

Abstract
Recent advances in next-genetic sequencing technology have facilitated an expansion in the use of exome and genome sequencing in the research and clinical settings. While this has aided in the genetic diagnosis of individuals with atypical clinical presentations, there has been a marked increase in the number of incidentally identified variants of uncertain diagnostic significance (VUS) in genes identified as “clinically actionable” by the American College of Medical Genetics guidelines. ~20 of these genes are associated with cardiac diseases which carry a significant risk of sudden cardiac death (SCD). While identification of at-risk individuals is paramount, increased discovery of incidental VUS has placed a burden on the clinician tasked with determining the diagnostic significance of these findings. Herein, we describe the scope of this emerging problem using cardiovascular genetics to illustrate the challenges associated with VUS interpretation. We review the evidence for diagnostic weight of these variants, discuss the role of clinical genetics providers in patient care, and put forward general recommendations regarding the interpretation of incidentally identified variants found with clinical genetic testing.
Keywords: Genetics, Cardiomyopathy, Arrhythmias, Sudden Cardiac Death, Cardiomyopathies, channelopathies, genetic testing, incidental variants, secondary findings, variant of uncertain significance, exome sequencing
Expansion of Genetic Testing in the Clinical Setting
The rapid advancement and automation of genetic sequencing platforms over recent years has decreased the cost and time associated with genetic analysis. Clinical testing capabilities have expanded from disease-specific gene panels to include exome sequencing (ES) which interrogates coding regions of the genome. ES is frequently used when preliminary genetic testing is non-diagnostic. Genome sequencing (GS), sequencing of both coding and non-coding regions of the genome, has recently become increasingly utilized clinically and, represents the next frontier of clinical genetic testing1.
ES and GS can provide sensitive diagnostic information for cases with heterogeneous clinical presentations of suspected genetic disease which do not clearly represent a known syndrome or phenotype2. When combined with family history, ES results from patients and biological parents have been shown to facilitate a genetic diagnosis in up to a third of previously undiagnosed patients with suspected genetic disease3, 4. Such results may be clinically actionable, prompting redirection of care, and may facilitate cascade genetic screening of family members, thereby informing proper post-test care and genetic counseling3, 5. Despite the clear diagnostic utility of genetic testing, variants identified incidentally, are found with increasing frequency. Such findings pose a significant diagnostic challenge to the clinician.
Reporting of Incidental Variants
Recognizing the growing interpretation challenge posed by incidental variants, there has been an effort to categorize these variants based on both likelihood of pathogenicity and, if pathogenic, the “actionability” of the variant in altering medical care. The American College of Medical Genetics and Genomics (ACMG) guidelines for variant classification provide guidance on reporting of variants and, in particular, secondary or incidental findings from genetic testing6. The guidelines outline standardization of the interpretation and reporting of genetic test results by segregating variants into one of five categories (pathogenic, P; likely pathogenic, LP; uncertain significance, VUS; likely benign, LB; and benign, B) based on weighted criteria regarding variant characteristics, frequency in the population, familial segregation, and functional data if available (Table 1). In addition to reporting variants related to the indication for testing, the guidelines recommend reporting of secondary findings in 59 ‘medically actionable’ genes (also known as the ACMG-59), 20 of which are genes associated with channelopathies or cardiomyopathies7, 8 (Supplemental Table I6, 8, 9). A gene is deemed ‘medically actionable’ if the discovery of a LP/P variant would prompt medical intervention, therapy initiation, alter screening practices, or inform lifestyle modifications and/or anticipatory guidance. The ACMG recommends reporting of LP/P variants in these genes, regardless of patient characteristics or indication for genetic testing. This recommendation is based on the clear association between these presumptive disease-associated variants and diseases with a high risk of morbidity and/or mortality that can be mitigated clinically. Patients may choose to opt out of receiving these results; however, this recommendation forms much of the diagnostic strength of ES and GS.
Table 1:
Variant Classification Definitions
| Term | Definition |
|---|---|
| Pathogenic Variant | There is strong evidence that the variant directly contributes to the development of disease. |
| Likely Pathogenic Variant | There is a high likelihood (greater than 90% certainty) that the variant is disease-causing |
| Variant of Uncertain Significance (VUS) | There is insufficient evidence to support a more definitive classification of the variant as either likely pathogenic or benign. |
| Likely Benign Variant | There is a high likelihood (greater than 90% certainty) that the variant is not disease-causing. |
| Benign Variant | There is strong evidence that the variant does not cause disease. |
| Incidental finding | Genetic testing results which are unrelated to the initial diagnostic indication for testing. Variants identified incidentally, may be classified as pathogenic, likely pathogenic, VUS, likely benign, or benign. |
| Incidental VUS | Incidentally found variant classified as a variant of uncertain significance. |
| Secondary finding | Incidental variant/finding that localizes to a gene within the ACMG-59 |
| ACMG-59 | 59 genes associated with a variety of conditions, which the ACMG recommends reporting when found in any context. All of the ACMG-59 genes are associated with diseases which have a clear set of clinical characteristics, can be diagnosed early, and have effective treatment and/or intervention options. |
In addition to ACMG variant classifications, the Centers for Disease Control and Prevention (CDC) Office of Public Health Genomics classifies incidentally identified variants into ‘tiers’ based on their clinical implications. Tier 1 genes are those felt to be associated with a clinically actionable, highly penetrant disease with an alternative means of diagnostic confirmation. Variants in Tier 2 genes may be actionable, and variants in Tier 3 genes have insufficient evidence for recommendations on clinical action10. CDC Tier 1 variants and genes are associated with highly prevalent diseases which are poorly recognized and for which early detection and intervention is likely to significantly decrease morbidity and mortality. Examples of these diseases include hereditary breast and ovarian cancer, Lynch Syndrome, and familial hypercholesterolemia. Together, the ACMG and CDC classification of variants form the basis for the assessment of variants found routinely through genetic testing and for the reporting of clinically actionable variants found incidentally.
While reporting LP/P variants in the clinically actionable ACMG-59 genes not associated with the patient’s phenotype is recommended, reporting of variants of uncertain significance (VUS) in the ACMG-59 is not recommended by the ACMG due to the unclear diagnostic weight attributable to the variants, and the potential for either over- or under-ascribing disease risk to the variant11. Despite this recommendation, both secondarily- and incidentally- identified VUS still make their way to the referring physician through either direct or indirect means. This occurs through a number of mechanisms, highlighted in Table 27, 8. While the discovery of an incidental LP/P variant in the absence of overt clinical disease may not universally warrant medical treatment, it may inform decisions regarding follow-up and anticipatory guidance. The ethical issues regarding incidental VUS disclosures and the potential psychological risks to patients have been widely debated12, 13 and patients may choose to opt out of receiving this information.
Table 2:
Sources of Incidentally Identified VUS Reporting
| Sources of Incidentally Identified VUS Reporting |
|---|
| Referring physician requests expanded variant list |
| Interpretation of variant pathogenicity evolves over time |
| Discrepancies in interpretation between genetic testing companies/laboratories |
| VUS identified on gene panel sent for incorrect clinical indication |
| Non-CLIA-approved genetic testing, direct to consumer/consumer-initiated panels |
| Research-based genetic testing |
Interpreting Incidental Variants of Uncertain Significance
In contrast to incidental variants determined to be LP/P, incidental VUS, the approach to interpretation of incidentally found VUS is complex and demands consideration of a number of factors which may indicate pathogenicity including 1) association between the gene in question and disease phenotype, 2) absence of the variant in ostensibly healthy individuals, 3) localization of the gene to key protein domains, and 4) high evolutionary conservation of the affected amino acid across species. However, our understanding of the diagnostic weight of these factors continues to evolve. Recent studies of large population-based aggregate genome databases, such as Genome Aggregation Database (gnomAD), have revealed that some channelopathy- and cardiomyopathy-associated variants are present in the population, sometimes with minor allele frequencies (MAF) exceeding the frequency of the disease14, 15. Whether these variants represent non-penetrant disease-causing alleles within the population, which in the setting of other predisposing variables, may manifest into disease, is unclear16.
ACMG guidelines and genotype-phenotype correlations, documented in large databases such as ClinVar17 and in large cohorts from commercial genetic testing companies, are frequently taken into account when determining incidental variant pathogenicity. In practice, the determination of pathogenicity is dependent upon the reporting laboratory or institution. Cohort databases are fluid and may differ in their interpretation of variant pathogenicity predictions18. Variant predictions are subject to change over time and are frequently promoted or demoted in their pathogenicity classification as new information is reported. Additionally, laboratories and clinicians often disagree on variant classifications19, 20. A recent study found a very low rate of concordance (Cohen k=0.26) across experienced laboratories when evaluating the potential pathogenicity of incidentally identified KCNH2 and SCN5A variants and a review of clinical data showed no difference in the phenotypes between patients with variants designated as LP/P versus patients without such variants21.
VUS interpretation identified during direct-to-consumer genetic testing, which has become increasingly available and utilized, may pose additional challenges. Such testing is obtained by the individual, potentially without rigorous consultation with a cardiovascular genetics expert and may be undertaken in the absence of disease22. Therefore, a pre-test probability is not established prior to ordering the test, and a health care provider is not available to interpret the results of the test in the context of the patient’s clinical and family history. Further, when tests return an incidental VUS, patients may not have access to counseling from a genetics provider who can advise the next course of action, if any.
Bayes’ Theorem and a Probabilistic Approach to Variant Interpretation
Genetic testing is optimally approached as a probabilistic test with non-binary results. Given the significant variability of disease prevalence and frequency with which pathogenic variants are detected, it is essential that any approach to variant interpretation take this into account. Bayes’ theorem by Reverend Thomas Bayes can be a useful construct for determining the diagnostic weight of a genetic test (Figure 1)23. Prior to ordering a genetic test, consideration should be given to the pre-test probability of disease and the strength of the selected genetic test. In children with concern for cardiomyopathy or channelopathy, pre-test probability is a clinical assessment of the likelihood of the disease in question, considering prior variables such as the prevalence of the disease in the general population, personal and family history, and clinical evaluation. For example, given the low prevalence of long QT syndrome (LQTS) in the general population, it is unlikely that an identified incidental variant in a seemingly healthy individual is truly disease-causing a priori. Further, if after careful review of the individual’s past medical history, family history, and relevant clinical testing suspicion for LQTS is low, then pre-test probability for genetic testing remains low. Given the importance in determining this pre-test probability prior to ordering a genetic test, a comprehensive evaluation should be conducted by clinical experts knowledgeable about the disease or process in question to determine the clinical utility of a genetic test.
Figure 1:
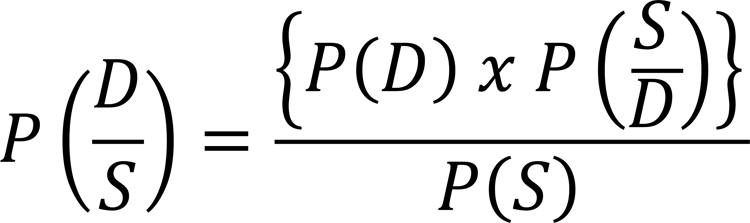
Equation of Bayes’ theorem. Bayes’ theorem can be applied to inform the post-test probability of a genetic diagnosis given the probability of the disease, probability of the genotype/phenotype in the general population, and the results from a genetic test. P is probability; D is disease; S is sign, symptom, and pattern of the disease.
Once the pre-test probability is determined, it is modified by the strength of the genetic test which can be further informed by a signal to noise ratio (SNR) analysis. SNR is a measure which has been used in science and engineering to compare the level of a desired signal to the level of background noise. Normalizing pathogenic variant frequency among individuals with disease against the “background” frequency of rare variants (generally from MAF<0.01 to <0.001, depending on the disease) yields a disease-specific SNR, which can aid in both variant classification and the likelihood of a genetic test returning a disease-associated variant. The “signal” in this context is the ability of the genetic test to accurately identify a patient with disease based on the prevalence of LP/P variants, while the “noise” indicates the background rate of rare population variants in ostensibly healthy individuals. A higher SNR reflects a higher diagnostic strength of associating a variant in question with a particular disease.
SNR can be measured at the disease level, gene level, or amino acid level. For example, a study of LQTS reported the frequency of disease-associated variants in KCNQ1, KCNH2, and SCN5A genes as 45%. Given the population frequency of rare (MAF<0.001) variants in these same genes of ~10%, the so-called SNR of pathologic variant to rare population variant was ~4:1. Gene-specific normalized SNR for KCNQ1, KCNH2, and SCN5A genes in the cases compared to the Genome Aggregation Database (gnomAD)20, a summary large-scale database of exome and genome sequencing data from healthy and diseased individuals, was 6.3, 7.0, and 0.76 respectively. This can be taken one step further, to the amino acid level of the protein in question. For example, the KCNQ1-encoded Kv7.1 potassium channel demonstrates high SNR along the pore domain, particularly elevated within the S5 and S6 transmembrane domain, which comprise the pore, and the channel selectivity filter24. In contrast, incidental VUS found by ES demonstrated no elevated areas along the protein topology, suggesting that these variants are indistinguishable from “noise”24. Supplemental Table II24–42 summarizes what is known about pathologic, healthy, and incidental variant frequencies for various channelopathies and cardiomyopathies.
When SNR is introduced into Bayes’ theorem, and is used to modify the strength of a genetic finding, it offers a method for refining pre-test clinical suspicion based on the diagnostic strength of the genetic finding to determine the post-test probability. This is particularly helpful when attempting to interpret an incidentally found VUS. Thus, if there is a moderate to high pre-test suspicion of disease, and an incidental VUS in a gene is identified to localize to a region with high SNR, then the post-test probability of variant pathogenicity is increased. Conversely, should an incidental VUS be identified in a gene or locus, with low SNR, a low pre-test clinical suspicion of disease is likely unchanged with identification of this variant. Post-test probability may change over time if the variant is found to segregate with disease on familial cascade screening or if the variant is reclassified based on new evidence. Figure 2 illustrates how pre-test probability and a high, intermediate, or low SNR could be used to inform diagnostic certainty of an incidental variant. Two cases are provided below to illustrate the application of this method.
Figure 2:
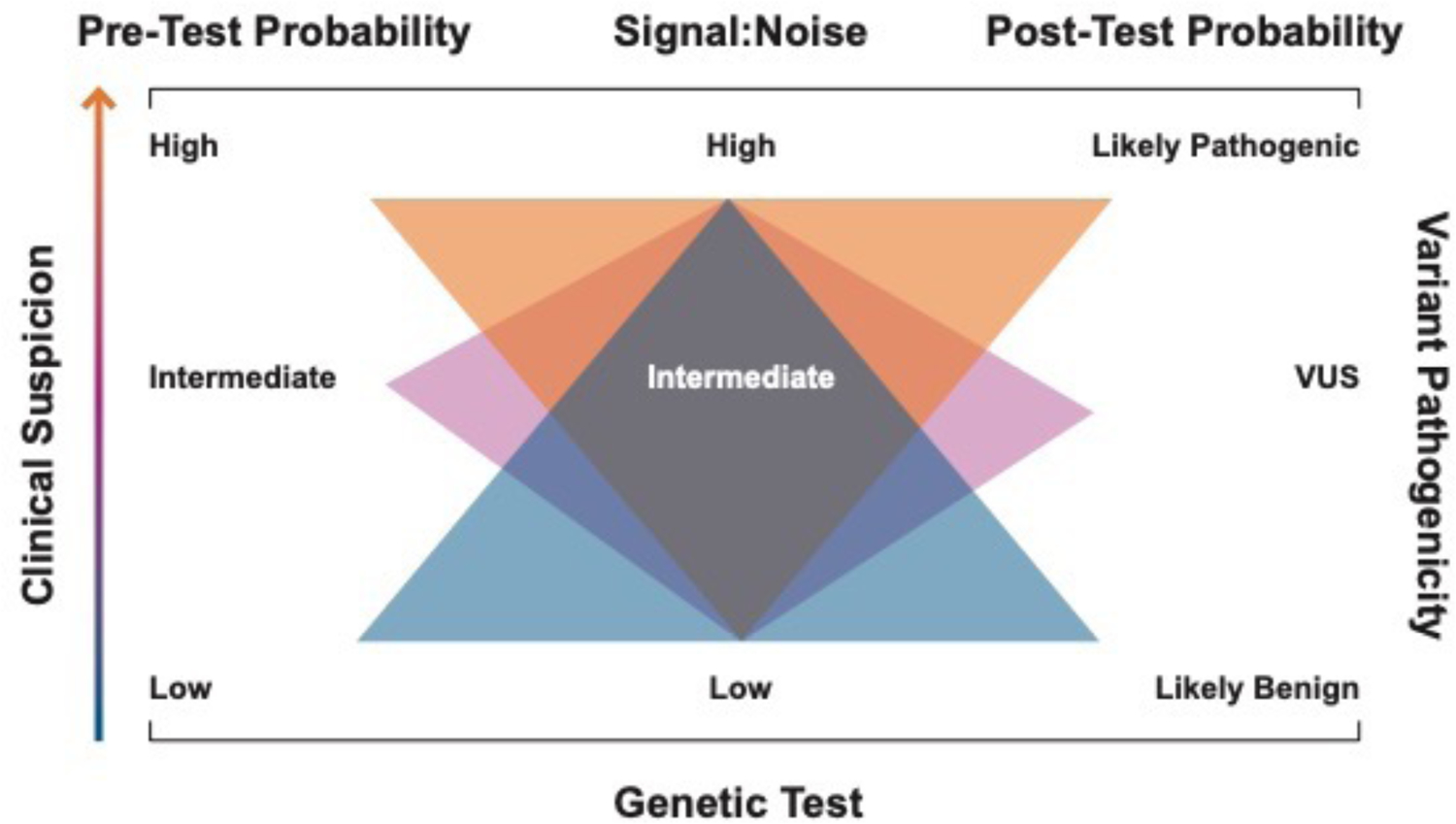
Schematic illustrating a probabilistic approach to variant interpretation. A pre-test probability of disease is established following a detailed, individualized evaluation for the disease in question. This pre-test probability is then modified by the strength of the genetic test based on signal-to-noise, among other measures to create a post-test probability of disease.
Case 1:
A 3 yo male with a history of developmental delay, mild facial dysmorphisms, static encephalopathy, and leukopenia undergoes trio whole ES evaluation. He is found to have a frameshift variant in KCNH2, KCNH2-Leu1100Profs*19. Family history is negative for syncope, seizures, and sudden death. The variant was absent in both parents suggesting a de novo inheritance pattern. On cardiac evaluation, there is no history of syncope. He is found to have a borderline prolonged QTc of 475 ms. An event monitor is placed which reports no concerning events. KCNH2, one of the 59 medically actionable genes identified by the ACMG, is recommended to be reported when found regardless of indication for testing. The KCNH2 gene has a high SNR and a strong association with LQTS type 2. The Leu110Profs*19 variant was not found in ClinVar and is predicted to have a loss of function mechanism, matching a known disease mechanism of LQTS type 2. The patient’s clinical finding of QT prolongation, the high signal to noise association of KCNH2 variants in LQTS, and the fulfilment of key pathogenicity assignment criteria by ACMG guidelines7 lead to a conclusion that this patient’s variant is likely pathogenic. Treatment with beta blocker and avoidance of QT prolonging medications was advised. No cascade screening is indicated in this case due to the de novo inheritance pattern.
Case 2:
A 17 yo female undergoes whole ES during an extensive evaluation for dysautonomia symptoms. A ANK2-Glu458Gly missense variant is found raising concern for LQTS. On cardiac evaluation, she does not have syncope or other cardiac symptoms. Family history is negative for syncope, long QT syndrome, and other sudden cardiac death-related illnesses. Her QTc is normal on ECG and an exercise treadmill test is normal. This variant was found in ClinVar. ANK2 demonstrates a low signal-to-noise ratio in relationship to LQTS with recent ClinGen evidence suggesting a loose correlation with LQTS. Using ACMG variant interpretation guidelines, the variant was assessed as being unlikely to be associated with disease. This patient should have follow-up evaluations periodically to reassess her clinical findings and the variant pathogenicity interpretation in the context of any new clinical findings.
Disease manifestation and subsequent sudden cardiac death risk are variable, even among individuals who share a common diagnosis and among individuals with variants in the same gene43. The finding of a variant in a disease-associated gene in the absence of clinical disease may be due to incomplete penetrance. While there is emerging evidence that variants in some genes may have prognostic relevance, for example desmosomal and LMNA variants in dilated cardiomyopathy (DCM) carry a risk of life-threatening ventricular arrhythmias and SCD44, the risk of sudden death is largely not genotype driven. Thus, the prognostic implications of an incidental VUS, and any subsequent treatment decisions, are based on the family history and the patient clinical phenotype, if present.
The Genetics of Sudden Cardiac Death-Predisposing Diseases: Genetic Testing for Channelopathies and Cardiomyopathies
Cardiac channelopathies such as long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), Brugada syndrome (BrS), and short QT syndrome (SQTS) are molecular defects in ion channels or channel-interacting proteins of the heart which cause electrophysiologic alterations that significantly increase the risk of arrhythmic events and SCD. Channelopathies are classically viewed as genetic disorders. Cardiomyopathies are primary diseases of the myocardium affecting the ability of the heart to contract or relax and include hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and dilated cardiomyopathy (DCM), all of which are also classically believed to have a genetic etiology45. While each of the channelopathy and cardiomyopathic syndromes manifest in different clinical phenotypes, the associated predisposition to sudden cardiac death makes the reporting and correct interpretation of variants in genes associated with these diseases critical. It is standard of care to only obtain gene panel testing in individuals with clinical diagnosis of disease or likely disease or in those with high risk of developing disease due to a previously identified pathogenic variant in their family46; however, secondary variants found on broad genetic testing and genetic testing which circumvents a clinical diagnosis, make variant interpretation challenging.
Here, we review the diagnostic weight of disease associated variants and put forward general recommendations regarding the interpretation of incidentally identified VUS which can be utilized in conjuction with consultation with medical genetics professionals.
Long QT Syndrome
LQTS is characterized by prolongation of the QT interval on resting electrocardiogram (ECG), based on age-related normal values with a structurally normal heart47. The clinical presentation varies from asymptomatic to syncope and SCD due to torsade de pointes48. The prevalence of LQTS is estimated to be as high as 1 in 200049. The three major genes associated with LQTS are KCNQ1 which encodes the IKs potassium channel (Kv7.1)50, KCNH2 which encodes the IKr potassium channel (Kv11.1)51 and SCN5A which encodes the INa sodium channel (NaV1.5)52. These channels orchestrate normal cardiac repolarization53. Variants in these three genes account for 65–75% of all LQTS variants and most disease-associated variants are missense mutations54. To date, at least 14 other minor genes have been associated with LQTS with relatively low frequency, many of which have had their association with LQTS called into question55. The background rate of rare LQTS-associated variants in healthy individuals is ~10%, giving a SNR of ~7.5:1 suggesting a higher probability of an individual with LQTS hosting a disease pathogenic variant compared to a rare, non-disease associated variant40. Approximately 0.5% of individuals undergoing exome and genome sequencing for non-LQTS indications are found to have incidentally found LP/P variants while ~11% of individuals will have an incidentally found VUS24. This suggests an LP/P and VUS variant rate localizing to KCNQ1, KCNH2, and SCN5A that exceeds the prevalence of disease by 10- and 220-fold, respectively (Table 3, Supplemental Table II24–42).
Table 3:
Comparison of the prevalence of incidental LP/P variants and VUS associated with inherited cardiac channelopathies and cardiomyopathies
| Disease | SNR Pathogenic: Population |
Incidental Variant Prevalence vs Disease Prevalence* | References | |
|---|---|---|---|---|
| Incidental LP/P: Prevalence | Incidental VUS: Prevalence | |||
| Hypertrophic cardiomyopathy | 10:1 | 2.5-fold | 34-fold | 25–29 |
| Arrhythmogenic right ventricular cardiomyopathy | 3.8:1 | 20-fold | 700-fold | 30, 31 |
| Dilated cardiomyopathy | 2.14:1 | N/A | N/A | 32–36 |
| Catecholaminergic polymorphic ventricular tachycardia | 8.3:1 | 20-fold | 900-fold | 37–39 |
| Long QT syndrome | 7.5:1 | 10-fold † 24-fold‡ |
220-fold 740-fold‡ |
24, 40, 41 |
| Brugada syndrome | 4.2:1 | 6-fold§ | 126-fold§ | 42 |
Disease-specific SNR calculations based on the following disease-associated genes:
HCM: MYH7, MYL3, MYL3, MYBPC3, TNNT2, TNNTI3, TNNC1, TPM1, ACTC
ARVC: PKP2, DSP, DSG2, DSC2, JUP, TMEM43, TGFB3, PKP4, PERP
DCM: TTNtvs
CPVT: RYR2, CASQ2
LQTS: KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, SNTA1, KCNJ5, CALM1, CALM2, CALM3, TRDN
Brugada Syndrome: SCN5A
Calculation based on presumption that all individuals with disease are genotype positive.
Only ACMG-designated actionable genes associated with LQTS (KCNQ1, KCNH2, and SCN5A).
All LQTS-associated genes.
Based on SCN5A. N/A, not available.
Catecholaminergic Polymorphic Ventricular Tachycardia
CPVT is the most lethal cardiac channelopathy with a mortality reaching 30% by the third decade of life if untreated and an estimated prevalence of 1 in 10,00056. CPVT patients demonstrate a normal resting ECG with a structurally normal heart, however exercise or emotional stress can provoke ventricular ectopy and arrhythmias, classically bidirectional ventricular tachycardia, which can cause syncope and sudden death57. The genes RYR2, CASQ2 and KCNJ2 have been associated with CPVT. Variants in the RYR2-encoded cardiac ryanodine receptor 2 (RyR2), are the most common finding, with a prevalence of 47%58. Genetic testing to identify CPVT cases has a fairly high diagnostic yield of ~60%37, 39. Variants in CPVT-associated genes are found in ostensibly healthy individuals at a rate of 6–11%, yielding a high SNR ratio of ~8:1 similar to LQTS. Incidentally found CPVT-associated LP/P variants and VUSs are 0.2% and 9%, respectively39, resulting in a frequency that is 20- to 90-fold greater than the disease prevalence, respectively. (Table 3, Supplemental Table II24–42)
Brugada Syndrome
BrS is a rare channelopathy with a heterogeneous clinical presentation ranging from asymptomatic to SCD. The typical ECG presentation is a right bundle branch block with persistent ST-segment elevation in the right precordial leads (V1 and V2)59. The patient’s ECG abnormality can be manifest at rest or develop with fever or after pharmacological challenge60. The first gene identified to be associated with BrS was SCN5A61. SCN5A accounts for 20–25% of all BrS probands and is the most common gene associated with the disease62. Several additional genes have been associated with BrS, including GPD1L63 and SCN1B64; however, variants in the SCN5A gene remain the largest contribution to disease42, 61. Further, the association of non-SCN5A genes with BrS have been called into question65. The background rate of rare, disease-associated variants in SNC5A among healthy individuals is ~5% resulting in a moderate SNR of ~4:1. Incidentally found SCN5A LP/P variants and VUS are 0.3% and 6.3%, respectively24, resulting in a frequency that is 6- to 126-fold greater than the disease prevalence, respectively24, 42 (Table 3, Supplemental Table II24–42)
Hypertrophic Cardiomyopathy
HCM is the most common cause of SCD in children and young adults with a prevalence of 1 in 500 in the general population27. HCM is characterized by asymmetrical hypertrophy of the left ventricle with occasional involvement of the right ventricle. Cardiac hypertrophy impairs cardiac relaxation and ventricular filling and can obstruct the left ventricular outflow45. Lethal arrhythmias not necessarily associated with the observed degree of hypertrophy can develop66. HCM is a disease of the cardiac sarcomere, with variants in genes encoding components of the thick filament (MYH7, MYL2, and MYL3), the intermediate filament (MYBPC3), and thin filament (TNNT2, TNNI3, TNNC1, TPM1, and ACTC) being the major causes of disease67, 68. Syndromic causes include Pompe disease, Fabry disease, and other rare metabolic syndromes. In individuals with disease, the frequency of LP/P variants is ~50%, depending on the cohort analyzed26, 69. Of these, variants in MYH7 and MYBPC3 comprise ~20–50%70, 71.
While the genetic basis for HCM has been well established, there has been increasing awareness of a significant background rate of rare genetic variation in these genes that occur in ostensibly healthy individuals. At least 5% of healthy individuals demonstrate a rare variant in one of these sarcomeric genes but will not manifest disease27, 28. This yields a high SNR of ~10:1. Variants classified as LP/P are found incidentally VUS in HCM-associated genes at a rate of 0.5% and VUSs found in ~6.8% of individuals yielding a 2.5- to 34-fold higher prevalence than HCM in the population25, 27 (Table 3, Supplemental Table 24–42).
Arrhythmogenic Right Ventricular Cardiomyopathy
ARVC is defined by loss of cardiac myocytes, particularly in the right ventricle, and replacement with fibrofatty tissues which can cause cardiac dysfunction and arrhythmic events. The estimated prevalence of ARVC is 1 in 1,000 to 5,000, although the true prevalence is challenging to determine72–75. Variants in genes encoding proteins in the cardiac desmosome have been associated with ARVC and include PKP2, DSP, DSG2, DSC2, JUP, TMEM43, and TGFB3. ~30–50% of patients with clinical ARVC host a single causative variant in one of these genes73. Causative variants in PKP2 account for 50% of patients with ARVC, and ~70% of patients with familial ARVC76. ARVC-associated variants are predominantly missense mutations, and the background rate of ARVC-associated variants found in healthy individuals is ~16%77 yielding a SNR ratio of ~3.8:1 The rate of incidental LP/P variants incidentally is ~0.4%, while incidental VUS are found in 14% of ES or GS cases resulting in an incidentally identified variant rate 20- to 700-fold higher than the prevalence of ARVC (Table 3, Supplemental Table II24–42). An analysis of SNR of ARVC-associated variants is optimal in cases with a single causative gene. Work by several groups suggest that compound heterozygosity can contribute to ARVC disease manifestation and is a significant risk factor for arrhythmic events and SCD78–80. SNR analyses have been conducted in cases with compound heterozygosity81, however due to the more significant contribution of compound heterozygosity in ARVC, SNR analyses may be difficult to interpret and may have limited utility in accurately modifying post-test probability.
Dilated Cardiomyopathy
DCM is a cardiomyopathy characterized by ventricular dilatation with systolic dysfunction. While the etiology of DCM can be diverse, including ischemic, infectious, rheumatic, toxic, metabolic, and inflammatory causes, idiopathic DCM is most common, identified at a rate of 1 in 250 in the general population36. Non-ischemic idiopathic DCM is heritable and has a prevalence of at least 1 in 250036 with approximately 20–50% cases of idiopathic DCM occurring in a heritable fashion, as familial DCM82–84. In pediatric cases, the incidence of DCM is 0.6/100,000 individuals per year and 35–45% of these cases are familial85, 86. The diagnostic yield of genetic tests in identifying idiopathic DCM in the general population is estimated at 12–40%32–34 in adult populations and ~50% in pediatric populations34, 87. The spectrum of genetic etiology is broad with over 40 genes associated with DCM; however, truncating variations localizing to TTN-encoded titin (TTNtvs) are the most common and are found in up to 25% of familial DCM and 18% of sporadic DCM88. The remainder of genes associated with DCM are rare variants, each accounting for <5% of DCM cases individually. A recent, large cohort study found that these non-TTN rare variants are typically not associated with DCM89. The background rate of variants in idiopathic DCM-associated genes found in healthy individuals is ~14%, giving a SNR of ~2:135. New, incidentally identified variants in DCM genes are emerging (Table 3, Supplemental Table II24–42) and our understanding of the genetics of DCM continues to evolve. Recent expert consensus ACMG guidelines have suggested a clear role for systematic interpretation of incidental variants in DCM-associated genes including comprehensive phenotypic evaluation and genetic variant interpretation90. Emerging evidence has suggested that SNR analyses can be leveraged in determining the likelihood of cardiomyopathy in TTNtv variants91. Specifically, incidentally identified TTNtvs localizing to areas of TTN with high S:N as well as areas with a high exon percent spliced in, were predictive of development of cardiomyopathy in the pediatric age range when ACMG guidelines were used.
Incidental Variants Identified in Cardiovascular Genes
While the overall frequency of LP/P variants is low, the rates of secondary findings of LP/P variants in clinically actionable genes may be higher than the prevalence of the disease in the population92. Recent studies have suggested that in the absence of supportive clinical findings, VUS likely represent rare, normal variants found within the healthy population which are not associated with disease24, 81. For this reason, the ACMG supports withholding these variants from standard clinical reporting. However, these variants may be discovered on expanded genetic testing and in such cases will require clinical interpretation. Thus, careful consideration of clinical factors in addition to reported genetic testing results is prudent.
While incidental findings in ACMG clinically actionable genes were originally predicted to affect ~1% of individuals undergoing ES9, recent evidence indicates that the rate of incidental findings in genes identified as clinically actionable may be higher24, 39. Additionally, ES and GS studies from racially and ethnically diverse cohorts have indicated that the prevalence of incidental findings of LP/P variants varies widely between populations, ranging from 0.5% to 14.4% for ES81, 93, 94 and 0.5% to 21% for GS95, 96. Identification of a high prevalence of incidentally found LP/P variants casts uncertainty on the ACMG prediction of a low rate of these variants, and with it, the diagnostic certainty when finding such a variant. Similarly, the rates of secondary variants identified as LP/P variants in ACMG-59 cardiovascular genes are also likely higher than previous estimates24, 39.
This relatively high burden of incidentally identified LP/P and VUS variants has several implications. Should LP/P variants represent true disease-associated risk alleles, they may represent either non- or very low-penetrant alleles. Further, the exceedingly high prevalence of VUS strongly suggests that the vast majority of these variants do not represent penetrant disease alleles. This poses a diagnostic dilemma and highlights the potential challenge awaiting the field of pediatric and adult cardiology with the rapid expansion of ES and GS. Should the majority of even LP/P variants never manifest disease, distinguishing probands who are at-risk of developing disease will be key in minimizing unnecessary clinical interventions (such as implantable cardiac defibrillator placement), appropriate cascade screening, and family counseling. Finally, this high prevalence underscores the importance of approaching interpretation of genetic testing in a probabilistic manner which takes into account a pre-test probability defined during a detailed clinical evaluation, the SNR of the disease/genes involves, and an individualized approach to applying the post-test probability of disease.
Limitations of Genomic Medicine
The use of SNR analyses can be helpful for guiding clinicians in incidental variant interpretation, particularly when disease- or gene-level SNR analyses are employed. However, challenges and inherent limitations exist. This is most acute in the application of amino acid-level SNR. As, SNR is a probabilistic association based on a relative frequency of variants described cases versus those in a population, this number is based on only the cases that come to medical attention and is therefore likely biased towards severe cases leaving less penetrant disease alleles less represented. Additionally, there may be other rare variants in other associated proteins which may have a risk-modulating effect or extrinsic factors, such as patient co-morbidities, that may alter the effect of a variant.
As illustrated in the cases above, SNR analyses are optimal for diseases, such as LQTS, which have a single-gene etiology. The utility of this approach in diseases with multigenic etiologies, those involving multiprotein complexes, and those with a sizeable burden of compound heterozygous disease, such as ARVC80, is unclear. Multiple groups have attempted to determine the clinical utility of SNR analyses in these types of disease, but results have been mixed, particularly in cases where SNR is applied to the molecular or amino acid level. For example, in HCM, recent independent studies have identified large domains of amino acids in genes encoding components of the cardiac troponin complex, as potential “hot spots” for increased pathogenicity and risk of sudden cardiac death97, 98. While these studies serve to illustrate the potential role a probabilistic approach to variant pathogenicity interpretation, a single gene variant remains only one variable out of many that influences presentation of disease. Indeed, even among in vitro models, the location of gene variants in “hot spot” domains does not fully explain variability in the functional impact on the protein. Thus, SNR-identified molecular “hot spots” are not solely predictive of disease expression or prognostic outcomes.
Age-Related Challenges and Long-Term Follow-up of Patients with Incidental VUS
There are inherent complexities in the analysis and interpretation of incidentally identified VUS associated with channelopathies and cardiomyopathies. Variant interpretation can be particularly difficult when identified in an asymptomatic young child, since it is impossible to know with complete certainty whether the child will go on to develop disease later in life. Conversely, interpreting the likelihood of pathogenicity of an incidental VUS can also be quite challenging in the adult population, particularly in the setting of comorbid disease which can confound manifestations of genetic disease. Genetic cardiomyopathies in adults can be more clinically variable in presentation and progressive in course which complicates both the assessment of SNR and clinical phenotyping. Clinical manifestations of disease can be age-dependent and therefore finding an incidental VUS, in a patient of any age, makes deep phenotyping on the multidisciplinary level critical.
During the initial evaluation, collaboration between multidisciplinary specialists should be the goal in order to optimally determine whether the individual demonstrates evidence of cardiovascular disease and to establish a pre-test probability. This evaluation should take into account patient age, history, family history, and a cardiac evaluation tailored to the variant identified. This pre-test probability is then integrated with the strength of the variant’s association with cardiovascular disease which can incorporate SNR as a relative risk of variant pathogenicity. This may change the overall assessment of the impact of the variant at the time of genetic testing, yielding the post-test probability. Long-term follow-up is important to refine these assessments over time as both patient evaluations and interpretations of the likelihood of variant pathogenicity change over time.
Summary of Recommendations and Clinical Implications
With ongoing advances in genetic sequencing, incidental VUS are increasingly found. The diagnostic relevance of such variants is best determined in the context of the pre-test probability and the strength of the genetic findings to allow for estimation of the probability that the variant is truly disease associated. This approach facilitates the identification of patients with disease, while minimizing unnecessary intervention and follow up of individuals without disease.
As noted above, in certain scenarios, genetic test results are optimally viewed as probabilistic and not as diagnostic, binary “positive” or “negative” tests. When confronted with an unexpected finding from an genetic test, either an incidental VUS or secondary finding, we recommend a stepwise approach to variant interpretation based on Bayes’ Theorem as illustrated in the algorithm in Figure 3, consisting of: 1) determining the pre-test probability of disease, 2) determining the diagnostic strength of the gene using SNR, 3) determining the post-test probability of disease, based on modification of the pre-test clinical suspicion and variant diagnostic strength, 4) incorporation of information from functional studies, as available, and 5) incorporation of variant segregation information from the clinical pedigree. This methodology can be effectively applied to the evaluation of most incidental variants associated with a channelopathy or cardiomyopathy.
Figure 3:
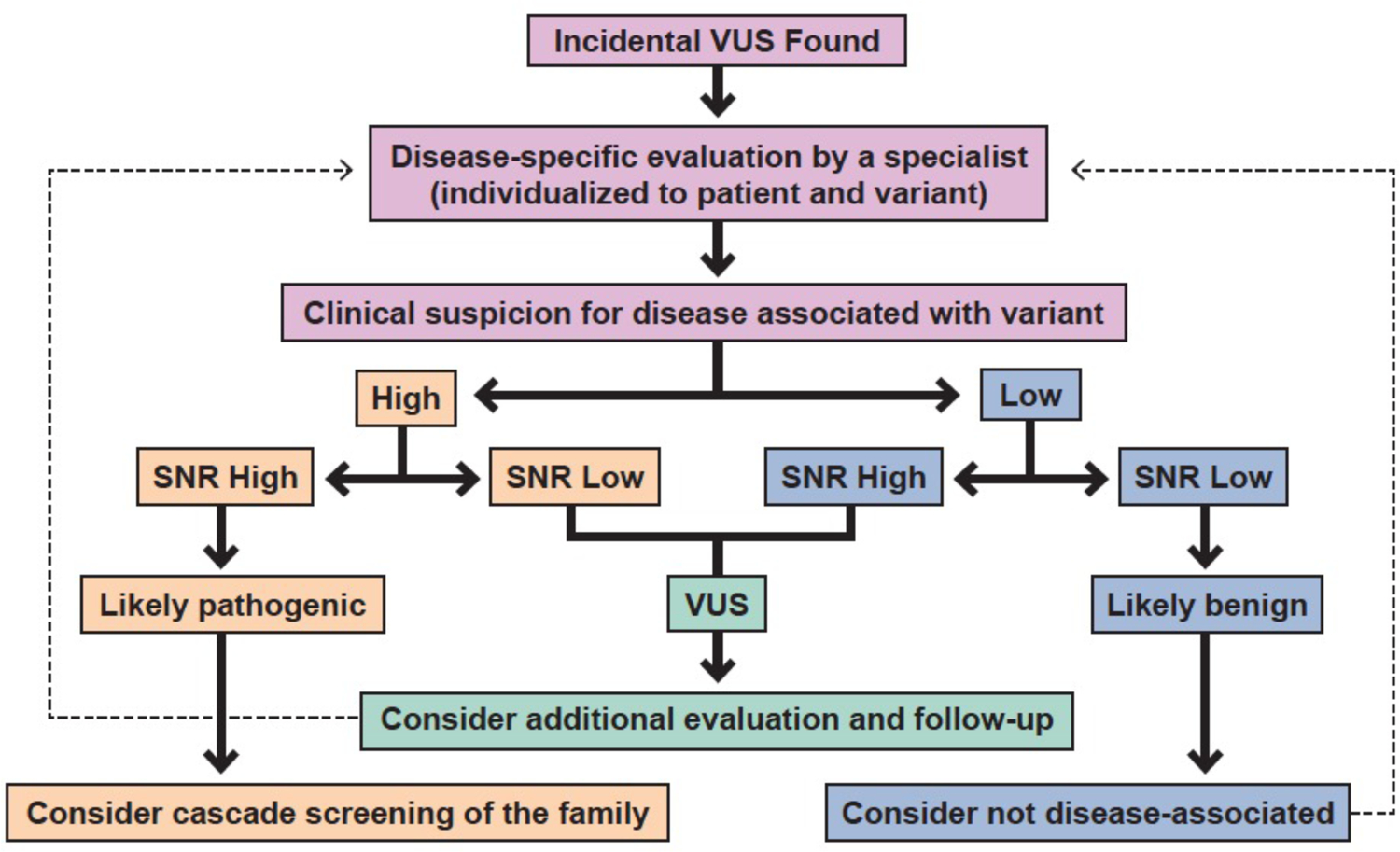
Algorithm summarizing recommendations for interpretation of incidental variants found in genes associated with cardiomyopathy or channelopathy diseases. Signal-to-noise ratio, SNR, based on disease-associated variant frequency.
Collaboration between both patients and their health care providers and among cardiology and genetics providers is vital. Determination of the utility of a genetic test, in addition to the interpretation of results often involves, and is enhanced by, communication between cardiologists and genetics specialists. Additionally, an established collaboration can be helpful when the findings of a genetic test necessitate cascade familial screening and long term follow up46, 99.
Supplementary Material
Acknowledgments:
We gratefully acknowledge the contributions of Perathu Kannu Rahesh Manivannan, MBBS in bringing together literature for this review.
Sources of Funding: JEE is supported by the NIH Clinical and Translational Science Award (UL1TR002553). PK is funded by NIH R21HG010747. APL is supported by the NIH K08-HL136839, Centers for Disease Control and Prevention (5NU50-DD004933), and Duke MEDx.
Nonstandard Abbreviations and Acronyms
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- B
benign
- BrS
Brugada syndrome
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- DCM
dilated cardiomyopathy
- ES
exome sequencing
- GS
genome sequencing
- HCM
hypertrophic cardiomyopathy
- LB
likely benign
- LP
likely pathogenic
- LQTS
long QT syndrome
- MAF
minor allele frequency
- P
pathogenic
- SNR
signal to noise ratio
- SQTS
short QT syndrome
- SCD
sudden cardiac arrest
- TTNtvs
TTN-encoded titin truncating variants
- VUS
variants of uncertain diagnostic significance
Footnotes
Disclosures: None
References:
- 1.Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet 2016;135:359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N. What can exome sequencing do for you? J Med Genet 2011;48:580–9. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, Wei C, Indugula SR, Zou F, Meng H, et al. Clinical Impact and Cost-Effectiveness of Whole Exome Sequencing as a Diagnostic Tool: A Pediatric Center’s Experience. Front Pediatr 2015;3:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ACMG Board of Directors. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med 2015;17:68–9. [DOI] [PubMed] [Google Scholar]
- 7.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249–255. [DOI] [PubMed] [Google Scholar]
- 9.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khoury MJ, Coates RJ, Evans JP. Evidence-based classification of recommendations on use of genomic tests in clinical practice: dealing with insufficient evidence. Genet Med 2010;12:680–3. [DOI] [PubMed] [Google Scholar]
- 11.Katz AE, Nussbaum RL, Solomon BD, Rehm HL, Williams MS, Biesecker LG. Management of Secondary Genomic Findings. Am J Hum Genet 2020;107:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vears DF, Senecal K, Clarke AJ, Jackson L, Laberge AM, Lovrecic L, Piton A, Van Gassen KLI, Yntema HG, Knoppers BM, et al. Points to consider for laboratories reporting results from diagnostic genomic sequencing. Eur J Hum Genet 2018;26:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weck KE. Interpretation of genomic sequencing: variants should be considered uncertain until proven guilty. Genet Med 2018;20:291–293. [DOI] [PubMed] [Google Scholar]
- 14.Kaltman JR, Evans F, Fu YP. Re-evaluating pathogenicity of variants associated with the long QT syndrome. J Cardiovasc Electrophysiol 2018;29:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giudicessi JR, Rohatgi RK, Tester DJ, Ackerman MJ. Variant Frequency and Clinical Phenotype Call Into Question the Nature of Minor, Nonsyndromic Long-QT Syndrome-Susceptibility Gene-Disease Associations. Circulation 2020;141:495–497. [DOI] [PubMed] [Google Scholar]
- 17.Sundaram L, Gao H, Padigepati SR, McRae JF, Li Y, Kosmicki JA, Fritzilas N, Hakenberg J, Dutta A, Shon J, et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 2018;50:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diebold I, Schon U, Scharf F, Benet-Pages A, Laner A, Holinski-Feder E, Abicht A. Critical assessment of secondary findings in genes linked to primary arrhythmia syndromes. Hum Mutat 2020;41:1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Safarova MS, Klee EW, Baudhuin LM, Winkler EM, Kluge ML, Bielinski SJ, Olson JE, Kullo IJ. Variability in assigning pathogenicity to incidental findings: insights from LDLR sequence linked to the electronic health record in 1013 individuals. Eur J Hum Genet 2017;25:410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, Berg JS, Biswas S, Bowling KM, Conlin LK, et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet 2016;99:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Driest SL, Wells QS, Stallings S, Bush WS, Gordon A, Nickerson DA, Kim JH, Crosslin DR, Jarvik GP, Carrell DS, et al. Association of Arrhythmia-Related Genetic Variants With Phenotypes Documented in Electronic Medical Records. JAMA 2016;315:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Council NR. Direct-To-Consumer Genetic Testing: Summary of a Workshop Washington (DC): The National Academies Press; 2010. [PubMed] [Google Scholar]
- 23.Spicer CC. Test reduction: II--Bayes’s theorem and the evaluation of tests. Br Med J 1980;281:592–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landstrom AP, Fernandez E, Rosenfeld JA, Yang Y, Dailey-Schwartz AL, Miyake CY, Allen HD, Penny DJ, Kim JJ. Amino acid-level signal-to-noise analysis of incidentally identified variants in genes associated with long QT syndrome during pediatric whole exome sequencing reflects background genetic noise. Heart Rhythm 2018;15:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc 2005;80:739–44. [DOI] [PubMed] [Google Scholar]
- 26.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003;107:2227–32. [DOI] [PubMed] [Google Scholar]
- 27.Kapplinger JD, Landstrom AP, Bos JM, Salisbury BA, Callis TE, Ackerman MJ. Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J Cardiovasc Transl Res 2014;7:347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bick AG, Flannick J, Ito K, Cheng S, Vasan RS, Parfenov MG, Herman DS, DePalma SR, Gupta N, Gabriel SB, et al. Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am J Hum Genet 2012;91:513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen PS, Havndrup O, Hougs L, Sorensen KM, Jensen M, Larsen LA, Hedley P, Thomsen AR, Moolman-Smook J, Christiansen M, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat 2009;30:363–70. [DOI] [PubMed] [Google Scholar]
- 30.Haggerty CM, James CA, Calkins H, Tichnell C, Leader JB, Hartzel DN, Nevius CD, Pendergrass SA, Person TN, Schwartz M, et al. Electronic health record phenotype in subjects with genetic variants associated with arrhythmogenic right ventricular cardiomyopathy: a study of 30,716 subjects with exome sequencing. Genet Med 2017;19:1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carruth ED, Young W, Beer D, James CA, Calkins H, Jing L, Raghunath S, Hartzel DN, Leader JB, Kirchner HL, et al. Prevalence and Electronic Health Record-Based Phenotype of Loss-of-Function Genetic Variants in Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Genes. Circ Genom Precis Med 2019;12:e002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care M, et al. Genetic testing for dilated cardiomyopathy in clinical practice. J Card Fail 2012;18:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 2014;16:601–8. [DOI] [PubMed] [Google Scholar]
- 34.Mak TSH, Lee YK, Tang CS, Hai JSH, Ran X, Sham PC, Tse HF. Coverage and diagnostic yield of Whole Exome Sequencing for the Evaluation of Cases with Dilated and Hypertrophic Cardiomyopathy. Sci Rep 2018;8:10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nouhravesh N, Ahlberg G, Ghouse J, Andreasen C, Svendsen JH, Haunso S, Bundgaard H, Weeke PE, Olesen MS. Analyses of more than 60,000 exomes questions the role of numerous genes previously associated with dilated cardiomyopathy. Mol Genet Genomic Med 2016;4:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–47. [DOI] [PubMed] [Google Scholar]
- 37.Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol 2009;2:6–15. [DOI] [PubMed] [Google Scholar]
- 38.Jabbari J, Jabbari R, Nielsen MW, Holst AG, Nielsen JB, Haunso S, Tfelt-Hansen J, Svendsen JH, Olesen MS. New exome data question the pathogenicity of genetic variants previously associated with catecholaminergic polymorphic ventricular tachycardia. Circ Cardiovasc Genet 2013;6:481–9. [DOI] [PubMed] [Google Scholar]
- 39.Landstrom AP, Dailey-Schwartz AL, Rosenfeld JA, Yang Y, McLean MJ, Miyake CY, Valdes SO, Fan Y, Allen HD, Penny DJ, et al. Interpreting Incidentally Identified Variants in Genes Associated With Catecholaminergic Polymorphic Ventricular Tachycardia in a Large Cohort of Clinical Whole-Exome Genetic Test Referrals. Circ Arrhythm Electrophysiol 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 2009;120:1752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol 2006;47:764–8. [DOI] [PubMed] [Google Scholar]
- 42.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res 2013;161:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol 2019;74:1480–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landstrom AP, Tester DJ, Ackerman MJ. Role of Genetic Testing for Sudden Death Predisposing Heart Conditions in Athletes. In: C L, ed. Sports Cardiology Essentials New York, NY: Springer; 2011. [Google Scholar]
- 46.Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 2020;13:e000067. [DOI] [PubMed] [Google Scholar]
- 47.Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957;54:59–68. [DOI] [PubMed] [Google Scholar]
- 48.Sarquella-Brugada G, Campuzano O, Iglesias A, Sanchez-Malagon J, Guerra-Balic M, Brugada J, Brugada R. Genetics of sudden cardiac death in children and young athletes. Cardiol Young 2013;23:159–73. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, et al. Prevalence of the congenital long-QT syndrome. Circulation 2009;120:1761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17–23. [DOI] [PubMed] [Google Scholar]
- 51.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995;80:795–803. [DOI] [PubMed] [Google Scholar]
- 52.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805–11. [DOI] [PubMed] [Google Scholar]
- 53.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005;294:2975–80. [DOI] [PubMed] [Google Scholar]
- 54.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005;2:507–17. [DOI] [PubMed] [Google Scholar]
- 55.Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, Feilotter H, Amenta S, Mazza D, Bikker H, et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020;141:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995;91:1512–9. [DOI] [PubMed] [Google Scholar]
- 57.Baltogiannis GG, Lysitsas DN, di Giovanni G, Ciconte G, Sieira J, Conte G, Kolettis TM, Chierchia GB, de Asmundis C, Brugada P. CPVT: Arrhythmogenesis, Therapeutic Management, and Future Perspectives. A Brief Review of the Literature. Front Cardiovasc Med 2019;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002;106:69–74. [DOI] [PubMed] [Google Scholar]
- 59.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992;20:1391–6. [DOI] [PubMed] [Google Scholar]
- 60.Brugada R, Campuzano O, Sarquella-Brugada G, Brugada J, Brugada P. Brugada syndrome. Methodist Debakey Cardiovasc J 2014;10:25–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293–6. [DOI] [PubMed] [Google Scholar]
- 62.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, Bloise R, Giustetto C, De Nardis R, Grillo M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 2002;105:1342–7. [DOI] [PubMed] [Google Scholar]
- 63.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007;116:2260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 2008;118:2260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, Jamal SM, Szybowska M, Morel CF, Bowdin S, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 2018;138:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002;287:1308–20. [DOI] [PubMed] [Google Scholar]
- 67.Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis-Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 1989;321:1372–8. [DOI] [PubMed] [Google Scholar]
- 68.Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012;60:705–15. [DOI] [PubMed] [Google Scholar]
- 69.Morner S, Richard P, Kazzam E, Hellman U, Hainque B, Schwartz K, Waldenstrom A. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol 2003;35:841–9. [DOI] [PubMed] [Google Scholar]
- 70.Colombo MG, Botto N, Vittorini S, Paradossi U, Andreassi MG. Clinical utility of genetic tests for inherited hypertrophic and dilated cardiomyopathies. Cardiovasc Ultrasound 2008;6:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol 2009;54:201–11. [DOI] [PubMed] [Google Scholar]
- 72.Norman MW, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: perspectives on disease. Z Kardiol 1999;88:550–4. [DOI] [PubMed] [Google Scholar]
- 73.Peters S, Trummel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol 2004;97:499–501. [DOI] [PubMed] [Google Scholar]
- 74.Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation 2006;113:1634–7. [DOI] [PubMed] [Google Scholar]
- 75.Haggerty CM, Murray B, Tichnell C, Judge DP, Tandri H, Schwartz M, Sturm AC, Matsumura ME, Murray MF, Calkins H, et al. Managing Secondary Genomic Findings Associated With Arrhythmogenic Right Ventricular Cardiomyopathy: Case Studies and Proposal for Clinical Surveillance. Circ Genom Precis Med 2018;11:e002237. [DOI] [PubMed] [Google Scholar]
- 76.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119–24. [DOI] [PubMed] [Google Scholar]
- 77.Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, Cox MG, Bhuiyan Z, Bikker H, Wiesfeld AC, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol 2011;57:2317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakajima T, Kaneko Y, Irie T, Takahashi R, Kato T, Iijima T, Iso T, Kurabayashi M. Compound and digenic heterozygosity in desmosome genes as a cause of arrhythmogenic right ventricular cardiomyopathy in Japanese patients. Circ J 2012;76:737–43. [DOI] [PubMed] [Google Scholar]
- 79.Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, Migliore F, Marra MP, Lorenzon A, De Bortoli M, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet 2013;6:533–42. [DOI] [PubMed] [Google Scholar]
- 80.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2010;55:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Headrick AT, Rosenfeld JA, Yang Y, Tunuguntla H, Allen HD, Penny DJ, Kim JJ, Landstrom AP. Incidentally identified genetic variants in arrhythmogenic right ventricular cardiomyopathy-associated genes among children undergoing exome sequencing reflect healthy population variation. Mol Genet Genomic Med 2019;7:e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989;80:564–72. [DOI] [PubMed] [Google Scholar]
- 83.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992;326:77–82. [DOI] [PubMed] [Google Scholar]
- 84.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2005;45:969–81. [DOI] [PubMed] [Google Scholar]
- 85.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006;296:1867–76. [DOI] [PubMed] [Google Scholar]
- 86.Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, Ramani F, Di Lenarda A, Mestroni L, Sinagra G. Natural History of Dilated Cardiomyopathy in Children. J Am Heart Assoc 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Long PA, Evans JM, Olson TM. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. J Cardiovasc Dev Dis 2017;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, Marvao Ad, Dawes TJW, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020;141:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2018;20:899–909. [DOI] [PubMed] [Google Scholar]
- 91.Connell PS, Berkman AM, Souder BM, Pirozzi EJ, Lovin JJ, Rosenfeld JA, Liu P, Tunuguntla H, Allen HD, Denfield SW, et al. Amino Acid-Level Signal-to-Noise Analysis Aids in Pathogenicity Prediction of Incidentally Identified TTN-Encoded Titin Truncating Variants. Circ Genom Precis Med 2021;14:e003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Group eCAW. Frequency of genomic secondary findings among 21,915 eMERGE network participants. Genet Med 2020;22:1470–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lawrence L, Sincan M, Markello T, Adams DR, Gill F, Godfrey R, Golas G, Groden C, Landis D, Nehrebecky M, et al. The implications of familial incidental findings from exome sequencing: the NIH Undiagnosed Diseases Program experience. Genet Med 2014;16:741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jang MA, Lee SH, Kim N, Ki CS. Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet Med 2015;17:1007–11. [DOI] [PubMed] [Google Scholar]
- 95.Yamaguchi-Kabata Y, Yasuda J, Tanabe O, Suzuki Y, Kawame H, Fuse N, Nagasaki M, Kawai Y, Kojima K, Katsuoka F, et al. Evaluation of reported pathogenic variants and their frequencies in a Japanese population based on a whole-genome reference panel of 2049 individuals. J Hum Genet 2018;63:213–230. [DOI] [PubMed] [Google Scholar]
- 96.Jain A, Gandhi S, Koshy R, Scaria V. Incidental and clinically actionable genetic variants in 1005 whole exomes and genomes from Qatar. Mol Genet Genomics 2018;293:919–929. [DOI] [PubMed] [Google Scholar]
- 97.Tadros HJ, Life CS, Garcia G, Pirozzi E, Jones EG, Datta S, Parvatiyar MS, Chase PB, Allen HD, Kim JJ, et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J Mol Cell Cardiol 2020;142:118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Medicine 2019;11:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ahmad F, McNally EM, Ackerman MJ, Baty LC, Day SM, Kullo IJ, Madueme PC, Maron MS, Martinez MW, Salberg L, et al. Establishment of Specialized Clinical Cardiovascular Genetics Programs: Recognizing the Need and Meeting Standards: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 2019;12:e000054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.