

Abstract
Recent research on genomic profiling of pancreatic ductal adenocarcinoma (PDAC) has identified many potentially actionable alterations. However, the feasibility of using genomic profiling to guide routine clinical decision making for PDAC patients remains unclear. We retrospectively reviewed PDAC patients between October 2013 and December 2017, who underwent treatment at the Johns Hopkins Hospital and had clinical tumor next-generation sequencing (NGS) through commercial resources. Ninety-two patients with 93 tumors tested were included. Forty-eight (52%) patients had potentially curative surgeries. The median time from the tissue available to the NGS testing ordered was 229 days (interquartile range 62–415). A total of three (3%) patients had matched targeted therapies based on genomic profiling results. Genomic profiling guided personalized treatment for PDAC patients is feasible, but the percentage of patients who receive targeted therapy is low. The main challenges are ordering NGS testing early in the clinical course of the disease and the limited evidence of using a targeted approach in these patients. A real-time department level genomic testing ordering system in combination with an evidence-based flagging system for potentially actionable alterations could help address these shortcomings.
Keywords: Clinical genomic testing, Actionable alteration, Matched therapy
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy with a 5-year overall survival rate of 9% for all stage patients [1]. The dismal 5-year survival is a result of the advanced stage of disease at diagnosis and it being refractory to treatment [2]. Many clinicians have turned to the genomic profiling of PDAC to identify potentially actionable alterations and hope the possible directed treatment can improve patient outcomes [3–7].
Despite the progress of genomic profiling in the clinical practice of many solid tumors [8,9], the benefit of genomic profiling for PDAC patients is still limited. These limitations are multi-factorial, including lack of effective targeted therapy in common driver alterations (KRAS, etc.), low prevalence of potentially actionable alterations [10], the genetic background complexity on which the rare targetable somatic mutations occur in PDAC.
Several clinical trials have prospectively demonstrated the feasibility of real-time genomic profiling for PDAC patients. However, only a small percentage are found to have potentially actionable alterations with clinical benefits [11–13]. Compared to well-designed clinical trials, real-time genomic testing in routine practice for PDAC patient care can be much more challenging. Although genomic testing is not standard clinical practice for PDAC patients so far, the most recently updated ASCO guideline for metastatic pancreatic cancer strongly recommends obtaining genomic testing for all treatment eligible patients to select patients for recommended therapies [14].
This study aimed to summarize the past use of clinical genomic profiling in PDAC patients at a single institution. The feasibility of utilizing this approach was assessed, challenges faced with its implementation identified, and future directions suggested.
2. Materials and methods
2.1. Study population
A retrospective study was performed to identify all PDAC patients who were managed at the Johns Hopkins Medical Institutions and underwent clinically directed next-generation sequencing (NGS) of their primary or metastatic tumor through commercial resources between October 2013 and December 2017. Approximately 3000 patients with PDAC were evaluated at the institution during this period. All genomic alteration information was obtained from the NGS test reports. General demographics and clinical data were obtained from a prospectively maintained institutional registry.
2.2. Genetic analysis
The NGS testing for all tumor tissue were done through commercial resources, including Foundation Medicine, Perthera, and Personal and Genome Diagnostics (PGDx) and with their panels. DNA was extracted from unstained slides or formalin-fixed paraffin-embedded (FFPE) for library preparation. Either Foundation Medicine Panel Version 1 (coding exons of 236 genes and introns of 19 genes involved in rearrangements) [7] or Foundation Medicine Panel Version 2 (coding exons of 315 genes and introns of 28 genes involved in rearrangements) [7] was used by Foundation Medicine and Perthera in the study cohort [15]. Both alterations marked as clinically relevant alterations and variants of uncertain significance were included in our analyses. CancerSelect™ panel, which included sequence analyses for 76 genes, copy number analyses for 13 genes, and rearrangement analyses for 13 genes, were used by PGDx in the study cohort. Since all patients except five were tested and reported with only tumor tissue (Foundation Medicine and Perthera), some reported alterations may be unappreciated germline variants. ClinVar [16] database was referred for functional significance of variants (last checked date 7/17/2019). The list of potentially actionable alterations in Table 1 was used to determine whether the sequenced tumor had any potentially actionable alteration. The potentially actionable alteration was defined as an alteration with US Food and Drug Administration (FDA) approved targeted therapies for any cancer type between Oct 2013 and Dec 2017, which was the period for patients included in this study.
Table 1.
List of potentially actionable alteration and matched therapy screened for all 92 patients.
| Potential actionable alteration | Matched therapy | Patient found in the study, N (%) | Patient received matched therapy, N (%) |
|---|---|---|---|
| BRCA1/2 mutation | PARP inhibitor | 7 (8) | 3 (3) |
| ATM mutation | PRAP inhibitor | 3 (3) | 0 |
| Microsatellite instability - high | Anti-PD-1 Antibodies | 0 | 0 |
| BRAF mutation | MEK and ERK inhibitors | 0 | 0 |
| ALK fusion | Crizotinib, Ceritinib | 0 | 0 |
| ROS1 fusion | Crizotinib | 0 | 0 |
| HER2 amplification | Trastuzumab or Neratinib | 0 | 0 |
| IDH2 mutation | Enasidenib | 0 | 0 |
| EGFR mutation | Erlotinib, Gefitinib, Afatinib | 0 | 0 |
2.3. Statistical analysis
All categorical variables were reported as frequencies and percentages, and all continuous variables were reported as means and standard deviations or medians and interquartile ranges (IQR) as deemed necessary. Chi-squared or Fisher test was used for categorical variables, as appropriate. Overall survival (OS) was calculated from the date of biopsy-confirmed diagnosis to date of death or censored at the last date when the patient was known to be alive. Kaplan-Meier curve and log-rank test were used to compare survival distributions between different groups. Progression-free survival (PFS) was calculated from the initiation of therapy to disease progression or censored at the time of change because of intolerance, surgery (used as neoadjuvant treatment) without progression. Cox model was used for survival analysis. P values from multiple testing were adjusted using the Benjamini-Hochberg method at level 0.05. All analyses were performed using R version 3.5.3 (R Foundation, Vienna, Austria). Package GenVisR (Version 4.0) was used for the genomic alteration landscape plot.
The Johns Hopkins Institutional Review Board approved the study for human research.
3. Results
3.1. Patient information
We included 92 patients in the study. The median age was 63 years (IQR: 55–70) and approximately half were male (N = 47, 51%). There were 48 (52%) patients underwent potentially curative surgeries, and a majority of these patients (81%) had pancreaticoduodenectomy. The remaining patients (48%) had metastasis or locally advanced disease (Table 2).
Table 2.
General demographic and clinicopathologic features of 92 patients.
| Variables | N (%) |
|---|---|
| Age (yrs), median (IQR) | 63 (55–70) |
| Gender | |
| Female | 45 (49) |
| Male | 47 (51) |
| Race | |
| White | 79 (86) |
| African American | 9 (10) |
| Asian | 3 (3) |
| Unknown | 1 (1) |
| Treatment | |
| Curative surgical resection | 48 (52) |
| Non-surgical resection | 44 (48) |
All the commercially conducted clinical NGS tests were ordered by patients’ oncology care providers. Of the patients undergoing surgical resection, 44 NGS tests were conducted on primary tumors (42 on surgery specimens and two on pre-surgery endoscopic ultrasound with fine-needle aspiration/fine needle biopsy (EUS-FNA/FNB)), two on liver metastases as a recurrence, one on lung metastasis as a recurrence, one on peritoneal metastases as a recurrence and one on right rectal muscle metastasis as a recurrence. For non-surgical patients, 16 NGS tests were done on primary tumors through EUS-FNA/FNB, 22 on metastatic liver lesions, one on lymph node metastasis, and five on metastatic peritoneal lesions.
3.2. Most common somatic alterations
In all 93 tumor tissues, the highest mutation prevalence was reported in KRAS (N = 86, 93%), consistent with 93% KRAS mutation of PDAC reported in using The Cancer Genome Atlas (TCGA) network [17]. Other highly mutated genes were TP53 (N = 63, 68%), SMAD4 (N = 21, 23%), and CDKN2A (N = 19, 20%). The landscape of genomic mutations and copy number variations were shown in Fig. 1. The associated between most common somatic mutations and clinicopathologic variables were shown in Table 3, and the OS of patients with common somatic mutations were calculated in Table 4. Considering the sample size for testing, we included all genes having more than five patients with mutations.
Fig. 1.
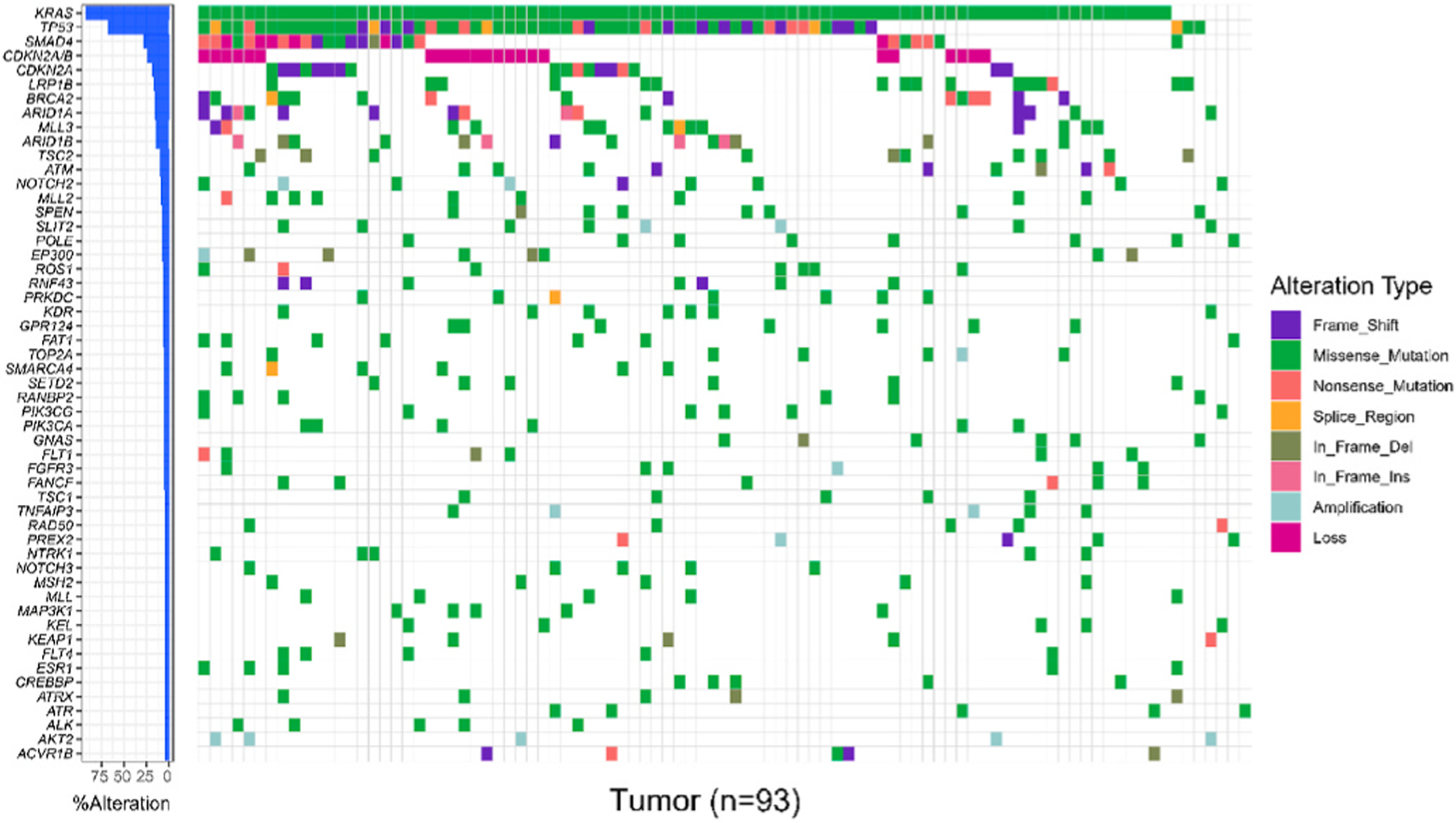
The landscape of genomic alteration and frequency of all 93 tumors.
Table 3.
The associations between clinicopathologic factors and genomic mutations.
| Gene | Age (years) | Gender | Tumor source | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≤63 N (%) | >63 N (%) | P | Pa | Female N (%) | Male N (%) | P | Pa | Metastasis N (%) | Primary N (%) | P | Pa | |
| KRAS | 41 (91) | 44 (94) | 0.71 | 1 | 42 (93) | 43 (91) | 1 | 1 | 30 (91) | 55 (93) | 0.7 | 1 |
| TP53 | 30 (67) | 33 (70) | 0.89 | 1 | 35 (78) | 28 (60) | 0.1 | 0.73 | 24 (73) | 39 (66) | 0.67 | 1 |
| SMAD4 | 6 (13) | 14 (30) | 0.1 | 1 | 13 (29) | 7 (15) | 0.17 | 0.73 | 9 (27) | 11 (19) | 0.48 | 1 |
| CDKN2A | 8 (18) | 10 (21) | 0.87 | 1 | 11 (24) | 7 (15) | 0.37 | 0.74 | 4 (12) | 14 (24) | 0.27 | 1 |
| LRP1B | 9 (20) | 7 (15) | 0.71 | 1 | 7 (16) | 9 (19) | 0.86 | 1 | 10 (30) | 6 (10) | 0.03 | 0.93 |
| ARID1A | 6 (13) | 9 (19) | 0.64 | 1 | 10 (22) | 5 (11) | 0.22 | 0.73 | 6 (18) | 9 (15) | 0.94 | 1 |
| BRCA2 | 6 (13) | 9 (19) | 0.64 | 1 | 6 (13) | 9 (19) | 0.64 | 0.96 | 4 (12) | 11 (19) | 0.56 | 1 |
| ARID1B | 4 (9) | 10 (21) | 0.15 | 1 | 8 (18) | 6 (13) | 0.7 | 0.96 | 3 (9) | 11 (19) | 0.36 | 1 |
| MLL3 | 7 (16) | 7 (15) | 1 | 1 | 9 (20) | 5 (11) | 0.34 | 0.74 | 6 (18) | 8 (14) | 0.77 | 1 |
| ATM | 3 (7) | 7 (15) | 0.32 | 1 | 3 (7) | 7 (15) | 0.32 | 0.74 | 3 (9) | 7 (12) | 1 | 1 |
| TSC2 | 4 (9) | 6 (13) | 0.74 | 1 | 8 (18) | 2 (4) | 0.05 | 0.73 | 5 (15) | 5 (8) | 0.52 | 1 |
| MLL2 | 6 (13) | 3 (6) | 0.31 | 1 | 5 (11) | 4 (9) | 0.74 | 0.96 | 5 (15) | 4 (7) | 0.27 | 1 |
| POLE | 5 (11) | 3 (6) | 0.48 | 1 | 6 (13) | 2 (4) | 0.15 | 0.73 | 3 (9) | 5 (8) | 1 | 1 |
| SPEN | 4 (9) | 4 (9) | 1 | 1 | 3 (7) | 5 (11) | 0.71 | 0.96 | 4 (12) | 4 (7) | 0.45 | 1 |
| EP300 | 5 (11) | 2 (4) | 0.26 | 1 | 5 (11) | 2 (4) | 0.26 | 0.73 | 2 (6) | 5 (8) | 1 | 1 |
| FAT1 | 4 (9) | 3 (6) | 0.71 | 1 | 3 (7) | 4 (9) | 1 | 1 | 5 (15) | 2 (3) | 0.09 | 0.93 |
| GPR124 | 3 (7) | 4 (9) | 1 | 1 | 3 (7) | 4 (9) | 1 | 1 | 3 (9) | 4 (7) | 0.7 | 1 |
| KDR | 4 (9) | 3 (6) | 0.71 | 1 | 5 (11) | 2 (4) | 0.26 | 0.73 | 2 (6) | 5 (8) | 1 | 1 |
| NOTCH2 | 3 (7) | 4 (9) | 1 | 1 | 4 (9) | 3 (6) | 0.71 | 0.96 | 1 (3) | 6 (10) | 0.41 | 1 |
| PRKDC | 2 (4) | 5 (11) | 0.44 | 1 | 3 (7) | 4 (9) | 1 | 1 | 2 (6) | 5 (8) | 1 | 1 |
| RNF43 | 1 (2) | 6 (13) | 0.11 | 1 | 5 (11) | 2 (4) | 0.26 | 0.73 | 3 (9) | 4 (7) | 0.7 | 1 |
| ROS1 | 3 (7) | 4 (9) | 1 | 1 | 4 (9) | 3 (6) | 0.71 | 0.96 | 3 (9) | 4 (7) | 0.7 | 1 |
| FANCF | 3 (7) | 3 (6) | 1 | 1 | 4 (9) | 2 (4) | 0.43 | 0.74 | 1 (3) | 5 (8) | 0.41 | 1 |
| FLT1 | 3 (7) | 3 (6) | 1 | 1 | 4 (9) | 2 (4) | 0.43 | 0.74 | 2 (6) | 4 (7) | 1 | 1 |
| GNAS | 3 (7) | 3 (6) | 1 | 1 | 1 (2) | 5 (11) | 0.2 | 0.73 | 1 (3) | 5 (8) | 0.41 | 1 |
| PIK3CA | 5 (11) | 1 (2) | 0.11 | 1 | 4 (9) | 2 (4) | 0.43 | 0.74 | 3 (9) | 3 (5) | 0.66 | 1 |
| PIK3CG | 3 (7) | 3 (6) | 1 | 1 | 4 (9) | 2 (4) | 0.43 | 0.74 | 6 (18) | 6 (10) | 0.08 | 0.93 |
| RANBP2 | 1 (2) | 5 (11) | 0.2 | 1 | 3 (7) | 3 (6) | 1 | 1 | 2 (6) | 4 (7) | 1 | 1 |
| SETD2 | 1 (2) | 5 (11) | 0.2 | 1 | 5 (11) | 1 (2) | 0.11 | 0.73 | 4 (12) | 2 (3) | 0.18 | 1 |
| SLIT2 | 3 (7) | 3 (6) | 1 | 1 | 3 (7) | 3 (6) | 1 | 1 | 2 (6) | 4 (7) | 1 | 1 |
| SMARCA4 | 3 (7) | 3 (6) | 1 | 1 | 5 (11) | 1 (2) | 0.11 | 0.73 | 4 (12) | 2 (3) | 0.18 | 1 |
Pa, adjusted p value
Table 4.
The associations between overall survival and genomic mutations.
| GENE | Mutation Positive | Mutation Negative | HR (95% CI) | P | Pa | ||
|---|---|---|---|---|---|---|---|
| N | OS median (95% CI) | N | OS median (95% CI) | ||||
| KRAS | 85 | 24.5 (21.3, 28.3) | 7 | 36 (22.7, NA) | 2.2 (0.8, 6) | 0.12 | 0.87 |
| TP53 | 63 | 24.5 (21.2, 29.5) | 29 | 27.3 (22.7, 44) | 1.2 (0.7, 2) | 0.43 | 0.87 |
| SMAD4 | 20 | 24.1 (18.9, 35) | 72 | 24.7 (21.3, 30.1) | 1.1 (0.7, 2) | 0.63 | 0.93 |
| CDKN2A | 18 | 24.2 (15.5, NA) | 74 | 24.7 (22.3, 28.4) | 0.9 (0.5, 1.6) | 0.66 | 0.93 |
| LRP1B | 16 | 25 (18.4, NA) | 76 | 24.7 (21.8, 30.1) | 1 (0.5, 1.9) | 1 | 1 |
| ARID1A | 15 | 18.9 (14.6, NA) | 77 | 25.3 (23, 29.5) | 1 (0.5, 2) | 0.93 | 0.99 |
| BRCA2 | 15 | 24.7 (18.4, 29.3) | 77 | 25.3 (21.6, 30.6) | 1.3 (0.7,2.4) | 0.44 | 0.9 |
| ARID1B | 14 | 24.1 (18.7, NA) | 78 | 25 (21.6, 29.3) | 0.8 (0.4, 1.6) | 0.58 | 0.87 |
| MLL3 | 14 | 25.7 (18, NA) | 78 | 24.7 (21.6, 29.3) | 1.1 (0.6, 2.1) | 0.69 | 0.93 |
| ATM | 10 | 22.8 (15.1, NA) | 82 | 25 (22.3, 29.3) | 1 (0.5, 2.1) | 0.96 | 0.99 |
| TSC2 | 10 | 31.8 (12.7, NA) | 82 | 24.7 (21.8, 28.4) | 0.8 (0.3, 1.8) | 0.53 | 0.9 |
| MLL2 | 9 | 18.4 (13, NA) | 83 | 25 (22.7, 29.5) | 1.6 (0.8, 3.3) | 0.21 | 0.87 |
| POLE | 8 | 28.4 (23, NA) | 84 | 24.5 (21.3, 27.5) | 0.7 (0.3, 1.7) | 0.45 | 0.87 |
| SPEN | 8 | 21.1 (15.5, NA) | 84 | 25 (22.3, 29.3) | 1.3 (0.6, 2.7) | 0.49 | 0.89 |
| EP300 | 7 | 34.2 (24.5, NA) | 85 | 24.7 (21.6, 28.4) | 0.5 (0.2, 1.6) | 0.24 | 0.87 |
| FAT1 | 7 | 18.7 (13, NA) | 85 | 25 (22.7, 29.5) | 1.5 (0.7, 3.5) | 0.32 | 0.87 |
| GPR124 | 7 | 27.3 (14.6, NA) | 85 | 24.7 (21.8, 29.3) | 1 (0.5, 2.4) | 0.92 | 0.99 |
| KDR | 7 | 24.5 (16.2, NA) | 85 | 24.7 (21.6, 29.5) | 1 (0.5, 2.4) | 0.91 | 0.99 |
| NOTCH2 | 7 | 37 (22.3, NA) | 85 | 24.7 (21.3, 28.4) | 0.7 (0.3, 1.5) | 0.34 | 0.85 |
| PRKDC | 7 | 21.3 (15.7, NA) | 85 | 25.3 (22.3, 30.1) | 1.5 (0.7, 3.5) | 0.33 | 0.85 |
| RNF43 | 7 | 23 (16.2, NA) | 85 | 24.7 (21.8, 29.3) | 1.1 (0.5, 2.8) | 0.78 | 0.98 |
| ROS1 | 7 | 18.9 (13.1, NA) | 85 | 24.7 (22.3, 29.5) | 1.5 (0.6, 3.4) | 0.38 | 0.88 |
| FANCF | 6 | 19.9 (16.7, NA) | 86 | 25 (22.7, 29.5) | 1.9 (0.8, 4.8) | 0.17 | 0.85 |
| FLT1 | 6 | 17.1 (11.5, NA) | 86 | 25.3 (22.3, 29.5) | 2.1 (0.9, 4.8) | 0.09 | 0.85 |
| GNAS | 6 | 27.8 (14.4, NA) | 86 | 24.7 (21.6, 28.4) | 1 (0.4, 2.4) | 0.94 | 0.99 |
| PIK3CA | 6 | 18.8 (13, NA) | 86 | 25 (21.8, 29.5) | 2.1 (0.8, 5.9) | 0.15 | 0.85 |
| PIK3CG | 6 | 29.3 (23, NA) | 86 | 24.7 (21.3, 28.4) | 0.8 (0.3, 1.9) | 0.57 | 0.92 |
| RANBP2 | 6 | 17.9 (16.2, NA) | 86 | 25 (22.3, 29.3) | 1.6 (0.7, 4.1) | 0.29 | 0.85 |
| SETD2 | 6 | 28.4 (24.7, NA) | 86 | 24.5 (21.3, 29.3) | 0.5 (0.2, 1.6) | 0.22 | 0.85 |
| SLIT2 | 6 | 22.9 (16.2, NA) | 86 | 25.3 (21.6, 29.5) | 1.8 (0.7, 4.5) | 0.23 | 0.85 |
| SMARCA4 | 6 | 23.2 (18.4, NA) | 86 | 24.7 (21.6, 29.5) | 1.1 (0.5, 2.9) | 0.76 | 0.98 |
CI, confidence interval; NA, not applicable; OS, overall survival; HR, hazard ratio; Pa, adjusted p value
Copy number alterations most frequently identified were CDKN2A/B loss (N = 23, 25%), SMAD4 loss (N = 6, 6%), and AKT2 amplification (N = 5, 5%). Other copy number alterations identified in at least 2% of tumors include GATA6 (N = 3, 3%), CCND3 (N = 3, 3%), MYC (N = 2, 2%), BARD1 (N = 2, 2%), SLIT2 (N = 2, 2%), ERBB2 (N = 2, 2%). Since the loss of CDKN2A/B were reported together in genomic test reports of most patients, CDKN2A/B loss was listed separately from CDKN2A mutation in Fig. 1.
Seven tumors did not have any alteration in KRAS. One of these tumors had a BRAF (V600_K601 > E). Other genetic alterations which have been previously reported in KRAS wild-type PDACs, including MYC, ERBB, and different RTKs amplifications [11,17], as well as ROS1 [12], ALK [18], RET [19], and NTRK1 [20] fusions, were not observed in these KRAS wild-type tumors.
3.3. Time from tumor tissue available to genomic testing ordered
Of the 92 patients tested, 91 were still alive at the time of the genomic result reported. The median time from tissue available (biopsy or surgical resection) to the genomic testing ordered by the providers was 229 days (IQR: 62–415). In a further subgroup analysis, the median time was 361 days (IQR 91–567) for surgical patients and 92 days (IQR 43–289) for non-surgical patients.
3.4. Actionable alteration and matched therapy
A majority (N = 82, 88%) of the tumors were tested with Foundation Medicine Panel Version 2 [7], followed by six patients with Foundation Medicine Panel Version 1 [7] and five patients with CancerSelect™ panel. A total of 10 (11%) patients were found to have potentially targetable alterations, and 3 (30%) of them received matched therapy. The details of patients with potentially actionable alterations were summarized in Table 1.
3.5. Microsatellite status and tumor mutation burden
A total of 49 tumors underwent microsatellite instability testing, and all of them were microsatellite stable (MSS). Of the 35 patients with tumor mutation burden (TMB) determined, six patients had intermediate TMB (definition: 5 < Muts/Mb < 21) with a median of 6.5 Muts/Mb (IQR 6–9). The other 29 patients had low TMB (definition: mutations/Mb < 6), with a median of 4 Muts/Mb (IQR 3–4). POLE mutations were observed in eight patients. Five of them had TMB tested, and all these five patients were TMB-low.
3.6. Homologous recombination deficiency (HRD) pathway gene alterations
Several mutations were found in HRD genes, including ATM, BRCA2, BRCA1, and PALB2 [17]. We classified all mutations in the four genes into 1) pathogenic: mutations reported as pathogenic/likely pathogenic in the ClinVar database or which were predicted to result in a truncated protein product (nonsense, frameshift, and splice-site mutations); 2) variants of uncertain significance (VUS): missense or inframe indel mutations reported as uncertain significance in the ClinVar database. Pathogenic mutations were detected in 10 tumors (11%), and were most frequently identified in BRCA2 (N = 6, 6%), followed by ATM (N = 3, 3%), BRCA1 (N = 1, 1%). Detailed information of mutations classified as pathogenic and VUS was given in Table 5 and Table 6, respectively. Inactivation of BRCA2 or BRCA1 has been reported to correlate positively with platinum-based chemotherapy and PARP inhibitor sensitivity in some PDAC patients [21–24]. Out of seven patients with BRCA2 or BRCA1 pathogenic mutations, six (86%) received platinum-based chemotherapy in the whole treatment process. One patient who was given platinum-based chemotherapy as adjuvant treatment had recurrence after 12 months. Of the other five evaluable patients on platinum-based chemotherapy, four (80%) patients had a partial response (PR), one (20%) had stable disease (SD) based on RECIST 1.1. Details were listed in Table 5.
Table 5.
Patients with pathogenic mutation in ATM, BRCA1, and BRCA2.
| Patient ID | Gene symbol | Amino acid change | Function | ClinVar clinical significance | History of other cancer | Platinum-based therapy | Response to platinum-based therapy (PFS in months) | PARP inhibitor | Response to PARP inhibitor |
|---|---|---|---|---|---|---|---|---|---|
| 17 | ATM | C117fs × 17 | Frameshift | – | No | Yes (first-line) | PR (11.2) | No | – |
| 46 | ATM | E343fs × 2 | Frameshift | Pathogenic | Gastric cancer, renal cell cancer | No (loss of follow-up) | – | No | – |
| 57 | ATM | E343fs × 2 | Frameshift | Pathogenic | Prostate cancer, colon cancer | Yes (first-line) | PD (2.1) | No | – |
| 28 | BRCA1 | Truncation intron 12 | Splice site | – | No | Yes (first-line) | SD (11.9) | No | – |
| 5 | BRCA2 | K2162fs × 5 | Frameshift | Pathogenic | No | Yes (neoadjuvant) | PR (5.6)a | Yes (neoadjuvant) | PR (18.5) |
| 8 | BRCA2 | T1566fs × 9 | Frameshift | Pathogenic | Breast cancer | Yes (adjuvant) | – | Yes (first-line) | PD (1.0) |
| 9 | BRCA2 | E2677*; Y2215fs × 13 | Nonsense; Frameshift | Pathogenic | No | Yes (first-line) | SD (6.8) | No | – |
| 53 | BRCA2 | Loss exon 26–27 | splice site | – | Testicular cancer | Yes (first-line) | PR (16.2) | No | – |
| 62 | BRCA2 | V1283fs × 2; | Frameshift | Pathogenic; | No | Yes (neoadjuvant) | PR (5.5)b | Yes (neoadjuvant and adjuvant) | PR (17.6) |
| 87 | BRCA2 | S1982fs × 22 | Frameshift | Pathogenic | No | Yes (neoadjuvant) | PR (6.0)c | No | – |
PR, partial response; SD, stable disease; PD, progressive disease
Platinum-based therapy and PARP inhibitor were used together and only PARP inhibitor was used as maintenance therapy after.
Platinum-based therapy was used alone and PARP inhibitor was used as maintenance therapy after.
Treatment was discontinued because of surgery other than disease progression
Table 6.
Patients with VUS in ATM, BRCA1, BRCA2, PALB2.
| Patient ID | Gene symbol | Amino acid change | Function | ClinVar clinical significance | History of other cancer | Platinum-based therapy | Response to platinum-based therapy (PFS in months) | PARP inhibitor |
|---|---|---|---|---|---|---|---|---|
| 54 | ATM | R586I | Missense | VUS | No | Yes (first-line) | SD (7.9) | No |
| 70 | ATM | L236V | Missense | VUS | No | Yes (three-line) | PD (2.3) | No |
| 71 | ATM | S2860del | Inframe deletion | VUS | No | Yes (first-line) | PD (1.5) | No |
| 93 | ATM | S978P | Missense | VUS | No | Yes (adjuvant) | – | No |
| 68 | BRCA2 | N108H | Missense | VUS | No | No | – | No |
| 19 | BRCA2 | S976I | Missense | VUS | No | Yes (second-line) | PD (2.4) | No |
| 62a | BRCA2 | D820E | Missense | VUS | No | Yes (neoadjuvant) | PR (5.5)c | Yes |
| 43 | BRCA2 | I1831T | Missense | VUS | No | No | – | No |
| 73 | BRCA2 | G2353R | Missense | VUS | No | Yes (neoadjuvant) | SD (13.1) | No |
| 76 | BRCA2 | A2632T | Missense | VUS | No | Yes (first-line) | PR (8.6) | No |
| 40 | PALB2 | G1021R | Missense | VUS | No | Yes (first-line) | SD (5.0)b | No |
VUS, variant of unknown significance; PR, partial response; SD, stable disease; PD, progressive disease
This patient has another pathogenic BRCA2 mutation, listed in Table 3.
Treatment was discontinued because of intolerance other than disease progression.
Platinum-based therapy was used alone and PARP inhibitor was used as maintenance therapy after.
Additionally, three patients with BRCA2 mutations were given a PARP inhibitor basing on NGS test results, with two recruited to clinical trials.
The first patient had two BRCA2 mutations, one classified as pathogenic (V1283fs × 2) and one classified as VUS (D820E), and was treated with off-label PARP inhibitor. The patient was initially diagnosed with resectable PDAC with elevated CA199 (205 U/mL). Gemcitabine/Abraxane was recommended after the Pancreas Multidisciplinary Cancer Clinic discussion, with a genomic testing ordered concurrently. New liver lesions were found after two cycles of Gemcitabine/Abraxane (Fig. 2A–C). Then, modified FOLFIRINOX (mFFX) was given with stable disease for primary tumor but decreasing liver lesions (Fig. 2D–F). A PARP inhibitor was given as maintenance treatment after seven cycles of mFFX. Computerized Tomography (CT) scan three months after the PARP inhibitor showed no lesions in the liver with CA199 continuing to trend down (Fig. 2G–I). The patient underwent pancreaticoduodenectomy 11 months after starting mFFX and five months after beginning PARP inhibition. PARP inhibitor was continued as adjuvant therapy after surgery and continued for a total of 17 months by the time of the last follow-up, which was 25 months after diagnosis and 12 months after surgery.
Fig. 2.

Computed tomography (CT) before and after PARP inhibitor treatment of a patient with mutations in BRCA2. A–C. CT before modified FOLFIRINOX treatment. D–F. CT after modified FOLFIRINOX before PARP inhibitor treatment. G–I. CT after PARP inhibitor treatment. Note: the yellow arrows indicate metastatic liver lesions; the orange arrows indicate primary lesions.
The second patient with a BRCA2 mutation was diagnosed with metastatic disease with a liver lesion found during surgical exploration. The patient was subsequently found to have a pathogenic BRCA2 mutation (K2162fs × 5). The patient was enrolled in a clinical trial with a PARP inhibitor plus FOLFOX for 19 months with a PR. CT scan at 19 months after treatment showed no obvious evidence of metastasis disease. Exploration was suggested after the pancreatic cancer tumor board discussion. The patient then had a pancreaticoduodenectomy with several liver segments biopsied negative for adenocarcinoma. PARP inhibitor was given as adjuvant therapy until a new lung lesion was found four months after surgery.
The third patient with a BRCA2 mutation was treated with a PARP inhibitor in a clinical trial. The patient was initially diagnosed with resectable PDAC and underwent pancreaticoduodenectomy. Five cycles of Gemcitabine/Abraxane was given as adjuvant therapy. The patient was found to have recurrence 12 months after surgery. The primary tumor was then sequenced, and a pathogenic BRCA2 mutation (T1566fs × 9) was reported. The patient received a PARP inhibitor in a clinical trial but unfortunately progressed after only one cycle.
The ATM gene codes for an integral component protein of double-strand DNA repairing in response to ionizing radiation. However, the response of cancers with ATM mutations to platinum-based chemotherapy and PARP inhibition is less well established [24–29]. Of the three patients with pathogenic ATM mutations, two received platinum-based chemotherapy. One patient had PR, and the other patient had PD. One patient had adjuvant stereotactic body radiation therapy (SBRT). None of the patients with a pathogenic ATM mutation received a PARP inhibitor. Details of pathogenic ATM mutations identified in sequenced patients are listed in Table 5.
4. Discussion
Despite the rapidly increasing volume of genomic testing in clinical practice, utilization of this approach in the management of patients with PDAC remains limited. The utilization of tumor genomic testing in our study increased from 6 in 2014 to 44 in 2017, which reflected the increasing awareness of the potential value of molecular profiling in pancreatic cancer treatment. Three (3%) patients with pathogenic mutations in the HRD pathway had a change of clinical decision to PARP inhibitor because of the genomic test results.
One patient, who had an NGS for primary tumor without a potentially actionable mutation, was sequenced again for a recurred tumor in the liver. Unfortunately, no new actionable alterations were found in the recurred lesion. The utility of genomic testing on multiple or recurrent tumors for discordant potential actionable alternations is unknown, and further studies are warranted. Interestingly, one patient with two different BRCA2 pathogenic mutations (E2677 × and Y2215fs × 13) had SD on FFX and progressed after 6.8 months, while another patient with one pathogenic BRCA2 mutation and one VUS BRCA2 mutation (V1283fs × 2, D820E) had PR on FFX and PRAP inhibitor for 17.6 months.
Based on the results, we found two major challenges that still need to be overcome before widespread adoption of genomic profiling in clinical practice.
First, during the routine clinical practice, even in a high-volume pancreatic cancer center, the medium time from when tumor tissue became available to when a clinical NGS tests was ordered is unacceptably long at 229 days (IQR 62–415). By contrast, the median time from the receiving of tissues to the reporting of the results was only 12 days (IQR 10–13). This result suggests that most of the NGS tests were not ordered when the tissues were available. This result can also explain why few patients had receive targeted therapies matched to their tumor mutations. This is likely due to the lack of an institutional system to facilitate the testing. Not surprisingly, the medium time from when the tissue became available to when the result was reported is much longer than the real-time genomic sequencing clinical trial by Lowery et al. [11] with a median of 45 days from patients were consented for the trial to the result were available. Future efforts shall be made toward getting the sequencing tests ordered sooner in the non-clinical trial, routine clinic setting after the tissues have become available.
Second, the purpose of the clinical tumor genomic testing is to find and use potential targeted therapies for the patient using genomic profiling results. However, we do not have enough evidence to find matched treatment for most alterations. Similarly, Lowery et al. [11] reported 3 of 225 (1%) patients were given a matched therapy based on the sequencing result, and Aguirre et al. [12] reported that 11 of 71 (15%) patients enrolled were treated with an experimental agent with direction provided from the genomic testing. Apart from this, the percentage of patients with potentially targetable alterations who received the match therapies in the clinical trial could not represent the condition in routine clinical practice. Two recently published large cohort studies have provided important information on screening patients of potentially targetable alterations and matched therapies [7,30]. Singhi et al. [7] defined the targetable alteration in different pathways and found 609 (17%) patients with potentially targetable alterations. Pishvaian et al. [30] reported 282 (26%) sequenced tumors tissue in the Know Your Tumor program to have targetable alterations. Besides, they demonstrated the survival benefit of patients who received matched therapies. Both studies showed more actionable mutation than our cohort, especially in fusion, amplification, MSI-H and/or TMB-H. The lower rate of targetable alteration in our cohort could come from the small sample size comparing the two cohort studies and the definition of targetable alterations. Results from the two studies delivered a promising signal that future efforts shall be made toward developing a system to help identify potentially targetable mutations from the patients’ reports in the non-clinical trial, routine clinic setting.
Outside of BRCA phenotype or Mismatch Repair deficiency (MMRd) tumor, there are no clinically relevant biomarker strategies in PDAC, although trials are ongoing. The COMPASS trial [31] recently reported and presented data on a modified Moffitt gene signature, demonstrating that the basal-like subgroup was associated with a poor prognosis. Furthermore, it was determined that GATA6, highly expressed in the tumors with the ‘classical’ phenotype, could separate these two molecular subgroups. Notably, the basal-like group was more likely to be resistant to oxaliplatin, and therefore mFFX and the classical group (GATA6 high) aligned with oxaliplatin-sensitivity. In the group tested, GATA6 high expression constituted only a small percentage (3 or 3%)
In light of the observations of the current study, the Johns Hopkins Pancreatic Cancer Precision Medicine Program (PMCoE) is in the process of implementing multiple changes to address the challenges identified (Fig. 3). Firstly, a system for well-coordinated and timely ordering of genomic profiling and delivery of results to the clinicians has been developed. Integration of this system into the clinical pathways will allow clinicians, who treat these patients at the multidisciplinary pancreatic clinics, to order genetic profiling in real-time, i.e., as early as within a day of tumor tissue available (biopsy or surgical resection). The genomic profiling will be performed in-house using a CLIA certified John Hopkins Molecular Lab with a Solid Tumor Panel, as well as MS and TMB. This reporting will be uploaded to the EMR as soon as results become available.
Fig. 3.

Schematic diagram of the pancreatic cancer precision medicine program.
As a follow-up for genomic results and further treatment, a flagging system will be programmed to notify providers of potentially actionable alterations in these patients. Furthermore, the flagging system will receive feedback from providers on treatment response to administration of the specific treatment, especially the alterations that are not reported in any database yet or have been reported but are still classified as having uncertain significance. The relay of this information back into the platform will allow updates to be made to the Johns Hopkins Pancreatic Cancer PMCoE for a genomic database. Accumulation of these data could potentially help identify associations between genetic alterations and tumor response to unique therapeutics. This system, in particular the development of a feedback loop, will help establish a robust, feasible, and effective program that can provide real-time precision medicine guided care to patients with PDAC.
This study has several limitations. First, as the majority of patients had genomic profiling of tumor tissue only, some genetic alterations could be germline and not somatic. The probability of germline alteration could be much higher for HRD pathway genes. Indeed, a recent update to NCCN guidelines recommends consideration of germline testing for all patients diagnosed with PDAC. Although an approach based on germline testing may be more feasible [32], the presence of somatic mutations in the HRD pathway in the absence of germline alterations indicates that a combination of germline testing and tumor genomic profiling remains important and clinically relevant [24]. In particular, future studies will determine the utility of germline testing together with or without tumor genomic profiling in patients with PDAC. Second, as a retrospective study, selection bias was unavoidable. Patients included in our study had genomic profiling ordered by providers at our institution using Foundation Medicine, Perthera, and PGDx, but not from other commercial sources or by providers at outside institutions. Therefore, the rates of actionable mutations and response to targeted therapy observed in our study may not represent unselected patients with PDAC. Third, our study had a small sample size, which provides insufficient power to perform statistical testing that most statistical analyses can only be descriptive. Even with these limitations, this is one of the largest studies to report a single institution’s experience of integrating genomic profiling in the management of patients with PDAC in a non-clinical trial setting.
In conclusion, a precision medicine approach to the management of patients with PDAC using genomic profiling is feasible. However, the percentage of patients who benefit from this approach remains extremely low. We need to develop a real-time ordering system for genomic profiling at the departmental or institutional level supplemented by a flagging system and an evidence-based reference database to address current challenges. In the future, a well-organized system could lead to an increase in the adoption of a precision medicine approach to the management of patients with PDAC and potentially result in improved long-term outcomes for patients.
Funding
This work was supported by the Johns Hopkins Pancreatic Cancer Precision Medicine Program.
Footnotes
Declaration of competing interest
Lei Zheng receives grant support from Bristol-Meyer Squibb, Merck, iTeos, Amgen, NovaRock, Inxmed, and Halozyme, and received the royalty for licensing GVAX to Aduro Biotech. LZ is a paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Akrevia, Sound Biologics, FusunBiopharmaceutical, Foundation Medicine, Data-revive, and Mingruzhiyao. LZ holds shares at Alphamab and Mingruzhiyao.
The abstract has been presented as abstract in Pancreas 2018, Baltimore, MD, US.
References
- [1].Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER 9 Regs Research Data, Nov 2018 Sub (1975–2016) <Katrina/Rita Population Adjustment> - Linked to County Attributes - Total U.S., 1969–2017 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, released April 2019, based on the November 2018 submission.
- [2].Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH, Recent progress in pancreatic cancer, CA: a cancer journal for clinicians 63 (2013) 318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek KJN, Whole Genome. Redefine Mutational Landsc. Pancreat. Canc 518 (2015) 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin W-C, Mansour J, Mollaee M, Wagner K-U, Koduru P, Yopp A.J.N.c., Whole-exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets, vol. 6, 2015, p. 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang S-B, Brown AM, Kim JCJN, A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns 538 (2016) 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aguirre AJ, Oncogenic NRG1 Fusions: A New Hope for Targeted Therapy in Pancreatic Cancer, (2019) Clincanres, 2019, p. 1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, Klempner SJ, Hendifar A, Milind JM, Golan TJG, Real-time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations that Might Be Targeted with Existing Drugs or Used as Biomarkers, 2019. [DOI] [PubMed]
- [8].Larsen JE, Cascone T, Gerber DE, Heymach JV, Minna J.D.J.C.j., Targeted therapies for lung cancer: clinical experience and novel agents 17 (2011) 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].El-Deiry WS, Goldberg RM, Lenz HJ, Shields AF, Gibney GT, Tan AR, Brown J, Eisenberg B, Heath EI, Phuphanich S, Kim E, Brenner AJ, Marshall JL, The current state of molecular testing in the treatment of patients with solid tumors, Ca - Cancer J. Clin 69 (2019) 305–343, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz GJN, Discovery and saturation analysis of cancer genes across 21 tumour types 505 (2014) 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, Leach T, Herbst B, Askan G, Maynard HJCCR, Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype 23 (2017) 6094–6100. [DOI] [PubMed] [Google Scholar]
- [12].Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, Raghavan S, Kim J, Brais LK, Ragon D.J.C.d., Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine, vol. 8, 2018, pp. 1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, Mead S, Jones MD, Samra JS, Gill AJ, Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy, IMPACT) trial 21 (2015) 2029–2037. [DOI] [PubMed] [Google Scholar]
- [14].Sohal DPS, Kennedy EB, Cinar P, Conroy T, Copur MS, Crane CH, Garrido-Laguna I, Lau MW, Johnson T, Krishnamurthi S, Moravek C, O’Reilly EM, Philip PA, Pant S, Shah MA, Sahai V, Uronis HE, Zaidi N, Laheru D, Metastatic pancreatic cancer: ASCO guideline update, J.Clin. Oncol JCO.20.01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Petricoin E, Bender RJ, Halverson DC, Rahib L, Hendifar AE, Mikhail S, Precision medicine for pancreatic cancer patients: preliminary results from the know your tumor program, J. Clin. Oncol 36 (2018). [Google Scholar]
- [16].Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR, ClinVar: public archive of relationships among sequence variation and human phenotype, Nucleic Acids Res. 42 (2014) D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G.J.C.c., Integrated genomic characterization of pancreatic ductal adenocarcinoma 32 (2017) 185–203, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Singhi AD, Ali SM, Lacy J, Hendifar A, Nguyen K, Koo J, Chung JH, Greenbowe J, Ross JS, Nikiforova MN, Identification of Targetable ALK Rearrangements in Pancreatic Ductal Adenocarcinoma, vol. 15, 2017, pp. 555–562. [DOI] [PubMed] [Google Scholar]
- [19].Heining C, Horak P, Uhrig S, Codo PL, Klink B, Hutter B, Fröhlich M, Bonekamp D, Richter D, Steiger K.J.C.d., NRG1 fusions in KRAS wild-type pancreatic cancer 8 (2018) 1087–1095. [DOI] [PubMed] [Google Scholar]
- [20].Pishvaian MJ, Garrido-Laguna I, Liu SV, Multani PS, Chow-Maneval E, Rolfo CJJPO, Entrectinib in TRK and ROS1 Fusion-Positive Metastatic Pancreatic Cancer, vol. 2, 2018, pp. 1–7. [DOI] [PubMed] [Google Scholar]
- [21].Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, Kurtz RC, Olson SH, Rustgi AK, Schwartz AG, Stoffel E, Syngal S, Zogopoulos G, Ali SZ, Axilbund J, Chaffee KG, Chen YC, Cote ML, Childs EJ, Douville C, Goes FS, Herman JM, Iacobuzio-Donahue C, Kramer M, Makohon-Moore A, McCombie RW, McMahon KW, Niknafs N, Parla J, Pirooznia M, Potash JB, Rhim AD, Smith AL, Wang Y, Wolfgang CL, Wood LD, Zandi PP, Goggins M, Karchin R, Eshleman JR, Papadopoulos N, Kinzler KW, Vogelstein B, Hruban RH, Klein AP, Whole Genome sequencing defines the genetic heterogeneity of familial pancreatic cancer, Canc. Discov 6 (2016) 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sahin IH, Iacobuzio-Donahue CA, O’Reilly EM, Molecular signature of pancreatic adenocarcinoma: an insight from genotype to phenotype and challenges for targeted therapy, Expert Opin. Ther. Targets 20 (2016) 341–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jackson SP, Bartek J, The DNA-damage response in human biology and disease, Nature 461 (2009) 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, Goble S, Lin KK, Biankin AV, Giordano H, Vonderheide RH, Domchek SM, Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation, JCO Precis Oncol. (2018) 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Choi M, Kipps T, Kurzrock R, ATM mutations in cancer: therapeutic implications, Mol. Canc. Therapeut 15 (2016) 1781–1791. [DOI] [PubMed] [Google Scholar]
- [26].Teng PN, Bateman NW, Darcy KM, Hamilton CA, Maxwell GL, Bakkenist CJ, Conrads TP, Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells, Gynecol. Oncol 136 (2015) 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, Agnew KJ, Pritchard CC, Scroggins S, Garcia RL, King MC, Swisher EM, Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas, Clin. Canc. Res 20 (2014) 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rigakos G, Razis E, BRCAness: finding the Achilles heel in ovarian cancer, Oncol. 17 (2012) 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, Moss PA, Taylor AM, Stankovic T, The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo, Blood 116 (2010) 4578–4587. [DOI] [PubMed] [Google Scholar]
- [30].Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, Mikhail S, Chung V, Sahai V, Sohal DP, Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial, The Lancet Oncol. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, Southwood B, Liang SB, Chadwick D, Zhang A, O’Kane GM, Albaba H, Moura S, Grant RC, Miller JK, Mbabaali F, Pasternack D, Lungu IM, Bartlett JMS, Ghai S, Lemire M, Holter S, Connor AA, Moffitt RA, Yeh JJ, Timms L, Krzyzanowski PM, Dhani N, Hedley D, Notta F, Wilson JM, Moore MJ, Gallinger S, Knox JJ, Genomics-driven precision medicine for advanced pancreatic cancer: early results from the COMPASS trial, Clin. Canc. Res 24 (2018) 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, Reinacher-Schick A, Tortora G, Algul H, O’Reilly EM, McGuinness D, Cui KY, Schlienger K, Locker GY, Kindler HL, Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer, N. Engl. J. Med 381 (2019) 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]