

Abstract
23-O-(1,4′-Bipiperidine-1-carbonyl)betulinic acid (BBA), a synthetic derivative of 23-hydroxybetulinic acid (23-HBA), shows a reversal effect on multidrug resistance (MDR) in our preliminary screening. Overexpression of ATP-binding cassette (ABC) transporters such as ABCB1, ABCG2, and ABCC1 has been reported in recent studies to be a major factor contributing to MDR. Our study results showed that BBA enhanced the cytotoxicity of ABCB1 substrates and increased the accumulation of doxorubicin or rhodamine123 in ABCB1 overexpressing cells, but had no effect on non ABCB1 substrate, such as cisplatin; what’s more, BBA slightly reversed ABCG2-mediated resistance to SN-38, but did not affect the ABCC1-mediated MDR. Further studies on the mechanism indicated that BBA did not alter the expression of ABCB1 at mRNA or protein levels, but affected the ABCB1 ATPase activity by stimulating the basal activity at lower concentrations and inhibiting the activity at higher concentrations. In addition, BBA inhibited the verapamil-stimulated ABCB1 ATPase activity and the photolabeling of ABCB1 with [125I] iodoarylazidoprazosin in a concentration-dependent manner, indicating that BBA directly interacts with ABCB1. The docking study confirmed this notion that BBA could bind to the drug binding site(s) on ABCB1, but its binding position was only partially overlapping with that of verapamil or iodoarylazidoprazosin. Importantly, BBA increased the inhibitory effect of paclitaxel in ABCB1 overexpressing KB-C2 cell xenografts in nude mice. Taken together, our findings suggest that BBA can reverse ABCB1-mediated MDR by inhibiting its efflux function of ABCB1, which supports the development of BBA as a novel potential MDR reversal agent used in the clinic.
Keywords: BBA, multidrug resistance, ABCB1, ABCG2
Graphical Abstract
INTRODUCTION
Multidrug resistance (MDR) of cancer is described as the phenomenon of cancer cells developing resistance to various structurally and functionally unrelated drugs simultaneously; it is believed to be the major contributor to chemotherapy failure.1 The underlying mechanisms and causes responsible for MDR developed in cancer cells are complicated and not completely understood. Overexpression of ATP-binding cassette (ABC) transporters in cancer cells has been reported as one of the primary factors associated with MDR.2 ABC transporters are a family of large, membrane-bound proteins. ABC transporter family includes 49 members and is divided into seven subfamilies (A–G) based on sequence similarities.3 In humans, three major proteins found to be involved in cancer MDR are P-glycoprotein (P-gp/ABCB1/MDR1), multidrug resistance protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2/MXR/ABCP).4–7 Accumulated evidence shows that these transporters pump a wide range of structurally and functionally diverse amphipathic anticancer drugs out of tumor cells, thereby reducing intracellular drug retention, resulting in chemotherapeutic drug resistance.8 ABCB1/P-gp is a classical and the best-studied multidrug resistance related protein, which pumps out a variety of anticancer drugs, such as anthracyclines, topoisomerase inhibitors, vinca alkaloids, and taxanes.9 Therefore, inhibition of ABCB1 may resensitize the MDR cancer cells to the treatment of anticancer drugs. Currently, there are three generations of ABCB1 inhibitors classified by the time and the strategy employed in the discovery. The first generation ABCB1 inhibitors are identified as substrates of ABCB1 such as verapamil, cyclosporine A, and several calmodulin antagonists.10–13 Their low binding affinities require high doses to inhibit the efflux of anticancer drugs, which lead to side effects.14 The second generation ABCB1 inhibitors are mostly the derivatives of the first generation ABCB1 inhibitors, including PSC-833 and R-verapamil.15,16 This generation inhibitors have improved efficacy and reduced side effects compared with the first generation, however, they affect the activity of CYP3A4, which leads to a decrease in the metabolism of various anticancer drugs, and cause unacceptable toxicity.13 The third generation ABCB1 inhibitors include LY335979, CBT-1, and XR9576.17–19 These inhibitors are designed specifically for high transport affinity and low pharmacokinetic interaction, and show advantages compared to the previous generations. Nevertheless, to search for novel MDR modulators with higher efficacy and lower toxicity is still an urgent problem to be solved.
In China, the combination of anticancer drugs with traditional Chinese medicines (TCMs) has a long history in clinical cancer therapy, which shows good efficacy and low toxicity.20 In recent years, investigation on active ingredients of TCMs clinically used for cancer treatment has been becoming one of the promising pathways to obtain novel MDR modulators. For example, tetramethylpyrazine (TMP), a bioactive constituent isolated from the root of Ligusticum chuanxiong, can reverse the drug resistance of MCF-7/ADR and BEL-7402/ADR cells.21,22 Tetrandrine from Stephania tetrandra can prevent K562 cells from acquiring multidrug resistance, and its derivate H1 exhibits a more potent reversal effect by inhibiting the transport function and expression of ABCB1.23,24 Other natural active constituents from Chinese herbs, such as schisandrol A, emodin, rhynchophylline, jatrorrhizine, and indirulin, have been reported for their effects on reversing multidrug resistance of cancer.25–27
23-Hydroxybutulinic acid (23-HBA), a natural triterpene and major active ingredient isolated from a traditional Chinese herb, Pulsatilla chinensis, presents cytotoxicity toward several cancer cell lines.28,29 It has been reported that 23-HBA enhances sensitivity of doxorubicin (DOX, ADR) on MCF-7/ADR cell lines, indicating its potential to be developed as a novel MDR modulator.30 23-HBA has a low bioavailability due to its poor water solubility; structural modification has led to a series of derivatives aiming to enhance its bioactivity and improve pharmacological properties.31 Our preliminary screening results show that 23-O-(1,4′-bipiperidine-1-carbonyl)betulinic acid (BBA) had the most potential MDR reversal activity. This study has therefore been focused on the deep investigation of BBA’s MDR reversal activity and an attempt at elucidating its underlying mechanism.
EXPERIMENTAL SECTION
Reagents.
DOX, paclitaxel, (S)-camptothecin (SN-38), vinblastine, and mitoxantrone were purchased from Sigma. Cisplatin was obtained from Alfa Aesar. Vincristine (VCR) was provided by Hainan Periwinkle Pharmaceutical. The ABCB1 inhibitors verapamil (VRP) and cyclosporine A (CSA) were purchased from Merck Calbiochem and Beyotime Institute of Biotechnology, respectively. Antibodies against total ERK, total AKT, p-AKT(Thr308/Ser473) were supplied by Cell Signaling Technology. Anti-ABCB1/P-gp antibody was a product of Merck Calbiochem. Antibodies against Bcl-2 and p-ERK(Tyr204) were purchased from Santa Cruz. Alexa Fluor 488 goat anti-mouse IgG (H+L) was purchased from Life Technologies. [125I]Iodoarylazidoprazosin (IAAP) (2,200 Ci/mmol) was obtained from Perkin-Elmer Life Sciences. BBA was synthesized as previously described, and the chemical structure is shown in Figure 1A.31 Other chemicals were obtained from Sigma.
Figure 1.
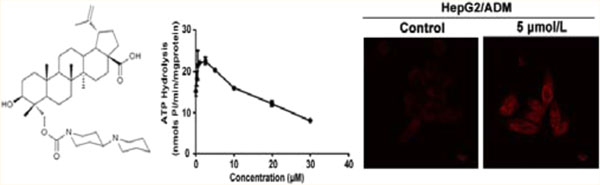
(A) The chemical structure of BBA. BBA inhibited the function of ABCB1. (B) The effect of BBA on the accumulation of DOX (top) and rhodamine123 (bottom) detected by laser scanning confocal microscope. Original amplification, 63; bar, 10 μm. (C) The accumulation of DOX (left) and rhodamine123 (right) in HepG2/ADM cells was analyzed by flow cytometry. (D) The effect of BBA on the efflux of rhodamine123 in HepG2/ADM cells. The mean values are plotted, and the error bars depict standard of deviation from at least three independent experiments performed in triplicate.
Cell Lines and Cell Culture.
Human hepatocellular carcinoma cell line HepG2 and its DOX-selected ABCB1-overexpressing derivate HepG2/ADM were kindly provided by Prof. Kowk-Pui Fung (The Chinese University of Hong Kong, Hong Kong).32 The human breast adenocarcinoma MCF-7 and its DOX-selected ABCB1-overexpressing derivate MCF-7/ADR were gifts from Prof. Li-Wu Fu (Sun Yat-Sen University, China). ABCB1 is overexpressed in HepG2/ADM and MCF-7/ADR cell lines compared with their parental cell lines (Supplementary Figure S1A in the Supporting Information).33 The human epidermoid carcinoma cell line KB-3–1, its colchicine-selected ABCB1-overexpressing drug-resistant cell line KB-C2, and its ABCC1-overexpressing cell subline were kindly provided by Dr. Shin-ichi Akiyama (Kagoshima University, Japan).34,35 The ABCC1 transfected KB-MRP1 cell line36 was provided by Dr. Kazumitsu Ueda (Kyoto University, Japan). These cell lines were cultured in RPMI-1640 or DMEM supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C in a humidified incubator containing 5% CO2. HEK293/pcDNA3.1, HEK/ABCG2–482-G2, HEK/ABCG2–482-R5, and HEK/ABCG2–482-T7 cells were established by selection with G418 after transfecting HEK293 with either empty pcDNA3.1 vector or pcDNA3.1 vector containing full length of ABCG2 coding arginine (R), glycine (G), or threonine (T) at 482 amino acid position.36 Similarly, HEK/ABCB1 cell line was generated by transfecting HEK293 with ABCB1 expressing vector. These transfected cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin, and 2 mg/mL G418 in a humidified incubator containing 5% CO2 at 37 °C.
MTT Assay.
Cells (5 × 103/well) were seeded in 96-well plates and cultured overnight. MDR modulator (BBA or verapamil) combined with various concentrations of a chemotherapeutic drug (DOX, paclitaxel, VCR, vinblastine, mitoxantrone, SN-38, or cisplatin) was added, and the cells were further cultured for 72 h. Cell viability was determined as previously described.37 The concentration required to inhibit cell growth by 50% (IC50) was calculated from survival curves using GraphPad Prism 4.0 software.
Confocal Microscopy Observation of Intracellular DOX and Rhodamine123.
Cells (1 × 105) were treated with various concentrations of BBA (0, 0.625, 1.25, 2.5, 5 μM) or verapamil (5 μM) for 2 h at 37 °C. For detection of DOX accumulation, 10 μM DOX was added and incubated for an additional 2 h in darkness at 37 °C; for detection of rhodamine123 accumulation, rhodamine123 (5 μM) was added and incubated for another hour. After washing cells thrice with cold PBS, images were acquired by a confocal microscope (LSM 510, Zeiss, Ger) at 488 nm extraction and 590 nm emission wavelength for DOX or 535 nm emission wavelength for rhodamine123.
Flow Cytometric Analysis of the Accumulation of DOX and Rhodamine123.
One milliliter of cell suspensions (1 × 106 cells/mL) was treated with various concentrations of BBA (0, 0.625, 1.25, 2.5, 5 μM) or verapamil (5 μM) for 2 h at 37 °C and then incubated with 10 μM DOX for an additional 2 h or 5 μM rhodamine123 for another 1 h in darkness at 37 °C, respectively. After that, cells were collected by centrifugation at 2,500 rpm and washed thrice with cold PBS. Finally, cells were resuspended in PBS buffer and analyzed by flow cytometry (Epics XL, Beckman, USA) equipped with excitation wavelength 488 nm and emission wavelength 590 or 535 nm respectively. Data were analyzed using WinMDI2.8 software.
Rhodamine123 Efflux Assay.
To measure drug efflux, 1 × 104 cells/well were seeded in 96-well black plates and cultured overnight. Then rhodamine123 (5 μM) with or without reversers (5 μM BBA or verapamil) was added and incubated for 2 h at 37 °C. Cells were sequentially washed thrice with cold PBS and placed in fresh medium or medium containing 5 μM of BBA or verapamil and incubated at 37 °C, and cells were washed twice with cold PBS at the desired time (0, 10, 20, 40 min) and measured spectrofluorometrically at 488 nm excitation and 535 nm emission wavelengths using a microplate reader (DTX880, Beckman).
Reverse Transcription PCR.
Cells (1 × 106) were treated with various concentrations of BBA for 24 h. Total cellular RNA was extracted by TRIzol Reagent (Invitrogen). The first strand cDNA was synthesized by Oligo dT primers using TIANScript RT kit (Tiangen Biotech) following the manufacturer’s instruction. The mRNA expression of ABCB1 in HepG2/ADM and MCF-7/ADR treated with BBA was detected as previously described.37
Western Blot Analysis.
Cells (1 × 106) were incubated with different concentrations of BBA for 48 h. Expression levels of ABCB1, AKT, p-AKTSer473, p-AKTThr308, ERK1/2, p-ERK1/2Tyr204 and Bcl-2 in total cell lysates were evaluated by Western blot as previously described.37
Immunofluorescence Assay.
Cells (1 × 105) were treated with 5 μM BBA for 24 h, then washed thrice with cold PBS, and fixed with 4% paraformaldehyde for 15 min at room temperature. After being washed thrice with cold PBS, cells were blocked with 3% BSA in PBST (1× PBS containing 0.2% Tween-80) for 30 min, followed by incubation with ABCB1 monoclonal antibody (1:100 dilution) at 4 °C overnight. After rinsing thrice with cold PBS, cells were stained with Alexa Fluor 488 conjugated goat anti-mouse secondary antibody (1:500 dilution) for 1 h at room temperature in darkness and rinsed thrice with cold PBS. DNA specific dye DAPI (1 μg/μL) was used for nuclear counterstaining. Immunofluorescence images were acquired by a confocal microscope (LSM 510, Zeiss) at 495 nm excitation and 519 nm emission wavelength (FITC) and 340 nm excitation and 488 nm emission wavelength (DAPI).
ATPase Assay.
The vanadate (Vi)-sensitive ATPase activity of ABCB1 was determined with crude membrane fractions from High-Five insect cells expressing human ABCB1 as described.38 The membrane fractions (100 μg of protein/mL) were incubated in ATPase assay buffer (50 mM MES-Tris pH 6.8, 50 mM KCl, 5 mM sodium azide, 1 mM EGTA, 1 mM ouabain, 10 mM MgCl2 and 2 mM DTT), containing BBA at various concentrations and Vi (0 or 0.3 mM), for 5 min at room temperature. The ATPase reaction was started by addition of 5 mM ATP and immediately transferring the reaction to 37 °C water bath for 20 min. The reaction was stopped by addition of 100 μL of 5% SDS solution. The liberated inorganic phosphate was determined as previously described.38
Photoaffinity Labeling of ABCB1 with [I125]IAAP.
The crude membrane fractions from High-Five insect cells expressing human ABCB1 were prepared following the protocol as previously described.39 Total 50–70 μg of crude membrane protein was incubated with BBA at various concentrations in 50 mM Tris-HCl, pH 7.5 at room temperature. Samples were further incubated for 5 min with [I125]IAAP (5–7 nM) under subdued light. Photo-cross-linking was initiated by exposing solution to UV 365 nM for 5 min. The reaction was quenched with addition of 5×-SDS–PAGE sample buffer. Protein (18 μg) was loaded per lane on a 7% Tris-acetate gel and ran at 150 V for 80 min. The gels were dried and exposed to Bio-Max MR film (Eastman Kodak) at −70 °C for 6–12 h. The radioactivity incorporated in ABCB1 was quantified using STORM 860 PhosphorImager System (GE Healthcare) and software Image Quant.
Docking Protocol.
We employed the crystal structure of mouse ABCB1 reported previously (PDB entry code: 3G5U) to complete this docking experiment.40 BBA, verapamil, and IAAP were docked into the binding site of ABCB1. The 3D structures of BBA, verapamil, and IAAP were built using the Sketch Molecule module in SYBYL 8.1 program package of Tripos, Inc., and the structural energy minimization was performed using the standard Tripos molecular mechanics force field and Gasteiger–Hückel charge. The max iterations for the minimization was set to 2,000, and the minimization was terminated when the energy gradient convergence criterion of 0.05 kcal/mol·Å was reached. In present work, the residues method was applied. Other parameters were established by default in software.
Animals and Tumor Xenograft Experiments.
Male nu/nu NCR nude mice from Taconic (Albany, NY), 5 to 6 weeks old and weighing 18 to 22 g, were housed at the animal facility of St. John’s University. All animals were housed in barrier facilities and received sterilized food and water. All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee of St. John’s University. The KB-C2-inoculated nude xenograft model was established as described by Fu and colleagues.41 Briefly, 1 × 107 KB-C2 cells were implanted sc under the shoulder of the nude mice. The mice were randomized into four groups after the tumors reached a mean diameter of 0.5 cm, and then received various regimens: (a) control (saline, q3d × 6, ip); (b) paclitaxel (q3d × 6, ip, 18 mg/kg); (c) BBA (q3d × 6, po, 15 mg/kg); (d) BBA (q3d × 6, po, 15 mg/kg) and paclitaxel (q3d × 6, ip, 18 mg/kg) (BBA was dissolved in 10% N-methylpyrrolidinone and 90% polyethylene glycol 300, and was given 2 h before paclitaxel was injected). The mouse weight, tumor volumes, feeding behavior, and activity were recorded every 3 days. When the mean tumor weight reached about 1 g, the mice were euthanized and xenografts were removed and measured. Tumor volumes and the ratio of growth inhibition were estimated as previously described.42
Data Analysis.
Each experiment was performed at least three times, and data are expressed as mean ± standard deviation (SD). Significant difference was determined using Student’s t-test (two-tailed, α = 0.05).
RESULTS
BBA Sensitizes ABCB1-Overexpressing Cells to Chemotherapeutic Drugs.
The cytotoxic and modulating effects of BBA in ABCB1-, ABCC1-, and ABCG2-overexpressing cell lines were determined by MTT assay. The subcytotoxic concentration of BBA with 85% of cell survival was calculated to be 5 μM for the sensitive and resistant cell lines (Supplementary Figure S1B in the Supporting Information). Concentrations of BBA at 0.625, 1.25, 2.5, and 5 μM were therefore used in the following experiments to determine its MDR reversal activity and to explore underlying mechanisms. Investigation of BBA’s modulating effect on MDR was first conducted in both parental (HepG2, MCF-7, and KB-3–1) and resistant (HepG2/ADM, MCF-7/ADR, and KB-C2) cell lines. BBA greatly enhanced the cytotoxicity of these chemotherapeutic drugs in MDR cells but not in parental cells (Table 1). Similar experiments were then performed in ABCB1-transfected HEK293 cells. Consistently, BBA sensitized HEK293/ABCB1 cells to the treatment of VCR, vinblastine, and paclitaxel, but not cisplatin, which is not a substrate of ABCB1. To further study whether BBA is a specific ABCB1 reversal agent, we carried out experiments in the ABCG2- and ABCC1-overexpressing cells. It is known that mutations at amino acid 482 in ABCG2 affect the specific property of ABCG2 substrates, therefore, we treated wild-type and mutant ABCG2-overexpressing HEK293-ABCG2 cells with 5 μM BBA in the presence of mitoxantrone or SN-38; the ABCC1-overexpressing cell lines KB-CV60 and KB/MRP1 were treated with VCR or vinblastine.36,43 It was found that BBA slightly decreased the sensitivity of ABCG2–482-R5 cells (wild-type ABCG2) to mitoxantrone, but no significant difference was observed between ABCG2–482-T7 and ABCG2–482-G2 cells (mutant types) treated with or without BBA (Table 2). In addition, BBA at 5 μM significantly increased the sensitivity of three ABCG2-overexpressing cell lines to SN-38. However, BBA could not reverse or reduce the drug resistance of KB-CV60 and KB/MRP1 cells (Table 2). These results indicate that BBA specifically reverses ABCB1-mediated drug resistance, and slightly reduces wild-type ABCG2-mediated drug resistance to SN-38, but has no effect on ABCC1-mediated drug resistance.
Table 1.
Effect of BBA on Reversing P-gp-Mediated Drug Resistance
| IC50 ± SDa (μM) (fold-reversal)b |
||||
|---|---|---|---|---|
| HepG2 | HepG2/ADM | MCF-7 | MCF-7/ADR | |
| doxorubicin | 0.3809 ± 0.0333 (1.00) | 85.3891 ± 2.9511 (1.00) | 0.2437 ± 0.0267 (1.00) | 17.1000 ± 0.8839 (1.00) |
| + BBA 0.625 μM | 0.3611 ± 0.0030 (1.05) | 21.6396 ± 1.2168 (3.95) | 0.2414 ± 0.0106 (1.01) | 4.1837 ± 0.4922 (4.09) |
| + BBA 1.25 μM | 0.3255 ± 0.0228 (1.17) | 6.0008 ± 0.0541 (14.23) | 0.2220 ± 0.0393 (1.10) | 2.3363 ± 0.3107 (7.32) |
| + BBA 2.5 μM | 0.3261 ± 0.0326 (1.17) | 3.6789 ± 0.3552 (23.21) | 0.2472 ± 0.0251 (0.99) | 1.2590 ± 0.1754 (13.58) |
| + BBA 5 μM | 0.3190 ± 0.0079 (1.19) | 1.5932 ± 0.0180 (53.60) | 0.1997 ± 0.0231 (1.22) | 0.6982 ± 0.0754 (24.49) |
| + verapamil 5 μM | 0.2989 ± 0.0345 (1.27) | 4.6455 ± 0.0487 (18.38) | 0.1693 ± 0.0343 (1.44) | 2.6930 ± 0.2222 (6.35) |
| vincristine | 0.0149 ± 0.0093 (1.00) | 2.6260 ± 0.1231 (1.00) | 0.0094 ± 0.0001 (1.00) | 0.7423 ± 0.0183 (1.00) |
| + BBA 0.625 μM | 0.0168 ± 0.0086 (0.88) | 0.7426 ± 0.0564 (3.54) | 0.0093 ± 0.0007 (1.01) | 0.0898 ± 0.0062 (8.23) |
| + BBA 1.25 μM | 0.0127 ± 0.0064 (1.17) | 0.2677 ± 0.0357 (9.81) | 0.0088 ± 0.0008 (1.06) | 0.0326 ± 0.0026 (22.80) |
| + BBA 2.5 μM | 0.0170 ± 0.0081 (0.87) | 0.0676 ± 0.0086 (38.88) | 0.0094 ± 0.0008 (1.00) | 0.0119 ± 0.0013 (62.59) |
| + BBA 5 μM | 0.0128 ± 0.0049 (1.16) | 0.0199 ± 0.0037 (132.29) | 0.0065 ± 0.0002 (1.45) | 0.0049 ± 0.0004 (151.04) |
| + verapamil 5 μM | 0.0108 ± 0.0077 (1.38) | 0.1193 ± 0.0407 (22.01) | 0.0050 ± 0.0006 (1.88) | 0.0252 ± 0.0028 (29.48) |
| paclitaxel | 0.0019 ± 0.0002 (1.00) | 1.5010 ± 0.0447 (1.00) | 0.0023 ± 0.0005 (1.00) | 0.7186 ± 0.0465 (1.00) |
| + BBA 0.625 μM | 0.0019 ± 0.0002 (1.01) | 0.9099 ± 0.0337 (1.65) | 0.0023 ± 0.0008 (1.00) | 0.1273 ± 0.0117 (5.65) |
| + BBA 1.25 μM | 0.0022 ± 0.0002 (0.86) | 0.3705 ± 0.0298 (4.05) | 0.0020 ± 0.0007 (1.15) | 0.0319 ± 0.0018 (22.54) |
| + BBA 2.5 μM | 0.0014 ± 0.0002 (1.35) | 0.0862 ± 0.0097 (17.42) | 0.0020 ± 0.0003 (1.15) | 0.0168 ± 0.0023 (42.72) |
| + BBA 5 μM | 0.0011 ± 0.0001 (1.72) | 0.0191 ± 0.0016 (78.59) | 0.0019 ± 0.0005 (1.25) | 0.0048 ± 0.0004 (151.07) |
| + verapamil 5 μM | 0.0016 ± 0.0002 (1.18) | 0.1419 ± 0.0150 (10.58) | 0.0022 ± 0.0004 (1.05) | 0.0121 ± 0.0021 (59.32) |
| cisplatin | 5.2585 ± 0.6399 (1.00) | 7.1703 ± 0.7670 (1.00) | 10.5700 ± 1.5712 (1.00) | 9.0415 ± 0.1110 (1.00) |
| + BBA 5 μM | 5.5675 ± 0.0799 (1.29) | 7.5537 ± 1.0330 (0.95) | 11.9433 ± 1.0310 (0.89) | 9.8800 ± 1.7112 (0.92) |
| + verapamil 5 μM | 5.5405 ± 0.3613 (1.29) | 7.4770 ± 0.8122 (0.96) | 10.2740 ± 1.0080 (1.03) | 9.1560 ± 1.2785 (0.99) |
| IC50 ± SDa (nM) (fold-reversal)b |
||||
| KB-3–1 | KB-C2 | HEK293 | HEK293/ABCB1 | |
| vincristine | 8.95 ± 0.69 (1.00) | 897.10 ± 80.45 (1.00) | 10.20 ± 0.89 (1.00) | 755.35 ± 60.57 (1.00) |
| + BBA 1.25 μM | 9.52 ± 0.81 (0.94) | 645.38 ± 117.05 (1.39) | 10.15 ± 0.90 (1.00) | 293.56 ± 28.76 (2.57) |
| + BBA 2.5 μM | 8.49 ± 0.11 (1.05) | 266.28 ± 93.54 (3.37) | 9.53 ± 0.71 (1.07) | 42.67 ± 3.76 (17.70) |
| + BBA 5 μM | 8.34 ± 0.74 (1.07) | 71.73 ± 13.62 (12.51) | 9.21 ± 1.12 (1.11) | 24.65 ± 2.73 (30.64) |
| + verapamil 5 μM | 5.91 ± 0.46 (1.51) | 74.07 ± 5.17 (12.11) | 8.90 ± 0.87 (1.15) | 17.91 ± 1.89 (42.17) |
| vinblastine | 53.26 ± 10.09 (1.00) | 1808.59 ± 18.55 (1.00) | 13.87 ± 1.28 (1.00) | 301.58 ± 27.22 (1.00) |
| + BBA 1.25 μM | 55.32 ± 8.74 (0.96) | 1761.98 ± 112.44 (1.03) | 13.15 ± 1.39 (1.05) | 106.12 ± 13.89 (2.84) |
| + BBA 2.5 μM | 49.57 ± 3.52 (1.07) | 796.84 ± 18.70 (2.27) | 12.89 ± 1.37 (1.08) | 34.93 ± 4.21 (8.63) |
| + BBA 5 μM | 70.09 ± 7.59 (0.76) | 146.22 ± 18.16 (12.37) | 13.32 ± 1.06 (1.04) | 16.82 ± 1.71 (17.93) |
| + verapamil 5 μM | 25.19 ± 2.64 (2.11) | 86.05 ± 5.33 (21.02) | 11.37 ± 0.95 (1.22) | 16.30 ± 1.60 (18.50) |
| paclitaxel | 6.79 ± 0.11 (1.00) | 2840.71 ± 549.87 (1.00) | 4.19 ± 0.32 (1.00) | 157.20 ± 13.07 (1.00) |
| + BBA 1.25 μM | 6.39 ± 0.13 (1.06) | 2377.54 ± 340.53 (1.19) | 3.99 ± 0.41 (1.05) | 40.45 ± 4.64 (3.89) |
| + BBA 2.5 μM | 6.93 ± 0.10 (0.98) | 1971.65 ± 275.33 (1.44) | 3.79 ± 0.35 (1.11) | 21.75 ± 1.96 (7.23) |
| + BBA 5 μM | 7.72 ± 0.15 (0.88) | 277.48 ± 15.12 (10.24) | 3.91 ± 0.37 (1.07) | 12.11 ± 1.28 (12.98) |
| + verapamil 5 μM | 6.49 ± 0.47 (1.05) | 131.89 ± 9.89 (21.54) | 3.52 ± 0.25 (1.19) | 15.93 ± 1.34 (9.87) |
| cisplatin | 3056.33 ± 323.51 (1.00) | 2840.44 ± 358.52 (1.00) | 2208.82 ± 82.24 (1.00) | 2526.16 ± 70.69 (1.00) |
| + BBA 5 μM | 4800.12 ± 208.38 (0.64) | 4939.01 ± 260.35 (0.58) | 2067.82 ± 93.44 (1.07) | 2370.29 ± 151.77 (1.07) |
| + verapamil 5 μM | 3035.92 ± 528.93 (1.01) | 3821.76 ± 249.19 (0.74) | 2511.15 ± 48.79 (0.88) | 2651.52 ± 99.08 (0.95) |
Data in the table are shown as the means ± SD of at least three independent experiments performed in triplicate.
The fold-reversals are calculated as IC50 for cells with the anticancer drug in the absence of inhibitors divided by that in the presence of inhibitors.
Table 2.
Effect of BBA on Reversing BCRP- and MRP1-Mediated Drug Resistance
| IC50 ± SDa (nM) (fold-reversal)b |
|||||
|---|---|---|---|---|---|
| HEK 293/pcDNA-3.1 | ABCG2–482-R5 | ABCG2–482-T7 | ABCG2–482-G2 | ||
| mitoxantrone | 7.57 ± 0.35 (1.00) | 84.31 ± 8.37 (1.00) | 442.33 ± 182.04 (1.00) | 448.26 ± 103.04 (1.00) | |
| + BBA 2.5 μM | 7.13 ± 0.24 (1.06) | 164.57 ± 6.28 (0.51) | 546.76 ± 76.75 (0.81) | 528.61 ± 62.61 (0.85) | |
| + BBA 5 μM | 6.86 ± 0.26 (1.10) | 143.63 ± 15.77 (0.59) | 545.32 ± 134.39 (0.81) | 511.33 ± 60.32 (0.88) | |
| + nilotinib 5 μM | 6.72 ± 0.29 (1.13) | 7.31 ± 0.34 (11.53) | 8.29 ± 0.25 (53.36) | 19.30 ± 10.07 (23.23) | |
| SN-38 | 8.32 ± 0.66 (1.00) | 260.60 ± 16.93 (1.00) | 121.32 ± 10.50 (1.00) | 176.26 ± 7.63 (1.00) | |
| + BBA 1.25 μM | 8.50 ± 0.72 (0.98) | 262.83 ± 12.62 (0.99) | 111.07 ± 12.46 (1.09) | 168.67 ± 17.09 (1.04) | |
| + BBA 2.5 μM | 8.34 ± 0.34 (1.00) | 245.40 ± 17.95 (1.06) | 108.28 ± 7.26 (1.12) | 133.99 ± 14.81 (1.32) | |
| + BBA 5 μM | 7.64 ± 0.60 (1.09) | 120.10 ± 11.01 (2.17) | 64.12 ± 7.76 (1.89) | 69.79 ± 5.71 (2.53) | |
| + FTC 2.5 μM | 8.36 ± 0.65 (1.00) | 12.61 ± 1.42 (20.67) | 15.37 ± 1.40 (7.89) | 11.87 ± 0.63 (14.85) | |
| IC50 ± SDa (nM) (fold-reversal)b |
|||||
| KB-3–1 | KB-CV60 | KB-MRP1 | |||
| vincristine | 8.75 ± 0.39 (1.00) | 336.25 ± 75.21 (1.00) | 182.05 ± 18.28 (1.00) | ||
| + BBA 5 μM | 9.65 ± 1.33 (0.91) | 404.33 ± 30.80 (0.83) | 171.17 ± 12.12 (1.06) | ||
| + PAK104P 10 μM | 6.11 ± 0.23 (1.43) | 21.93 ± 0.42 (15.33) | 19.70 ± 0.20 (9.24) | ||
| vinblastine | 57.68 ± 8.06 (1.00) | 484.57 ± 96.32 (1.00) | 296.50 ± 14.87 (1.00) | ||
| + BBA 5 μM | 68.58 ± 11.23 (0.84) | 568.67 ± 54.87 (0.85) | 344.89 ± 72.18 (0.86) | ||
| + PAK104P 10 μM | 53.33 ± 7.51 (1.08) | 41.93 ± 2.07 (11.56) | 94.72 ± 19.37 (3.13) | ||
Data in the table are shown as the means ± SD of at least three independent experiments performed in triplicate.
The fold-reversal is calculated as the IC50 for cells with the anticancer drug in the absence of inhibitors divided by that in the presence of inhibitors.
BBA Modulates ABCB1-Mediated Transport.
In order to investigate whether BBA-induced improvement of sensitivities of chemotherapeutic drugs in ABCB1-overexpressing cells is due to its inhibitory effect on the transporter function of ABCB1, we measured accumulation of DOX in HepG2/ADM cells using confocal microscopy and flow cytometry assay. Parental HepG2 cells and the corresponding MDR HepG2/ADM cells treated with verapamil (5 μM) were used as positive controls. Untreated MDR cells (Control, shown in the second panel of Figure 1B) have much lower levels of accumulation of DOX in cytosol; in a dose-dependent manner, BBA treatment induced a remarkable increase in intracellular accumulation of DOX which was indicated by stronger red fluorescence intensity (top row of Figure 1B). Then we tested the effect of BBA on accumulation of a specific ABCB1 substrate rhdoamine123. BBA enhanced the intracellular accumulation of rhdomaine123 in a dose-dependent manner (bottom row of Figure 1B). BBA at 5 μM (purple color) had the strongest effect on the accumulation of DOX and outperformed verapamil (turquoise color) at the same concentration, azure blue peak (left panel of Figure 1C). Similar phenomena were observed in rhodamine123 accumulation experiments in BBA-treated resistant cells (right panel of Figure 1C). Both experimental results demonstrate that BBA promotes the accumulation of ABCB1 substrates. To better understand BBA-induced intracellular accumulation of anticancer drugs, we further quantitatively measured intracellular concentrations of rhodamine123 after treating HepG2/ADM cells, with 5 μM of BBA or verapamil, or without treatment, for 2 h, and then washed at 10, 20, and 40 min, respectively. BBA treated MDR cells had significantly higher intracellular levels of rhodamine123, indicating that BBA attenuated rhodamine123 efflux (Figure 1D, –■–) at all tested time points; its effect was more potent than that of verapamil (Figure 1D, –▲–). Collectively, our results suggest that BBA modulates MDR by suppressing efflux of ABCB1 substrates, thereby increasing their intracellular accumulation. Similar results were found in the other ABCB1-overexpressing cell line MCF-7/ADR (Supplementary Figure S2A–C in the Supporting Information).
BBA Does Not Affect mRNA and Protein Expression Levels of ABCB1.
A possible underlying mechanism of BBA’s effect in reducing drug efflux is that BBA downregulates ABCB1 protein expression. We therefore measured mRNA and protein expression levels of ABCB1 in HepG2/ADM cell before and after BBA treatment. The expression levels of MDR1 mRNA and ABCB1 protein of HepG2/ADM cell with BBA treatment did not show significant change (Figure 2A). To further investigate if there was any change in expression of membrane-bound ABCB1 protein, the immunofluorescence analysis using Alexa Fluor 488-conjugated anti-mouse IgG antibody was conducted; consistent with the above results, BBA treatment for 24 h at concentration even up to 5 μM could not alter the expression of ABCB1 located on membrane and cytosol of cells, and it also could not trigger translocation of ABCB1 from membrane to cytosol (Figure 2B). Similar results were found in another ABCB1-overexpressing cell line, MCF-7/ADR (Supplementary Figure S2D,E in the Supporting Information).
Figure 2.
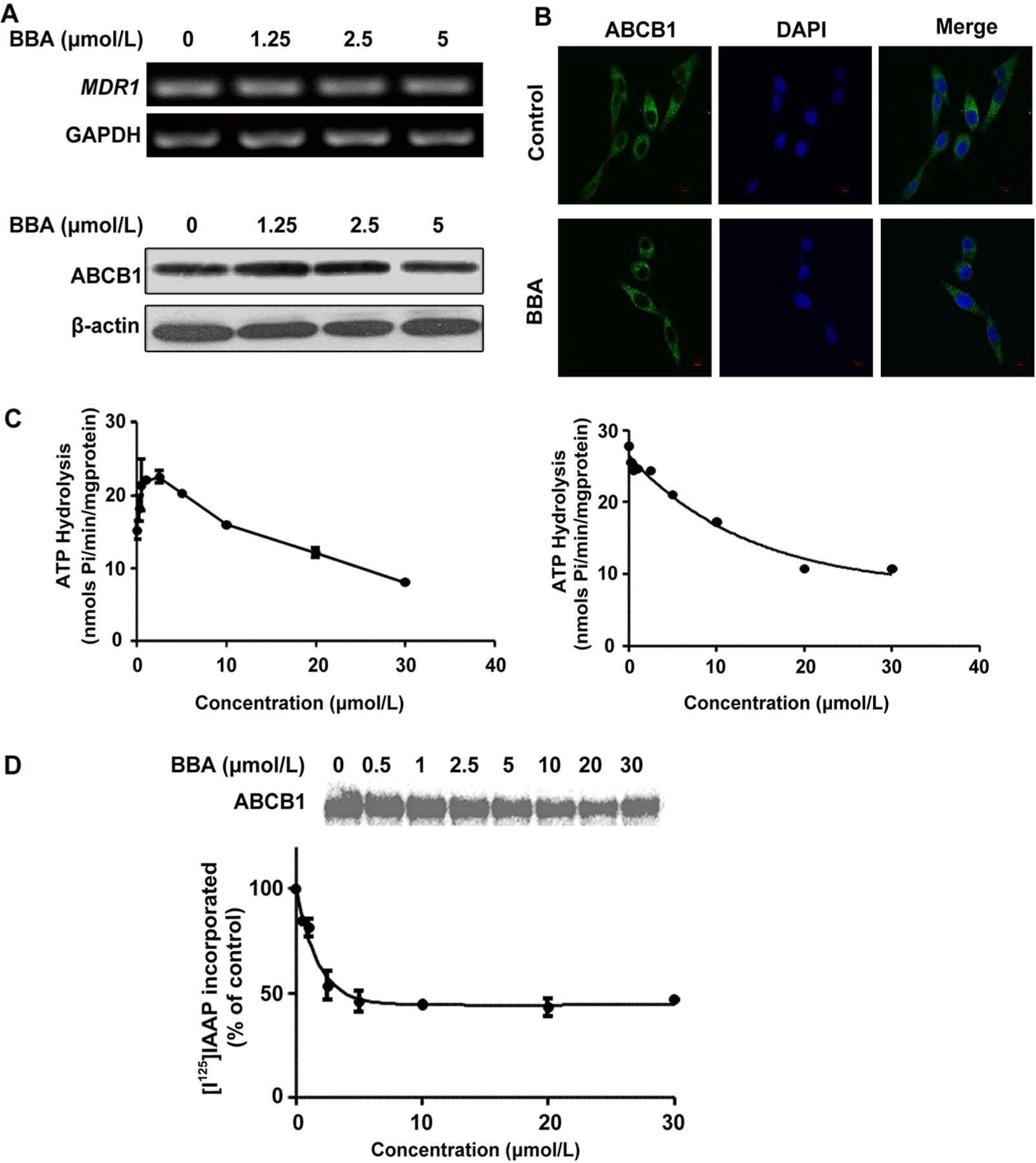
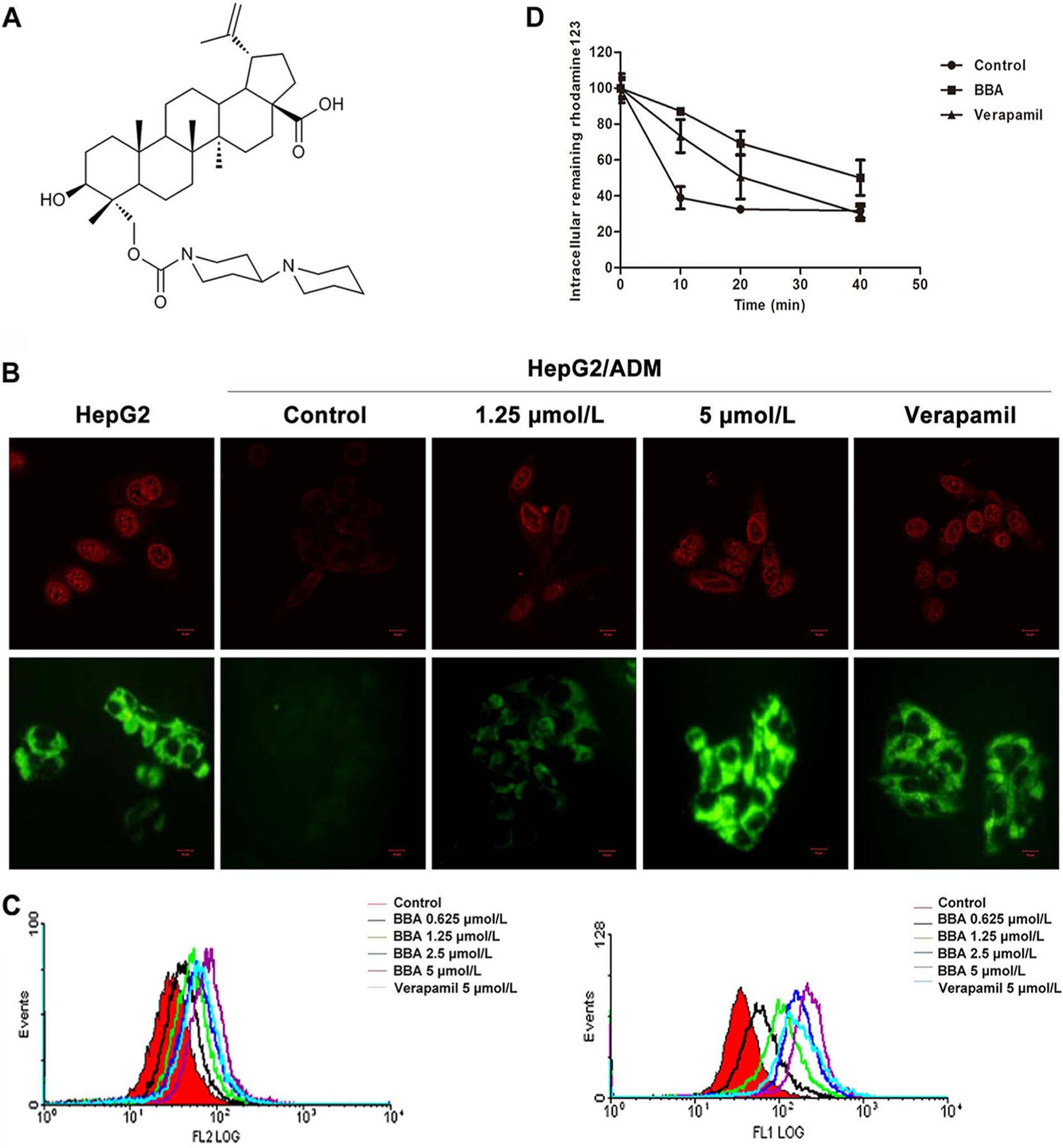
BBA affected the ATP activity, and partly inhibited the photoaffinity labeling of ABCB1 with [I125]IAAP. (A) BBA did not affect the mRNA (top) and protein expression (bottom) of multidrug-resistant proteins in HepG2/ADM cells. (B) The localization of ABCB1 at membrane and cytosol of HepG2/ADM cells. Immunofluorescence staining of cells with primary antibody against ABCB1 and Alexa Fluor 488-conjugated secondary antibody was observed by confocal microscope. Original amplification, 63; bar, 10 μm. No immunostaining occurred when the primary antibody was omitted from the procedure (data not shown). (C) The Vi-sensitive ATPase activity of ABCB1 was determined in membrane vesicles as a function of concentration of BBA in the absence (left) and presence of 5 μM verapamil (right). The values and the error bars represent the mean values and standard of deviation from three independent experiments in plot A, while the values in plot B are averages of three independent experiments. (D) The top panel shows the autoradiogram of incorporation of IAAP into the ABCB1 band from a representative experiment. The radioactivity associated with the ABCB1 band was quantified and represented in the lower panel using a phosphorimager and the curve-fitting software GraphPad PRISM. The mean values are plotted, and the error bars depict standard of deviation from at least three independent experiments.
BBA Affects the ATPase Activity.
ABCB1 is an ATP-dependent membrane transporter, and its transport function is regulated by ATP hydrolysis. As shown in the results described in the previous section, BBA did not alter ABCB1 expression. Therefore, we speculate that reversal activity of BBA is due to the effect of ATPase activity. The ATPase assay results (left panel of Figure 2C) showed that BBA at lower concentrations up to 2.5 μM stimulated the basal activity of ABCB1 by 1.5-fold but at higher concentrations (up to 30 μM) it consistently reduced the activity to near basal levels. At 30 μM, BBA also inhibited the verapamil-stimulated ATPase activity with the maximum inhibition of 60% (in relation to control) (right panel of Figure 2C).
BBA Inhibits [I125]IAAP Photolabeling of ABCB1.
The results of the [125I]IAAP photoaffinity labeling assay showed that increasing concentrations of BBA consistently reduced the amount of [125I]IAAP incorporated into ABCB1, suggesting that BBA inhibits [125I]IAAP binding at the drug binding site of ABCB1 (Figure 2D). At 30 μM of BBA, the inhibition of [125I]IAAP binding was 50%, which was much lower compared to near 95% inhibition with known ABCB1 inhibitors such as cyclosporine A at 10 μM concentration. The incomplete inhibition with BBA even at high concentration suggests that the BBA binding site on ABCB1 may only partially overlap with that of [125I]IAAP.
BBA Is Not a Transported Substrate of ABCB1 and Has Persistent Reversal Activity.
BBA itself has an equally moderate inhibitory effect on the proliferation of HepG2 and HepG2/ADM cell lines in a concentration-dependent manner (Figure 3A). These data indicate that overexpression of ABCB1 does not confer cell resistance to BBA, and BBA may not be a substrate of ABCB1. To further support this notion, the effects of two ABCB1 inhibitors, verapamil and cyclosporine A, on BBA cytotoxicity toward ABCB1-overexpressing resistant cells were determined by MTT assay. As shown in Figure 3B, neither verapamil nor cyclosporine A treatment affected the sensitivity of BBA in HepG2/ADM cells. These data indicate that BBA is not a transport substrate of ABCB1, or only an inhibitor of ABCB1. Furthermore, we studied the duration of BBA reversal activity using the method described previously.44 It was found that washout of BBA at different concentrations (1.25 and 5 μM) after 24 h treatment still could increase the cytotoxicity of DOX toward resistant cells, although this effect was weaker than for the group with BBA existing in the medium (Figure 3C). In contrast, the MDR reversal activity of verapamil was almost completely lost when it was washed out with fresh medium.
Figure 3.
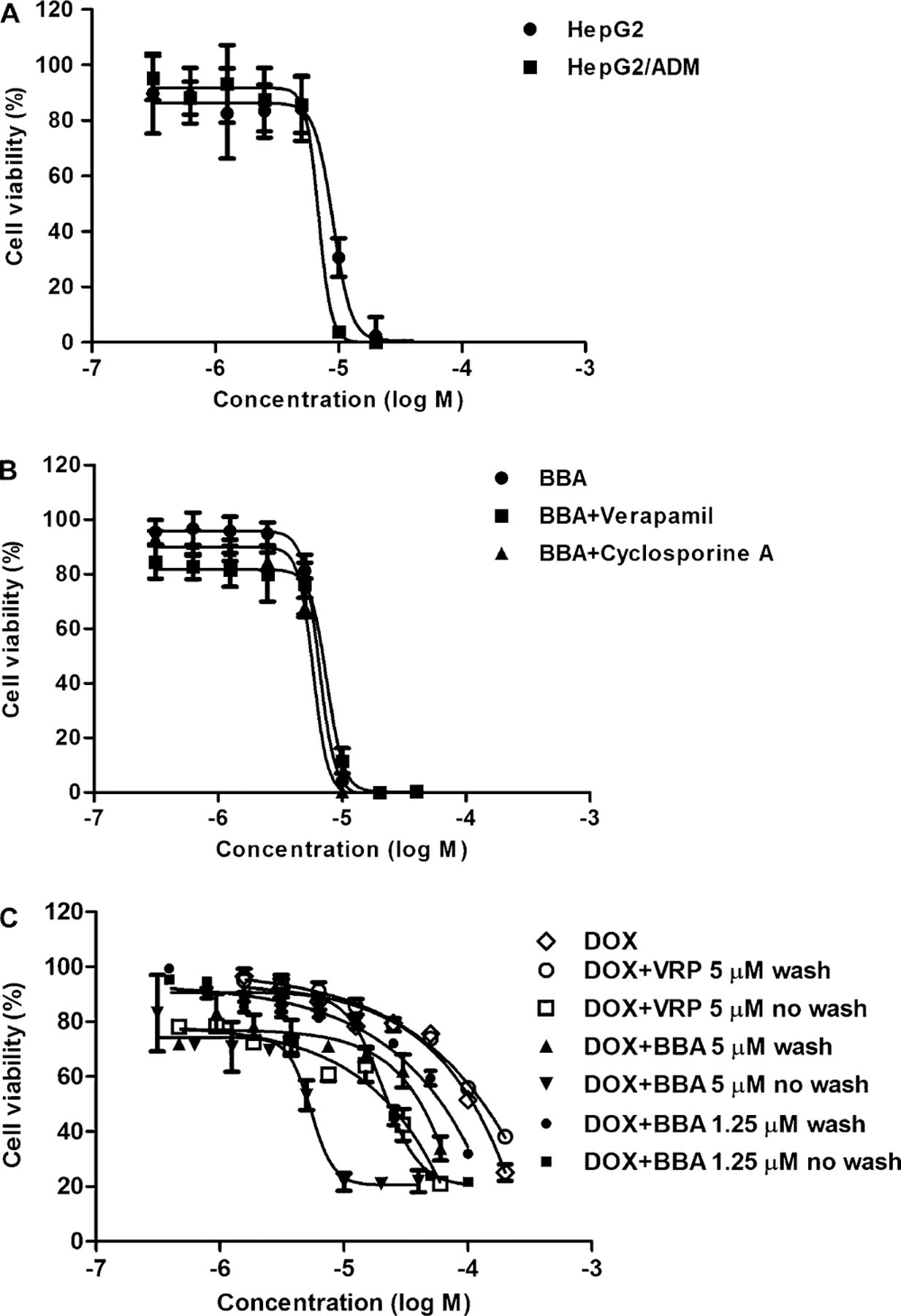
BBA was not a transported substrate of ABCB1 and had persistent reversal activity. (A) Cytotoxicity of BBA on drug-sensitive HepG2 and drug-resistant HepG2/ADM cells. (B) Effect of verapamil and cyclosporine A on the sensitivity of BBA in ABCB1-overexpressing HepG2/ADM cells. Cells were treated with various concentrations of BBA in the presence or absence of verapamil (5 μM) or cyclosporine A (5 μM) for 48 h, and cytotoxicity of BBA was detected by MTT assay. The mean values are plotted, and the error bars depict standard of deviation from at least three independent experiments. (C) Duration of BBA reversing DOX resistance in HepG2/ADM cells. Cells were treated with or without BBA (1.25 and 5 μM) or verapamil (5 μM) for 24 h before being washed out, then treated with DOX for 48 h. Cytotoxicity of DOX was detected by MTT assay. The mean values are plotted, and the error bars depict standard of deviation from at least three independent experiments.
Molecular Docking Model of BBA Binding to ABCB1.
BBA is for the first time reported for its reversal activity on ABCB1-mediated MDR, and the existing results have indicated that the effect of BBA on the ABCB1 transport function may be related to the direct interaction on ABCB1. Therefore, to better confirm our hypothesis, and to probe the probable binding site of BBA to ABCB1, we performed a virtual docking experiment. BBA (labeled in yellow) was able to bind to ABCB1 at the same site as verapamil (labeled in green) and IAAP (labeled in red), but did not completely overlap (Figure 4A; the right panel is the enlarged drawing). Among the residual amino acids of BBA, ALA951, SER218, PR0219, ALA981, PHE979, and ILE977 (Figure 4B,C) were the same as verapamil and IAAP. This docking result further confirmed that BBA binding to ABCB1 partly coincided with the site of verapamil and IAAP, but did not completely overlap.
Figure 4.
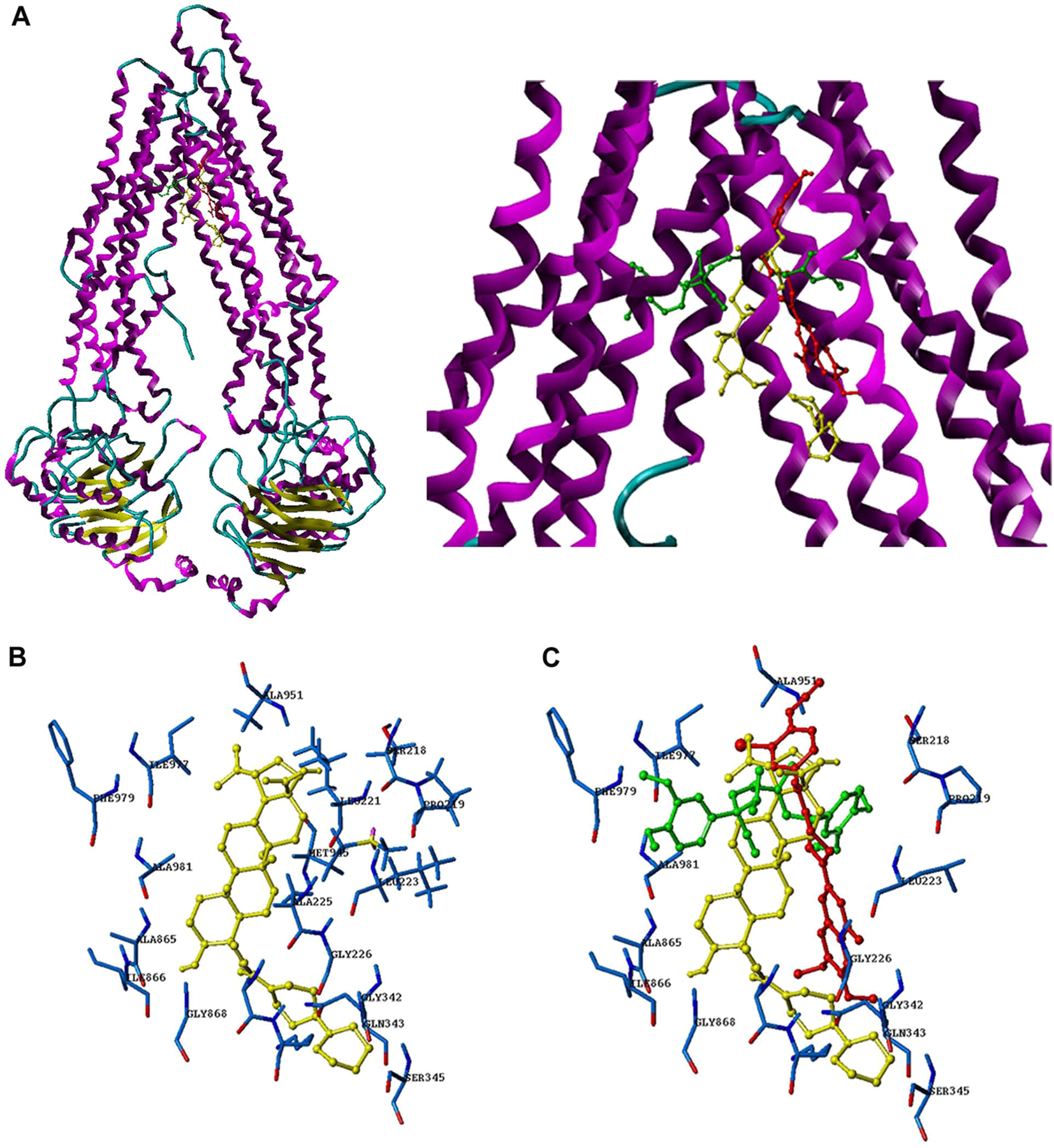
Docking analysis of BBA, verapamil, and IAAP toward ABCB1. (A) The binding mode and position of BBA, verapamil, and IAAP with ABCB1. (B) The residual amino acid around BBA. (C) The residual amino acid around BBA, verapamil, and IAAP; key residues were labeled.
BBA Suppresses the Phosphorylation of AKT and ERK1/2 in ABCB1-Expressing Resistant Cells.
To elucidate the underlying mechanism of BBA reversal activity, we determined AKT and ERK1/2 expression and phosphorylation levels in BBA-treated cells. It was shown that BBA treatment at the higher concentration (5 μM) led to a significant reduction in phosphorylation of AKT at Thr308 and Ser473, but had no effect on the total AKT level. Consequently, we found that BBA also inhibits total ERK1/2 level and its phosphorylation. Another important molecule, Bcl-2, which promotes resistance by prevention of chemotherapeutic drug-induced apoptosis, was detected in MDR cells, but there was no significant change observed in its expression level after BBA treatment.45 BBA has no effect on the total level of AKT and ERK1/2, phosphorylation of AKT and ERK1/2, or the expression level of Bcl-2 on HepG2 cell line (Supplementary Figure S3 in the Supporting Information). Taken together, our results suggest that the mechanism by which BBA sensitizes resistant cells to chemotherapeutic drugs is at least partially linked to inhibition of phosphorylation of AKT and ERK1/2.
BBA Reverses ABCB1-Mediated MDR in Nude Mouse Xenograft Model.
KB-C2 cell xenograft model in nude mice was used to investigate the efficacy of BBA to reverse resistance to paclitaxel in vivo. No significant difference was observed in tumor size between experimental animals treated with saline and BBA. However, mean tumor size in the combination group of BBA and paclitaxel was significantly smaller than that of the other three groups (P < 0.05; Figure 5A,B). The tumor weight in combination group was also significantly lower than that of the other groups (P < 0.05; Figure 5C). These results suggest that BBA had a significant reversal effect on ABCB1-mediated MDR in vivo. Moreover, at the doses tested, no significant change in the body weight was observed in the combination group (Figure 5D), indicating that the combination treatment probably did not lead to increase side effect.
Figure 5.
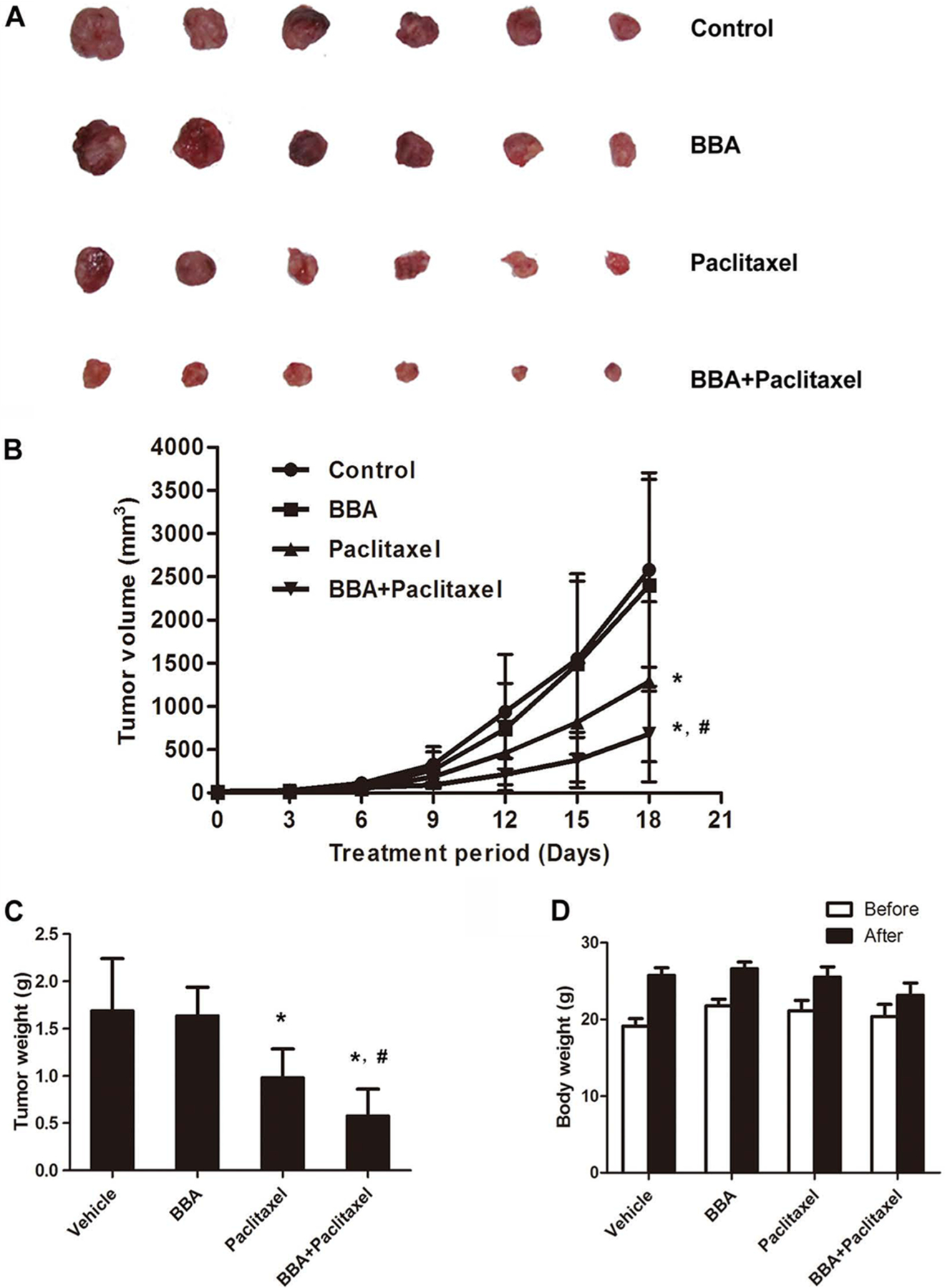
The MDR reversal effect of BBA on KB-C2 xenograft model. Potentiation of antitumor effects of paclitaxel by BBA in ABCB1 overexpressing KB-C2 xenograft model is shown. (A) A representative picture of the excised KB-C2 tumor sizes from different mice is shown on the 18th day after implantation. (B) Changes in tumor volume over the time course of the experiment in ABCB1 overexpressing KB-C2 xenograft model are shown. The treatments were as follows: (a) control (saline, q3d × 6), (b) BBA (15 mg/kg, po, q3d × 6), (c) paclitaxel (18 mg/kg, ip, q3d × 6), and (d) BBA (15 mg/kg, po, q3d × 6) + paclitaxel (18 mg/kg, ip, q3d × 6), BBA was given 2 h before paclitaxel administration. Points represent mean of tumor volume for each group (n = 6) after implantation. Each point on line graph represents the mean of tumor volume (mm3) at a particular day after implantation, and each bar represents SD. *, P < 0.05 versus the control group; #, P < 0.05 versus paclitaxel alone group. (C) The bar graph represents the mean of tumor weights (mice, n = 6) of the excised KB-C2 tumor from different mice. Each column represents the mean of determinations, and the bar represents SD. *, P < 0.05 versus the control group; #, P < 0.05 versus the paclitaxel alone group. (D) Changes in the mean of body weight over the time course of the experiment in ABCB1 overexpressing KB-C2 xenograft model are shown. One pilot study and two independent experiments were carried out using athymic (nu/nu) NCR nude mice implanted sc with KB-C2 cells. A representative result is shown.
DISCUSSION
Natural sources are a fertile ground to find novel drugs; in recent years modification of plant-derived natural products became a promising road to novel drug discovery. In order to develop 23-hydroxybetulinic acid derivatives as MDR reversal agents, we carried out a list of structural modifications of 23-hydroxybetulinic acid systemically as previously shown and performed an extensive screening of new derivatives against their MDR reversal activity.31 We found that introducing a bulky and hydrophilic moiety into C-23 site yields potent compounds, such as BBA, exhibiting excellent MDR reversal activities (Supplementary Table 1 in the Supporting Information). Further study on the structure–activity relationship is an ongoing effort in our laboratory. This study, examining the BBA-induced changes in resistances to anticancer drugs, DOX, VCR, vinblastine, and paclitaxel, in both parental and resistant cancer cell lines (HepG2, HepG2/ADM, MCF-7, MCF-7/ADR, and KB-3–1, KB-C2), as well as ABCB1-transfected HEK293 cells, first demonstrated that BBA remarkably reduces resistance of MDR cells to ABCB1 substrates. Based on the above results, further experiments were conducted in vivo. The KB-C2 cell xenograft model was selected, and the effect of BBA was observed. In this study, BBA significantly enhanced the anticancer activity of paclitaxel, with no significant change in the body weight in the combination group. Therefore, we conclude that BBA selectively reverses ABCB1-mediated MDR in both in vitro and in vivo. In addition, it has been found that BBA has several distinguishing characteristics comparing to other MDR inhibitors as following: (i) BBA is a derivative of natural product 23-hydroxybetulinic acid existing in great quantities in the traditional Chinese herb Pulsatilla chinensis which has been frequently used as an adjuvant in cancer treatment;46 it is also easy to prepare by extraction and semisynthesis at relatively low cost as reported by our group.31,47 (ii) BBA has lower toxicity toward mice compared with other third generation MDR reversers, indicated by the fact that no significant toxicity was found after treatment with BBA at 5 g/kg/day in single dose acute toxicity test or 30 mg/kg/day for 40 consecutive days. (iii) BBA modulates the P-gp ATPase activity in a biphase way and cannot be transported by P-gp, which is different from the established third generation inhibitors. Collectively, these characteristics support the potential of BBA to be developed into an adjuvant to anticancer chemotherapy. However, we still need to further compare the MDR reversal activity of BBA with third generation MDR reversers and investigate toxicity and pharmacokinetics of BBA alone or combined with anticancer drugs.
The results from accumulation experiments indicated that BBA reduces anticancer drug resistance by increasing intracellular drug accumulation. We subsequently performed an efflux experiment using ABCB1 specific substrate rhodamine123 to determine whether this accumulation effect is directly associated with ABCB1 function. Our results showed that BBA suppresses ABCB1-modulated efflux, resulting in intracellular retention of the drug in HepG2/ADM and MCF-7/ADR cell lines.
The attenuated ABCB1 transporting function can result from downregulated protein expression. However, the RT-PCR and Western blot analysis of ABCB1 expression, prior to or after BBA treatment, in ABCB1-overexpressing cells showed that neither mRNA nor protein level of ABCB1 was decreased upon BBA treatment. Immunofluorescence analysis confirmed that there was no translocation of ABCB1 from membrane to cytosol/nuclei on HepG2/ADM cells. These findings support that BBA suppresses the process of pumping anticancer drug out of MDR cells by a mechanism other than downregulating the overexpressed ABCB1 transporter.
Next, we looked into the effect of BBA on the function of ABCB1. ABCB1 is a 170 kDa membrane protein, with two transmembrane domains (TMD) and two nucleotide binding domains (NBD). When a substrate is recognized by ABCB1, ATP hydrolysis and binding to NBDs and a large conformational change in TMD are consequentially induced, presenting the substrate and drug-binding site to the outer leaflet or extracellular space.40 The inhibition of drug transport could be resulting from the blockage of specific recognition of the substrate to weaken its interaction with ABCB1, or coupling of ATP hydrolysis for translocation of the substrate, which is related to the ATPase. Our result on basal ABCB1 ATPase activity assay showed a classic bell-shaped ATPase response curve varying with the concentrations of BBA, which is consistent with the observation reported by Muñoz-Martínez.48 At a lower concentration (<2.5 μM), BBA stimulated the ATPase activity; at a higher concentration (>2.5–30 μM), it inhibited ATPase activity. However, BBA had an inhibitory effect on verapamil stimulated ATPase activity, which indicates that BBA may compete with verapamil at the same binding site of ABCB1. The [125I]IAAP photoaffinity labeling experiment also showed that BBA may partly overlap with IAAP. These results suggest that BBA has direct interaction with ABCB1, and the molecular docking results further confirmed it. Nevertheless, the docking results also pointed out that there is no completely coincident binding site with verapamil, indicating a different mechanism of action of BBA from verapamil. Verapamil was identified as a substrate of ABCB1 competing with anticancer drugs for efflux, and one typical character of the substrate is that it can be transported by ABCB1.49 But our drug sensitivity study results showed that the cytotoxicity of BBA on both resistant and sensitive cell lines was almost the same, and the addition of ABCB1 inhibitors cyclosporine A or verapamil did not change its cytotoxicity on ABCB1 overexpressed cell line either, confirming that BBA is less likely transported by ABCB1. A nonsubstrate modulator could potentially have lower toxicity as lower concentration was applied and may have relatively sustained reversal activity. In fact, persistent reversal activity of BBA at different concentrations (1.25 and 5 μM) was observed after BBA washout post 24 h treatment. This not only further confirms that BBA is not a transported substrate of ABCB1 but also suggests its inhibitory property against ABCB1. The docking results also point out the different binding site from verapamil and IAAP, indicating that BBA may regulate the function of ABCB1 in a different way.
It has been well-known that aberrant activation of AKT and ERK1/2 pathways plays an importance role in the development of the MDR phenotype of tumor cells, and Kim reported that cancer cells either expressing constitutively active AKT or containing AKT gene amplification are also far more resistant to paclitaxel than cancer cells expressing low levels of AKT, thus suppression of AKT and ERK1/2 signaling pathways has been considered as an effective therapeutic approach to overcome drug resistance.50–52 Unchanged total AKT protein level, along with significantly reduced ERK1/2 protein level and phosphorylation levels in both protein kinases, indicates that the reversal activity of BBA may be associated with suppression of ERK1/2 by inhibited AKT phosphorylation. Additional experiments ought to be performed to confirm and further elucidate the underlying mechanism involved in the cell signaling pathway, which is beyond the scope of this study.
In the present study, it is interesting to find that BBA has a better MDR reversal effect than verapamil in HepG2/ADM and MCF-7/ADR cells, but a similar or less effect than verapamil in KB-C2 and HEK293/ABCB1 cells. As reported by our collaborators, DOX-induced ABCB1-overexpressing drug-resistant HepG2/ADM cells showed the upregulation of several drug transporters (ABCB8, ABCB4) and other drug-resistant factors (Sorcin, CDH1), which may be different with those drug resistance related molecules of colchicine-selected ABCB1-overexpressing drug-resistant KB-C2 cells and ABCB1-transfected HEK293/ABCB1 cells.53,54 On the other hand, our results also showed that BBA reversed drug resistance through more than one mechanism. In addition to attenuating ABCB1 transporter function, BBA also modulated other drug resistance-associated proteins, for example, inhibition of phosphorylation of Akt and ERK. Recently, our preliminary study demonstrated that BBA could also reverse ABCC10-mediated drug resistance (data not shown). These results indicate that BBA reverses multidrug resistance through not only inhibition of ABCB1 but also modulation of other drug-resistant factors, which still needs to be further studied. In addition, it is also found that BBA slightly decreases the sensitivity of ABCG2–482-R5 cells to mitoxantrone, possibly indicating that BBA itself as a weak substrate for ABCG2 stimulates the transporter function of ABCG2 to pump mitoxantrone out of cells. At the same time, BBA significantly increases the sensitivity of three ABCG2-overexpressing cells to SN-38. This further confirms BBA is a substrate of ABCG2, and increased sensitivity may be due to BBA binding to ABCG2 at the same site of SN-38, thereby competitively inhibiting the transportation of SN-38 out of the cells by ABCG2. These hypotheses will need to be further investigated by detection of ABCG2 transporter function before and after BBA treatment as well as analysis of the binding site of BBA to ABCG2.
In conclusion, we found that BBA, a derivate of 23-HBA, reverses ABCB1-mediated MDR by inhibition of its drug efflux function, and increases the intracellular accumulation of anticancer drugs in ABCB1-overexpressing cells. The reversal activity may also be partially associated with the suppression of ERK1/2 and its phosphorylation form and the inhibition of phosphorylation of AKT. These results provide solid molecular evidence in supporting the development of BBA to be a novel MDR modulator in clinical cancer therapy in the future.
Supplementary Material
ACKNOWLEDGMENTS
Special thanks to Prof. Kwok-Pui Fung (The Chinese University of Hong Kong, Hong Kong) for HepG2/ADM cells, Prof. Li-Wu Fu (Sun Yat-Sen University, China) for MCF-7/ADR cells, Prof. Michael M. Gottesman (National Cancer Institute, NIH, USA) for KB-3–1 cells, Dr. Shin-ichi Akiyama (Kagoshima University, Japan) for KB-C2, KB-CV60 cells, Dr. Kazumitsu Ueda (Kyoto University, Japan) for KB-MRP1 cells, and Drs. Susan E. Bates and Robert W. Robey (National Cancer Institute, NIH, USA) for HEK293, ABCB1, ABCG2, and ABCC1 transfected cells. This work was supported by Science and Technology Program of China (2009ZX09103–313 and 2012ZX09103101–053) and Guangzhou City (2009A1-E011–1, 2010U1-E00531–9, and 2011J2200045), National Science Foundation of China (No. 30901847 and 90913020), NIH R15 (No. 1R15CA143701), and Program for Changjiang Scholars and Innovative Research Team in University (IRT0965). J.B. and S.V.A. were supported by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, NIH.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Further information on MDR reversal activities of 23-HBA derivatives, in vivo MDR reversal effect of BBA, and the inhibitory effect of BBA on the ABCB1 function and the phosphorylation of AKT and ERK1/2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Baguley BC Multidrug resistance in cancer. Methods Mol. Biol 2010, 596, 1–14. [DOI] [PubMed] [Google Scholar]
- (2).Sodani K; Patel A; Kathawala RJ; Chen ZS Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dean M; Hamon Y; Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res 2001, 42, 1007–1017. [PubMed] [Google Scholar]
- (4).Cole S; Bhardwaj G; Gerlach J; Mackie J; Grant C; Almquist K; Stewart A; Kurz E; Duncan A; Deeley R. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [DOI] [PubMed] [Google Scholar]
- (5).Juliano RL; Ling VA surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [DOI] [PubMed] [Google Scholar]
- (6).Uchida S; Shimada Y; Watanabe G; Li ZG; Hong T; Miyake M; Imamura M. Motility-related protein (MRP-1/CD9) and KAI1/CD82 expression inversely correlate with lymph node meta-stasis in oesophageal squamous cell carcinoma. Br. J. Cancer 1999, 79, 1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Doyle LA; Yang W; Abruzzo LV; Krogmann T; Gao Y; Rishi AK; Ross D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sarkadi B; Homolya L; Szakács G; Váradi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol. Rev 2006, 86, 1179–1236. [DOI] [PubMed] [Google Scholar]
- (9).Coley HM Overcoming multidrug resistance in cancer: clinical studies of p-glycoprotein inhibitors. Methods Mol. Biol 2010, 596, 341–358. [DOI] [PubMed] [Google Scholar]
- (10).Sikic B. Pharmacologic approaches to reversing multidrug resistance. Semin. Hematol 1997, 34, 40–47. [PubMed] [Google Scholar]
- (11).Tsuruo T; Iida H; Tsukagoshi S; Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 1981, 41, 1967–1972. [PubMed] [Google Scholar]
- (12).Twentyman P; Fox N; White D. Cyclosporin A and its analogues as modifiers of adriamycin and vincristine resistance in a multi-drug resistant human lung cancer cell line. Br. J. Cancer 1987, 56, 55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Krishna R; Mayer LD Multidrug resistance (MDR) in cancer: Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. E ur. J. Pharm. Sci 2000, 11, 265–283. [DOI] [PubMed] [Google Scholar]
- (14).Tan B; Piwnica-Worms D; Ratner L. Multidrug resistance transporters and modulation. Curr. Opin. Oncol 2000, 12, 450–458. [DOI] [PubMed] [Google Scholar]
- (15).Baer MR; George SL; Dodge RK; O’Loughlin KL; Minderman H; Caligiuri MA; Anastasi J; Powell BL; Kolitz JE; Schiffer CA Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood 2002, 100, 1224–1232. [PubMed] [Google Scholar]
- (16).Nobili S; Landini I; Giglioni B; Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets 2006, 7, 861–879. [DOI] [PubMed] [Google Scholar]
- (17).Lee CH Reversing agents for ATP-binding cassette (ABC) transporters: application in modulating multidrug resistance (MDR). Curr. Med. Chem.: Anti-Cancer Agents 2004, 4, 43–52. [DOI] [PubMed] [Google Scholar]
- (18).Shukla S; Wu CP; Ambudkar SV Development of inhibitors of ATP-binding cassette drug transporters-present status and challenges. Expert Opin. Drug Metab. Toxicol 2008, 4, 205–223. [DOI] [PubMed] [Google Scholar]
- (19).Mistry P; Stewart AJ; Dangerfield W; Okiji S; Liddle C; Bootle D; Plumb JA; Templeton D; Charlton P. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res 2001, 61, 749–758. [PubMed] [Google Scholar]
- (20).Qi F; Li A; Inagaki Y; Gao J; Li J; Kokudo N; Li XK; Tang W. Chinese herbal medicines as adjuvant treatment during chemo- or radio-therapy for cancer. Biosci. Trends 2010, 4, 297–307. [PubMed] [Google Scholar]
- (21).Zhang Y; Liu X; Zuo T; Liu Y; Zhang JH Tetramethylpyrazine reverses multidrug resistance in breast cancer cells through regulating the expression and function of P-glycoprotein. Med. Oncol 2011, 1–5. [DOI] [PubMed]
- (22).Wang XB; Wang SS; Zhang QF; Ming L; Li HL; Ying L; Wang JN; Zheng F; Guo LY; Xiang JZ Inhibition of tetramethylpyrazine on P-gp, MRP2, MRP3 and MRP5 in multidrug resistant human hepatocellular carcinoma cells. Oncol. Rep 2010, 23, 211–215. [PubMed] [Google Scholar]
- (23).Zhu XL; Xu WL; Lu XJ; Luo WJ; Zhou LL; Chen QY Mechanisms of preventive effect of tetrandrine on acquired multidrug resistance in K562 cells. J. Exp. Hematol 2011, 19, 363–366. [PubMed] [Google Scholar]
- (24).Wei N; Sun H; Wang F; Liu G. H1, a novel derivative of tetrandrine reverse P-glycoprotein-mediated multidrug resistance by inhibiting transport function and expression of P-glycoprotein. Cancer Chemother. Pharmacol 2011, 67, 1017–1025. [DOI] [PubMed] [Google Scholar]
- (25).Huang M; Jin J; Sun H; Liu GT Reversal of P-glycoprotein-mediated multidrug resistance of cancer cells by five schizandrins isolated from the Chinese herb Fructus Schizandrae. Cancer Chemother. Pharmacol 2008, 62, 1015–1026. [DOI] [PubMed] [Google Scholar]
- (26).Zeng R; Zhou ZW; Wu CF; Zhou YL Reversal effect of aloe emodin liposomes on cisplatin resistance line A549/DDP human lung adenocarcinoma cells. China J. Chin. Mater. Med 2008, 33, 1443–1445. [PubMed] [Google Scholar]
- (27).Zhang H; Yang L; Liu S; Ren L. Study on active constituents of traditional Chinese medicine reversing multidrug resistance of tumor cells in vitro. J. Chin. Med. Mater 2001, 24, 655–657. [PubMed] [Google Scholar]
- (28).Ji ZN; Ye WC; Liu GG; Hsiao W. 23-Hydroxybetulinic acid-mediated apoptosis is accompanied by decreases in bcl-2 expression and telomerase activity in HL-60 Cells. Life Sci 2002, 72, 1–9. [DOI] [PubMed] [Google Scholar]
- (29).Zhou JP; Li D; Wu XM; Ye WC; Zhang LY Synthesis and antitumor activity of derivatives of 23-hydroxybetulinic acid. Chin. Chem. Lett 2007, 18, 1195–1198. [Google Scholar]
- (30).Zheng Y; Zhou F; Wu X; Wen X; Li Y; Yan B; Zhang J; Hao G; Ye W; Wang G. 23-Hydroxybetulinic acid from Pulsatilla chinensis (Bunge) Regel synergizes the antitumor activities of doxorubicin in vitro and in vivo. J. Ethnopharmacol 2010, 128, 615–622. [DOI] [PubMed] [Google Scholar]
- (31).Lan P; Wang J; Zhang DM; Shu C; Cao HH; Sun PH; Wu XM; Ye WC; Chen WM Synthesis and antiproliferative evaluation of 23-hydroxybetulinic acid derivatives. Eur. J. Med. Chem 2011, 217, 2490–2502. [DOI] [PubMed] [Google Scholar]
- (32).Cheung JY; Ong RC; Suen YK; Ooi V; Wong HN; Mak TC; Fung KP; Yu B; Kong SK Polyphyllin D is a potent apoptosis inducer in drug-resistant HepG2 cells. Cancer Lett 2005, 217, 203–211. [DOI] [PubMed] [Google Scholar]
- (33).Fu L; Liang Y; Deng L; Ding Y; Chen L; Ye Y; Yang X; Pan Q. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother. Pharmacol 2004, 53, 349–356. [DOI] [PubMed] [Google Scholar]
- (34).Akiyama SI; Fojo A; Hanover JA; Pastan I; Gottesman MM Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somatic Cell Mol. Genet 1985, 11, 117–126. [DOI] [PubMed] [Google Scholar]
- (35).Taguchi Y; Yoshida A; Takada Y; Komano T; Ueda K. Anti-cancer drugs and glutathione stimulate vanadate-induced trapping of nucleotide in multidrug resistance-associated protein (MRP). FEBS Lett 1997, 401, 11–14. [DOI] [PubMed] [Google Scholar]
- (36).Robey R; Honjo Y; Morisaki K; Nadjem T; Runge S; Risbood M; Poruchynsky M; Bates S. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br. J. Cancer 2003, 89, 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dai C; Tiwari AK; Wu CP; Su X; Wang SR; Liu D; Ashby CR; Huang Y; Robey RW; Liang Y. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res 2008, 68, 7905–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ambudkar SV Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol 1998, 292, 504–514. [DOI] [PubMed] [Google Scholar]
- (39).Kerr KM; Sauna ZE; Ambudkar SV Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in human P-glycoprotein. J. Biol. Chem 2001, 276, 8657–8664. [DOI] [PubMed] [Google Scholar]
- (40).Aller SG; Yu J; Ward A; Weng Y; Chittaboina S; Zhuo R; Harrell PM; Trinh YT; Zhang Q; Urbatsch IL; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fu L; Zhang Y; Liang Y; Yang X; Pan Q. The multidrug resistance of tumour cells was reversed by tetrandrine in vitro and in xenografts derived from human breast adenocarcinoma MCF-7/adr cells. Eur. J. Cancer 2002, 38, 418–426. [DOI] [PubMed] [Google Scholar]
- (42).Mi Y; Liang Y; Huang H; Zhao H; Wu CP; Wang F; Tao L; Zhang C; Dai CL; Tiwari AK Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res 2010, 70, 7981–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Honjo Y; Hrycyna CA; Yan QW; Medina-Perez WY; Robey RW; van de Laar A; Litman T; Dean M; Bates SE Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res 2001, 61, 6635–6639. [PubMed] [Google Scholar]
- (44).Newman MJ; Rodarte JC; Benbatoul KD; Romano SJ; Zhang C; Krane S; Moran EJ; Uyeda RT; Dixon R; Guns ES; Mayer LD Discovery and characterization of OC144–093, a novel inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Res 2000, 60, 2964–2972. [PubMed] [Google Scholar]
- (45).Reed JC; Miyashita T; Takayama S; Wang HG; Sato T; Krajewski S; Aimé-Sempé C; Bodrug S; Kitada S; Hanada M. BCL-2 family proteins: Regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J. Cell. Biochem 1996, 60, 23–32. [DOI] [PubMed] [Google Scholar]
- (46).Wang Y; Wang GC; Zhao HN; Fan CL; Ye WC RP-HPLC determination of 23-hydroxybetulinic acid in the roots of Pulsatilla chinensis. Chin. J. Pharm. Anal 2007, 27, 13–15. [Google Scholar]
- (47).Zhao HN; Wang Y; Su WK; Zhang XQ; Ye WC Preparation of 23-hydroxybetulinic Acid from the Roots of Pulsatilla chinensis. J. Chin. Med. Mater 2007, 30, 170–172. [PubMed] [Google Scholar]
- (48).Munoz-Martinez F; Lu P; Cortes-Selva F; Perez-Victoria JM; Jimenez IA; Ravelo AG; Sharom FJ; Gamarro F; Castanys S. Celastraceae sesquiterpenes as a new class of modulators that bind specifically to human P-glycoprotein and reverse cellular multidrug resistance. Cancer Res 2004, 64, 7130–7138. [DOI] [PubMed] [Google Scholar]
- (49).Thomas H; Coley HM Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165. [DOI] [PubMed] [Google Scholar]
- (50).West KA; Sianna Castillo S; Dennis PA Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updates 2002, 5, 234–248. [DOI] [PubMed] [Google Scholar]
- (51).Katayama K; Yoshioka S; Tsukahara S; Mitsuhashi J; Sugimoto Y. Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol. Cancer Ther 2007, 6, 2092–2102. [DOI] [PubMed] [Google Scholar]
- (52).Kim D; Dan HC; Park S; Yang L; Liu Q; Kaneko S; Ning J; He L; Yang H; Sun M. Akt/PKB signaling mechanisms in cancer and chemoresistance. Front. Biosci 2005, 10, 975–987. [DOI] [PubMed] [Google Scholar]
- (53).Wang J; Chan JY; Fong CC; Tzang CH; Fung KP; Yang M. Transcriptional analysis of doxorubicin-induced cytotoxicity and resistance in human hepatocellular carcinoma cell lines. Liver Int 2009, 29, 1338–1347. [DOI] [PubMed] [Google Scholar]
- (54).Jiang L; Chan JY; Fung KP Epigenetic loss of CDH1 correlates with multidrug resistance in human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun 2012, 422, 739–744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.