

Abstract
Familial British dementia (FBD) and familial Danish dementia (FDD) are two autosomal dominant neurodegenerative diseases caused by mutations in the BRI2 gene. FBD and FDD are characterized by widespread cerebral amyloid angiopathy (CAA), parenchymal amyloid deposition, and neurofibrillary tangles. Transgenic mice expressing wild-type and mutant forms of the BRI2 protein, Bri2 knock-in mutant mice, and Bri2 gene knock-out mice have been developed. Transgenic mice expressing a human FDD-mutated form of the BRI2 gene have partially reproduced the neuropathological lesions observed in FDD. These mice develop extensive CAA, parenchymal amyloid deposition, and neuroinflammation in the central nervous system. These animal models allow the study of the molecular mechanism(s) underlying the neuronal dysfunction in these diseases and allow the development of potential therapeutic approaches for these and related neurodegenerative conditions. In this review, a comprehensive account of the advances in the development of animal models for FBD and FDD and of their relevance to the study of Alzheimer disease is presented.
Keywords: FBD, FDD, Mouse models, Neurodegeneration, Amyloid, CAA, Neuroinflammation
Introduction
Two early onset autosomal dominant diseases first reported by Worster-Drought et al. (1933) and Strömgren et al. (1970) are known as familial British dementia (FBD) and familial Danish dementia (FDD), respectively (Vidal et al. 1999, 2000b). These diseases are characterized clinically by gradually progressive dementia and ataxia and neuropathologically by amyloid angiopathy and neurofibrillary tangles (NFTs) in the hippocampus. These basic neuropathological features are also shared by Alzheimer disease (AD) and some forms of prion disease (Giaccone et al. 2008). Although different amyloid peptides are deposited in these conditions, the tau deposits are antigenically, ultrastructurally, and biochemically indistinguishable (Ghetti et al. 1989; Giaccone et al. 1990; Holton et al. 2001, 2002; Giaccone et al. 2008). This finding of similar pathological lesions seems to support a unifying pathologic mechanism in which accumulation of amyloidogenic peptides could trigger a complex pathological cascade leading to neurodegeneration (Golde 2003). However, it remains to be determined whether abnormal tau phosphorylation is the consequence of cerebral amyloid deposition or a different disease characteristic, given that hyper-phosphorylated tau can lead to dementia in the absence of amyloid plaques (Goedert et al. 2000). Since the discovery that mutations in a novel gene, BRI2, are the genetic abnormalities associated with the development of FBD and FDD (Vidal et al. 1999, 2000b), several attempts have been made to generate animal models for these neurodegenerative diseases. Mutations in the amyloid β precursor protein (AβPP), the presenilin-1 (PSEN-1), and presenilin-2 (PSEN-2) genes that are associated with familial forms of AD have provided a potential means for the generation of rodent models of AD. These models have played an important role in the investigation of the mechanism(s) involved in amyloid formation, neurofibrillar degeneration, and cell death in the brain in AD (Duyckaerts et al. 2008; Kokjohn and Roher 2009). The study of new animal models of amyloid deposition such as those for FBD and FDD may provide important clues for the identification of the pathologic mechanism(s) by which amyloidogenic peptides, regardless of their primary amino acid sequence, may lead to neurodegeneration. Furthermore, these in vivo model systems may be crucial in the development and testing of novel therapeutic approaches (Vidal et al. 2000a). This review will focus on our current knowledge of the development of animal models for FBD and FDD and their relevance to the study of AD.
Familial British and Danish dementia
In 1933, Worster-Drought first described a British kindred with presenile dementia and spastic paralysis (Worster-Drought et al. 1933, 1944), and subsequently an identical condition was reported in two siblings (Griffiths et al. 1982). Both families were later shown to be part of the same pedigree. The disease was called familial cerebral amyloid angiopathy with nonneuritic plaque formation (Plant et al. 1990) or FBD (Vidal et al. 1999, 2000b). Clinical symptoms in patients with FBD appear during the fifth decade of life and include progressive dementia, spastic tetraparesis, and cerebellar ataxia. The majority of affected patients succumb in the sixth decade of life (Plant et al. 1990). Neuropathological studies of patients with FBD revealed the presence of hippocampal extracellular perivascular amyloid plaques and intraneuronal formation of NFTs within the limbic regions (Plant et al. 1990; Revesz et al. 1999). Amyloid plaques were primarily localized to the hippocampus and cerebellum and were rarely present in the neocortex (Plant et al. 1990; Revesz et al. 1999; Holton et al. 2001, 2002). The NFTs found in FBD, like the amyloid plaques, were numerous in the hippocampus. Biochemical analysis of NFTs isolated from a single FBD case revealed that the insoluble tau had a Western blot pattern that was indistinguishable to that seen in AD (Holton et al. 2001, 2002) implying the same or similar composition of tau species in both diseases. In addition, extensive cerebral amyloid angiopathy (CAA) was observed in the leptomeningeal vessels and in gray and white matter vessels within the cerebrum, cerebellum, brainstem, and spinal cord of patients with FBD (Plant et al. 1990; Revesz et al. 1999; Holton et al. 2001, 2002). Although FBD has been considered a familial form of CAA, cerebral hemorrhage rarely occurs in these patients (Plant et al. 1990; Mead et al. 2000).
Strömgren and collaborators described in 1970 a Danish kindred from the Djursland peninsula of Denmark with predominant clinical features of vision impairment, hearing loss, and progressive dementia. The disease was named heredopathia ophthalmo-oto-encephalica (Strömgren et al. 1970) or FDD (Vidal et al. 2000b). Clinically, FDD is characterized by the development and progression of cataracts during the third decade of life (Strömgren et al. 1970). Hearing impairment was reported to appear 10–20 years following the development of ocular problems. Severe to total perceptive hearing loss developed by the fifth decade of life, with vestibular reflex disturbances, slurred speech, and a swaying gait (Strömgren et al. 1970; Strömgren 1981). Patients with FDD developed cerebellar ataxia with intention tremor in their fourth decade of life, however, unlike FBD cases, did not develop spastic para-paresis. Progressive dementia probably begins in the sixth decade and may be associated with paranoid reactions and temporal disturbances of consciousness. The majority of patients succumb within the fifth or sixth decade of life. Neuropathological findings of FDD closely resemble those of FBD; however, parenchymal deposits found in the hippocampus of patients with FDD were Congo red and Thioflavine S (ThS) negative (Vidal et al. 2000b; Holton et al. 2002). Another noteworthy neuropathological difference between FBD and FDD was the finding of amyloid β (Aβ) deposition and co-deposition with the Danish amyloid in all patients with FDD (Vidal et al. 2000b). Aβ deposition, mostly Aβ42, was observed either in combination with ADan or alone in blood vessel walls and in brain parenchyma (Holton et al. 2002; Tomidokoro et al. 2005). The insoluble tau found in the NFTs in FDD was, as that observed in FBD, composed of paired helical filaments. Biochemical analyses revealed a Western blot banding profile similar to that found in FBD as well as AD (Holton et al. 2002).
BRI2 and generation of amyloid peptides
The characterization of amyloid fibrils isolated from FBD and FDD individuals (Vidal et al. 1999, 2000b) led to the cloning of the BRI2 gene (also known as ITM2b, Deleersnijder et al. 1996) and the identification of the genetic defects associated with these pathological conditions (Vidal et al. 1999, 2000b). The BRI2 gene encodes a protein of 266 amino acids (Fig. 1a) that belongs to a family of integral type II transmembrane domain proteins (Deleersnijder et al. 1996; Vidal et al. 1999, 2000b, 2001). Cleavage between its ectodomain (residue Arg243 and Glu244, KGIQKR↓EAS) by pro-protein convertases (PCs) (Kim et al. 1999, 2002; Choi et al. 2004) releases a 23 amino acid propeptide (Bri2-23) from the wild-type precursor (Fig. 1b). In addition, a large part of the remaining ectodomain, the BRICHOS domain (Sanchez-Pulido et al. 2002) is also shed (Choi et al. 2004) by ADAM10 (Martin et al. 2008) and released into the extracellular space (Fig. 1a). The remaining membrane associated N-terminal fragment (NTF) undergoes intramembrane proteolysis mediated by SPPL2a or SPPL2b. This cleavage generates an intracellular domain (ICD), which is liberated into the cytosol and a secreted C domain (Fig. 1a) (Martin et al. 2008, 2009). Patients with FBD had a single base A to T transversion (799A > T) at position 1 of the stop codon TGA that segregated with the disease in affected members of the family. The abolition of the endogenous stop codon and the creation of the AGA codon for arginine resulted in the read-through to the next in-frame stop codon 30 bases downstream generating a longer-than-normal protein of 277 amino acids (p.X267ArgextX11) instead of the normal 266 amino acid protein. The 34 amino acid carboxy-terminal (C-terminal) peptide of the mutant protein was isolated from amyloid deposits from FBD patients and was referred to as British amyloid (ABri) (Vidal et al. 1999, 2000b). Analysis of the BRI2 gene in individuals with FDD showed the presence of a 10 nucleotide duplication (787_796dupTTTAATTTGT) one codon upstream of the stop codon. This insertion caused a frameshift and read-through of the endogenous stop codon to the next in-frame stop codon, generating a protein with an additional 11 amino acids in the C-terminus with a length of 277 amino acids (p.Ser266PhefsX11). The 34 amino acid C-terminal peptide of the mutant protein was isolated from amyloid deposits from FDD patients and was referred to as Danish amyloid (ADan) (Vidal et al. 2000b). Cleavage between residues Arg243 and Glu244 by PCs (Kim et al. 1999, 2002; Choi et al. 2004) releases the 34 amino acid ABri and ADan peptides from the 277 amino acid mutant forms of BRI2 associated with FBD and FDD (Fig. 1b).
Fig. 1.
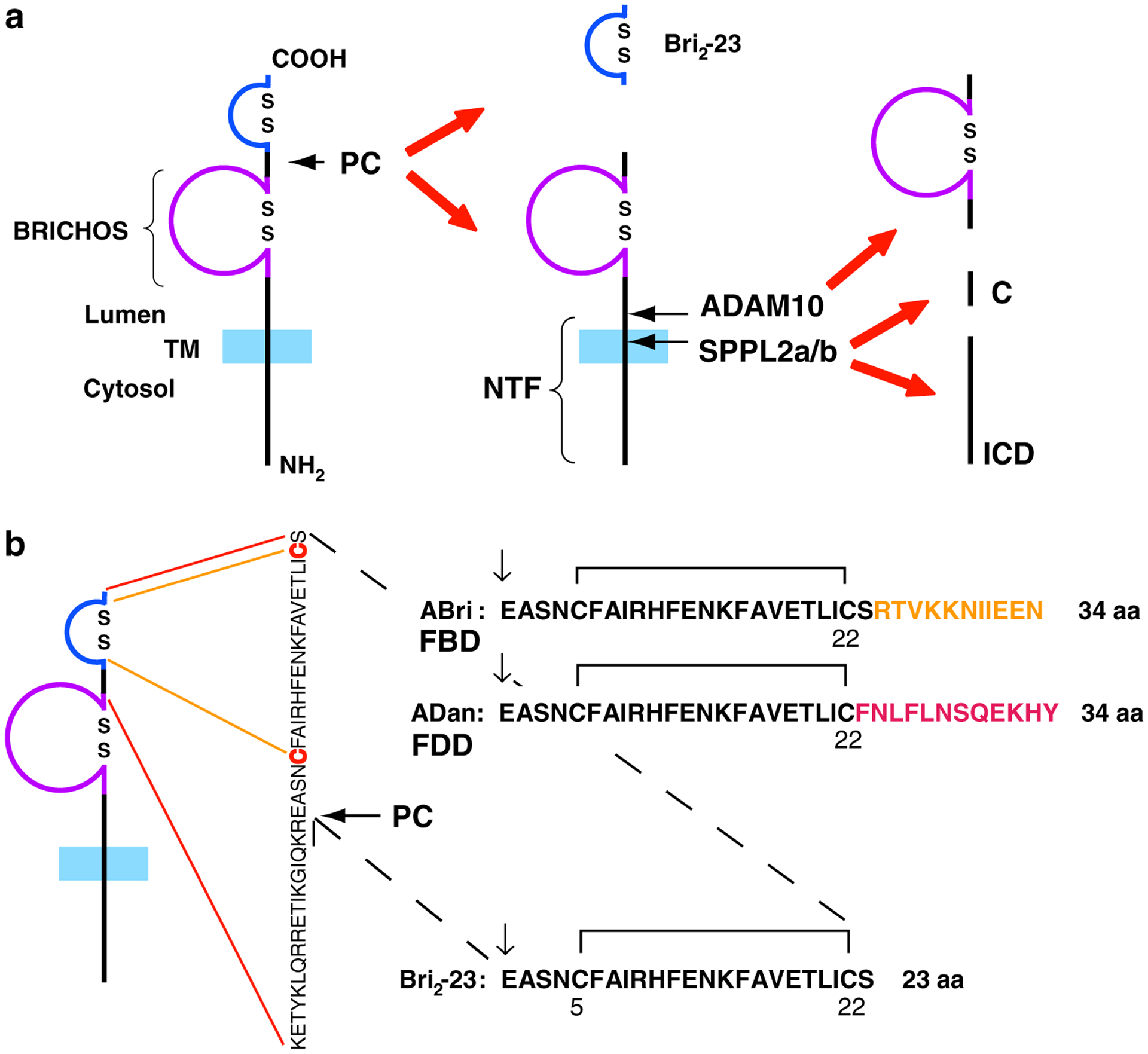
Schematic diagram of wild-type and mutated forms of the type-II transmembrane protein BRI2 as originally proposed by Vidal et al. (2004). a Processing by pro-protein convertases (PCs) releases a 23 amino acid peptide (Bri2-23). Processing by ADAM10 in the ectodomain of BRI2 releases the BRICHOS domain and an N-terminal fragment (NTF). Further processing of the membrane bound fragment by SPPL2a/SPPL2b releases an intracellular domain (ICD) and a secreted low molecular weight C-terminal peptide (C). Disulfide bonded loops in the BRICHOS domain and in the carboxy-terminus of BRI2 are indicated. b Processing by PCs of the mutated precursor proteins releases the 34 amino acid ABri and ADan peptides
BRI2 has been found expressed in all human and mouse tissues tested (Pittois et al. 1998; Vidal et al. 1999, 2000b; Pickford et al. 2003), with high levels of expression in heart, placenta, kidney, pancreas, and liver. The BRI2 gene is ubiquitously expressed in the white and gray matter of the brain and the pattern of expression partially correlates with the distribution of amyloid deposits in FBD and FDD (Holton et al. 2001, 2002; Akiyama et al. 2004). The BRI2 gene was mapped to chromosome 13q14.3 in humans and to chromosome 14 in the mouse (Pittois et al. 1998; Vidal et al. 1999, 2000b). Although the function of BRI2 is unknown, experimental evidence supports a role for BRI2 in neuronal differentiation (Choi et al. 2004). Given its chromosomal localization in a hotspot for sporadic prostate cancer, BRI2 has been proposed as a tumor suppressor gene (Latil et al. 2003). A shorter isoform lacking the sequence encoded by exon 1 has been reported to be induced by IL-2 deprivation and to have a BH-3 domain, which is important for the dimerization mediating proapoptotic functions (Fleischer et al. 2002; Fleischer and Rebollo 2004).
Interestingly, during the search for regulators of AβPP cleavage, it was found that BRI2 binds AβPP and inhibits Aβ formation (Fotinopoulou et al. 2005; Matsuda et al. 2005), suggesting that BRI2 plays a significant role in AβPP processing. BRI2 has been shown to physically associate with AβPP, and that this interaction serves to suppress AβPP processing by secretases and the production of Aβ in vivo (Matsuda et al. 2008). Increasing BRI2 levels in the mouse brain by transgenic expression reduces AβPP cleavage by α- and β-secretase as well as amyloid plaque formation. Furthermore, reducing BRI2 expression in cell lines by RNAi and by gene targeting in mice increases AβPP processing. The finding that sAβPPα, sAβPPβ, Aβ40, and Aβ42 levels are increased in BRI2 knockdown cells and are augmented in murine heterozygous and homozygous knock-out mice ( and ) strongly supports the notion that BRI2 is an important physiological regulator of AβPP processing (Matsuda et al. 2008).
Transgenic models relevant to FBD and FDD
Expression of a cDNA encoding the human FBD mutant form of BRI2 and wild-type BRI2 transgenes in mice was performed (Pickford et al. 2006) using the pan-neuronal heterologous mouse prion promoter (moPrnp) (Borchelt et al. 1996) and mouse Thy-1.2 promoter (Aigner et al. 1995). Analysis of wild-type and mutant BRI2 moPrnp lines, initially generated on an outbred B6/D2/SW background, revealed high levels of transgene expression in the hippocampus, cerebellum, cortex, and specific nuclei in the thalamus, hypothalamus, and pons (Pickford et al. 2006). FBD-mutant BRI2 was detected in the mutant lines using an antibody specific to the ABri peptide (epitope located on the C-terminus of the BRI2 precursor protein carrying the FBD mutation). However, the ~4 kDa ABri peptide was not detected in hetero- or homozygous transgenic mice up to 24 months of age. Similar results were obtained upon analysis of Thy-1.2-FBD mutant transgenic mice on a B6D2 background, although low levels of the ABri peptide were reported after immunoprecipitation from plasma and whole brain extracts of 12-month-old mice, suggesting that processing of the precursor by PCs and ABri secretion can occur in this model, but at very low levels (Pickford et al. 2006). The two mutant transgenic mice (moPrnp and Thy-1.2) had specific accumulation of intracellular mutant BRI2 immunoreactivity, but no extracellular deposits were detected. To promote ABri production, elevated levels of the PC furin in the brains of moPrnp-FBD transgenic mice were induced using recombinant adeno-associated virus (AAV) vectors. No full-length BRI2 protein was detected in moPrnp-FBD transgenic mice, suggesting that mutant BRI2 was cleaved when human furin was locally over-expressed. However, no ABri peptides or extracellular deposits were detected (Pickford et al. 2006). To test whether BRI2 can modify AβPP processing and AD pathology in vivo, Matsuda et al. (2008) generated transgenic mice expressing human wild-type BRI2 under the control of the calcium–calmodulin-dependent kinase II (αCaMKII) promoter (Mayford et al. 1996), which drives the expression of the transgene in the postnatal forebrain. At 2 months of age, transgenic expression of BRI2 correlated with decreased amounts of sAβPPβ and total sAβPP fragments. Double-transgenic mice carrying a mutated human AβPP gene and transgenic BRI2 (TgCRND8/BRI2) had significantly reduced sAβPPα, sAβPPβ, Aβ40, and Aβ42 levels compared with littermate TgCRND8 controls (Matsuda et al. 2008).
Transgenic animals were also developed that express a human cDNA encoding the FBD and FDD mutant forms of BRI2 under the control of the Syrian hamster prion promoter (SHaPrnp) (Hsiao et al. 1996) in C57BL/6J background (Coomaraswamy et al. 2006, 2008). Initial neuropathological characterization of FDD transgenic mice showed an age-dependant deposition of ADan in the brain. In addition, these mice showed a noteworthy microglial and astrocytic response. Beginning at 2.5 months of age, ADan was found predominantly on the walls of cerebral vessels as CAA. Parenchymal plaques were also observed. At 2 months of age, these mice showed significantly decreased levels of endogenous mouse Aβ40 and Aβ42 peptides (Coomaraswamy et al. 2008).
A transgenic mouse model of FDD (Tg-FDD) was generated with a cDNA encoding the FDD mutant form of human BRI2 under the control of the moPrnp promoter (Vidal et al. 2009). Transgenic mice developed amyloid accumulation in the brain with similar regional deposition (Figs. 2, 3, 4). In 7-month-old Tg-FDD mice, the ADan peptide was detected in hetero- and homozygous transgenic mice. The ADan peptide was present in the formic acid soluble fraction mostly as a ~4 kDa monomer, but polymers, in particular trimers and tetramers, were also seen by western blot analysis using antibodies specific for ADan. In these mice, there is also evidence of the presence of ADan oligomers (Vidal et al. 2009), which may play an important role in the pathogenesis of FDD since in AD it has been suggested that oligomers and protofibrils rather than fibrillar structures are responsible for the pathogenesis of the disease (Walsh and Selkoe 2007). By 12 months of age, Tg-FDD mice started to show an abnormal grooming behavior. Animals also showed an arched back and walked with a wide-based gait and shorter steps. Feet clasping was observed upon suspension of the mice by their tails. In contrast, wild-type mice demonstrated normal limb posture when suspended by their tails. At neuropathological examination, no obvious changes were observed up to 6 months of age. At 7 months of age, Tg-FDD mice showed CAA primarily in pial (leptomeningeal) cerebellar vessels (Figs. 4, 5). As the animals aged, large and medium-sized parenchymal and penetrating vessels of the cerebrum (neocortex, hippocampus, thalamus, and olfactory bulb), the brain stem, and the spinal cord also showed CAA (Figs. 2, 5, 6). The walls of the vessels were thickened with different amounts of amyloid deposition, sometimes seen in the form of focal globular clumps (Figs. 5, 6). Vessel walls that were positive for ADan also showed a loss of smooth muscle cells (SMCs). In concurrence with the neuropathological finding of no hemorrhages in patients with FDD (Holton et al. 2002), the extensive and severe CAA observed in Tg-FDD mice did not lead to vascular hemorrhages in any of the mice studied up to the age of 21 months. Parenchymal amyloid deposition was seen in brain areas affected by CAA with the exception of the cerebellum, where it was relatively uncommon to observe the presence of ThS fluorescent material that was not associated with vessels. In the hippocampus, amyloid deposition was most prominent in the CA3 and CA2 regions and the hilus (Fig. 3). Amyloid deposits were seen as small plaques (7–10 μm in diameter) that sometimes showed a weak ThS fluorescence surrounding, and in the form of punctate structures, which were immunolabeled using anti-ADan antibodies. Immunohistochemical analysis using anti-ADan also showed the presence of abundant ADan diffuse deposits. Aged Tg-FDD mice had extensive vascular amyloid deposition, parenchymal ADan deposition, and amyloid-associated gliosis. In addition to astrocytes, activated microglia were observed in the close vicinity of vascular and parenchymal amyloid deposits (Vidal et al. 2009). Microglial cell processes were seen surrounding amyloid deposits but were not observed in association with diffuse deposits (Fig. 6). Amyloid deposits were also immunostained using antibodies against P-component and ApoE, but preliminary attempts to identify Aβ deposits were unsuccessful. The presence of CAA and the localized loss of SMCs seen in the Tg-FDD mouse model strongly suggest that neurons are the source of the cerebrovascular amyloid in FDD and denote the vasculotropic nature of the ADan peptide (Vidal et al. 2009). Immunohistochemistry using antibody AT8 (an antibody against the microtubule associated protein tau phosphorylated at Ser202/Thr205) revealed the presence of numerous tau-positive structures throughout the cerebral cortex and hippocampus. No NFTs were observed by ThS fluorescence. Tg-FDD mice were crossed with a P301S tau transgenic strain (Tg-tau) (Murrell et al. 2006). In these mice, the smallest tau four-repeat isoform (383 aa) with the P301S mutation is expressed under the control of the moPrnp promoter. This model exhibits tau-induced neurodegeneration in the hippocampus, piriform cortex, brain stem, and spinal cord where argentophilic and ThS fluorescent deposits are observed. These deposits are also immunoreactive with phosphorylation-dependent anti-tau monoclonal antibodies. Double homozygous transgenic (Tg-Tau × Tg-FDD) mice in the C57BL/6J background have been generated. Preliminary analysis of Tg-Tau × Tg-FDD showed the presence of ADan amyloid and phosphorylated tau, as well as a microglial and astrocytic response (Fig. 7). Although mutations in the gene encoding tau (MAPT) have been found in patients with Frontotemporal Dementia and Parkinsonism linked to chromosome 17 (FTDP-17), expression of FTDP-17 mutant tau together with the Danish mutant form of human BRI2 resulted in a more complete FDD-like pathology in mice, which could not be achieved by over-expression of the Danish mutant form of human BRI2 alone. This animal model may provide insights onto whether ADan can modify tau pathology, in the identification of toxic species and in the molecular pathways involved in neurodegeneration in FDD.
Fig. 2.
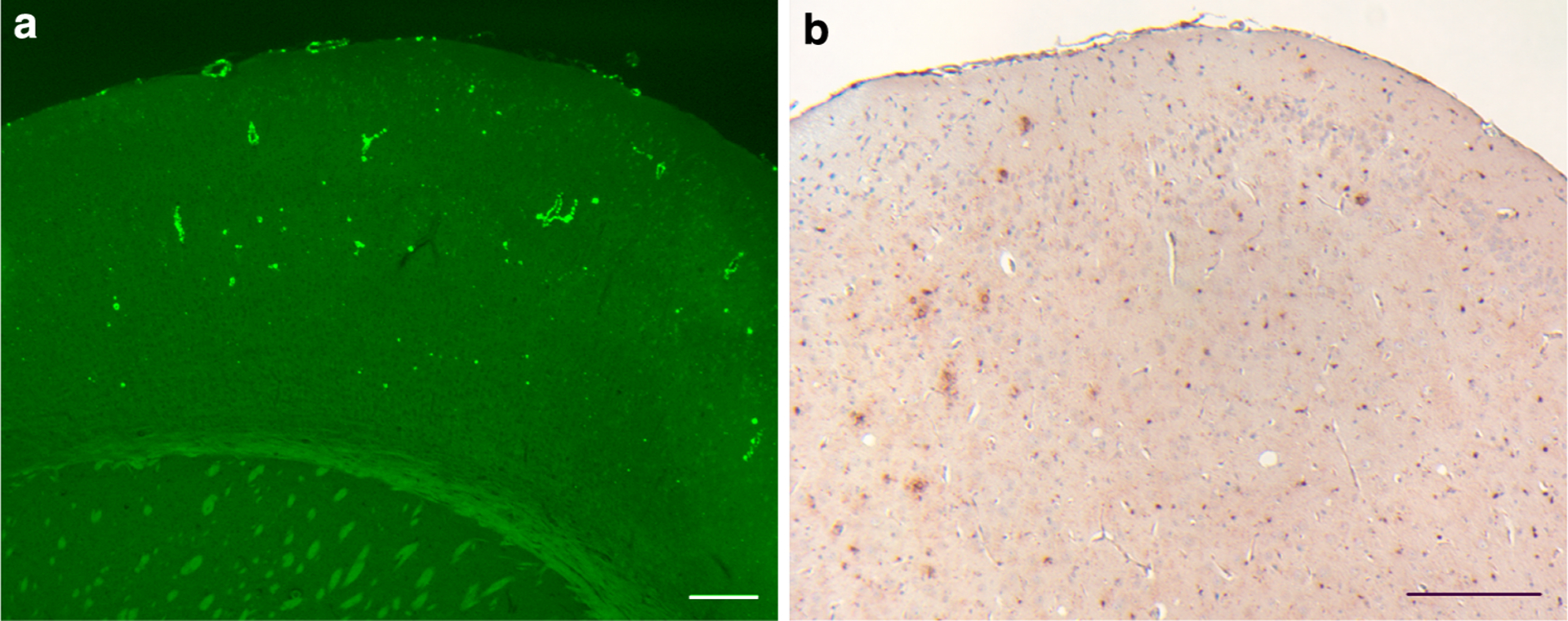
ADan deposition in the cerebral cortex. a All cerebral cortical layers of Tg-FDD mice show ThS-fluorescent amyloid deposition. b Amyloid deposits were immunoreactive with anti-ADan antibodies (Ab 1700), which also stained the subpial region of the cerebral cortex and diffuse parenchymal deposits. Sections were from a 21 month-old Tg-FDD mice. Staining with ThS (a). Immunohistochemistry using Ab 1700 was done as described (Vidal et al. 2009) (b). Scale bars a, b 200 μm
Fig. 3.
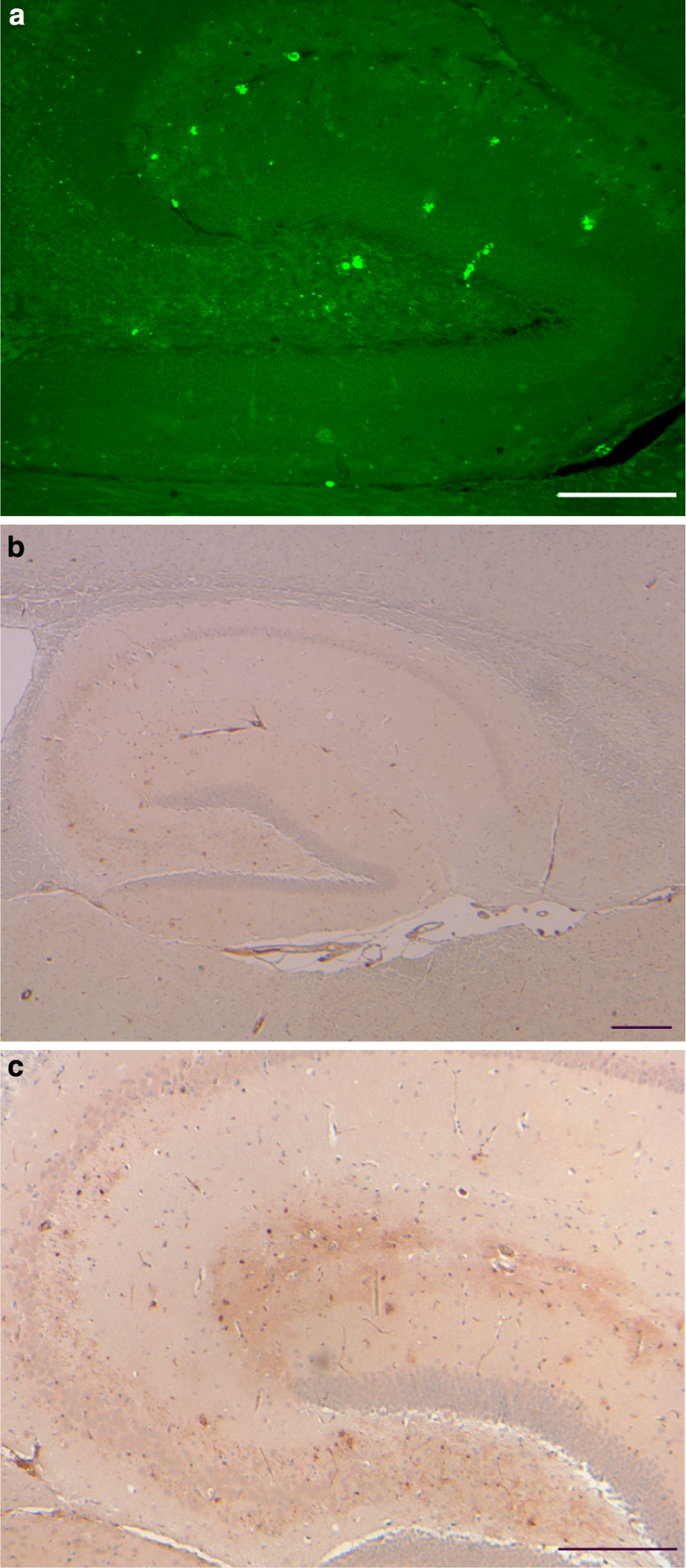
Vascular and parenchymal ADan deposition in the hippocampus. a Amyloid deposition was observed throughout the hippocampal formation of Tg-FDD mice by ThS. Fluorescent small, punctate deposits, ~3 μm in diameter were also observed. b ADan-immunopositive structures outlined the hippocampus. ADan was found in the walls of large and medium size vessels and in the walls of vessels of the hippocampal fissure. Sections were from a 21-month-old Tg-FDD mice. Staining with ThS (a). Immunohistochemistry using Ab 1700 (b, c). Scale bars a 100 μm, b, c 200 μm
Fig. 4.
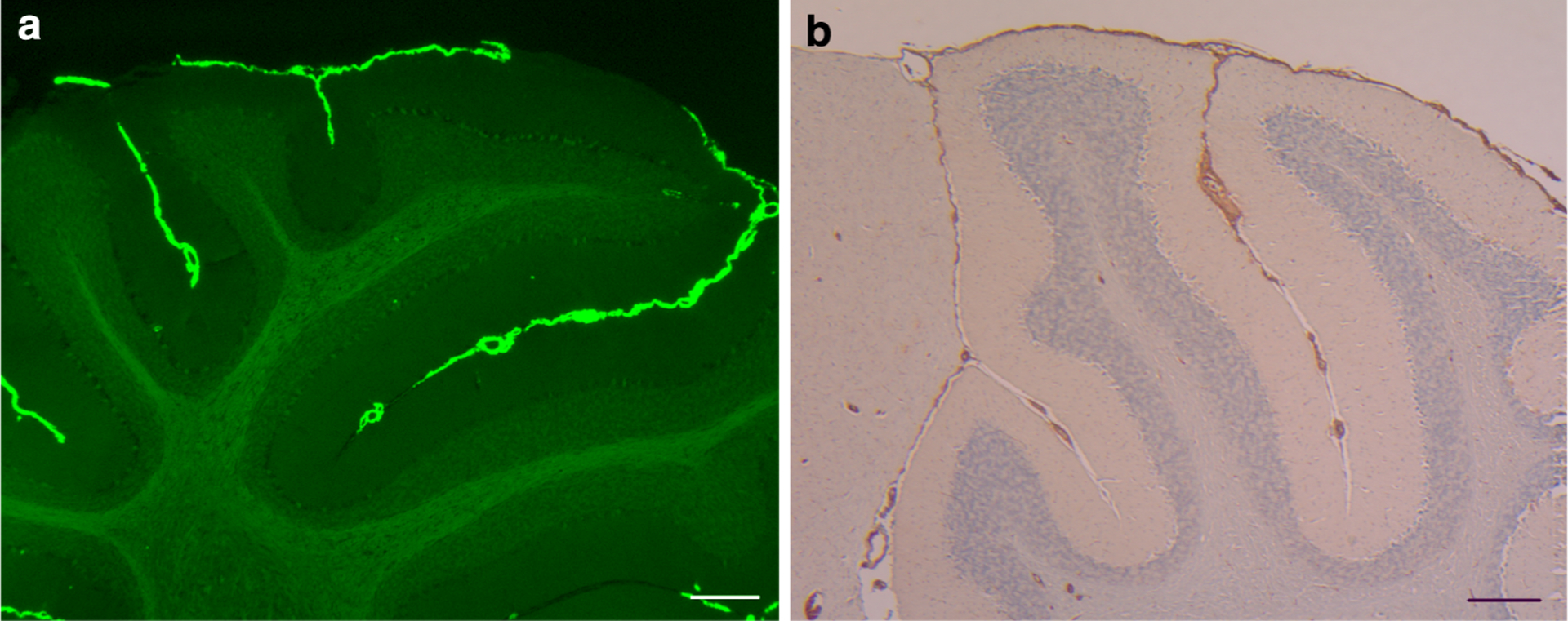
ADan deposition in Cerebellar leptomeninges. a ADan deposition was observed in pial (leptomeningeal) vessels of the cerebellum of Tg-FDD mice. b Antibodies against ADan immunolabel leptomeningeal and cortical blood vessels, and the subpial region of the cerebellar cortex of Tg-FDD mice. Sections were from a 21-month-old Tg-FDD mice. Staining with ThS (a). Immunohistochemistry using Ab 1700 (b). Scale bars a 100 μm, b 200 μm
Fig. 5.
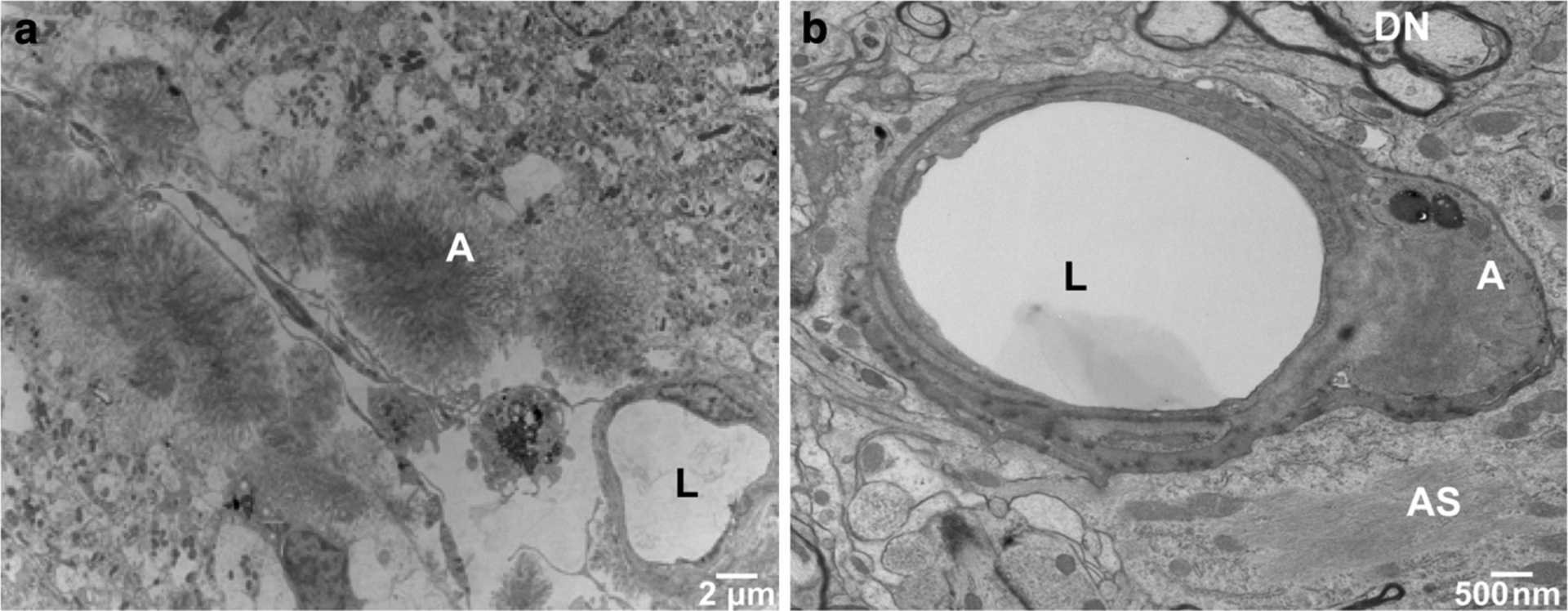
Ultrastructural analysis of cerebral vascular ADan deposition in Tg-FDD mice. a Amyloid deposits in leptomeningeal vessels of the cerebellum were observed within the vessel wall or appeared as circumscribed deposits with radially oriented fibrils. b Abnormal thickening of the basal lamina as well as degeneration and death of endothelial cells was observed in hippocampal vessel walls, in some cases with penetrance of amyloid into the vascular basement membrane. Transmission electron microscopy (TEM) of a 18-month-old Tg-FDD mouse. Amyloid (A), vessel lumen (L), dystrophic neurites (DN) and astrocytic processes (AS) were identified
Fig. 6.
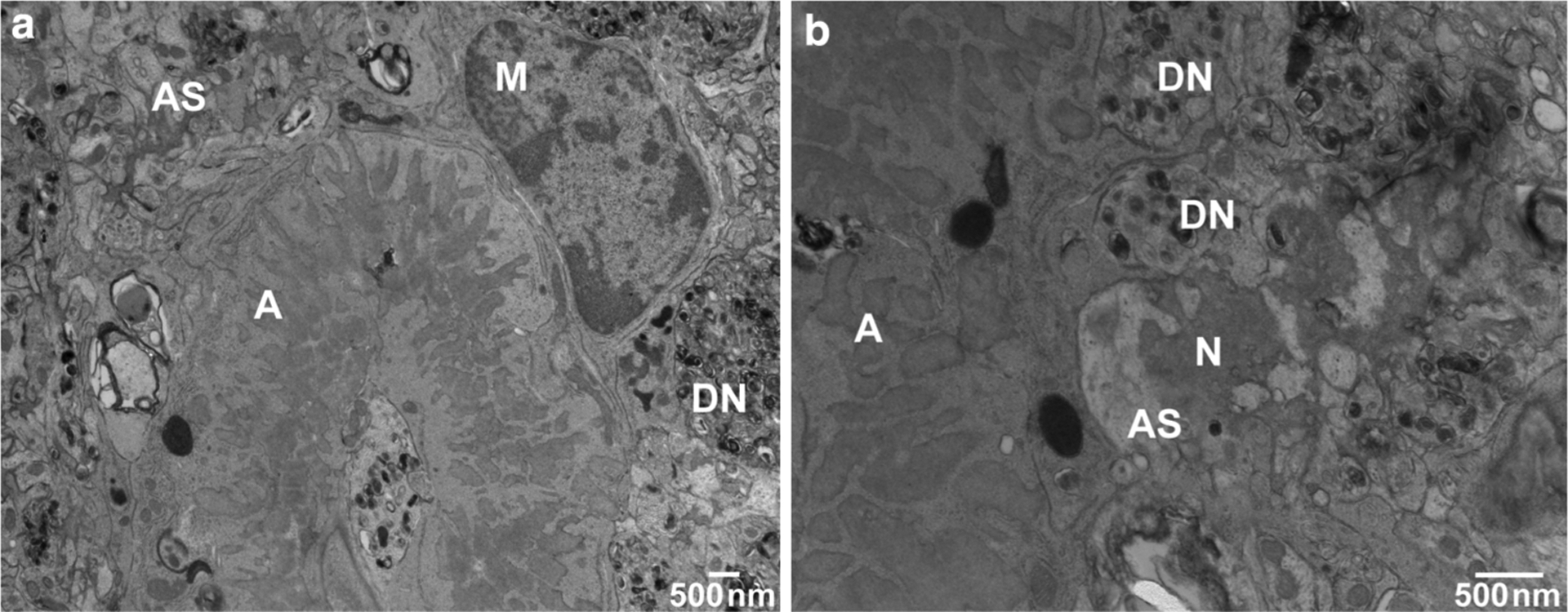
Ultrastructure of hippocampal parenchymal amyloid deposits. a Bundles of fibrillar amyloid are seen within microglial cells. Swollen astroglial processes containing glial filaments are seen in close proximity to the amyloid deposit as well as degenerating neuronal processes and dystrophyc neurites. b Neurons adjacent to amyloid deposits, particularly in the hippocampus, showed morphological evidence of degeneration, and often ultrastructural features of cellular death. Transmission electron microscopy (TEM) of an 18-month-old Tg-FDD mouse. Amyloid (A), dystrophic neurites (DN), microglia (M), neurons (N), and astrocytic processes (AS) were identified
Fig. 7.
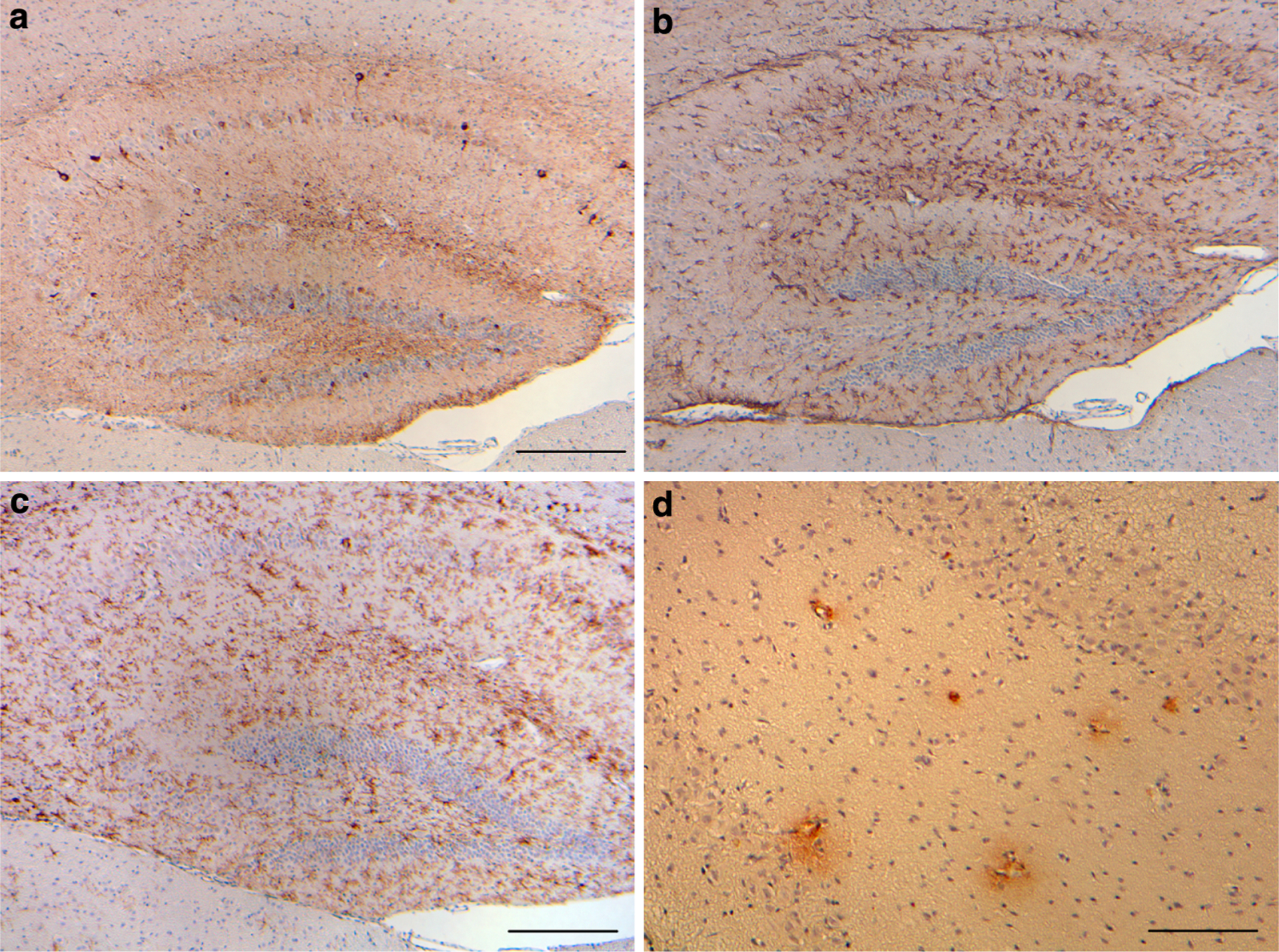
Tg-Tau × Tg-FDD mice. a Phosphorylated tau, b GFAP-positive reactive astrocytes, and c keratan sulfate-positive activated microglia in the hippocampus and motor cortex of Tg-Tau × Tg-FDD mice. d Antibodies against ADan immunolabel hippocampal blood vessels and perivascular deposits in Tg-Tau × Tg-FDD mice. Sections were from a 12-month-old homozygous Tg-Tau × Tg-FDD mice. Immunohistochemistry using AT8 (a), anti-GFAP (b), 5D4 (c), and anti-ADan (1700) (d). Scale bars a–c 200 μm, d 100 μm
Bri2 knock-in
In addition to transgenic models, a knock-in (KI) mouse model was developed for FDD (Giliberto et al. 2009) using a targeting strategy in which exon 6 of the murine Bri2 was replaced by a mutated exon 6 carrying the FDD mutation (Vidal et al. 2000b). No detectable ADan deposition in blood vessels or parenchyma was observed in animals up to 18 months of age. This novel murine model of FDD will allow further studies of the pathogenesis of FDD, apart from over-expression artifacts that are present in transgenic models (Duyckaerts et al. 2008). Moreover, it may provide important clues on the interaction between ADan and Aβ peptides as seen in individuals with FDD (Vidal et al. 2000b) and the interaction between BRI2 and AβPP (Matsuda et al. 2005, 2008; Fotinopoulou et al. 2005).
BRI2-fusion proteins for the expression of Aβ peptides
The knowledge gained on the processing and metabolism of BRI2 led to the development of a novel expression system that selectively expresses Aβ40 or Aβ42 in the secretory pathway. This system utilizes fusion constructs in which the sequence encoding the 23-amino-acid propeptide (Fig. 1a) at the C-terminus of BRI2 is replaced with a sequence encoding either Aβ40 or Aβ42 (Lewis et al. 2001). The system was used to generate AAV vectors to target expression of individual Aβ peptides to the hippocampus of adult rats, allowing the examination of the role of each peptide in the pathology and cognitive deficits seen in AD models in the absence of AβPP over-expression (Lawlor et al. 2007). In addition, transgenic mice were also developed in which the moPrnp promoter was used to drive the expression of the BRI2–Aβ40 and BRI2–Aβ42 fusion proteins (McGowan et al. 2005). Interestingly, mice expressing high levels of Aβ40 did not develop overt amyloid pathology, but in contrast, mice expressing lower levels of Aβ42 accumulated insoluble Aβ42 and developed compact amyloid plaques, CAA, and diffuse Aβ deposits. When mice expressing BRI2–Aβ42 were crossed with mutant AβPP (Tg2576) transgenic mice, there was also a massive increase in amyloid deposition, establishing that Aβ42 is essential for amyloid deposition in the parenchyma and also in vessels (McGowan et al. 2005). In addition, after crossing the BRI2–Aβ40 mice that selectively express high levels of Aβ40 with both Tg2576 mice and BRI2–Aβ42 mice, it was found that these mice had a decreased Aβ deposition, suggesting that Aβ42 and Aβ40 have opposing effects on amyloid deposition: Aβ42 promotes amyloid deposition but Aβ40 inhibits it (Kim et al. 2007). To study the anti-amyloidogenic effect of Aβ40 in another AβPP mouse model, recombinant AAV1 (rAAV1)-mediated gene transfer was used to deliver the BRI2–Aβ40 and BRI2 transgenes to the brains of postnatal day 0 (P0) TgCRND8 hAPPKM670/671NL+V717F AβPP mice (Kim et al. 2008). In these studies, it was shown that BRI2 suppressed Aβ deposition in TgCRND8 mice to an extent equivalent to Aβ40. Although it was not possible to completely rule out subtle effects on Aβ generation that could influence deposition, the authors reported that the suppressive effect of BRI2 was likely to be mediated by inhibition of Aβ aggregation by the secreted peptide (Bri2–23) (Fig. 1a) since expression of a deletion construct lacking the secreted peptide sequence had no effect on Aβ deposition after expression in vivo and that Bri2–23 had an inhibitory effect on Aβ42 aggregation in vitro (Kim et al. 2008).
Bri2 knock-out
Bri2-null mice were generated by Cre recombinase-mediated deletion of exon 2 of the murine Bri2 gene which encodes the transmembrane region (Matsuda et al. 2008). Both heterozygous and homozygous mice were viable, fertile with no obvious abnormal phenotype. AβPP processing in the brains of 11-month-old mice showed significant increase in both endogenous sAβPPα and sAβPPβ levels. In addition, an increase in sAβPPα production in brain homogenates of double (TgAPP695/PSEN1DE9 double transgenic mice (called here AβPP-PS1) expressing two familial AD mutant genes (AβPP-Swedish and PS1ΔE9) (Savonenko et al. 2003) compared with control mice was observed, with a statistically significant increase of both Aβ40 and Aβ42 in compared with mice (Matsuda et al. 2008). These experiments clearly demonstrated that reducing Bri2 expression by gene targeting increased AβPP processing and Aβ formation.
Conclusion
Animal models have been invaluable in AD for elucidating the pathophysiological mechanisms associated with this disease. Transgenic models have allowed the in vivo study of Aβ deposition and the development of therapeutic screens. To investigate the pathologic mechanism(s) involved in the process of neurodegeneration in FBD and FDD, animal models for these diseases have been generated (Pickford et al. 2006; Coomaraswamy et al. 2006, 2008; Matsuda et al. 2008; Vidal et al. 2009). Available experimental models of amyloid deposition include transgenic mice over-expressing human mutant AβPP as single AβPP transgenic or double AβPP/PSEN transgenic mice. On the other hand, transgenic mice for FBD and FDD are excellent in vivo systems for the study of cerebral amyloidosis, for the identification of pathologic pathways shared by different amyloid peptides (such as Aβ and ADan) and for the development of therapeutic strategies for these neurodegenerative diseases (Vidal et al. 2000a). In addition to the transgenic models, the Bri2-KI models of FBD and FDD, which although they do not reproduce the neuropathological features of the disease, are important systems in which to study the molecular basis of these diseases without over-expression of the protein.
Although the function of BRI2 remains largely unknown, it has been found to interact with AβPP, the precursor of Aβ, decreasing the efficiency of AβPP processing to generate Aβ (Fotinopoulou et al. 2005; Matsuda et al. 2005, 2008). In addition, the 23 amino acid C-terminal peptide released from wild-type BRI2 by normal processing was reported to inhibit Aβ aggregation (Kim et al. 2008). Thus, Bri2-KI and Bri2-KO mouse models will be important tools in delineating the role of BRI2 on AβPP processing in AD, and may help elucidate the role of BRI2 in the pathologic process leading to neurodegeneration, not only in FBD and FDD, but also in AD. In summary, these novel mouse models offer new opportunities for the recognition of therapeutic agents that can be effective in the prevention and treatment of these devastating human diseases.
Acknowledgments
The authors are grateful to Debra Lucas, Janice Pennington, and Rose Richardson for their technical assistance. This study was supported by grants from the National Institute of Health NS050227, AG10133, AG027139, and AG033007, by the Alzheimer’s Association (IIRG-05-14220), and by the American Health Assistance Foundation (A2008-304).
References
- Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F, Brenner HR, Caroni P (1995) Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 83:269–278 [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kondo H, Arai T, Ikeda K, Kato M, Iseki E, Schwab C, McGeer PL (2004) Expression of BRI, the normal precursor of the amyloid protein of familial British dementia, in human brain. Acta Neuropathol 107:53–58 [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal 13:159–163 [DOI] [PubMed] [Google Scholar]
- Choi SI, Vidal R, Frangione B, Levy E (2004) Axonal transport of British and Danish amyloid peptides via secretory vesicles. FASEB J 18:373–375 [DOI] [PubMed] [Google Scholar]
- Coomaraswamy J, Herzig MC, Kaeser SA, Ghiso J, Jucker M (2006) Transgenic mouse models of familial British and Danish dementias. ICAD S116:P1–P072 [Google Scholar]
- Coomaraswamy J, Woelfing H, Schaefer C, Kaeser SA, Akiyama H, Goedert M, Mathews PM, Ghiso J, Jucker M (2008) Modeling familial Danish dementia. ICAD:P1–076 [Google Scholar]
- Deleersnijder W, Hong G, Cortvrindt R, Poirier C, Tylzanowski P, Pittois K, Van Marck E, Merregaert J (1996) Isolation of markers for chondro-osteogenic differentiation using cDNA library subtraction. Molecular cloning and characterization of a gene belonging to a novel multigene family of integral membrane proteins. J Biol Chem 271:19475–19482 [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B (2008) Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:5–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer A, Rebollo A (2004) Induction of p53-independent apoptosis by the BH3-only protein ITM2Bs. FEBS Lett 557:283–287 [DOI] [PubMed] [Google Scholar]
- Fleischer A, Ayllon V, Dumoutier L, Renauld JC, Rebollo A (2002) Proapoptotic activity of ITM2B(s), a BH3-only protein induced upon IL-2-deprivation which interacts with Bcl-2. Oncogene 21:3181–3189 [DOI] [PubMed] [Google Scholar]
- Fotinopoulou A, Tsachaki M, Vlavaki M, Poulopoulos A, Rostagno A, Frangione B, Ghiso J, Efthimiopoulos S (2005) BRI2 interacts with amyloid precursor protein (APP) and regulates amyloid beta (Abeta) production. J Biol Chem 280:30768–30772 [DOI] [PubMed] [Google Scholar]
- Ghetti B, Tagliavini F, Masters CL, Beyreuther K, Giaccone G, Verga L, Farlow MR, Conneally PM, Dlouhy SR, Azzarelli B, Bugiani O (1989) Gerstmann–Sträussler–Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology 39:1453–1461 [DOI] [PubMed] [Google Scholar]
- Giaccone G, Tagliavini F, Verga L, Frangione B, Farlow MR, Bugiani O, Ghetti B (1990) Neurofibrillary tangles of the Indiana kindred of Gerstmann–Sträussler–Scheinker disease share anti-genic determinants with those of Alzheimer disease. Brain Res 530:325–329 [DOI] [PubMed] [Google Scholar]
- Giaccone G, Mangieri M, Capobianco R, Limido L, Hauw JJ, Haïk S, Fociani P, Bugiani O, Tagliavini F (2008) Tauopathy in human and experimental variant Creutzfeldt–Jakob disease. Neurobiol Aging 29:1864–1873 [DOI] [PubMed] [Google Scholar]
- Giliberto L, Matsuda S, Vidal R, D’Adamio L (2009) Generation and initial characterization of BRI2-ADan Knock In mice. PLoS One (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Ghetti B, Spillantini MG (2000) Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann N Y Acad Sci 920:74–83 [DOI] [PubMed] [Google Scholar]
- Golde TE (2003) Alzheimer disease therapy: can the amyloid cascade be halted? J Clin Invest 111:11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths RA, Mortimer TF, Oppenheimer DR, Spalding JM (1982) Congophilic angiopathy of the brain: a clinical and pathological report on two siblings. J Neurol Neurosurg Psychiatry 45:396–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton JL, Ghiso J, Lashley T, Rostagno A, Guerin CJ, Gibb G, Houlden H, Ayling H, Martinian L, Anderton BH, Wood NW, Vidal R, Plant G, Frangione B, Revesz T (2001) Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol 158:515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton JL, Lashley T, Ghiso J, Braendgaard H, Vidal R, Guerin CJ, Gibb G, Hanger DP, Rostagno A, Anderton BH, Strand C, Ayling H, Plant G, Frangione B, Bojsen-Moller M, Revesz T (2002) Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J Neuropathol Exp Neurol 61:254–267 [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102 [DOI] [PubMed] [Google Scholar]
- Kim SH, Wang R, Gordon DJ, Bass J, Steiner DF, Lynn DG, Thinakaran G, Meredith SC, Sisodia SS (1999) Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nat Neurosci 2:984–988 [DOI] [PubMed] [Google Scholar]
- Kim SH, Creemers JW, Chu S, Thinakaran G, Sisodia SS (2002) Proteolytic processing of familial British dementia-associated BRI variants: evidence for enhanced intracellular accumulation of amyloidogenic peptides. J Biol Chem 277:1872–1877 [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E (2007) Abeta40 inhibits amyloid deposition in vivo. J Neurosci 27:627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Miller VM, Levites Y, West KJ, Zwizinski CW, Moore BD, Troendle FJ, Bann M, Verbeeck C, Price RW, Smithson L, Sonoda L, Wagg K, Rangachari V, Zou F, Younkin SG, Graff-Radford N, Dickson D, Rosenberry T, Golde TE (2008) BRI2 (ITM2b) inhibits Abeta deposition in vivo. J Neurosci 28:6030–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokjohn TA, Roher AE (2009) Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement 5:340–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latil A, Chene L, Mangin P, Fournier G, Berthon P, Cussenot O (2003) Extensive analysis of the 13q14 region in human prostate tumors: DNA analysis and quantitative expression of genes lying in the interval of deletion. Prostate 57:39–50 [DOI] [PubMed] [Google Scholar]
- Lawlor PA, Bland RJ, Das P, Price RW, Holloway V, Smithson L, Dicker BL, During MJ, Young D, Golde TE (2007) Novel rat Alzheimer’s disease models based on AAV-mediated gene transfer to selectively increase hippocampal Abeta levels. Mol Neurodegener 2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PA, Piper S, Baker M, Onstead L, Murphy MP, Hardy J, Wang R, McGowan E, Golde TE (2001) Expression of BRI-amyloid beta peptide fusion proteins: a novel method for specific high-level expression of amyloid beta peptides. Biochim Biophys Acta 1537:58–62 [DOI] [PubMed] [Google Scholar]
- Martin L, Fluhrer R, Reiss K, Kremmer E, Saftig P, Haass C (2008) Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J Biol Chem 283:1644–1652 [DOI] [PubMed] [Google Scholar]
- Martin L, Fluhrer R, Haass C (2009) Substrate requirements for SPPL2b-dependent regulated intramembrane proteolysis. J Biol Chem 284:5662–5670 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Giliberto L, Matsuda Y, Davies P, McGowan E, Pickford F, Ghiso J, Frangione B, D’Adamio L (2005) The familial dementia BRI2 gene binds the Alzheimer gene amyloid-beta precursor protein and inhibits amyloid-beta production. J Biol Chem 280:28912–28916 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Giliberto L, Matsuda Y, McGowan EM, D’Adamio L (2008) BRI2 inhibits amyloid beta-peptide precursor protein processing by interfering with the docking of secretases to the substrate. J Neurosci 28:8668–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274:1678–1683 [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T (2005) Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47:191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S, James-Galton M, Revesz T, Doshi RB, Harwood G, Pan EL, Ghiso J, Frangione B, Plant G (2000) Familial British dementia with amyloid angiopathy: early clinical, neuropsychological and imaging findings. Brain 123:975–991 [DOI] [PubMed] [Google Scholar]
- Murrell JR, Gnezda AG, Green A, Greally J, Grahame N, Ghetti B (2006) Pathology of a transgenic mouse expressing human P301S tau. Brain Pathol 16:S44 [Google Scholar]
- Pickford F, Onstead L, Camacho-Prihar C, Hardy J, McGowan E (2003) Expression of mBRI2 in mice. Neurosci Lett 338:95–98 [DOI] [PubMed] [Google Scholar]
- Pickford F, Coomaraswamy J, Jucker M, McGowan E (2006) Modeling familial British dementia in transgenic mice. Brain Pathol 16:80–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittois K, Deleersnijder W, Merregaert J (1998) cDNA sequence analysis, chromosomal assignment and expression pattern of the gene coding for integral membrane protein 2B. Gene 217: 141–149 [DOI] [PubMed] [Google Scholar]
- Plant GT, Revesz T, Barnard RO, Harding AE, Gautier-Smith PC (1990) Familial cerebral amyloid angiopathy with nonneuritic amyloid plaque formation. Brain 113:721–747 [DOI] [PubMed] [Google Scholar]
- Revesz T, Holton JL, Doshi B, Anderton BH, Scaravilli F, Plant GT (1999) Cytoskeletal pathology in familial cerebral amyloid angiopathy (British type) with non-neuritic amyloid plaque formation. Acta Neuropathol 97:170–176 [DOI] [PubMed] [Google Scholar]
- Sanchez-Pulido L, Devos D, Valencia A (2002) BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci 27:329–332 [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Xu GM, Price DL, Borchelt DR, Markowska AL (2003) Normal cognitive behavior in two distinct congenic lines of transgenic mice hyperexpressing mutant APP SWE. Neurobiol Dis 12(3):194–211 [DOI] [PubMed] [Google Scholar]
- Strömgren E (1981) Heredopathia ophthalmo-oto-encephalica. In: Vinken PJ, Bruyn GW (eds) Handbook of clinical neurology, vol 42. North-Holland Publishing Company, Amsterdam, pp 150–152 [Google Scholar]
- Strömgren E, Dalby A, Dalby MA, Ranheim B (1970) Cataract, deafness, cerebellar ataxia, psychosis and dementia—a new syndrome. Acta Neurol Scand 46(S43):261. [DOI] [PubMed] [Google Scholar]
- Tomidokoro Y, Lashley T, Rostagno A, Neubert TA, Bojsen-Møller M, Braendgaard H, Plant G, Holton J, Frangione B, Révész T, Ghiso J (2005) Familial Danish dementia: co-existence of Danish and Alzheimer amyloid subunits (ADan and Aβ) in the absence of compact plaques. J Biol Chem 280(44):36883–36894 [DOI] [PubMed] [Google Scholar]
- Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J (1999) A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399:776–781 [DOI] [PubMed] [Google Scholar]
- Vidal R, Ghiso J, Frangione B (2000a) New familial forms of cerebral amyloid and dementia. Mol Psychiatry 5:575–576 [DOI] [PubMed] [Google Scholar]
- Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen-Moller M, Braendgaard H, Plant G, Ghiso J, Frangione B (2000b) A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA 97:4920–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Calero M, Revesz T, Plant G, Ghiso J, Frangione B (2001) Sequence, genomic structure and tissue expression of Human BRI3, a member of the BRI gene family. Gene 266:95–102 [DOI] [PubMed] [Google Scholar]
- Vidal R, Delisle MB, Ghetti B (2004) Neurodegeneration caused by proteins with an aberrant carboxyl-terminus. J Neuropathol Exp Neurol 63:787–800 [DOI] [PubMed] [Google Scholar]
- Vidal R, Barbeito AG, Miravalle L, Ghetti B (2009) Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol 19:58–68 Epub 2008 Apr 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ (2007) A beta oligomers—a decade of discovery. J Neurochem 101:1172–1184 [DOI] [PubMed] [Google Scholar]
- Worster-Drought C, Hill TR, McMenemey WH (1933) Familial presenile dementia with spastic paralysis. J Neurol Psychopathol 14:27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worster-Drought C, Greenfield JG, McMenemey WH (1944) A form of familial presenile dementia with spastic paralysis. Brain 67:38–43 [DOI] [PMC free article] [PubMed] [Google Scholar]