

Abstract
Recently discovered “Trim-Away” mechanism opens a new window for fast and selective degradation of endogenous proteins. However, the in vivo and clinical application of this approach is stuck by the requirement of special skills and equipment needed for the intracellular delivery of antibodies. Hereby, an antibody conjugated polymer nanogel system, Nano-ERASER, for intracellular delivery and release of antibody, and degradation of a specific endogenous protein has been developed. After being delivered into cells, the antibody is released and forms complex with its target protein, and subsequently binds to the Fc receptor of TRIM21. The resulted complex of target protein/antibody/TRIM21 is then degraded by the proteasome. The efficacy of Nano-ERASER has been validated by depleting GFP protein in a GFP expressing cell line. Furthermore, Nano-ERASER successfully degrades COPZ1, a vital protein for cancer cells, and kills those cells while sparing normal cells. Benefit from its convenience and targeted delivery merit, Nano-ERASER technique is promising in providing a reliable tool for endogenous protein function study as well as paves the way for novel antibody-based Trim-Away therapeutic modalities for cancer and other diseases.
Keywords: Protein degradation, antibody delivery, intracellular release, nanogel, Trim21
Graphical Abstract
An antibody-loaded nanogel system (Nano-ERASER) is developed for the intracellular delivery and release of antibodies and degradation of a specific endogenous protein through a TRIM21-mediated degradation by the proteasome. Nano-ERASER provides a reliable tool for endogenous protein function study as well as paves the way for novel antibody-based therapeutic modalities for cancer and other diseases.
1. Introduction
Proteins play crucial roles in the human body, including enzymes, carriers, structure building blocks, hormone signaling, defense, and storage. The malfunction of protein causes various diseases, such as Alzheimer’s disease, amyotrophic lateral sclerosis, cystic fibrosis, type 2 diabetes, and cancer.[1] Contrast to gene therapy, protein therapy is free of permanent and/or erratically genetic alterations.[2] Thanks to its high specificity and low side effects, protein therapy has been considered as a safe and reliable method for the treatment of diseases. Antibody, a functional member of the protein family that can bind to proteins with high specificity, has been extensively explored for protein therapy,[3] especially in cancer immunotherapy.
Recently, Clift et al. discovered a so-called “Trim-Away” method, which utilizes antibodies to degrade endogenous proteins in mammalian cells without prior modification of the genome or mRNA.[4] The mechanism of Trim-Away involves the intracellular antibody receptor TRIM21, which is an E3 ubiquitin ligase that binds to the Fc domain of antibodies,[5] and TRIM21 is commonly expressed in various cell types because of its indispensable physiological role.[6] During Trim-Away, the target protein is bound by the antibody, followed by TRIM21-mediated ubiquitination to generate a protein complex, which is subsequently degraded in the proteasome. Since Trim-Away is a highly efficient technique in depleting a target protein inside cells, it opens a window to be applied to a broad spectrum of intracellular proteins with the prerequisite of the availability of its corresponding antibody. However, the wide application of Trim-Away is severely hindered by the cell membrane impermeability of antibodies. To overcome the barrier, Clift et al. delivered antibodies into cells by microinjection and electroporation techniques, which require special skills or equipment and are difficult for in vivo applications, not to mention for future clinical translation.[7] Since the debut of Trim-Away technology, it has been extensively explored in biomedical research. However, due to the difficulty of available approaches, thus far most of the successful applications of Trim-Away were limited to embryos and oocytes.[8] Therefore, there is an urgent need for the develop a convenient Trim-Away approach.
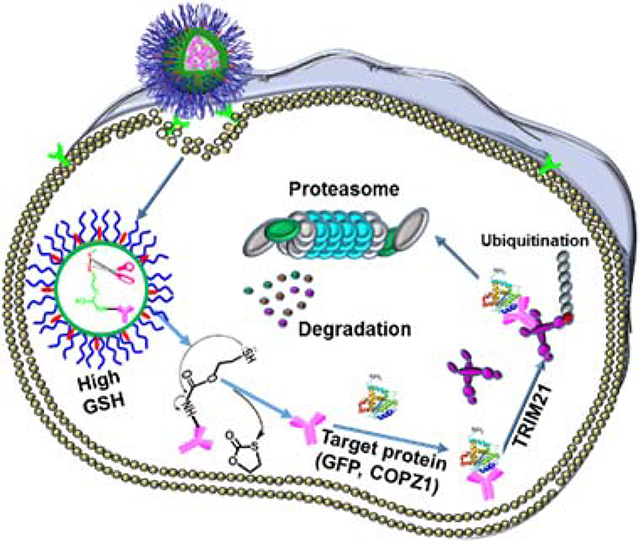
With the rapid development of drug delivery systems, nanoparticulate delivery vehicles designed for intracellular delivery of protein/antibody surged in the past decade,[9] including inorganic nanoparticles,[10] liposomes,[11] and polymeric nanocarriers.[12] Our group has been devoting to investigating drug-loaded polymeric micelles and nanogels for cancer and central nervous system related diseases.[13] Herein, we aim to develop a convenient and safe version of Trim-Away by employing polymer nanogels as an alternative antibody intracellular delivery for microinjection and electroporation. In this design, the antibody is encapsulated inside the nanogel by conjugation with the help of a redox-sensitive traceless linker (Figure 1A). The antibody-loaded nanogel enters cells through receptor-mediated endocytosis (Figure 1B) and subsequently releases its payload due to the elevated intracellular glutathione (GSH). After that, the antibody binds to its target protein and TRIM21 to yield a protein/antibody/TRIM21 complex, which can be degraded by a proteasome-mediated cellular protein degradation machinery. It has been confirmed that the Nanogel-facilitated protein intracellular specific degradation (Nano-ERASER) technique can selectively and effectively degrade endogenous proteins, green fluorescence protein (GFP) and coatomer subunit zeta-1(COPZ1), in cancer cells. Furthermore, it was revealed that the depletion of COPZ1 results in the selective killing of cancer cells. We expect the success of Nano-ERASER could pave the road for the in vivo and clinical application of Trim-Away.
Figure 1.

Schematic illustration of (A) the fabrication of antibody-loaded nanogels from antibody-polymer conjugates, and (B) the mechanism of Nano-ERASER in transporting and intracellularly traceless releasing of antibody, and subsequent degrading target proteins through Trim-Away pathway.
2. Results and discussion
Poly[(2-(pyridin-2-yldisulfanyl)ethyl acrylate)-co-[poly(ethylene glycol)]] (PDA-PEG) polymer bearing p-nitrophenylcarbonate (NPC) moieties in side chains was synthesized by attaching NPC to beta-mercaptoethanol modified PDA-PEG (PDA-PEG-BME) (Scheme S1), which was prepared as we previously reported (Figure S1).[13d] The protein/antibody was reacted with PDA-PEG-NPC to produce protein/antibody conjugated polymers, in which the NPCs are replaced by the reactive lysine groups of the protein/antibody (Scheme S1). The redox-sensitive linker between the antibody and the polymer backbone is self-immolative and readily cleaved by reducing agents such as GSH.[9b] As illustrated in Figure 1A, the resulted polymers were fabricated into polymer nanogels via the crosslinking reaction induced by tris(2-carboxyethyl)phosphine (TCEP). The polymer nanogels were further modified with a tumor-targeting ligand, cyclic Arg-Gly-Asp-D-Phe-Cys (RGD) peptide, to facilitate it entering cancer cells. After being taken up by cells, the self-immolative linker would be cleaved by the intracellular elevated GSH, leading to the traceless release of the antibody (Figure 1B), which makes the nanogels more advantageous for delivering protein/antibody into cells since the intracellular level of GSH (2–10 mM) is much higher than that in the blood stream (2–20 μM).[14]
To validate our strategy, bovine serum albumin (BSA) was utilized as a model protein to investigate the conjugation and subsequent release of protein/antibody from the polymer by gel electrophoresis using SDS-PAGE gel. As displayed in Figure 2A, the protein band of free BSA disappeared in the lane loaded with polymer PDA-PEG-BSA, which indicates the success of protein-polymer conjugation. However, the appearance of the BSA band in the lane loaded with GSH-pretreated PDA-PEG-BSA suggests the redox-responsive release of the protein. Then polymer PDA-PEG-BSA was crosslinked by TCEP to fabricate polymeric nanogel NG-BSA, and the unreacted NPCs were eliminated by the addition of ethylenediamine. The amount of BSA contained in NG-BSA was determined using BCA protein assay kit according to the reported method.[12] The loading efficiency (LE) and loading content (LC) of BSA were calculated to be 77% and 3.4%, respectively. Dynamic light scattering (DLS) determined that NG-BSA had a hydrodynamic size of 135.4 nm with a dispersity of 0.12 (Figure S2), and a slightly negative zeta potential (Figure S3). Transmission electron microscopy (TEM) showed that NG-BSA had a spherical shape and a diameter of 109.3 nm in a dry state (Figure S4).
Figure 2.
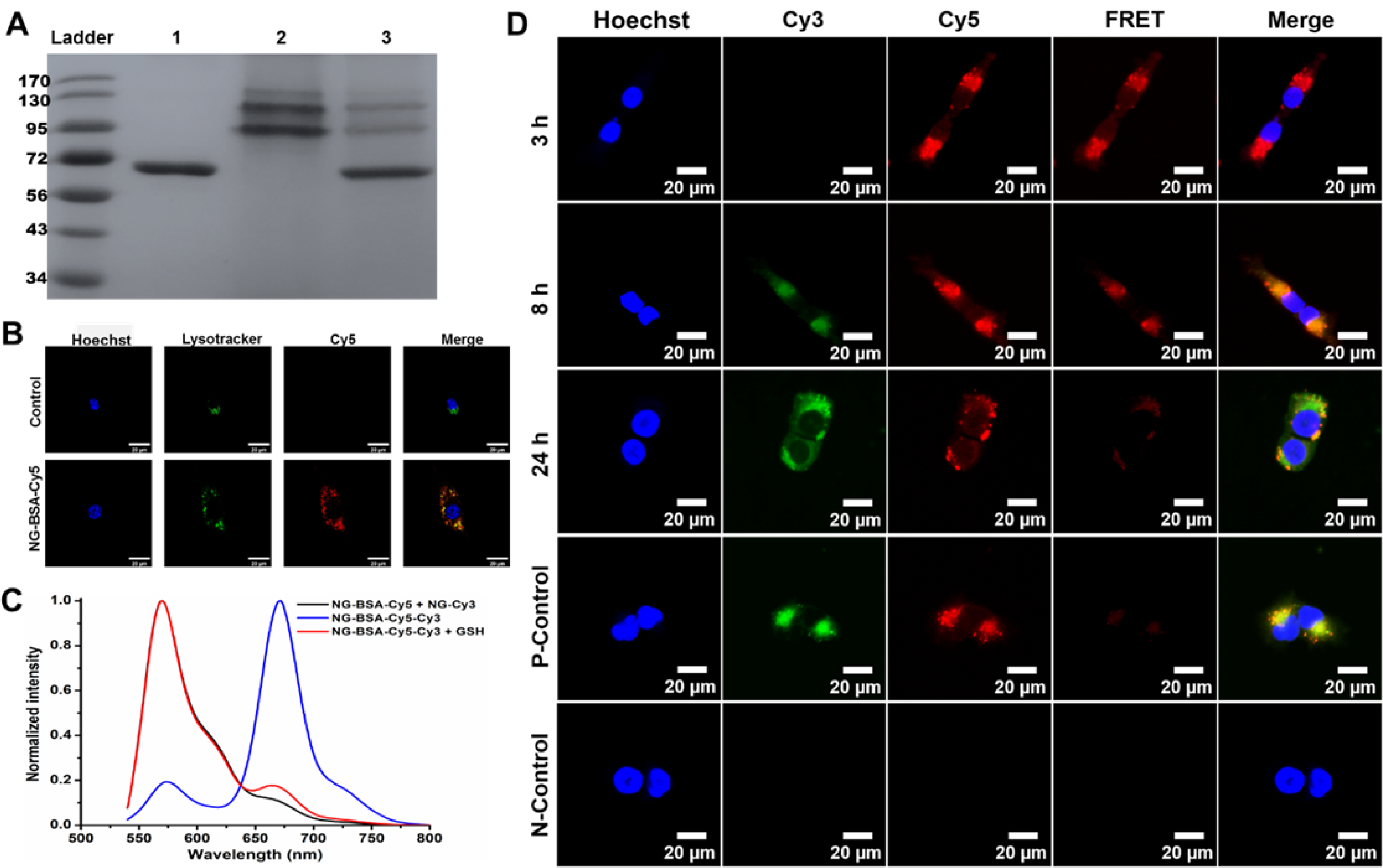
Characterization of the NG-BSA. (A) Gel electrophoresis of (1) free BSA, (2) polymer PDA-PEG-BSA, and (3) GSH treated PDA-PEG-BSA. (B) Sub-cellular colocalization of NG-BSA-Cy5 and lysosome after 3 h of incubation. (C) Fluorescence emission spectra of a mixture of nanogels NG-BSA-Cy5 and NG-Cy3, and nanogel NG-BSA-Cy5-Cy3 in the absence and presence of GSH (λex = 525 nm). (D) Confocal fluorescence images of MCF-7 cells after being incubated with NG-BSA-Cy5-Cy3 for 3, 8, and 24 h. Cells with no treatment and those treated with the mixture of NG-BSA-Cy5 and NG-Cy3 were utilized as a negative control (N-Control) and positive control (P-Control), respectively, and imaged after 24 h of treatment.
In order to study the cellular uptake and intracellular release of the protein from the nanogel fluorescence resonance energy transfer (FRET) technique was employed. BSA was labeled with a fluorescent dye Cyanine5 (Cy5) to generate BSA-Cy5 with the absorption and fluorescent emission wavelengths of 648 nm and 670 nm, respectively (Figure S5). Polymer PDA-PEG-BSA-Cy5 was synthesized in the same way as PDA-PEG-BSA except for replacing BSA with BSA-Cy5 to fabricate a Cy5 labeled nanogel NG-BSA-Cy5. To evaluate the cellular uptake and lysosomal escaping capacity of the nanogel, NIH3T3 cells were incubated with NG-BSA-Cy5 and monitored with confocal microscopy with the help of lysotracker green. The abundant red signals inside the cells proved that NG-BSA-Cy5 could effectively enter the cells. After 3 h of incubation, only about 50% of the signals from the nanogels (red) were overlapped with lysotracker signals (green), suggesting that around half of the nanogels had escaped from the lysosome, as shown in Figure 2B, which paved the road for the subsequent releasing of the protein/antibody and protein-antibody binding. Since the PDA groups of the nanogel could be protonated under an acidic condition, we attribute this quick lysosomal escaping capacity to the proton sponge effect of the nanogel system.
Since fluorescence probes, Cy3 and Cy5, are a well-established FRET pair,[15] the PDA-PEG-BSA-Cy5 polymer was mixed with a Cyanine3 (Cy3, λabs = 555 nm, λem = 570 nm) labeled polymer PDA-PEG-Cy3, which was prepared as we previously reported,[16] to fabricate a Cy5 and Cy3 co-labeled nanogel NG-BSA-Cy5-Cy3. A mixture of nanogels NG-BSA-Cy5 and NG-Cy3, which were prefabricated from polymers PDA-PEG-BSA-Cy5 and PDA-PEG-Cy3 alone, respectively, was prepared as non-FRET control system. As shown in Figure 2C, when excited at 525 nm, a wavelength can excite fluorophore Cy3 while sparing Cy5, intense fluorescence of Cy3 (568 nm) and negligible fluorescence of Cy5 (670 nm) were observed from the above mixture of nanogels. On the contrary, nanogel NG-BSA-Cy5-Cy3 emitted weak fluorescence of Cy3 and a strong fluorescence of Cy5 under the same condition, which demonstrates that efficient FRET phenomenon occurred between Cy3 and Cy5 fluorophores of NG-BSA-Cy5-Cy3. However, in the presence of GSH, the fluorescence of Cy3 emitted by NG-BSA-Cy5-Cy3 was significantly enhanced while that of Cy5 decreased simultaneously, which is similar to the fluorescence emission pattern of the mixed nanogels of NG-BSA-Cy5 and NG-Cy3. These observations suggested that BSA-Cy5 was released from the nanogel by GSH so that the two fluorophores were no longer in proximity, and thus the FRET phenomenon was diminished.
To investigate whether the GSH-induced FRET-inhibition phenomenon could occur inside cells, we incubated nanogel NG-BSA-Cy5-Cy3 with human breast cancer MCF-7 cells and recorded the fluorescence emission from cells using confocal fluorescence microscopy. First of all, as shown in Figure 2D, the intense fluorescence inside the cells confirmed that the protein-loaded nanogels could be efficiently taken up by cells. Moreover, upon the excitation of a 555 nm laser, an apparent FRET phenomenon was observed after 3 h of incubation with NG-BSA-Cy5-Cy3, as evidenced by the very weak fluorescence of Cy3 (green) and strong fluorescence of Cy5 (red). At 8 h post-incubation, the significantly increased fluorescence of Cy3 and declined fluorescence of Cy5 indicated that some of the Cy5-labelled protein BSA-Cy5 had been liberated, and the FRET efficiency decreased accordingly. After 24 h of incubation, the FRET phenomenon nearly disappeared, and the fluorescence of cells was almost the same as that of cells co-incubated with nanogels NG-BSA-Cy5 and NG-Cy3 in the positive control group, which suggests that most of the loaded BSA-Cy5 had already been released from NG-BSA-Cy5-Cy3. Taken together, the results shown in Figure 2 confirmed the effectiveness of our design strategy that the polymer nanogel can deliver a conjugated protein into cells and release it intracellularly.
To validate that the bioactivity of the intracellular delivered protein/antibody is conserved, anti-GFP, an antibody for green fluorescence protein (GFP), was conjugated to the polymer by reacting with polymer PDA-PEG-NPC to yield PDA-PEG-aGFP, and subsequently fabricated into NG-aGFP nanogel following the same procedures of NG-BSA production. DLS analysis found that NG-aGFP has a hydrodynamic diameter of 125.9 nm (dispersity: 0.20) (Figure 3A), and carries negative surface charge (Figure S3). TEM imaging revealed a spherical morphology of NG-aGFP with a diameter of 103.3 nm in the dry state (Figure 3B). The loading content and loading efficiency of anti-GFP in NG-aGFP were determined to be 7.6% and 85%, respectively. As shown in Figure 3C, gel electrophoresis demonstrated that the antibody loaded into NG-aGFP could be effectively recovered upon GSH treatment. The fragmented protein bands that appeared in the right panel of Figure 3C was due to the cleavage of the disulfide bonds in the antibody and the nanogels.
Figure 3.
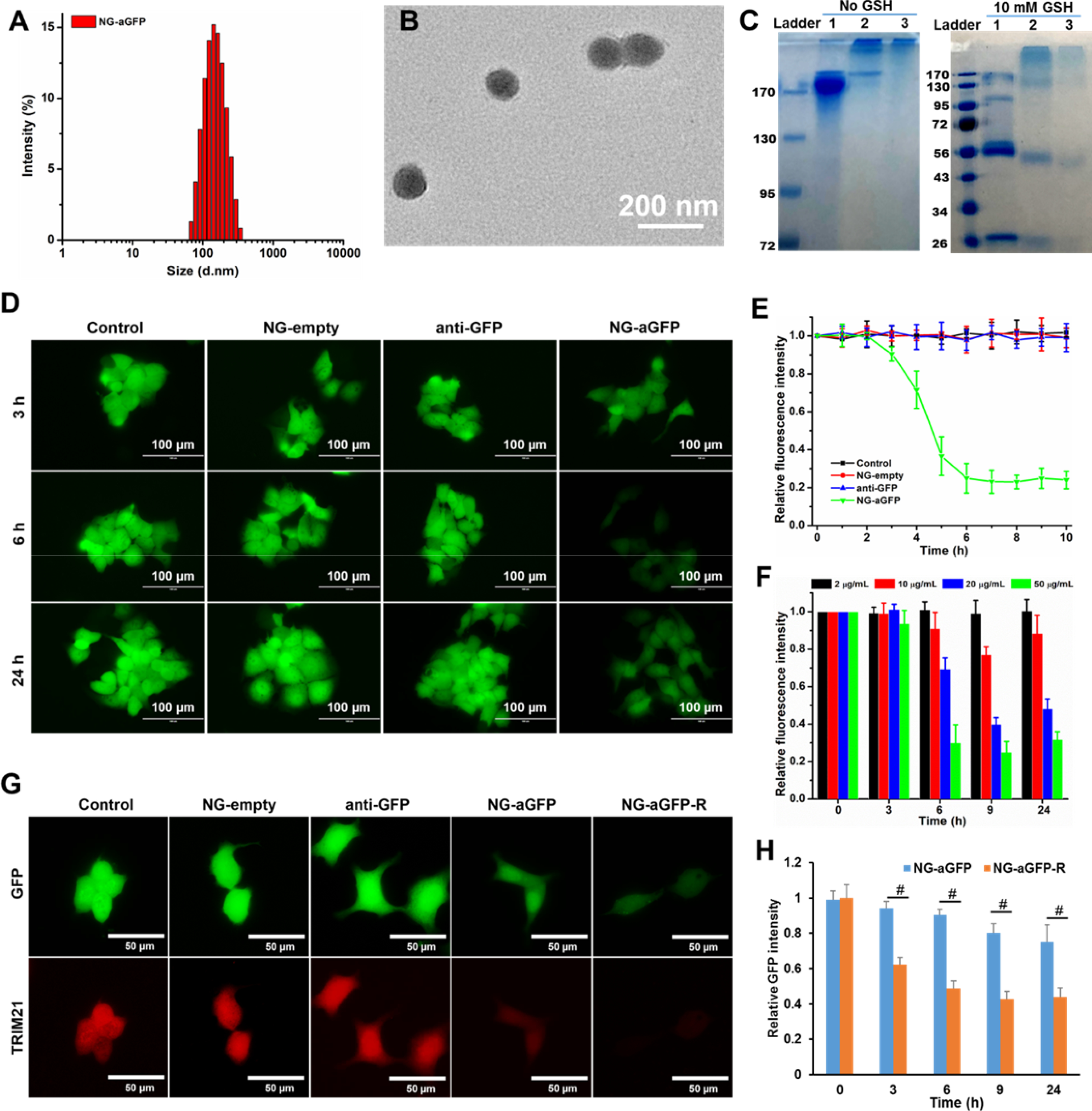
Characterization and activity evaluation of NG-aGFP. (A) Size distribution and (B) TEM image of nanogel NG-aGFP. (C) Gel electrophoresis of (1) free anti-GFP antibody, (2) PDA-PEG-aGFP, and (3) NG-aGFP. Samples in the right panel were treated with 10 mM GSH for 4 hours before electrophoresis. TRIM21-transfected MCF-7/GFP cells were used in (D-F). (D) Fluorescence images and (E) relative fluorescence intensity data of cells after incubation with empty nanogel (NG-empty), free anti-GFP antibody (anti-GFP), and NG-aGFP nanogel. (F) Relative fluorescence intensity of cells after being incubated with free anti-GFP and NG-aGFP at different anti-GFP equivalent concentrations. (G) Fluorescence images of cells, GFP (green) and TRIM21 (red) channels, after being incubated with free anti-GFP and nanogels at the anti-GFP equivalent concentration of 20 μg/mL for 9 h. (H) Relative GFP fluorescence intensity of cells after being incubated with NG-aGFP and NG-aGFP-R at anti-GFP equivalent concentrations of 20 μg/mL. Data represent the means ± SD, n=5. #P <0.01.
Since TRIM21 protein is a crucial component for Trim-Away technique, we transfected GFP-expressing human breast cancer MCF-7/GFP cells with pmCherry-C1-mTRIM21 plasmid as described in the literature,[4] to generate TRIM21 overexpressing MCF-7/GFP cells. The success of the transfection was evidenced by the mCherry fluorescence emission in the cells (Figure S6). Subsequently, the cells were treated with free anti-GFP antibody, empty nanogel (NG-empty), and NG-aGFP at an anti-GFP equivalent concentration of 100 μg/mL. As shown in Figure 3D, the GFP fluorescence significantly decreased in the cells incubated with NG-aGFP after 6 h of incubation. In contrast, no noticeable fluorescence intensity change was observed in other treatment groups. The above observation was further confirmed with a quantitative analysis of the fluorescence intensity of the cells (Figure 3E), which also proved that the intracellular delivery of anti-GFP via NG-aGFP effectively degraded the endogenous GFP protein. It was further revealed that higher concentrations of NG-aGFP yielded better protein degradation efficiency (Figure 3F), suggesting that protein degradation is in a dose-dependent manner. It has been reported that the Trim-Away method relies on the overexpression of protein TRIM21.[4] To verify that, the same set of experiments were conducted in MCF-7/GFP cells without TRIM21-transfection. As expected, the protein degradation efficiency in these cells was considerably limited in comparison to that in TRIM21-transfected cells because of the low endogenous TRIM21 level (Figure S7–8), which confirms the necessity of TRIM21 in the Nano-REASER.
To enhance the cellular uptake of the nanogel, NG-aGFP was decorated with a thiol-containing RGD peptide, which can bind to the αvβ3 integrin, a receptor overexpressed on a broad spectrum of cancer cells,[13a, 17] to yield RGD-modified nanogel NG-aGFP-R. NG-aGFP-R has a hydrodynamic size of 130.4 nm (dispersity: 0.19) (Figure S9) and carries a less negative surface charge than that of NG-aGFP (Figure S3). In addition, the NG-aGFP-R is stable in both PBS and PBS supplemented with serum for more than one week (Figure S10). Both confocal microscopy (Figure S11A) and flow cytometry (Figure S11B) validated that the functionalization of RGD peptide significantly enhanced the cellular uptake of the nanogels. As shown in Figure 3G, a weaker GFP fluorescence signal was observed in the cells treated with NG-aGFP-R than that in the NG-aGFP treated cells, demonstrating that the antibody-loaded nanogel is more efficient in degrading target protein after RGD modification. The quantified analysis of GFP fluorescence intensity further verified the enhanced protein degradation efficiency of NG-aGFP-R nanogel (Figure 3H). Moreover, the simultaneously decreased fluorescence of both GFP and mCherry (Figure 3G and Figure S12) indicated the depletion of TRIM21 in the process of protein degradation, which further evidenced the essential role of TRIM21 in the Trim-Away technique. Importantly, cell viability assay of free anti-GFP and nanogels in TRIM21-transfected MCF-7/GFP cells showed that NG-empty and NG-aGFP nanogels exhibited negligible cytotoxicity at the same conditions (Figure S13), which demonstrated that the nano-ERASER induced no additional damage to cells while degrading a target protein.
In order to probe the role of proteasomes in intracellular protein degradation, bortezomib, a proteasome inhibitor, was employed. As shown in Figure S14A, the addition of bortezomib effectively inhibited proteasome’ function, evidenced by the accumulation of ubiquitin in Figure S14A. As expected, the amount of ubiquitin-linked GFP in the NG-aGFP treated group was also boosted. Most importantly, the inhibition of proteasome also prevented the selective degradation of GFP in the NG-aGFP treated cells (Figure S14B). Taken together, Figure S14 proved that Nano-ERASER degraded specific protein through a ubiquitin proteasome-mediated pathway.
To validate whether the nano-ERASER can be a potential therapeutic tool, coatomer protein complex ζ1 (COPZ1) protein was selected as our target. COPZ1 and COPZ2 are the two isoforms of coatomer protein complex 1 (COPI), which play a vital role in intracellular protein trafficking.[18] Normal cells express both COPZ1 and COPZ2 proteins, whereas COPZ2 is down-regulated in most cancer cells. As a result, cancer cells depend heavily on COPZ1 to carry out the protein sorting between the Golgi apparatus and endoplasmic reticulum (ER), which is essential for their proliferation.[19] Therefore, the depletion of COPZ1 would kill cancer cells, while sparing normal cells. The antibody of COPZ1, anti-COPZ1, was loaded into nanogels to yield NG-aCOPZ1 through the same procedures used for NG-aGFP fabrication. NG-aCOPZ1 has a hydrodynamic size of 140.8 nm with a dispersity of 0.21 (Figure S15) and carries a negative zeta potential (Figure S3). The LC and LE values of anti-COPZ1 in NG-aCOPZ1 nanogel were determined to be 3.1% and 81%, respectively. Owing to the significantly enhanced protein degradation efficiency after the RGD peptide modification, NG-aCOPZ1 was decorated with RGD as well to yield NG-aCOPZ1-R nanogel, which has a hydrodynamic diameter of 151.3 nm (dispersity: 0.22) (Figure 4A) and a spherical shape with a dry-state size of 121.0 nm (Figure 4B). The zeta potential of NG-aCOPZ1-R was less negative in comparison with NG-aCOPZ1 (Figure S3) due to the conjugation of the positively charged RGD peptide. Similar to NG-aGFP-R, NG-aCOPZ1-R showed outstanding stability in PBS and PBS supplemented with serum (Figure S16). It has been reported that silencing the expression of COPZ1 could inhibit the proliferation of cancer cells while sparing normal ones.[19] To probe whether anti-COPZ1 loaded nanogels can induce the degradation of COPZ1 protein, and subsequently results in the death of cancer cells, cell viability assay was adopted. In MCF-7 cells without TRIM21 transfection, all nanogels, including NG-empty, NG-aCOPZ1, and NG-aCOPZ1-R, induced negligible inhibitory effect on cell growth even when the concentration was high (Figure S17). However, in TRIM21-transfected MCF-7 cells, both NG-aCOPZ1 and NG-aCOPZ1-R nanogels inhibited cell growth in a dose-dependent manner (Figure 4C). At an anti-COPZ1 equivalent concentration of 40 μg/mL, the viability of cells was decreased to 44.7% and 27.9% by NG-aCOPZ1 and NG-aCOPZ1-R, respectively. As expected, free anti-COPZ1 induced no effect towards both TRIM21-transfected and non-transfected cells. The higher Trim-Away efficiency of NG-aCOPZ1-R should be ascribed to the targeting effect of RGD ligand. Meanwhile, the western blotting analysis showed that the amount of endogenous COPZ1 was considerably diminished in cells incubated with both nanogels (Figure 4D), as additionally evidenced by the quantified data (Figure 4E). Due to the lack of COPZ2 expression in cancer cells, the protein sorting between ER and Golgi apparatus solely relies on COPZ1. The selective degradation of COPZ1 by NG-aCOPZ1-R in the MCF-7 cells disrupted a crucial protein modification process required for cell proliferation. Therefore, it is the Trim-Away of COPZ1 protein that resulted in the death of the cancer cells. Moreover, fluorescence images of mCherry-TRIM21-transfected MCF-7/GFP cells before and after incubation with NG-aCOPZ1-R nanogel showed that the mCherry signal was almost completely diminished. In contrast, GFP signal was barely affected in the process of the Trim-Away COPZ1 protein (Figure 4F), demonstrating the high specificity of the Nano-ERASER technique in degrading a target protein. Taken together, these results suggest that the NG-aCOPZ1-R effectively delivered anti-COPZ1 antibody into cancer cells, degraded the targeted COPZ1 protein through the Trim-Away pathway, and killed the cancer cells. To assess the safety of the anti-COPZ1 Nano- ERASER, TRIM21-transfected NIH-3T3 cells were treated with the above anti-COPZ1-loaded nanogels. The non-significant cell death in Figure S18 suggests that the anti-COPZ1 Nano-ERASER is safe to normal cells (NIH3T3 cells), which was due to the existence of COPZ2, the counterpart of COPZ1, in the normal cells. All the results collectively confirmed that the Nano-ERASER technique can efficiently and specifically degrade an endogenous protein and has the potential to be exploited as a protein therapy modality for cancer treatment and other biomedical applications.
Figure 4.

Characterization and activity evaluation of NG-aCOPZ1-R. (A) Size distribution and (B) TEM image of nanogel NG-aCOPZ1-R. (C) Cell viability of TRIM21-transfected MCF-7 cells after being incubated with free anti-COPZ1 antibody, NG-empty, NG-aCOPZ1, and NG-aCOPZ1-R nanogels at different anti-COPZ1 equivalent concentrations. (D) The COPZ1 protein expression in TRIM21-transfected MCF-7 cells measured by western blotting assay after being incubated with free anti-COPZ1 and relevant nanogels, and (E) the corresponding quantitative analysis. Data represent the means ± SD, n=3. *P<0.05, #P <0.01. (F) Fluorescence of GFP (green) and TRIM21 (red) emitted from TRIM21-transfected MCF-7/GFP cells before and after being treated with NG-aCOPZ1-R nanogel.
3. Conclusion
In summary, an antibody covalently conjugated polymer nanogel system, Nano-ERASER, has been developed for intracellular delivery and release of antibodies and degradation of a specific endogenous protein. The antibodies are conjugated to the polymer via a redox-sensitive self-immolative linker, which can be cleaved by intracellular elevated GSH to release the bioactive payload. After being delivered into cells, the released antibody forms complex with its target protein. Subsequently, with the help of proteasome, the targeted protein can be efficiently depleted through a TRIM21-mediated degradation. Our study proved that Trim-Away could be achieved in a large number of cells through Nano-ERASER, without the requirement of any in vivo impractical skills, such as microinjection and electroporation, which is a significant step to translate the Trim-Away into a practical tool. Therefore, Nano-ERASER could be the third approach for depleting an endogenous protein in addition to RNAi-based gene silence and CRISPR-based gene deletion technologies. More importantly, Nano-ERASER specifically depletes a protein inside the cell without manipulating its genome. Notably, this new technique has been successfully employed to degrade a vital protein COPZ1 for cancer cells and kill them without causing damage to normal cells. Benefit from its low requirement for specific skills and equipment, Nano-ERASER technique is promising in providing a reliable and convenient tool for endogenous protein function study. Coupled with the targeted delivery merit of nanoparticles, Nano-ERASER paves the way for novel antibody-based Trim-Away therapeutic modalities for cancer and other diseases.
Supplementary Material
Acknowledgments
The authors want to thank National Institutes of Health (1R01AG054839–01A1, 1R41CA254500–01A1, and 1R21CA252360–01) for financial support of the research.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Binglin Sui, Department of Discovery and Biomedical Sciences, College of Pharmacy, University of South Carolina, 715 Sumter, Columbia, SC 29208 (USA).
Mingming Wang, Department of Discovery and Biomedical Sciences, College of Pharmacy, University of South Carolina, 715 Sumter, Columbia, SC 29208 (USA).
Chen Cheng, Department of Discovery and Biomedical Sciences, College of Pharmacy, University of South Carolina, 715 Sumter, Columbia, SC 29208 (USA).
Quanguang Zhang, Department of Neuroscience & Regenerative Medicine, Medical College of Georgia at Augusta University.
Jiajia Zhang, Department of Epidemiology and Biostatistics, University of South Carolina.
Daping Fan, Department of Cell Biology and Anatomy, University of South Carolina.
Peisheng Xu, Department of Discovery and Biomedical Sciences, College of Pharmacy, University of South Carolina, 715 Sumter, Columbia, SC 29208 (USA).
References
- [1].Leader B, Baca QJ, Golan DE, Nat. Rev. Drug Discov 2008, 7, 21. [DOI] [PubMed] [Google Scholar]
- [2].Gu Z, Biswas A, Zhao M, Tang Y, Chem. Soc. Rev. 2011, 40, 3638. [DOI] [PubMed] [Google Scholar]
- [3].a) Leavy O, Nat. Rev. Immunol 2010, 10, 297; [DOI] [PubMed] [Google Scholar]; b) Weiner GJ, Nat. Rev. Cancer 2015, 15, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC, Schuh M, Cell 2017, 171, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yoshimi R, Chang T-H, Wang H, Atsumi T, Morse HC, Ozato K, J. Immunol 2009, 182, 7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Shi J, Ma Y, Zhu J, Chen Y, Sun Y, Yao Y, Yang Z, Xie J, Molecules 2018, 23; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Y, Yu L-C, BioEssays 2008, 30, 606. [DOI] [PubMed] [Google Scholar]
- [8].a) Zhou CJ, Wang XY, Han Z, Wang DH, Ma YZ, Liang CG, Cell Cycle 2019, 18, 2784; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weir E, McLinden G, Alfandari D, Cousin H, Dev Biol 2021, 470, 74; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mehlmann LM, Uliasz TF, Lowther KM, Biol Reprod 2019, 101, 338; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Eisa AA, Bang S, Crawford KJ, Murphy EM, Feng WW, Dey S, Wells W, Kon N, Gu W, Mehlmann LM, Vijayaraghavan S, Kurokawa M, iScience 2020, 23, 101523; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chen X, Liu M, Lou H, Lu Y, Zhou MT, Ou R, Xu Y, Tang KF, Genome Biol 2019, 20, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Singh K, Ejaz W, Dutta K, Thayumanavan S, Bioconjugate Chem. 2019, 30, 1028; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sui B, Cheng C, Xu P, Adv. Therap 2019, 2, 1900062; [Google Scholar]; c) Yu M, Wu J, Shi J, Farokhzad OC, J. Controlled Release 2016, 240, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Fu J, Yu C, Li L, Yao SQ, J. Am. Chem. Soc 2015, 137, 12153; [DOI] [PubMed] [Google Scholar]; b) Yuan P, Zhang H, Qian L, Mao X, Du S, Yu C, Peng B, Yao SQ, Angew. Chem. Int. Ed 2017, 56, 12481. [DOI] [PubMed] [Google Scholar]
- [11].Lee Y, Ishii T, Kim HJ, Nishiyama N, Hayakawa Y, Itaka K, Kataoka K, Angew. Chem. Int. Ed 2010, 49, 2552. [DOI] [PubMed] [Google Scholar]
- [12].a) Dutta K, Hu D, Zhao B, Ribbe AE, Zhuang J, Thayumanavan S, J. Am. Chem. Soc 2017, 139, 5676; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dutta K, Kanjilal P, Das R, Thayumanavan S, Angew. Chem. Int. Ed. 2021, 60, 1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Bahadur K R C, Xu P, Advanced Materials 2012, 24, 6479; [DOI] [PubMed] [Google Scholar]; b) He H, Cattran AW, Nguyen T, Nieminen A-L, Xu P, Biomaterials 2014, 35, 9546; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Markoutsa E, Xu P, Mol. Pharmaceutics 2017, 14, 1591; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sui B, Cheng C, Wang M, Hopkins E, Xu P, Adv. Funct. Mater 2019, 29, 1906433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Taresco V, Alexander C, Singh N, Pearce AK, Adv. Therap 2018, 1, 1800030; [Google Scholar]; b) Yang HY, Li Y, Lee DS, Adv. Therap 2018, 1, 1800011. [Google Scholar]
- [15].Chen T, He B, Tao J, He Y, Deng H, Wang X, Zheng Y, Adv. Drug Delivery Rev 2019, 143, 177. [DOI] [PubMed] [Google Scholar]
- [16].He H, Markoutsa E, Li J, Xu P, Acta Biomater. 2018, 68, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang S, Yu G, Wang Z, Jacobson O, Tian R, Lin L-S, Zhang F, Wang J, Chen X, Adv. Mater 2018, 30, 1803926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Beck R, Ravet M, Wieland FT, Cassel D, FEBS Lett. 2009, 583, 2701; [DOI] [PubMed] [Google Scholar]; b) Dodonova SO, Diestelkoetter-Bachert P, von Appen A, Hagen WJH, Beck R, Beck M, Wieland F, Briggs JAG, Science 2015, 349, 195. [DOI] [PubMed] [Google Scholar]
- [19].Shtutman M, Baig M, Levina E, Hurteau G, Lim C.-u., Broude E, Nikiforov M, Harkins TT, Carmack CS, Ding Y, Wieland F, Buttyan R, Roninson IB, Proc. Natl. Acad. Sci. U. S. A 2011, 108, 12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.