2.5-fold recovery of the peak absorbance was observed for all conjugates. Fluorescent detection limits of trypsin were 1.2 nM for the dimer, 51 pM for the trimer, and 43 nM for the tetramer. PA imaging of the dimer revealed a 5-fold signal enhancement at 680 nm with nanomolar sensitivity to trypsin. The activated probe scaffold was imaged subcutaneously in mice and its signal was linearly correlated to the concentration. The probe could be tuned for other protease targets by changing the peptide substrate, and we demonstrated PA detection of Mpro associated with SARS-Cov-2 using [Cy5.5]2[GTSAVLQSGFRK]1. The performance of this dye–peptide scaffold demonstrates a readily synthesized and tunable platform for PA/fluorescence monitoring of protease activity with potential value in areas such as disease monitoring, tumor imaging, intraoperative imaging, in vitro diagnostics, and point-of-care sensing.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge funding from the National Institutes of Health via R01 DE031114, R21 AI157957–01, R21 DE029917–01, DP2 HL137187-S1, R21 AG065776-S1, and S10 OD021821. This publication was supported in part by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1650112 and the National Cancer Institute of the National Institutes of Health under the award number T32 CA153915. C.M. graciously acknowledges support from the ARCS (Achievement Reward for College Scientists) Foundation. J.V.J. also acknowledges the generous support of The Shiley Foundation.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssensors.1c00518.
Synthetic scheme and structures of conjugates, mass spectra, 1H NMR spectra of [Cy5.5]2[RRK]1 in D2O/DMSO-d6 mixtures, HPLC-MS monitoring of proteolysis for probes and controls, fluorescence titration spectra, PA sensitivity experiments, HPLC-MS of the Mpro-responsive probe, PA signal in blood and saliva, and in vivo image analysis (PDF)
Notes
The authors declare no competing financial interest.
Contributor Information
Colman Moore, Department of Nanoengineering, University of California, San Diego, La Jolla, California 92093, United States.
Raina M. Borum, Department of Nanoengineering, University of California, San Diego, La Jolla, California 92093, United States.
Yash Mantri, Department of Bioengineering, University of California, San Diego, La Jolla, California 92093, United States.
Ming Xu, Department of Nanoengineering, University of California, San Diego, La Jolla, California 92093, United States.
Pavla Fajtová, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, California 92093, United States.
Anthony J. O’Donoghue, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, California 92093, United States.
Jesse V. Jokerst, Department of Nanoengineering, Materials Science and Engineering Program, and Department of Radiology, University of California, San Diego, La Jolla, California 92093, United States.
REFERENCES
- (1).Wang LV Photoacoustic Imaging and Spectroscopy; CRC Press, 2017. [Google Scholar]
- (2).Moore C; Jokerst JV Strategies for image-guided therapy, surgery, and drug delivery using photoacoustic imaging. Theranostics 2019, 9, 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vu T; Razansky D; Yao J Listening to tissues with new light: recent technological advances in photoacoustic imaging. J. Opt 2019, 21, 103001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Frinking P; Segers T; Luan Y; Tranquart F Three decades of ultrasound contrast agents: a review of the past, present and future improvements. Ultrasound Med. Biol 2020, 46, 892–908. [DOI] [PubMed] [Google Scholar]
- (5).Ramirez DG; Abenojar E; Hernandez C; Lorberbaum DS; Papazian LA; Passman S; Pham V; Exner AA; Benninger RK Contrast-enhanced ultrasound with sub-micron sized contrast agents detects insulitis in mouse models of type1 diabetes. Nat. Commun 2020, 11, 2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cox B; Laufer JG; Arridge SR; Beard PC Quantitative spectroscopic photoacoustic imaging: a review. J. Biomed. Opt 2012, 17, 061202. [DOI] [PubMed] [Google Scholar]
- (7).Moore C; Jokerst JV Photoacoustic Ultrasound for Enhanced Contrast in Dental and Periodontal Imaging. In Dental Ultrasound in Periodontology and Implantology; Springer, 2021, pp 215–230. DOI: 10.1007/978-3-030-51288-0_11 [DOI] [Google Scholar]
- (8).de Leon A; Wei P; Bordera F; Wegierak D; McMillen M; Yan D; Hemmingsen C; Kolios MC; Pentzer EB; Exner AA Pickering Bubbles as Dual-Modality Ultrasound and Photoacoustic Contrast Agents. ACS Appl. Mater. Interfaces 2020, 12, 22308–22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hariri A; Wang J; Kim Y; Jhunjhunwala A; Chao DL; Jokerst JV In vivo photoacoustic imaging of chorioretinal oxygen gradients. J. Biomed. Opt 2018, 23, 036005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Guggenheim JA; Allen TJ; Plumb A; Zhang EZ; Rodriguez-Justo M; Punwani S; Beard PC Photoacoustic imaging of human lymph nodes with endogenous lipid and hemoglobin contrast. J. Biomed. Opt 2015, 20, 50504. [DOI] [PubMed] [Google Scholar]
- (11).Zhou Y; Tripathi SV; Rosman I; Ma J; Hai P; Linette GP; Council ML; Fields RC; Wang LV; Cornelius LA Noninvasive determination of melanoma depth using a handheld photoacoustic probe. J. Invest. Dermatol 2017, 137, 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Manohar S; Gambhir SS Clinical photoacoustic imaging. Photoacoustics 2020, 19, 100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Attia ABE; Balasundaram G; Moothanchery M; Dinish US; Bi R; Ntziachristos V; Olivo M A review of clinical photoacoustic imaging: Current and future trends. Photoacoustics 2019, 16, 100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Choi W; Park E-Y; Jeon S; Kim C Clinical photoacoustic imaging platforms. Biomed. Eng. Lett 2018, 8, 139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Huang J; Pu K Activatable Molecular Probes for Second Near-Infrared Fluorescence, Chemiluminescence, and Photoacoustic Imaging. Angew. Chem 2020, 132, 11813–11827. [DOI] [PubMed] [Google Scholar]
- (16).Miao Q; Pu K Emerging designs of activatable photoacoustic probes for molecular imaging. Bioconjugate Chem 2016, 27, 2808–2823. [DOI] [PubMed] [Google Scholar]
- (17).Ou H; Li J; Chen C; Gao H; Xue X; Ding D Organic/polymer photothermal nanoagents for photoacoustic imaging and photothermal therapy in vivo. Sci. China Mater 2019, 62, 1740–1758. [Google Scholar]
- (18).Weissleder R; Mahmood U Molecular imaging. Radiology 2001, 219, 316–333. [DOI] [PubMed] [Google Scholar]
- (19).López-Otín C; Bond JS Proteases: Multifunctional Enzymes in Life and Disease*. J. Biol. Chem 2008, 283, 30433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dominy SS; Lynch C; Ermini F; Benedyk M; Marczyk A; Konradi A; Nguyen M; Haditsch U; Raha D; Griffin C Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv 2019, 5, No. eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jiang T; Olson ES; Nguyen QT; Roy M; Jennings PA; Tsien RY Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 17867–17872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yan R; Ye D Molecular imaging of enzyme activity in vivo using activatable probes. Sci. Bull 2016, 61, 1672–1679. [Google Scholar]
- (23).Wang L; Yang P-P; Zhao X-X; Wang H Self-assembled nanomaterials for photoacoustic imaging. Nanoscale 2016, 8, 2488–2509. [DOI] [PubMed] [Google Scholar]
- (24).Dragulescu-Andrasi A; Kothapalli S-R; Tikhomirov GA; Rao J; Gambhir SS Activatable oligomerizable imaging agents for photoacoustic imaging of furin-like activity in living subjects. J. Am. Chem. Soc 2013, 135, 11015–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang D; Qi GB; Zhao YX; Qiao SL; Yang C; Wang H In situ formation of nanofibers from purpurin18-peptide conjugates and the assembly induced retention effect in tumor sites. Adv. Mater 2015, 27, 6125–6130. [DOI] [PubMed] [Google Scholar]
- (26).Yan R; Hu Y; Liu F; Wei S; Fang D; Shuhendler AJ; Liu H; Chen H-Y; Ye D Activatable NIR Fluorescence/MRI Bimodal Probes for in Vivo Imaging by Enzyme-Mediated Fluorogenic Reaction and Self-Assembly. J. Am. Chem. Soc 2019, 141, 10331–10341. [DOI] [PubMed] [Google Scholar]
- (27).Ye D; Shuhendler AJ; Cui L; Tong L; Tee SS; Tikhomirov G; Felsher DW; Rao J Bioorthogonal cyclization-mediated in situ self-assembly of small-molecule probes for imaging caspase activity in vivo. Nat. Chem 2014, 6, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Xia X; Yang M; Oetjen LK; Zhang Y; Li Q; Chen J; Xia Y An enzyme-sensitive probe for photoacoustic imaging and fluorescence detection of protease activity. Nanoscale 2011, 3, 950–953. [DOI] [PubMed] [Google Scholar]
- (29).Liu C; Li S; Gu Y; Xiong H; Wong W.-t.; Sun L Multispectral photoacoustic imaging of tumor protease activity with a gold nanocage-based activatable probe. Mol. Imag. Biol 2018, 20, 919–929. [DOI] [PubMed] [Google Scholar]
- (30).Yang K; Zhu L; Nie L; Sun X; Cheng L; Wu C; Niu G; Chen X; Liu Z Visualization of protease activity in vivo using an activatable photo-acoustic imaging probe based on CuS nanoparticles. Theranostics 2014, 4, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Morgounova E; Johnson SM; Shao Q; Hackel B; Ashkenazi S Lifetime-based photoacoustic probe activation modeled by a dual methylene blue-lysine conjugate. In Photons Plus Ultrasound: Imaging and Sensing 2014; International Society for Optics and Photonics, 2014, p 89435F.In [Google Scholar]
- (32).Morgounova E; Shao Q; Hackel BJ; Thomas DD; Ashkenazi S Photoacoustic lifetime contrast between methylene blue monomers and self-quenched dimers as a model for dual-labeled activatable probes. J. Biomed. Opt 2013, 18, 056004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li Y; Zhou Y; Yue X; Dai Z Cyanine conjugates in cancer theranostics. Bioact. Mater 2021, 6, 794–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sun W; Guo S; Hu C; Fan J; Peng X Recent development of chemosensors based on cyanine platforms. Chem. Rev 2016, 116, 7768–7817. [DOI] [PubMed] [Google Scholar]
- (35).Mishra A; Behera RK; Behera PK; Mishra BK; Behera GB Cyanines during the 1990s: a review. Chem. Rev 2000, 100, 1973–2012. [DOI] [PubMed] [Google Scholar]
- (36).Benson RC; Kues HA Absorption and fluorescence properties of cyanine dyes. J. Chem. Eng. Data 1977, 22, 379–383. [Google Scholar]
- (37).Kilian HI; Kang H; Nyayapathi N; Fukuda T; Adluru E; Zhang H; Quinn B; Xia J; Choi HS; Lovell JF Facile formulation of a long-wavelength cyanine for optical imaging in the second near-infrared window. Biomater. Sci 2020, 8, 4199–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Polom K; Murawa D; Rho Y. s.; Nowaczyk P; Hünerbein M; Murawa P Current trends and emerging future of indocyanine green usage in surgery and oncology: a literature review. Cancer 2011, 117, 4812–4822. [DOI] [PubMed] [Google Scholar]
- (39).Li Y; Zhou Y; Yue X; Dai Z Cyanine Conjugate-Based Biomedical Imaging Probes. Adv. Healthcare Mater 2020, 9, 2001327. [DOI] [PubMed] [Google Scholar]
- (40).Zhao X; Fan Z; Qiao Y; Chen Y; Wang S; Yue X; Shen T; Liu W; Yang J; Gao H; Zhan X; Shang L; Yin Y; Zhao W; Ding D; Xi R; Meng M AIEgens Conjugation Improves the Photothermal Efficacy and Near-Infrared Imaging of Heptamethine Cyanine IR-780. ACS Appl. Mater. Interfaces 2020, 12, 16114–16124. [DOI] [PubMed] [Google Scholar]
- (41).Mokrousov MD; Novoselova MV; Nolan J; Harrington W; Rudakovskaya P; Bratashov DN; Galanzha EI; Fuenzalida-Werner JP; Yakimov BP; Nazarikov G Amplification of photoacoustic effect in bimodal polymer particles by self-quenching of indocyanine green. Biomed. Opt. Express 2019, 10, 4775–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Miranda D; Huang H; Kang H; Zhan Y; Wang D; Zhou Y; Geng J; Kilian HI; Stiles W; Razi A Highly-soluble cyanine J-aggregates entrapped by liposomes for in vivo optical imaging around 930 nm. Theranostics 2019, 9, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hestand NJ; Spano FC Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev 2018, 118, 7069–7163. [DOI] [PubMed] [Google Scholar]
- (44).West W; Pearce S The dimeric state of cyanine dyes. J. Phys. Chem 1965, 69, 1894–1903. [Google Scholar]
- (45).Kasha M; Rawls H; El-Bayoumi MA The exciton model in molecular spectroscopy. Pure Appl. Chem 1965, 11, 371–392. [Google Scholar]
- (46).Geoghegan KF; Rosner PJ; Hoth LR Dye-Pair Reporter Systems for Protein–Peptide Molecular Interactions. Bioconjugate Chem 2000, 11, 71–77. [DOI] [PubMed] [Google Scholar]
- (47).Weissleder R; Tung C-H; Mahmood U; Bogdanov A In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat. Biotechnol 1999, 17, 375–378. [DOI] [PubMed] [Google Scholar]
- (48).Morales J; Pawle RH; Akkilic N; Luo Y; Xavierselvan M; Albokhari R; Calderon IAC; Selfridge S; Minns R; Takiff L; Mallidi S; Clark HA DNA-Based Photoacoustic Nanosensor for Interferon Gamma Detection. ACS Sens 2019, 4, 1313–1322. [DOI] [PubMed] [Google Scholar]
- (49).Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Mellott DM; Tseng C-T; Drelich A; Fajtová P; Chenna BC; Kostomiris DH; Hsu J; Zhu J; Taylor ZW; Kocurek KI A Clinical-Stage Cysteine Protease Inhibitor blocks SARS-CoV-2 Infection of Human and Monkey Cells. ACS Chem. Biol 2021, 16, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Arconada-Alvarez SJ; Lemaster JE; Wang J; Jokerst JV The development and characterization of a novel yet simple 3D printed tool to facilitate phantom imaging of photoacoustic contrast agents. Photoacoustics 2017, 5, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wang J; Chen F; Arconada-Alvarez SJ; Hartanto J; Yap L-P; Park R; Wang F; Vorobyova I; Dagliyan G; Conti PS; Jokerst JV A Nanoscale Tool for Photoacoustic-Based Measurements of Clotting Time and Therapeutic Drug Monitoring of Heparin. Nano Lett 2016, 16, 6265–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Boccia AC; Lukeš V; Eckstein-Andicsová A; Kozma E Solvent- and concentration-induced self-assembly of an amphiphilic perylene dye. New J. Chem 2020, 44, 892–899. [Google Scholar]
- (54).Nişancı B; Daştan A; Bozdemir ÖA Aromatic stacking of a perylenetetracarboxylic tetraester: Self-assembly in both water and chloroform. Tetrahedron Lett 2018, 59, 3558–3562. [Google Scholar]
- (55).Armbruster DA; Pry T Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev 2008, 29, S49–S52. [PMC free article] [PubMed] [Google Scholar]
- (56).Kaur J; Singh PK Trypsin Detection Strategies: A Review. Crit. Rev. Anal. Chem 2020, 1–19. [DOI] [PubMed]
- (57).Kuramochi H; Nakata H; Ishii S.-i. Mechanism of association of a specific aldehyde inhibitor, leupeptin, with bovine trypsin. J. Biochem 1979, 86, 1403–1410. [DOI] [PubMed] [Google Scholar]
- (58).Mukherjee P; Desai P; Ross L; White EL; Avery MA Structure-based virtual screening against SARS-3CL(pro) to identify novel non-peptidic hits. Bioorg. Med. Chem 2008, 16, 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Rut W; Groborz K; Zhang L; Sun X; Zmudzinski M; Pawlik B; Wang X; Jochmans D; Neyts J; Młynarski W SARS-CoV-2 M pro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol 2021, 17, 222–228. [DOI] [PubMed] [Google Scholar]
- (60).Beutler E; Waalen J The definition of anemia: what is the lower limit of normal of the blood hemoglobin concentration? Blood 2006, 107, 1747–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Robles FE; Chowdhury S; Wax A Assessing hemoglobin concentration using spectroscopic optical coherence tomography for feasibility of tissue diagnostics. Biomed. Opt. Express 2010, 1, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Guentsch A; Kramesberger M; Sroka A; Pfister W; Potempa J; Eick S Comparison of Gingival Crevicular Fluid Sampling Methods in Patients With Severe Chronic Periodontitis. J. Periodontol 2011, 82, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mozaffarzadeh M; Moore C; Golmoghani EB; Mantri Y; Hariri A; Jorns A; Fu L; Verweij MD; Orooji M; de Jong N Motion-compensated noninvasive periodontal health monitoring using handheld and motor-based photoacoustic-ultrasound imaging systems. Biomed. Opt. Express 2021, 12, 1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Moore C; Bai Y; Hariri A; Sanchez JB; Lin C-Y; Koka S; Sedghizadeh P; Chen C; Jokerst JV Photoacoustic imaging for monitoring periodontal health: A first human study. Photoacoustics 2018, 12, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.