

Spontaneous coronary artery dissection (SCAD) is a cause of acute coronary syndrome, myocardial infarction, and sudden death in predominantly young to middle-aged women with few atherosclerotic risk factors. Etiology of the pathognomonic medial hematoma or intimal tear of the vessel wall is likely multifactorial. A genetic basis for disease susceptibility has been discovered by whole-genome sequencing and genome-wide association studies (GWAS), revealing rare segregating variants of major effect in familial SCAD and common risk-conferring variants in sporadic SCAD, respectively.1,2 Pregnancy-associated SCAD (P-SCAD), occurring intrapartum or postpartum, is associated with more severe clinical presentation and outcomes.3 Its marked predilection for women and recognition of pregnancy as a risk factor implicate hormonal milieu in SCAD pathogenesis, yet genetic factors contributing to P-SCAD have not been reported.
We performed a GWAS comprised of the P-SCAD patients from our original SCAD GWAS, using the same methodology as previously described.2 Data in the current study are available from the corresponding author on reasonable request. Patients were enrolled in the Mayo Clinic SCAD Registry and provided informed written consent for genetic research under an Institutional Review Board-approved protocol. SCAD was diagnosed by coronary angiography and controls were individuals without known arteriopathies. P-SCAD was defined as SCAD during pregnancy, following miscarriage, or 0–52 weeks postpartum. Consecutively recruited discovery and replication cohorts were white females of European decent, comprised of 53 P-SCAD patients (36.4±4.2 years) and 1477 controls (64.0±14.5 years), and 32 P-SCAD patients (34.9±3.7 years) and 334 controls (51.0±15.3 years), respectively. Fibromuscular dysplasia was more common in the replication cohort (65% versus 33%; P = 0.02), but there were no significant differences in age, parity, nursing status, preeclampsia, or timing of SCAD in relation to pregnancy. Among the 85 women within both cohorts, four (4.7%) had SCAD during pregnancy (one in first-trimester, three in third-trimester), 56 (65.9%) within 12 weeks of delivery, and 25 (29.4%) within 12–52 weeks of delivery.
Approximately 5 million single nucleotide variants (SNVs) spanning all chromosomes were genotyped or imputed, followed by systematic quality control analysis and data harmonization. To detect chromosomal loci associated with SCAD susceptibility, all SNVs were assessed for genome-wide associations using logistic regression models assuming additive allele effects.2 Individuals with <90% Caucasian ancestry were excluded (STRUCTURE v2.2). The genomic inflation factor (λ = 1.009) implied a low possibility of false positive associations due to population stratification. A significant association with P-SCAD was identified at chromosome 4p16.1 (rs28533862-A: OR, 4.63; 95% CI, 2.73 – 7.89; P = 1.61 × 10−08; cases: 22.7%, controls: 7.4%; Figure 1A) after adjusting for the top 5 principal components and remained significant after false discovery rate testing (q value = 0.01). The Firth regression model (PLINK v2.0), used to account for case-control imbalance, yielded similar association findings for rs28533862. The five SCAD risk SNVs identified in our previous GWAS,2 not segregated by SCAD subtypes, did not reach genome-wide significance in the current P-SCAD analysis (rs4970935, P = 0.08; rs9349379, P = 0.001; rs11172113, P = 0.04; rs2015637, P = 0.02; rs28451064, P = 0.03). Conversely, the identified P-SCAD risk SNV did not reach genome-wide significance in the previous all-SCAD GWAS (OR, 1.67;95% CI, 1.29 2.16; P = 0.0001132). Targeted genotyping of rs28533862 in the replication cohort revealed the same directionality of A-allele risk for P-SCAD (OR, 2.14; 95% CI, 0.98 – 4.66; P = 0.05; cases: 14.1%, controls: 7.2%) and meta-analysis of discovery and replication results strengthened statistical significance of the association (OR, 3.62; 95% CI, 2.33 – 5.62; P = 9.07 × 10−09). Overall, the rs28533862-A risk allele was present in 19.4% of P-SCAD patients and 7.4% of controls. There were no significant differences in age, fibromuscular dysplasia, parity, nursing, and pre-eclampsia between risk allele carriers (n=31) and non-carriers (n=54), with the exception of the proportion who had SCAD during pregnancy or ≤12 weeks postpartum (84% versus 63%; P = 0.03).
Figure 1: Pregnancy-associated spontaneous coronary artery dissection genetic associations.
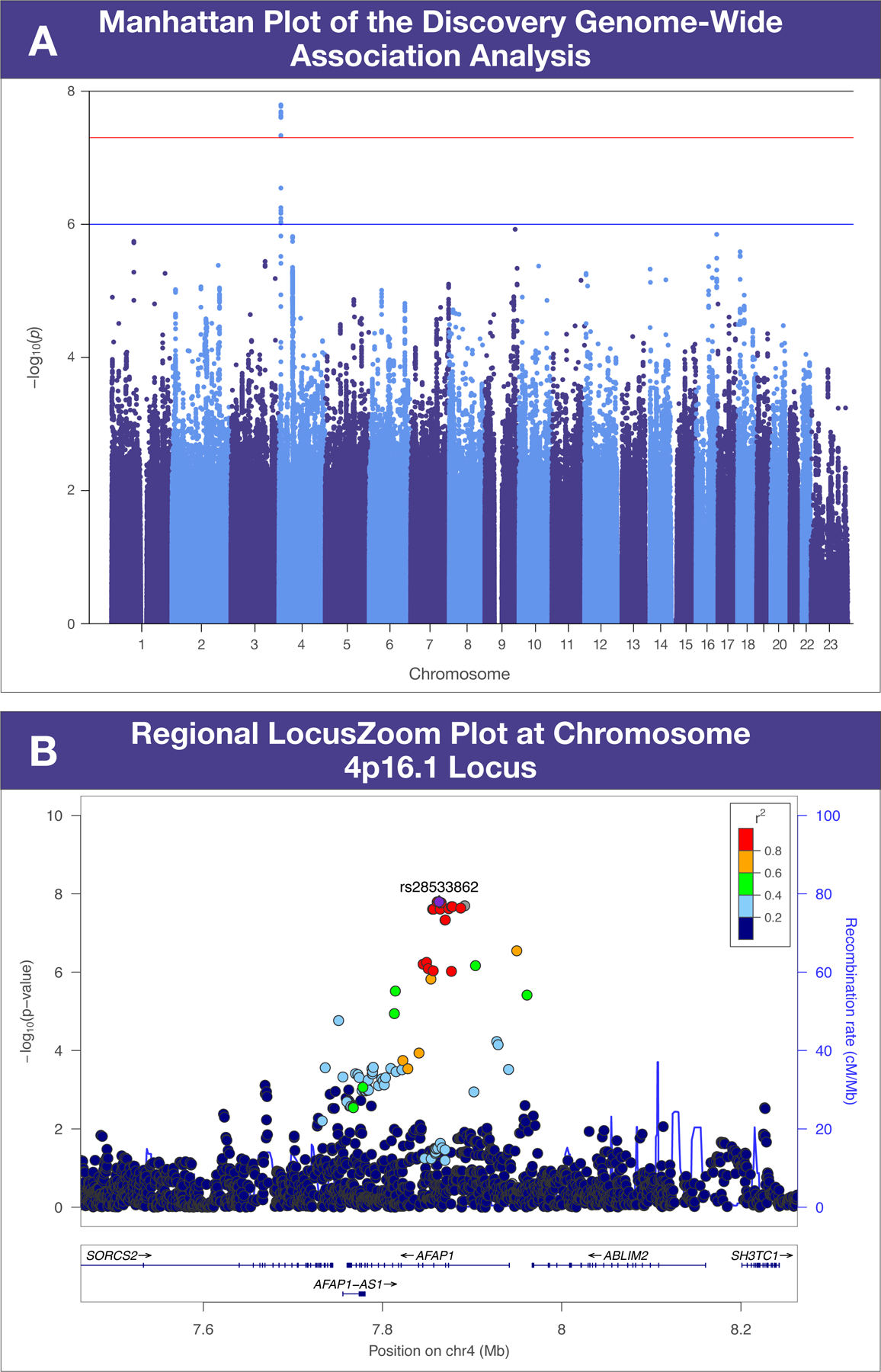
Chromosomal position and P-value −log10 scale are designated by x and y axes, respectively. (A). The horizontal red line indicates genome-wide significance (P < 5 × 10−08). (B). Eight hundred kilobase region surrounding rs28533862. Linkage disequilibrium estimates (r2) are color-coded.
The P-SCAD susceptibility locus on chromosome 4p16.1 encompasses five genes within an 800-kilobase region surrounding rs28533862 (Figure 1B). The index SNV is located within the third intron of AFAP1, encoding actin filament associated protein 1, and overlaps a high-confidence enhancer region (GH04J007852: University of California Santa Cruz Genome Browser). AFAP1 was the most plausible positional candidate for P-SCAD based on several lines of evidence including highest ranking for predicted gene-enhancer associations (GeneHancer) and high ranking of messenger RNA expression among arterial tissues (Genotype-Tissue Expression Portal). Loss of AFAP1 in cancer cells impairs focal adhesion formation,4 a cellular mechanism for actin cytoskeleton perturbation hypothesized in SCAD pathogenesis based on previous genetic investigations.1,2 Moreover, an AFAP1 duplication was identified in a patient with thoracic aortopathy.5 Relevant to hormonal milieu in P-SCAD, tyrosine phosphorylation of Afap1 by prolactin signaling is required for normal function of the lactating mammary gland in postpartum mice.4 P-SCAD occurs most frequently in the early postpartum period, a time when plasma prolactin levels are elevated and remain so in lactating women. Notably, 15 risk allele carriers (48.4%) described an onset of SCAD symptoms during or shortly after lactation. Collectively, these data identify a genetic risk allele for P-SCAD and implicate AFAP1 as a positional candidate gene. Our findings require independent replication and functional validation.
Acknowledgements
We thank Dr. Jeanne Theis and Rhianna Sundsbak for helpful discussions and critical review of the manuscript, Jill Boyum and Brenda Speltz for patient recruitment, and the patients who volunteered for this study. We acknowledge the invaluable genomic resources provided by the Mayo Clinic Genome Consortia and Biobank, Center for Individualized Medicine.
Sources of Funding
This work was supported by funding from SCAD Research Inc, NIH T32 GM72474, and resources from the Genome Consortia and Biobank, Mayo Clinic Center for Individualized Medicine.
Nonstandard abbreviations:
- SCAD
spontaneous coronary artery dissection
- GWAS
genome-wide association study
- P-SCAD
pregnancy-associated SCAD
- SNV
single nucleotide variant
Footnotes
Disclosures
None
References
- 1.Turley TN, Theis JL, Sundsbak RS, Evans JM, O’Byrne MM, Gulati R, Tweet MS, Hayes SN, Olson TM. Rare Missense Variants in TLN1 Are Associated With Familial and Sporadic Spontaneous Coronary Artery Dissection. Circ Genomic Precis Med. 2019;12:173–182. doi: 10.1161/CIRCGEN.118.002437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turley TN, O’Byrne MM, Kosel ML, de Andrade M, Gulati R, Hayes SN, Tweet MS, Olson TM. Identification of Susceptibility Loci for Spontaneous Coronary Artery Dissection. JAMA Cardiol. 2020;5:929–938. doi: 10.1001/jamacardio.2020.0872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tweet MS, Hayes SN, Codsi E, Gulati R, Rose CH, Best PJM. Spontaneous Coronary Artery Dissection Associated With Pregnancy. J Am Coll Cardiol. 2017;70:426–435. doi: 10.1016/j.jacc.2017.05.055 [DOI] [PubMed] [Google Scholar]
- 4.Cunnick JM, Kim S, Hadsell J, Collins S, Cerra C, Reiser P, Flynn DC, Cho Y. Actin filament-associated protein 1 is required for cSrc activity and secretory activation in the lactating mammary gland. Oncogene. 2015;34:2640–2649. doi: 10.1038/onc.2014.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prakash SK, LeMaire SA, Guo DC, Russell L, Regalado ES, Golabbakhsh H, Johnson RJ, Safi HJ, Estrera AL, Coselli JS, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010;87:743–756. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]