

Abstract
Background
Genetic factors alter the risk for nonalcoholic fatty liver disease (NAFLD). We sought to identify NAFLD-associated genes and elucidate gene networks and pathways involved in the pathogenesis of NAFLD.
Methods
Quantitative global hepatic gene expression analysis was performed on 53 morbidly obese Caucasian subjects undergoing bariatric surgery (27 with NAFLD and 26 controls). After standardization of data, gene expression profiles were compared between patients with NAFLD and controls. The set of genes that significantly correlated with NAFLD was further analyzed by hierarchical clustering and ingenuity pathways analyses.
Results
There were 25,643 quantitative transcripts, of which 108 were significantly associated with NAFLD (p<0.001). Canonical pathway analysis in the NAFLD-associated gene clusters showed that the hepatic fibrosis signaling was the most significant pathway in the up-regulated NAFLD gene cluster containing three (COL1A1, IL10, IGFBP3) significantly altered genes, whereas the endoplasmic reticulum stress and protein ubiquitination pathways were the most significant pathways in the down-regulated NAFLD gene cluster, with the first pathway containing one (HSPA5) and the second containing two (HSPA5, USP25) significantly altered genes. The four primary gene networks associated with NAFLD were involved in cell death, immunological disease, cellular movement, and lipid metabolism with several significantly altered “hub” genes in these networks.
Conclusions
This study reveals the canonical pathways and gene networks associated with NAFLD in morbidly obese Caucasians. The application of gene network analysis highlights the transcriptional relationships among NAFLD-associated genes and allows identification of hub genes that may represent high-priority candidates for NAFLD.
Keywords: Fatty liver, NAFLD, Genes, Gene networks, Gene expression, Pathogenesis
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the United States [1–3]. It has a wide spectrum of phenotypes starting with simple steatosis (SS) to steatosis with a variable mix of subphenotypes including ballooning, inflammation, and fibrosis, as diagnosed on liver biopsy [4]. Nonalcoholic steatohepatitis (NASH) represents an advanced phenotype where most of these subphenotypes are present [5]. Unlike SS, which rarely results in liver complications [6, 7], NASH can progress to cirrhosis, liver failure, and hepatocellular carcinoma [6, 7]. The prevalence of NAFLD and NASH will probably increase as part of the ongoing obesity epidemic and will likely add a huge burden to the healthcare system [8].
Although some of the molecular mechanisms involved in onset and progression of steatosis, necro-inflammation, and fibrosis in NAFLD have been well characterized [9], little is known about the underlying complex gene interaction networks involved in NAFLD.
Several observations point to a role for genetic factors in the pathogenesis of NAFLD. Liver biopsy findings in patients who share similar clinical risk factors for NAFLD (e.g., obesity, diabetes, hypertriglyceridemia) widely vary from normal to the entire histological spectrum of NAFLD [10, 11]. Family studies suggest that the risk for NAFLD is partly heritable [12–14]. Ethnicity influences the frequency and severity of NAFLD [15, 16]. Finally, emerging data link gene variants with NAFLD and its histological phenotypes [17–19].
Hepatic gene expression in NAFLD has been analyzed in several human studies[11, 20–23], with reported differential expression of genes involved in lipid metabolism, mitochondrial function, oxidative stress, insulin signaling, cell death, and hepatic fibrosis. There was however little or no concordance among these studies on the genes described to be associated with NAFLD, and although their functional classifications were described, the potential interactions among significantly altered genes were not characterized.
The aims of this study were to identify genes associated with NAFLD and elucidate gene networks and pathways involved in the pathogenesis of NAFLD using integrated bioinformatics analyses of global quantitative hepatic expression in a morbidly obese Caucasian cohort.
Methods
Recruitment and Phenotyping of Subjects
The study protocol has been reviewed and approved by the Medical College of Wisconsin's Institutional Review Board. Subjects gave written informed consent for participation in the study. Subjects were of northern European descent, morbidly obese (BMI≥40 kg/m2 or >35 kg/m2 with significant comorbidities) with documented unsuccessful dietary attempts to lose and maintain weight, and who underwent bariatric surgery at the MCW. A protocol intraoperative liver biopsy was performed on all patients for histological phenotyping. Patients with alcohol intake >20 g/d and those with other liver diseases (hepatitis B, hepatitis C, autoimmune hepatitis, primary biliary cirrhosis, Wilson's disease, alpha1 antitrypsin deficiency, or hemochromatosis) based on positive disease-specific serological tests and suggestive liver histology were excluded. Patients using drugs associated with NAFLD (systemic glucocorticosteroids, tamoxifen, tetracycline, amiodarone, methotrexate, valproic acid, anabolic steroids, estrogens at doses higher than those used for hormone replacement, or other known hepatotoxins) preceding the liver biopsy were also excluded. For all participants, clinical, and biochemical data were collected.
Collection of Biological Material
All subjects had fasting blood samples for serum extraction collected in the morning of planned surgery. In addition to the biopsy obtained for routine histological analysis and phenotyping, an additional intraoperative liver biopsy was done, and tissue was snap-frozen immediately in the operating room and used for RNA studies.
Histological Evaluation and Phenotypes Definitions
All liver biopsy samples were read by two expert pathologists (R.K. and D.E.K.) to define the NAFLD phenotype and semiquantitatively score the individual histological features/subphenotypes including steatosis, lobular and portal inflammation, hepatocellular ballooning, Mallory's hyaline, and fibrosis according to the scoring system suggested by the NIH NASH Clinical Research Network working group [24]. NAFLD was diagnosed when ≥5% macrosteatosis was present. Subjects with 0 to 5% macrosteatosis were diagnosed as non-NAFLD controls. Both pathologists confirmed the phenotype as NAFLD or non-NAFLD control on all included subjects. The analyses in this study focused on genes associated with NAFLD.
RNA Isolation from Liver Tissue
Frozen liver tissue samples were mixed with lysis buffer, homogenized and used for total RNA isolation following the Qiagen RNeasy Mini Kit protocol. The isolated RNA concentration (μg) and purity (260:280 nm) were measured using a NanoDrop spectrophotometer. The resulting RNA was reverse-transcribed, converted into cDNA, and subsequently amplified producing cRNA using the MessageAmp II RNA Amplification Kit (Ambion). This method has been shown to reliably amplify small amounts of messenger RNA (mRNA) obtained from limited tissue samples obtained by biopsy.
Transcriptional Profiling
We used commercially available Illumina HumanWG-6 Expression BeadChips for whole genome expression analysis. The BeadChips contain six arrays, each with approximately 48,000 probes derived from human genes in the National Center for Bioinformatic Information Reference Sequence (RefSeq) and UniGene databases. This system uses a “direct hybridization” assay, whereby gene-specific probes are used to detect labeled RNAs. Each bead in the array contains a 50-mer, sequence-specific oligo probe synthesized using Illumina's Oligator™ in-house technology. More than 1.6 million beads are available to quantitate RNA levels for each sample. The HumanWG-6 Expression BeadChips were scanned on an Illumina BeadArray™ reader. The BeadStudio software package included with the Illumina® BeadStation 500GX system extracted gene expression data from images collected from the Illumina BeadArray Reader. The application reports experiment performance based on built-in controls that accompany each experiment.
Identification of Detectable Transcripts
For each sample, approximately 48,000 transcripts were quantified using the BeadChip supplied by Illumina. A χ2 “tail” test was used to assess whether there is a significant excess of samples with transcript-specific expression values above the 95th percentile of the null distribution. We used this test because it allows “detection" of even those RNA molecules that are clearly present above baseline levels in only a subset of individuals. Using a false discovery rate of 5% [25], we identified expression phenotypes that exhibited significant expression by this criterion.
Standardization of Expression Values
For the analysis of transcriptional variation, we focused on those detectable transcripts that passed a tail test that determined if there is sufficient quantitative signal over that expected by chance. To minimize the influence of overall signal levels, which may reflect RNA quantity and quality rather than a true biological difference between individuals, abundance values of all retained transcripts were standardized by z-scoring within individuals (using decile percentage bins of transcripts, grouped by average log-transformed raw signals across individuals), followed by linear regression against individual-specific average log-transformed raw signal and its squared value. For each transcript, we directly normalized these residual expression scores by employing an inverse Gaussian transformation across individuals to ensure that the assumptions underlying variance components-based analyses were not violated. The normalized phenotypes were comparable between individuals and across transcripts. Current micro-arrays allow accurate detection of gene transcripts that correlate well with real-time PCR as shown in many studies [11, 20, 21, 26, 27]. Furthermore, the high heritability of housekeeping gene transcripts traditionally used in RT-PCR validation (e.g., heritability for B-actin expression is 0.373, p1:0 × 10−9, and for cyclophilin-D is 0.559, p1:0 × 10−9) [28] make them less than ideal as references for internal standardization. Lastly, the limited availability of tissue for mRNA extraction restricted our ability to run RT-PCR analysis.
Detection of NAFLD-Associated Gene Transcripts
The t test statistic was used to measure the difference between the sample means in units of the standard deviation for which the difference can be tested using certain p value. We used a significance cut-point of p<0.001.
Hierarchical Clustering
This was performed with two groups of samples (NAFLD vs. controls) using Genesis software [29]. Cluster analysis was performed to divide genes or samples into groups on the basis of similarities among their gene expression profiles, which is often indicative of similarity with respect to function.
Gene Network and Pathway Analyses
To describe the interactions between the altered gene expression levels in NAFLD, the Ingenuity Pathways Analysis (IPA) software program (Ingenuity Systems, version 6.1) was used. This software application allows identification of biological and network/pathway interactions between genes. After uploading a list of genes that are significantly differentially expressed in our samples, the program was used to uncover any interactions between the genes. IPA identifies the biological functions and pathways that are most relevant to the experimental datasets. The altered genes identified by expression profiling analysis were mapped to genetic networks available in the IPA database, ranked by score, and presented as a graph indicating the molecular relationships between genes.
Ingenuity Pathway Analysis Procedures
From the llumina HumanWG-6 data, we uploaded the ProbeID and the associated expression value to upload into IPA. Each ProbeID was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. These genes, called focus genes, were overlaid onto a global molecular network developed from information contained in the database. Networks of these focus genes were then algorithmically generated based on their types of interactions (direct and/or indirect). Scores were generated (based on Fisher's test) to rank networks according to how relevant they are to the genes in the input dataset. The score takes into account the number of focus genes (genes in our lists) in the network and the size of the network to approximate how relevant this network is to the original list of focus genes. The network is then presented as a graph indicating the molecular relationships between genes/gene products. Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). The connectivity of genes (nodes) is based on data in the IPA knowledge base, which is a large repository of gene-phenotype associations, molecular interactions, chemical knowledge, and regulatory events, pulled from the full text of scientific publications by Ph.D.-level scientists. The node color indicates the degree of up-regulation (red) or downregulation (green). Nodes are displayed using various shapes that represent the functional class of the gene product. A functional analysis of a network then identified the biological functions and/or diseases that were most significant to the genes in the network.
Results
Characteristics of Study Subjects
Fifty-three Caucasian subjects whose histological phenotype was confirmed by the two study pathologists were included in this analysis: 27 with NAFLD and 26 controls. The clinical and laboratory characteristics of these subjects are summarized in Table 1. As anticipated, subjects with NAFLD had higher mean fasting glucose, insulin, triglycerides, and ALT levels. The total cholesterol, HDL, LDL, and alkaline phosphatase levels were similar between the groups. There were no significant differences in age, BMI, percentage of females, or fractions of subjects with diabetes or hypertension between the two groups.
Table 1.
Characteristics of study subjects
| Variable | Control (n=26) | NAFLD (n=27) | p |
|---|---|---|---|
|
| |||
| Age (years) | 40.5±12.4 | 43.1±10.3 | ns |
| Female, n (%) | 25 (96) | 24 (89) | ns |
| BMI (kg/m2) | 48.8±5.9 | 49.6±7.4 | ns |
| Diabetes, n (%) | 7 (26.9) | 11 (40.7) | ns |
| Hypertension, n (%) | 13 (50) | 19 (70.3) | ns |
| Fasting glucose (mg/dL) | 103.9±43.2 | 131.0±50.8 | 0.04 |
| Fasting insulin (mu/mL) | 20.2±13.5 | 35.8±20.0 | 0.001 |
| Total cholesterol (mg/dL) | 175.8±29.1 | 178.3±38.6 | ns |
| Triglycerides (mg/dL) | 123.6±37.3 | 172.9±80.3 | 0.006 |
| HDL (mg/dL) | 45.4±9.1 | 41.1±7.9 | ns |
| LDL (mg/dL) | 105.5±26.2 | 103.3±32.2 | ns |
| ALT (U/L) | 18.1±11.6 | 30.0±16.3 | 0.003 |
| Total bilirubin (mg/dL) | 0.4±0.1 | 0.5±0.3 | ns |
| Alkaline phosphatase (U/L) | 74.3±31.1 | 73.3±23.2 | ns |
| Platelets (103/uL) | 309.3±73.6 | 298.9±72.7 | ns |
Data presented as frequency or mean ± standard deviation unless otherwise indicated
Hepatic Transcriptional Profile in NAFLD
A total of 25,643 quantitative transcripts were identified in liver using the proposed transcriptional profiling. Of these, 108 were significantly associated with NAFLD (p<0.001). The strongest 10 up- or down-regulated NAFLD-associated genes are shown in Table 2.
Table 2.
Strongest NAFLD-associated genes
| Molecules Exp. | Value Exp. Chart |
|---|---|
|
| |
| Log ratio up-regulated | |
| PPFIBP1 | 0.583 |
| ZAK | 0.527 |
| WNK4 | 0.526 |
| RGN | 0.524 |
| SMUG1 | 0.521 |
| MLLT10 | 0.505 |
| DCN | 0.497 |
| CYP4F22 | 0.497 |
| CSN2 | 0.495 |
| FABP4 | 0.491 |
| Log ratio down-regulated | |
| GABRB2 | −0.638 |
| CHERP | −0.600 |
| EBNA1BP2 | −0.579 |
| DYNC1I1 | −0.570 |
| RAB3IP | −0.562 |
| KLHL8 | −0.546 |
| TUBD1 | −0.507 |
| HEATR3 | −0.491 |
| OCM2 | −0.480 |
| MKX | −0.476 |
Cluster-Based Canonical Pathway Analysis
To gain further insights into the pathogenesis of NAFLD, we analyzed the NAFLD-correlated genes to elucidate dominant canonical pathways. Hierarchical clustering analysis was first performed to organize the 108 NAFLD-correlated transcripts based on similarity of function within the up- or down-regulated clusters (Fig. 1). These two clusters were then subjected to canonical pathway analysis, which showed that hepatic fibrosis signaling was the most significant pathway in the up-regulated NAFLD gene cluster containing three (COL1A1, IL10, IGFBP3) significantly altered genes. Endoplasmic reticulum (ER) stress and protein ubiquitination pathways were the most significant pathways in the down-regulated NAFLD gene cluster, with the first pathway containing one (HSPA5) and the second containing two (HSPA5, USP25) significantly altered genes. Remaining relevant pathways not reaching significance at p <0.05 in each cluster are shown in Fig. 2.
Fig. 1.
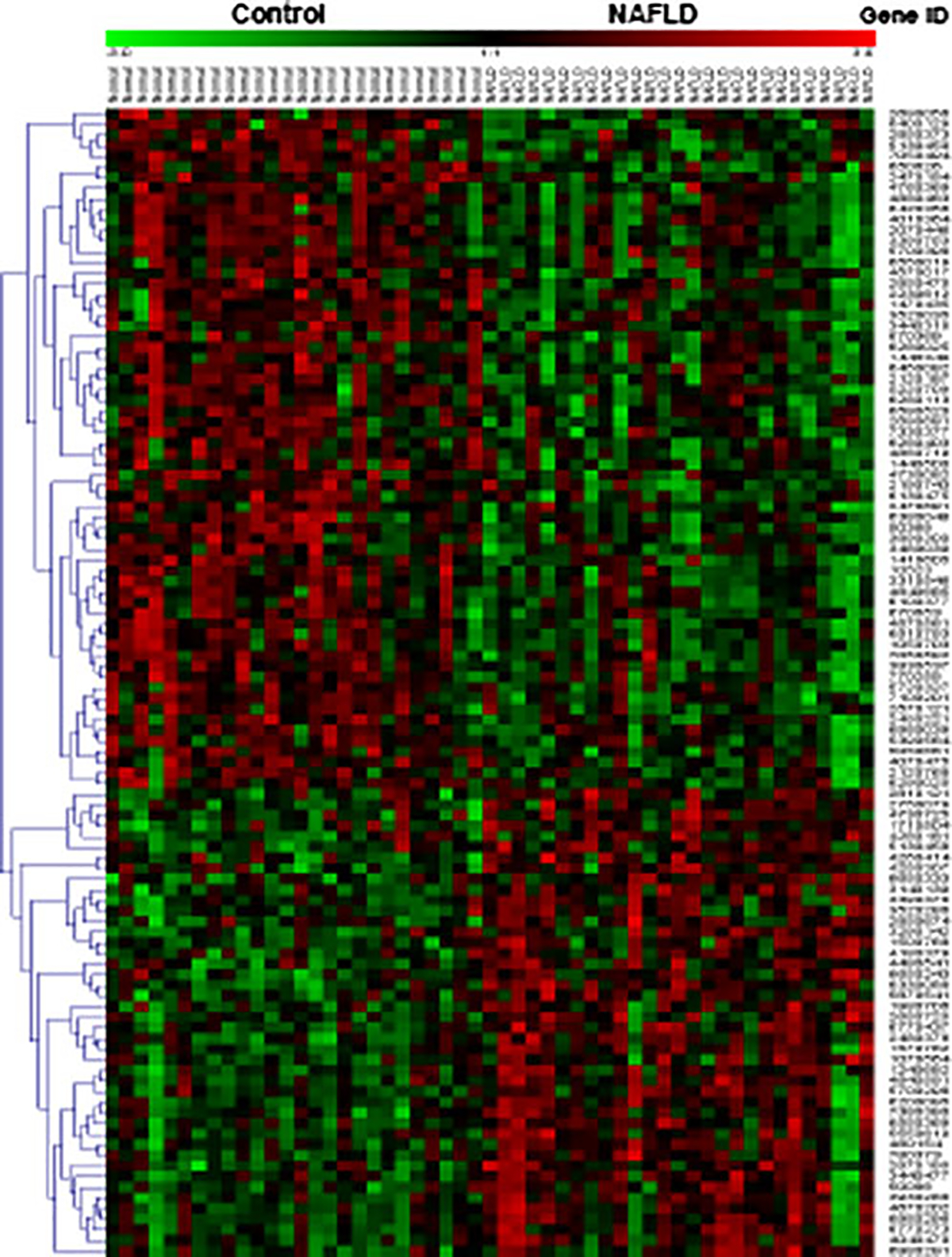
Hierarchal clustering of 108 NAFLD-correlated genes
Fig. 2.
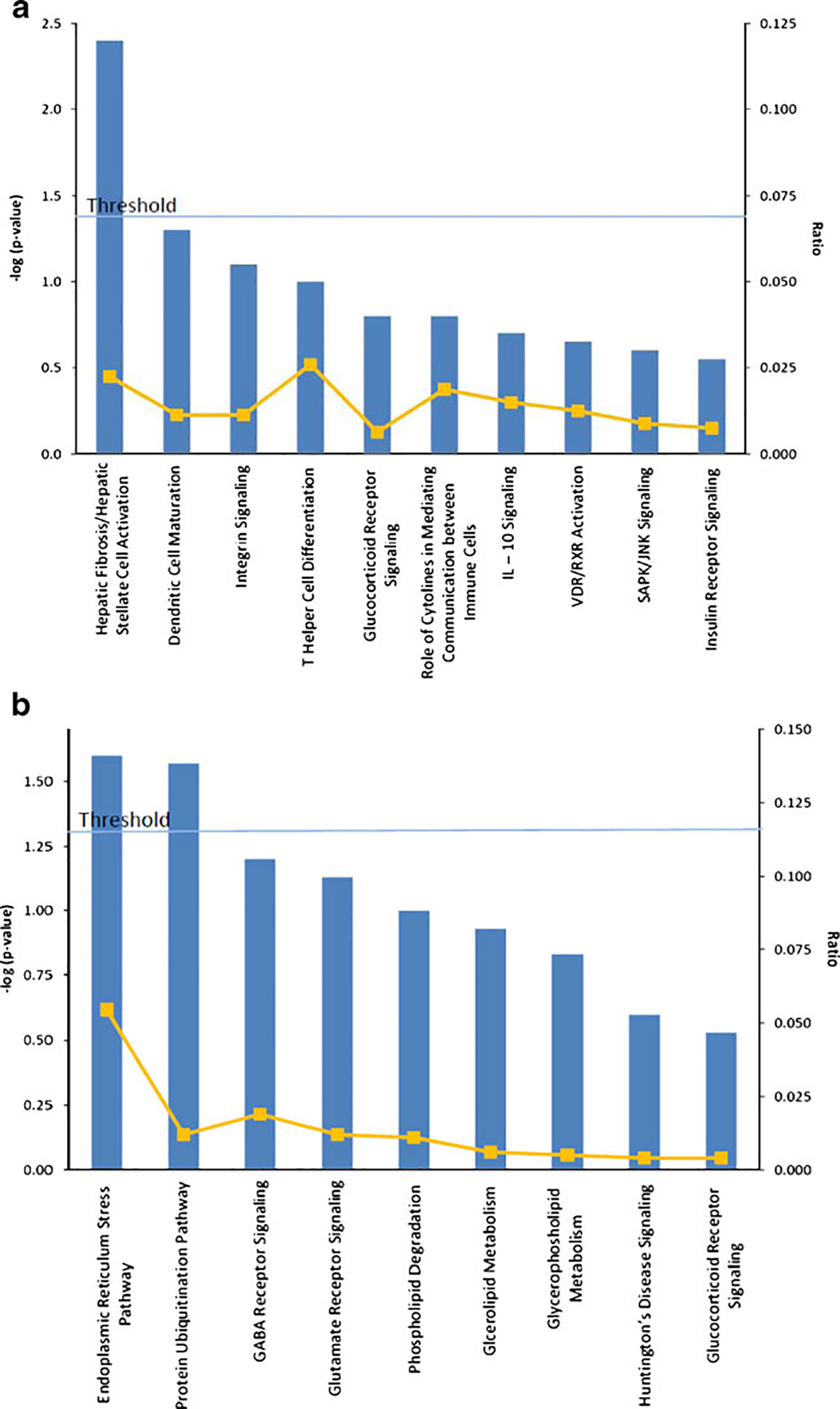
The canonical pathways detected in each NAFLD-associated gene cluster. a Up-regulated pathways and b Down-regulated pathways
Hepatic Gene Networks Analysis
To determine significant biological functions and to reveal transcriptional correlations among genes associated with NAFLD, the 108 significant genes were subjected to gene network analysis.
The most significant cellular and molecular biological functions associated with NAFLD-correlated genes were cell death, cellular movement, antigen presentation, cell morphology, and cellular development. Genes in each of these functional categories are listed in Table 3.
Table 3.
Gene biological functions associated with NAFLD
| p | Molecules | |
|---|---|---|
|
| ||
| Cell death | 3.43E-04–3.14E-02 | SMUG1, DDIT3, DPP3, IL10, DCN, EEF1A2, CXCL12, IGFBP3, ULBP2, RGN, MAFB, HSPA5 |
| Cellular movement | 5.46E-04–3.52E-02 | TSPAN3, COL1A1, IL10, ITGA9, DCN, CXCL12, IGFBP3, FABP4, MAFB |
| Antigen presentation | 9.93E-04–3.13E-02 | COL1A1, IL10, DCN, CXCL12, FABP4 |
| Cell morphology | 9.93E-04–3.48E-02 | RAB3IP, COL1A1, PSPN, MLLT10, IL10, KPNA2, CXCL12, NEFM, IGFBP3 |
| Cellular development | 1.57E-03–3.22E-02 | CD164, COL1A1, DDIT3, IL10, DCN, CXCL12, IGFBP3, MAFB, PRDM16 |
Of the 108 NAFLD-associated genes, 63 had connectivity data in IPA knowledge base and were eligible for network analysis. Four significant hepatic gene networks were noted in NAFLD (Table 4). Prominent gene functions within these networks included cell death, immunological disease, cancer, cellular growth and proliferation, cellular movement, lipid metabolism, molecular transport, and small molecule biochemistry.
Table 4.
The significant hepatic gene networks associated with NAFLD
| ID | Score | Top functions | Focus molecules | Molecules in network |
|---|---|---|---|---|
|
| ||||
| 1 | 39 | Cell death, renal necrosis/cell death, renal, and urological disease | 18 | Akt, AMOTL2, Ap1, Ck2, COL1A1, Collagen(s), CSN2, CXCL12, DCN, DDIT3, DGKZ, ERK, FABP4, FSH, IGFBP3, IL1, IL10, Insulin, ITGA9, Jnk, LDL, MAFB, Mapk, NFkB (complex), P38 MAPK, PI3K, PPFIBP1, RAB3IP, Ras, STAT5a/b, STX4, Tgf beta, ULBP2, UNC5CL, ZAK |
| 2 | 23 | Immunological disease, cancer, cellular growth, and proliferation | 12 | CD164, CD274, CD33 (includes EG:945), CHI3L2, CTSZ (includes EG:1522), DPP3, FOS, GPNMB, GPR109B, HRSP12, HSP, IFNG, KPNA2, LZTS1, MEFV, MIRN17 (includes EG:406952), MKX, MRPS6 (includes EG:64968), NAT6, NEFM, neuroprotectin D1, NFE2L2, OCM2, PRKACA, PSPN, RBP3, RIF1, SLC15A3, SLC1A4, TGFB1, TNF, TYRP1, USP4, USP25, VIM |
| 3 | 21 | Cellular movement, nervous system development and function, organismal functions | 11 | ATF6B, CLDN2, CLDN3, CLDN11, DGKD (includes EG:8527), EEF1A2, ERP29, GCHFR, GPR37, HDLBP, HEATR3, HNF4A, HSPA5, INS1, ITGB1, Mg2+, OS9, POLL, RBM42, RGN, SEL1L, SFRS12, SLC2A4, SMAD2, SMUG1, SNRPD3, SSBP1, STXBP4, THRAP3, TMBIM6, TRAF6, TSPAN, TSPAN3, VARS, WNK4 |
| 4 | 21 | Lipid metabolism, molecular transport, small molecule biochemistry | 11 | ALOX5AP, ASF1B, ATP1B1, BICD1, CCNE1, CEP76, CHERP, CLIP1, COIL, DOT1L, DYNC1I1, DYNLL2, EBNA1BP2, FAAH, GABRB2, HIST1H3E, Histone h4, HTR1A, IL4, KLC2, KLHL8, LEPROT, MLLT10, NFIL3, NOC2L, NR3C1, PPP5C, progesterone, SLC1A7, SMARCB1, SS18, TGFBI, TNFSF4, TP53BP1, TP53I3 |
To demonstrate the biological interactions of these genes within a network and highlight “hub” genes that interact with other NAFLD-correlated genes, gene networks involved in cell death, lipid metabolism, and molecular transport are shown in Fig. 3.
Fig. 3.
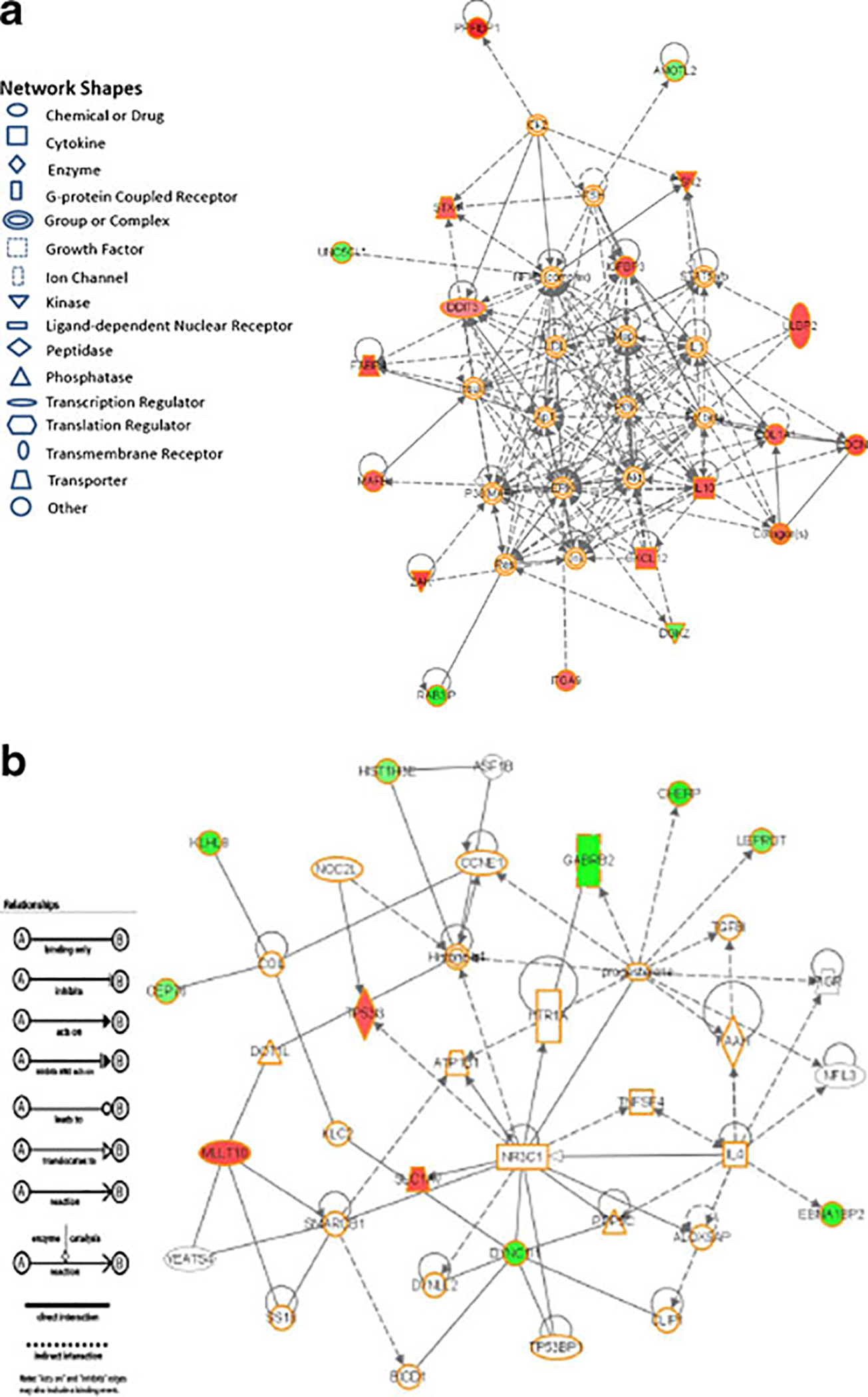
Significant hepatic gene networks in NAFLD. a This gene network is involved in cell death. Several significantly expressed hub genes such as COL1A1, IL10, and DCN can be identified. b This gene network is involved in lipid metabolism, molecular transport, and small molecules biochemistry. Red: up-regulated in NAFLD compared to control; green: down-regulated in NAFLD compared to control
Discussion
Several human studies have analyzed hepatic gene expression in NAFLD [11, 20–23]. These studies reported differential expression of genes involved in lipid metabolism, mitochondrial function, oxidative stress, insulin signaling, cell death, and hepatic fibrosis. There was however little or no concordance among these studies on the genes described to be associated with NAFLD. This may be due to small sample size, study of ethnically mixed cohorts or cohorts of different ethnicities, or use of different gene expression platforms with varying numbers of interrogated transcripts. Despite these limitations, these studies provided insights into the pathogenesis of NAFLD and highlighted a large number of NAFLD-correlated genes. They also demonstrated the need for additional bioinformatics analytical methods that can both provide a systematic approach to connect the data to biology and aid in prioritizing the often large number of NAFLD-associated genes.
In this study, we analyzed global hepatic gene expression in a clinically well characterized and ethnically homogeneous cohort. Using gene networks and pathways analyses, we confirmed a significant role of several previously described biological processes in the pathogenesis of NAFLD.
There were several interesting genes in our study that showed strong evidence of up-regulation including FABP4, PPFIBP1, ZAK, RGN, SMUG1, DCN, CYP4F22, and CSN2.
The FABP4 gene has been also reported to be up-regulated in a prior hepatic expression study in NAFLD [23]. It encodes for fatty acid binding protein 4, adipocyte, a member of the fatty acid-binding proteins. FABPs are lipid chaperones with high affinity to binding long chain fatty acids and are important mediators of inflammation and insulin signaling in glucophages and adipocytes [30, 31]. Genetic variation in FABP4 has been shown to influence triglycerides levels and the risk for diabetes in humans [32]. Indeed, mice deficient in FABP4 and another FABP (mal1) are protected from diet-induced obesity, insulin resistance, diabetes, and fatty liver [33]. Therefore, FABP4 represents a logical NAFLD candidate gene.
The protein encoded by liprin beta 1 (PPFIBP1) is a member of the LAR protein-tyrosine phosphatase-interacting protein (liprin) family. Tyrosine phosphorylation of proteins is important in coordinating cellular responses to extracellular stimuli. This protein was found to play a role in altering tumor invasiveness and metastasis [34, 35].
The ZAK gene encodes a cytoplasmic protein, which is a member of the MAPKKK family of signal transduction molecules. The protein has proapoptotic activity, plays a role in cell cycle checkpoint regulation, and mediates the effects of tumor growth factor-β (TGF-β) in inducing myocardial fibrosis [36–38]. Although TGF-β is a potent inducer of hepatic stellate cells production of collagen [39], the effects of its interaction with ZAK on hepatic fibrosis are currently unknown.
The gene encoding regucalcin (RGN) was also among the strongest up-regulated in our study. Regucalcin is a calcium-binding protein, which regulates the calcium effects on liver cell functions, modulates lipid metabolism in the adipose and liver tissues in aging rats by interacting with leptin or adiponectin, and suppresses apoptotic cell death [40–42].
The SMUG1 (single-strand-selective monofunctional uracil-DNA glycosylase 1) gene encodes a glycosylase that removes uracil from single- and double-stranded DNA in nuclear chromatin, thus contributing to base excision repair [43, 44]. DNA damage is a well-described phenomenon in NAFLD [45].
The DCN gene encodes for decorin, an extracellular matrix proteoglycan that binds to type I collagen fibrils and exerts several important biological effects. Decorin suppresses TGF-β activation of hepatic stellate cells and thus influences matrix assembly and remodulation, and suppresses different cancer cell lines by inducing apoptosis [46–49].
The CYP4F22 (cytochrome P450, family 4, subfamily F, polypeptide 22) gene encodes a member enzyme of the cytochrome P450 superfamily that are involved in drug metabolism and synthesis of cholesterol, steroids, and other lipids.
The protein encoded by CSN2, casein beta, has a role in regulating protein stability and degradation via the ubiquitin–proteasome system and is involved in cell transformation, tumorigenesis, and hepatic regeneration [50–52].
Less is known about the strongest down-regulated genes discovered in this study. The CHERP (calcium homeostasis ER protein) gene encodes a protein that modulate calcium homoeostasis, cell growth and proliferation [53], and the DYNC1I1 (dynein, cytoplasmic 1, intermediate chain 1) gene encodes for a molecular motor protein that is important for cell division and transport of intracellular organelles and cell migration [54, 55].
Beyond the identification of individual genes, our analysis focused on the identification and characterization of overarching biological functions associated with these genes. The most significant biological functions involving genes with significantly altered expression included cell death, cellular movement, antigen presentation, cell morphology, and cellular development. These data are consistent with findings of other studies revealing the biological significance of cell death [56, 57] and altered immunological function [58, 59] in NAFLD.
Canonical pathways analysis revealed that the hepatic fibrosis pathway was the only significant up-regulated pathway in NAFLD, with COL1A1, IL10, and IGFBP3 being the most significantly altered in this cohort. Increased collagen 1 deposition results in progressive hepatic fibrosis, which may ultimately lead to cirrhosis. The type I procollagen, alpha 1 (COL 1A1) gene is up-regulated in subjects with NAFLD. Whether variants in this gene are associated with altered risk for increased fibrosis with NAFLD needs to be explored. Interleukin 10 (IL10) is a cytokine with pleiotropic effects on immunoregulation and inflammation. It has been shown to improve insulin resistance and reduce hepatic steatosis [60]. The insulin-like growth factor binding protein 3 (IGFBP3) is one of six high-affinity binding proteins for IGFs that reduce the levels of free IGF and antagonize their insulin-like activity. It interferes with adipocyte differentiation [61] and also interferes with insulin and glucose homeostasis resulting in impaired glucose tolerance and insulin resistance [62, 63]. IGFBP3 has IGF-independent cellular activities such control of cell growth and apoptosis [64, 65]. It therefore represents another attractive NAFLD candidate gene.
The finding that the two significantly down-regulated canonical pathways in NAFLD were the ER stress and protein ubiquitination pathways is interesting. This finding highlights the importance of ER in the pathogenesis of NAFLD, whether it is with the unfolded protein response to accumulating misfolded/unfolded proteins (ER stress) or the ubiquitination of these misfolded proteins as a quality-control response [66, 67]. These data are in line with earlier studies demonstrating a role for ER stress in inducing apoptosis in hepatocytes after accumulation of fatty acids [68] and with human data showing ER stress and suboptimal unfolded protein response in NAFLD [69]. There were two genes in these pathways that were significantly expressed in the dataset: HSPA5 and USP25. The heat shock 70-kDa protein 5 (HSPA5 also known as BIP or GRP78) is a member of the glucose-regulated proteins (GRPs). It acts as an ER chaperone and sensor of ER stress [70]. Although a recent human study of ethnically mixed cohort did not show a significant difference in HSPA5 (BIP) between controls and subjects with NAFLD [69], HPSA5 has been shown to influence insulin biosynthesis [71] and its overexpression in mice reduced ER stress and SREBP-1c cleavage and resulted in improvement in insulin sensitivity and hepatic triglycerides [72]. Based on these data, HS represents a logical NAFLD candidate gene. The ubiquitin-specific peptidase 25 (USP25) is protease involved in the release of ubiquitin from degraded proteins by disassembly of the polyubiquitin chains [73, 74]. However, its biological functions are largely unknown.
About 55% of our subjects with NAFLD had portal or lobular inflammation on liver biopsy. Of the differentially expressed genes in NAFLD, FABP4 and IL10 have important roles in mediating inflammation and thus may contribute to the inflammatory findings in these patients.
Although statistical significance of expression level changes may be one way to select a candidate gene for a given disease, gene network analysis offers the advantage of understanding the interaction of significant genes associated with a disease and the ability to find hub genes within a network that interact with several other genes up- and downstream of them. The high interconnectivity of hub genes with other correlated genes within a biological network may imply functional and biological importance of these genes [75]. This approach to identifying hub genes via gene network analysis has been applied to expression analysis of different tissues in other rodent and human disease models, where hub genes were proposed as potential biomarkers for the disease or targets for drug therapy [76–78]. Thus, these hub genes may be viewed as high-priority disease candidates for further study. In this dataset, four significant gene networks were associated with NAFLD (Table 4), and the IL10 and COL1A1 are examples of significantly expressed hub genes in network A.
The described functions of these top networks fit well with the known major physiological events in NAFLD, such as lipid metabolism, organismal functions, and cell death. Similar to our findings in human NAFLD, network analysis of hepatic gene expression in a rat model of NAFLD showed importance of genes involved in cell death, immune function, and lipid metabolism [79].
Several points need to be noted: (1) the subjects characterized within this study were morbidly obese and undergoing bariatric surgery. Therefore, findings of this study may not be applicable to other patients with NAFLD and lesser degrees of obesity. (2) Most of the subjects were females, and thus, there may be male-specific changes in hepatic gene expression that our analysis would not have uncovered. (3) Although correlation of significantly expressed genes levels with laboratory variables such as insulin and ALT levels are of significant clinical interest, the limited size of the current study cohort does not allow for these analyses with sufficient statistical power. (4) It is not possible from the current data to decipher which altered gene transcripts are primary or secondary changes in relation to NAFLD pathogenesis. Ultimately, combining genomics, proteomics, and transcriptomics methods may help in dissecting these changes. This study, nonetheless, combined the strengths of recruitment of an ethnically homogeneous cohort with detailed clinical phenotypic characterization, use of whole-genome quantitative transcriptional profiling, and application of integrated bioinformatics analyses to allow immediate connection of transcriptional data to NAFLD biology.
In summary, this study reveals the gene biological functions and canonical pathways associated with NAFLD. The application of gene networks analysis highlights the transcriptional relationships among NAFLD-associated genes and allows identification of hub genes that may represent high priority candidates for NAFLD.
Acknowledgments
This work was funded by a grant from the Biotechnology and Bioengineering Center of the Medical College of Wisconsin (SG and MO) and grant HL74168 from the Heart, Lung, and Blood Institute of the National Institutes of Health (MO).
Contributor Information
Samer Gawrieh, Department of Medicine, Division of Gastroenterology and Hepatology, Medical College of Wisconsin, 9200 W. Wisconsin Ave, Milwaukee, WI 53212, USA; Zablocki VA Medical Center, Milwaukee, WI, USA.
Tesfaye M. Baye, Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI, USA Biotechnology and Bioengineering Center, Medical College of Wisconsin, Milwaukee, WI, USA.
Melanie Carless, Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, TX, USA.
James Wallace, Department of Surgery, Medical College of Wisconsin, Milwaukee, WI, USA.
Richard Komorowski, Department of Pathology, Medical College of Wisconsin, Milwaukee, WI, USA.
David E. Kleiner, Laboratory of Pathology, National Cancer Institute, Bethesda, MD, USA
Deborah Andris, Department of Surgery, Medical College of Wisconsin, Milwaukee, WI, USA.
Bassem Makladi, Division of Endocrinology, Medical College of Wisconsin, Milwaukee, WI, USA.
Regina Cole, Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI, USA; Biotechnology and Bioengineering Center, Medical College of Wisconsin, Milwaukee, WI, USA.
Michael Charlton, Department of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN, USA.
Joanne Curran, Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, TX, USA.
Thomas D. Dyer, Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, TX, USA
Jac Charlesworth, Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, TX, USA.
Russell Wilke, Division of General Internal Medicine, Medical College of Wisconsin, Milwaukee, WI, USA; Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI, USA.
John Blangero, Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, TX, USA.
Ahmed H. Kissebah, Division of Endocrinology, Medical College of Wisconsin, Milwaukee, WI, USA Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI, USA.
Michael Olivier, Human and Molecular Genetics Center, Medical College of Wisconsin, Milwaukee, WI, USA; Biotechnology and Bioengineering Center, Medical College of Wisconsin, Milwaukee, WI, USA; Department of Physiology, Medical College of Wisconsin, Milwaukee, WI, USA.
References
- 1.Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118 (4):1388–93. [DOI] [PubMed] [Google Scholar]
- 2.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98(5):960–7. [DOI] [PubMed] [Google Scholar]
- 3.Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol. 2004;2(12):1048–58. [DOI] [PubMed] [Google Scholar]
- 4.Sanyal AJ. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology. 2002;123(5):1705–25. [DOI] [PubMed] [Google Scholar]
- 5.Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24(1):3–20. [DOI] [PubMed] [Google Scholar]
- 6.Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116(6):1413–9. [DOI] [PubMed] [Google Scholar]
- 7.Ekstedt M, Franzen LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44(4):865–73. [DOI] [PubMed] [Google Scholar]
- 8.Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis. 2008;28(4):339–50. [DOI] [PubMed] [Google Scholar]
- 9.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114(2):147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon JB, Bhathal PS, O'Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121(1):91–100. [DOI] [PubMed] [Google Scholar]
- 11.Younossi ZM, Baranova A, Ziegler K, et al. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology. 2005;42(3):665–74. [DOI] [PubMed] [Google Scholar]
- 12.Struben VM, Hespenheide EE, Caldwell SH. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am J Med. 2000;108(1):9–13. [DOI] [PubMed] [Google Scholar]
- 13.Willner IR, Waters B, Patil SR, et al. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol. 2001;96(10):2957–61. [DOI] [PubMed] [Google Scholar]
- 14.Schwimmer JB, Celedon MA, Lavine JE, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136 (5):1585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387–95. [DOI] [PubMed] [Google Scholar]
- 16.Browning JD, Kumar KS, Saboorian MH, et al. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am J Gastroenterol. 2004;99(2):292–8. [DOI] [PubMed] [Google Scholar]
- 17.Miele L, Beale G, Patman G, et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology. 2008;135(1):282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoneda M, Hotta K, Nozaki Y, et al. Association between PPARGC1A polymorphisms and the occurrence of nonalcoholic fatty liver disease (NAFLD). BMC Gastroenterol. 2008;8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sreekumar R, Rosado B, Rasmussen D, et al. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology. 2003;38(1):244–51. [DOI] [PubMed] [Google Scholar]
- 21.Chiappini F, Barrier A, Saffroy R, et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab Invest. 2006;86(2):154–65. [DOI] [PubMed] [Google Scholar]
- 22.Yoneda M, Endo H, Mawatari H, et al. Gene expression profiling of non-alcoholic steatohepatitis using gene set enrichment analysis. Hepatol Res. 2008;38(12):1204–12. [DOI] [PubMed] [Google Scholar]
- 23.Greco D, Kotronen A, Westerbacka J, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol. 2008;294(5):G1281–7. [DOI] [PubMed] [Google Scholar]
- 24.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21. [DOI] [PubMed] [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 26.Elam MB, Cowan GS Jr, Rooney RJ, et al. Hepatic gene expression in morbidly obese women: implications for disease susceptibility. Obesity (Silver Spring). 2009;17(8):1563–73. [DOI] [PubMed] [Google Scholar]
- 27.Maiti AK. Gene network analysis of oxidative stress-mediated drug sensitivity in resistant ovarian carcinoma cells. Pharmacogenomics J. 2010;10(2):94–104. [DOI] [PubMed] [Google Scholar]
- 28.Goring HH, Curran JE, Johnson MP, et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet. 2007;39(10):1208–16. [DOI] [PubMed] [Google Scholar]
- 29.Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7(6):489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furuhashi M, Fucho R, Gorgun CZ, et al. Adipocyte/Macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118(7):2640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuncman G, Erbay E, Hom X, et al. A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc Natl Acad Sci U S A. 2006;103(18):6970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maeda K, Cao H, Kono K, et al. Adipocyte/Macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005;1(2):107–19. [DOI] [PubMed] [Google Scholar]
- 34.Serra-Pages C, Medley QG, Tang M, et al. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem. 1998;273(25):15611–20. [DOI] [PubMed] [Google Scholar]
- 35.Kriajevska M, Fischer-Larsen M, Moertz E, et al. Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J Biol Chem. 2002;277(7):5229–35. [DOI] [PubMed] [Google Scholar]
- 36.Yang JJ. Mixed lineage kinase ZAK utilizing MKK7 and not MKK4 to activate the c-Jun N-terminal kinase and playing a role in the cell arrest. Biochem Biophys Res Commun. 2002;297 (1):105–10. [DOI] [PubMed] [Google Scholar]
- 37.Jandhyala DM, Ahluwalia A, Obrig T, et al. ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol. 2008;10(7):1468–77. [DOI] [PubMed] [Google Scholar]
- 38.Cheng YC, Kuo WW, Wu HC, et al. ZAK induces MMP-2 activity via JNK/p38 signals and reduces MMP-9 activity by increasing TIMP-1/2 expression in H9c2 cardiomyoblast cells. Mol Cell Biochem. 2009;325(1–2):69–77. [DOI] [PubMed] [Google Scholar]
- 39.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275(4):2247–50. [DOI] [PubMed] [Google Scholar]
- 40.Yamaguchi M. Role of calcium-binding protein regucalcin in regenerating rat liver. J Gastroenterol Hepatol. 1998;13(Suppl): S106–12. [DOI] [PubMed] [Google Scholar]
- 41.Izumi T, Yamaguchi M. Overexpression of regucalcin suppresses cell death and apoptosis in cloned rat hepatoma H4-II-E cells induced by lipopolysaccharide, PD 98059, dibucaine, or Bay K 8644. J Cell Biochem. 2004;93(3):598–608. [DOI] [PubMed] [Google Scholar]
- 42.Fukaya Y, Yamaguchi M. Overexpression of regucalcin suppresses cell death and apoptosis in cloned rat hepatoma H4-II-E cells induced by insulin or insulin-like growth factor-I. J Cell Biochem. 2005;96(1):145–54. [DOI] [PubMed] [Google Scholar]
- 43.Matsubara M, Tanaka T, Terato H, et al. Action mechanism of human SMUG1 uracil-DNA glycosylase. Nucleic Acids Symp Ser (Oxf). 2005;49(49):295–6. [DOI] [PubMed] [Google Scholar]
- 44.Pettersen HS, Sundheim O, Gilljam KM, et al. Uracil-DNA glycosylases SMUG1 and UNG2 coordinate the initial steps of base excision repair by distinct mechanisms. Nucleic Acids Res. 2007;35(12):3879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seki S, Kitada T, Yamada T, et al. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. 2002;37(1):56–62. [DOI] [PubMed] [Google Scholar]
- 46.Khetani SR, Szulgit G, Del Rio JA, et al. Exploring interactions between rat hepatocytes and nonparenchymal cells using gene expression profiling. Hepatology. 2004;40(3):545–54. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z, Li XJ, Liu Y, et al. Recombinant human decorin inhibits cell proliferation and downregulates TGF-beta1 production in hypertrophic scar fibroblasts. Burns. 2007;33(5):634–41. [DOI] [PubMed] [Google Scholar]
- 48.Kalamajski S, Aspberg A, Lindblom K, et al. Asporin competes with decorin for collagen binding, binds calcium and promotes osteoblast collagen mineralization. Biochem J. 2009;423(1):53–9. [DOI] [PubMed] [Google Scholar]
- 49.Chen AA, Khetani SR, Lee S, et al. Modulation of hepatocyte phenotype in vitro via chemomechanical tuning of polyelectrolyte multilayers. Biomaterials. 2009;30(6):1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Lu C, Wei H, et al. Hepatopoietin interacts directly with COP9 signalosome and regulates AP-1 activity. FEBS Lett. 2004;572(1–3):85–91. [DOI] [PubMed] [Google Scholar]
- 51.Pearce C, Hayden RE, Bunce CM, et al. Analysis of the role of COP9 signalosome (CSN) subunits in K562; the first link between CSN and autophagy. BMC Cell Biol. 2009;10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14(11):1209–25. [DOI] [PubMed] [Google Scholar]
- 53.Laplante JM, O'Rourke F, Lu X, et al. Cloning of human Ca2+ homoeostasis endoplasmic reticulum protein (CHERP): regulated expression of antisense cDNA depletes CHERP, inhibits intracellular Ca2+ mobilization and decreases cell proliferation. Biochem J. 2000;348(Pt 1):189–99. [PMC free article] [PubMed] [Google Scholar]
- 54.Vallee RB, Williams JC, Varma D, et al. Dynein: an ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58(2):189–200. [DOI] [PubMed] [Google Scholar]
- 55.Gennerich A, Vale RD. Walking the walk: how kinesin and dynein coordinate their steps. Curr Opin Cell Biol. 2009;21(1):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–43. [DOI] [PubMed] [Google Scholar]
- 57.Malhi H, Bronk SF, Werneburg NW, et al. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281 (17):12093–101. [DOI] [PubMed] [Google Scholar]
- 58.Li Z, Oben JA, Yang S, et al. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 2004;40(2):434–41. [DOI] [PubMed] [Google Scholar]
- 59.Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005;42(4):880–5. [DOI] [PubMed] [Google Scholar]
- 60.den Boer MA, Voshol PJ, Schroder-van der Elst JP, et al. Endogenous interleukin-10 protects against hepatic steatosis but does not improve insulin sensitivity during high-fat feeding in mice. Endocrinology. 2006;147(10):4553–8. [DOI] [PubMed] [Google Scholar]
- 61.Chan SS, Schedlich LJ, Twigg SM, et al. Inhibition of adipocyte differentiation by insulin-like growth factor-binding protein-3. Am J Physiol Endocrinol Metab. 2009;296(4):E654–63. [DOI] [PubMed] [Google Scholar]
- 62.Silha JV, Gui Y, Murphy LJ. Impaired glucose homeostasis in insulin-like growth factor-binding protein-3-transgenic mice. Am J Physiol Endocrinol Metab. 2002;283(5):E937–45. [DOI] [PubMed] [Google Scholar]
- 63.Chan SS, Twigg SM, Firth SM, et al. Insulin-like growth factor binding protein-3 leads to insulin resistance in adipocytes. J Clin Endocrinol Metab. 2005;90(12):6588–95. [DOI] [PubMed] [Google Scholar]
- 64.Oh Y, Muller HL, Lamson G, et al. Insulin-like growth factor (IGF)-independent action of IGF-binding protein-3 in Hs578T human breast cancer cells. Cell surface binding and growth inhibition. J Biol Chem. 1993;268(20):14964–71. [PubMed] [Google Scholar]
- 65.Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem. 1997;272(18):12181–8. [DOI] [PubMed] [Google Scholar]
- 66.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirsch C, Gauss R, Horn SC, et al. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458 (7237):453–60. [DOI] [PubMed] [Google Scholar]
- 68.Wei Y, Wang D, Pagliassotti MJ. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol Cell Biochem. 2007;303(1–2):105–13. [DOI] [PubMed] [Google Scholar]
- 69.Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):568–76. [DOI] [PubMed] [Google Scholar]
- 70.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581(19):3641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang L, Lai E, Teodoro T, et al. GRP78, but Not Protein-disulfide isomerase, partially reverses hyperglycemia-induced inhibition of insulin synthesis and secretion in pancreatic {beta}-cells. J Biol Chem. 2009;284(8):5289–98. [DOI] [PubMed] [Google Scholar]
- 72.Kammoun HL, Chabanon H, Hainault I, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119 (5):1201–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valero R, Marfany G, Gonzalez-Angulo O, et al. USP25, a novel gene encoding a deubiquitinating enzyme, is located in the gene-poor region 21q11.2. Genomics. 1999;62(3):395–405. [DOI] [PubMed] [Google Scholar]
- 74.Valero R, Bayes M, Francisca Sanchez-Font M, et al. Characterization of alternatively spliced products and tissue-specific isoforms of USP28 and USP25. Genome Biol. 2001;2(10): RESEARCH0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carlson MR, Zhang B, Fang Z, et al. Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics. 2006;7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacLennan NK, Dong J, Aten JE, et al. Weighted gene co-expression network analysis identifies biomarkers in glycerol kinase deficient mice. Mol Genet Metab. 2009;98(1–2):203–14. [DOI] [PubMed] [Google Scholar]
- 77.Horvath S, Zhang B, Carlson M, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103 (46):17402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saris CG, Horvath S, van Vught PW, et al. Weighted gene co-expression network analysis of the peripheral blood from amyotrophic lateral sclerosis patients. BMC Genomics. 2009;10:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharma MR, Polavarapu R, Roseman D, et al. Transcriptional networks in a rat model for nonalcoholic fatty liver disease: a microarray analysis. Exp Mol Pathol. 2006;81(3):202–10. [DOI] [PubMed] [Google Scholar]