

Abstract
Telomeropathies are a group of phenotypically heterogeneous diseases molecularly unified by pathogenic mutations in telomere-maintenance genes causing critically short telomeres. X-linked dyskeratosis congenita (DC), the prototypical telomere disease, manifested with ectodermal dysplasia, cancer predisposition, and severe bone marrow failure, is caused by mutations in DKC1, encoding a protein responsible for telomerase holoenzyme complex stability. To investigate the effects of pathogenic DKC1 mutations on telomere repair and hematopoietic development, we derived induced pluripotent stem cells (iPSCs) from fibroblasts of a DC patient carrying the most frequent mutation: DKC1 p.A353V. Telomeres eroded immediately after reprogramming in DKC1-mutant iPSCs but stabilized in later passages. The telomerase activity of mutant iPSCs was comparable to that observed in human embryonic stem cells, and no evidence of alternative lengthening of telomere pathways was detected. Hematopoietic differentiation was carried out in DKC1-mutant iPSC clones that resulted in increased capacity to generate hematopoietic colony-forming units compared to controls. Our study indicates that telomerase-dependent telomere maintenance is defective in pluripotent stem cells harboring DKC1 mutation and unable to elongate telomeres, but sufficient to maintain cell proliferation and self-renewal, as well as to support the primitive hematopoiesis, the program that is recapitulated with our differentiation protocol.
Keywords: induced pluripotent stem cells (iPSCs), telomeres, telomerase, X-linked dyskeratosis congenita, DKC1, hematopoietic differentiation
Introduction
Telomeres are specialized ribonucleoprotein structures at the chromosome termini and are composed of hexameric repetitive nucleotide sequences and capping proteins, preventing both degradation of chromosome ends by deoxyribonucleases, or fusion with other DNA molecules. Telomeres shorten during cellular mitotic division (Harley et al., 1990) and critically short telomeres engage cell senescence, apoptosis, or chromosome instability (Blasco, 2007).
In cells with high proliferative demand, telomere length is maintained by reverse transcription facilitated by the telomerase enzyme complex, which consists of the reverse transcriptase (TERT) and the RNA component (TERC), the template for synthesis of telomeric DNA (Cong et al., 2002). Additionally, the functional telomerase complex requires other proteins, such as dyskerin (encoded by the DKC1 gene) for its efficient biogenesis and stability (Schmidt and Cech, 2015). The absence of functional dyskerin (e.g., due to loss-of-function mutations in DKC1) leads to reductions in the TERC steady-state levels and consequently reduced telomerase activity (Wong and Collins, 2006).
Germline mutations in genes involved in telomere biology have been etiologically linked to a group of human disorders with a broad clinical spectrum, termed telomeropathies. The clinical phenotype reflects defective cell regeneration and a proclivity for cancer development (Calado and Young, 2009). X-linked dyskeratosis congenita (DC) is the prototype and firstly described telomeropathy (Mitchell et al., 1999). The disease is caused by mutations in DKC1 (Heiss et al., 1998) that results in very short telomeres. DC affects multiple organs, most commonly the bone marrow (80% of cases), the lungs, the liver, and mucocutaneous tissues (Dokal, 1999; Kirwan and Dokal, 2008). The hypocellular bone marrow and pancytopenia may be fatal by the second or third decades of life (Calado and Young, 2009; Knight et al., 1999; Kirwan and Dokal, 2009).
Modeling telomeropathies is challenging, as the cells from the tissues of interest are severely reduced in number (e.g., aplastic marrow, fibrotic liver, and lung) and animal models such as mice, fail in adequately recapitulate the human diseases (Calado and Dumitriu, 2013). Advances in pluripotent stem cells (PSCs)-based models provide useful alternatives for the investigation of mechanisms in degenerative pathologies (Jung et al., 2015). Both embryonic stem cells (ESC) and induced PSCs (iPSCs) express telomerase, which is essential to maintain their pluripotent phenotype. During epigenetic reprogramming from somatic cells to iPSCs, TERT and other telomerase components such as TERC and DKC1 are up-regulated (Takahashi et al., 2007; Agarwal et al., 2010). Given the crucial role of telomerase for pluripotency as well as the ability of PSCs to differentiate into virtually any cell type, these cells represent an ideal model to study telomeropathies.
Previous studies on telomere dynamics in DKC1-mutant iPSCs were contradictory in some aspects of telomere regulation, even in iPSCs of the same genetic background. Agarwal et al. (2010) and Batista et al. (2011) derived iPSCs from DKC1-mutant fibroblasts (del37L) isolated from the same patient. Agarwal et al. have shown that telomeres erode immediately after derivation but elongate over time. They also found that TERC was up-regulated in the pluripotent state, which was suggested as sufficient to overcome the limitations imposed by the DKC1 mutation. In contrast, Batista et al. reported progressive telomere erosion and TERC down-regulation in five clones of DKC1[del37L] iPSCs compared to the parental fibroblasts, and eventual loss of self-renewal. The iPSCs could not be maintained in the undifferentiated state for more than 36 passages and underwent spontaneous differentiation.
These divergences in observations may be a consequence of clonal variability, heterogeneity of iPSCs in culture, and other stochastic events, such as the selection of clones that acquired de novo mutations or copy number variations (CNVs) during the reprogramming process (Agarwal and Daley, 2011). The contribution of alternative lengthening of telomeres (ALT) was not assessed in these studies. Telomere lengthening is mostly telomerase-dependent, but 15% of tumors maintain telomeres via telomerase-independent ALT mechanisms. These mechanisms are based on homologous recombination-mediated DNA replication (Bryan et al., 1997; Dunham et al., 2000) and may play a role in telomere maintenance in the iPSCs. In addition, the effect of mutated dyskerin on tissue development (i.e., hematopoiesis) was not addressed in these previous studies of DKC1-mutant iPSCs.
To further investigate telomere biology in DKC1-mutated iPSCs, we derived iPSCs from a patient harboring the most common DKC1 mutation in X-linked DC (p.A353V). The telomere length, telomerase activity, and ALT were assessed in three iPSC clones at several passages. Clones were carefully screened to exclude those exhibiting karyotype abnormalities or CNVs. We also asked whether dysfunctional dyskerin affects hematopoietic development and differentiation in our patient-derived model. To address this question, we performed hematopoietic differentiation of two clones.
Material and methods
Isolation of fibroblasts:
Patient dermal fibroblasts were obtained via punch biopsy from the upper medial arm. The biopsy was performed at the University Hospital, Ribeirão Preto School of Medicine, University of São Paulo (HC-FMRP-USP, Ribeirão Preto, SP, Brazil) after the approval by the local ethics committee (HCRP 8723/2006) following written informed consent of his legal guardian. The biopsy was cut into pieces of approximately one mm3 and placed into 10 mL 1×PBS supplemented with 100 mg/mL collagenase II, 2.5 U/mL dispase, and 10 U/μL DNase I (Gibco). The skin pieces were incubated for 45 minutes on an orbital shaker (200 rpm) at 37°C. Fragments were washed with DMEM medium containing 10% fetal bovine serum and 1% penicillin-streptomycin-gentamicin (Gibco) and cultured for ten days in 6-well tissue culture plates covered with glass slips. Fibroblasts that grew out of the tissue were enzymatically dissociated using 0.05% trypsin (Gibco) and passaged to tissue culture flasks for expansion. Skin fragments and fibroblasts were maintained in culture in an incubator at 37°C with ambient oxygen level and 5% CO2.
Derivation of iPSCs:
Induced pluripotent stem cells (iPSCs) were derived from dermal fibroblasts through exogenous expression of the four transcription factors OCT4, SOX2, KLF4, and MYC using the single polycistronic lentiviral vector STEMCCA as described by Sommer et al. (2009). The vector was provided by G. Mostoslavsky (Boston University, Boston, Massachusetts, USA). Patient fibroblasts at passage 7 were transduced with STEMCCA lentiviruses in a single round at a multiplicity of infection (MOI) of 5. Five days after transduction, fibroblasts were transferred to gelatin-coated culture dishes pre-seeded the day before with mouse embryonic fibroblasts (MEFs, from GlobalStem). Transduced fibroblasts were cultured in embryonic stem cell (ESC) medium: DMEM/F12, supplemented with 20% knockout serum replacement, 1 mM L-glutamine, 0.1 mM nonessential amino acids (all Gibco), 10 ng/mL recombinant human fibroblast-like growth factor-basic (Peprotech), and 0.1 mM 2-mercaptoethanol (Sigma-Aldrich). In the first seven days following transference to feeder cells (MEFs), transduced fibroblasts were cultured in ESC medium supplemented with 0.5 mM valproic acid (Stemgent). Cells were kept in an incubator (37°C) under normoxic condition. On day 29 post-infection, individual colonies presenting ESC-like morphology were mechanically collected and expanded on MEFs and ESC medium. The iPSC clones were cultured for several passages; for passaging, colonies were enzymatically dissociated into small clumps of cells using collagenase IV (Gibco) every seven days. To avoid interference of the MEFs in the experiments, the iPSCs were transferred to a feeder-free platform using Matrigel basement membrane matrix (Corning) and maintained in culture in mTeSR medium (Stem Cell Technologies). An iPSC clone derived from a subject presenting normal-for-age telomere length and no family history of telomere diseases was used as a control for experiments (provided by Winkler et al., 2013), as well as H1, a human ESC line (WiCell International Stem Cell Bank). The iPSCs and ESCs were maintained in culture in an incubator at 37°C with ambient oxygen level and 5% CO2.
Removal of reprogramming transgenes:
The excision of integrated STEMCCA was performed by Cre-LoxP mediated recombination (Somers et al., 2010). For transient Cre-recombinase expression, the iPSCs were transduced by the non-integrating vector pCL20i4rEF1-Puro-T2A-cre-GFP (provided by Harry L. Malech, National Institute of Allergy and Infectious Diseases, Bethesda, Maryland, USA). Cells expressing Cre-recombinase were selected by puromycin treatment. Briefly, iPSCs were transfected with the vector using Lipofectamine LTX & Plus Reagent (Invitrogen) according to the manufacturer recommendations. Transfected cells were positively selected for 3 days in conditioned ESC medium containing puromycin (Sigma-Aldrich): day 1 = 3 μg/mL, days 2 and 3 = 2 μg/mL. After selection, MEFs were added, and ESC medium was changed daily until individual cells formed colonies. After 15 days, colonies presenting ESC-like morphology were mechanically collected and expanded on MEFs and ESC medium for several passages and then transferred to feeder-free culture on Matrigel (Corning). A multiplex PCR was performed to assess the copy numbers of STEMCCA transgenes integrated into the genome of the reprogrammed cells and to confirm their removal (Supplementary methods).
Sequencing:
Exon 11 of the DKC1 gene (NM_001363.4) was amplified by PCR using Platinum PCR SuperMix High Fidelity kit (Invitrogen), 200 ng DNA, and 0.2 μM primers (Table S3). PCR products were purified with the QIAquick PCR purification kit (Qiagen) followed by sequencing reaction using the BigDye Terminator Sequencing kit (Applied Biosystems), and purification of sequencing reaction through DyeEx 2.0 spin kit (Qiagen) according to the manufacturers’ recommendations. Products were loaded in the sequence analyzer ABI Prism 3100 (Applied Biosystems); chromatograms were analyzed with the CLC Main Workbench software v5.7 (CLC bio).
Immunocytochemistry:
Endogenous pluripotency markers were stained in iPSC clones as well as in the H1 ESC line (positive control). Cells were cultured in a 96-well plate for two days in MEFs and ESC medium. Cells were fixed with 4% paraformaldehyde for 30 minutes at room temperature. Then, cells were permeabilized with 0.2% Triton X (Sigma-Aldrich) for 30 minutes at room temperature, followed by blocking (1×PBS containing 3% bovine serum albumin [Gibco] and 5% donkey serum [Jackson ImmunoResearch]) for 2 hours at room temperature. Primary antibodies diluted in blocking buffer (1:100) were added followed by incubation at 4°C overnight. The next day, primary antibodies were removed, and cells were incubated with diluted secondary antibodies (1:500) at 4°C for 3 hours. Secondary antibodies were removed, and cells were washed with 1×PBS. Nuclei were stained using Vectashield with DAPI (Vector Labs). The cells were visualized under a fluorescence microscope (AxioImager.M2, Carl Zeiss). Images were acquired in the Applied Spectral Imaging software (Applied Spectral Imaging Ltd). Primary and secondary antibodies are provided in the Table S2.
Karyotyping:
The karyotyping was performed by WiCell Research Institute (Madison, WI, USA). For each iPSC clone, a total of 20 metaphases were counted and eight analyzed; four metaphases were karyotyped in a band resolution of either 425–450 or 450–475.
Quantitative PCR for telomere length:
Total DNA was extracted from pellets containing around 1×106 cells using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s recommendations. The integrity was assessed loading 100 ng DNA in a 1% agarose gel. The quantitative PCR for telomere length was adapted from methods described by Cawthon (2009), according to Gutierrez-Rodrigues et al. (2014). PCR reactions consisted of SYBR Green PCR Master Mix (Qiagen), 1.6 ng of DNA per reaction, and primers forward and reverse. Primers for either telomere repeats (T) or the single-copy gene 36B4 (S) were added at 900 nM (T forward, T reverse, and S reverse) and 300 nM (S forward). Sequences are provided in Table S5. PCR reactions were pipetted in triplicate using the Qiagility robot (Qiagen). Amplification and quantification using the Rotor-Gene Q (Qiagen) were performed twice: first using primers for telomeric repeats and second using primers for the single-copy gene. Average of triplicates was calculated for each reaction. The telomere length of each sample was represented as a relative T/S ratio. The final T/S ratio for a given sample represents the mean and the standard deviation (SD) of the three assays. A standard curve was run in each experiment using a control DNA at serial concentrations (10, 5, 2.5, 1.25, and 0.625 ng per reaction). An experiment was only accepted if standard curve had a correlation coefficient (r2 value) ≥ 0.9 for both T and S runs. As validation samples, a control DNA, as well as DNA from umbilical cord blood, were run along with samples.
Southern blot:
TeloTAGGG Telomere Length Assay kit (Roche) was used according to the manufacturer’s protocols. A total amount of 800 ng of genomic DNA per sample was digested using restriction enzymes HinfI and RsaI (Fast Digest – Fermentas Life Sciences). All the steps of DNA digestion, gel electrophoresis, southern transfer, hybridization, and determination of mean Terminal Restriction Fragment (TRF) were performed according to protocols of TeloTAGGG kit.
RT-qPCR for TERT, TERC, and DKC1 expression:
Total RNA was extracted from cell pellets using the RNeasy Mini Kit (Qiagen) according to manufacturer’s recommendations. The RNA integrity was checked using the RNA 6000 Nano kit, then analyzed on a 2100 Bioanalyzer instrument (Agilent). RT-PCR was performed using 500 ng RNA and the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) according to the manufacturer’s recommendations. The qPCR reactions were prepared in duplicate, each one containing 2.5 μL cDNA, 5 μL 2× TaqMan Gene Expression Master Mix, 1 μL 10× TaqMan probe (Applied Biosystems), and 1.5 μL ultra-pure water. Conditions for reactions were: 50°C for 2 min, and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Data were acquired on a 7500 Real-Time PCR System. The gene expression was calculated using the 2−ΔΔCt method (Livak and Schmittgen, 2001), normalized to the GAPDH expression, and represented as relative to the H1 hESCs.
Telomeric repeat amplification protocol (TRAP):
In vitro telomerase activity was assessed by TRAP assay using the TRAPeze XL Telomerase Detection kit (Millipore) according to the manufacturer instructions. Following the PCR reactions, the fluorescence measurements were acquired in a spectrofluorometer (Versa Max, Molecular Devices). The fluorescence of each sample was measured using the excitation/emission parameters for fluorescein (495 nm/516 nm) and sulforhodamine (600 nm/620 nm), as manufacturer’s recommendations. Accordingly, to obtain valid quantitative results, a standard curve using serial dilutions of the TSR8 control template (from kit) was produced. Telomerase activity of HeLa cells (ATCC) was set as 100%. Telomerase activity of each sample was represented as the average and standard error of three independent assays.
APB assay:
The cells were fixed with 2% paraformaldehyde for 10 min at room temperature, permeabilized with 0.5% Triton X-100 for 10 min, and blocked (1% BSA in 1×PBS) for 1 h at 37°C. Then, cells were incubated overnight at 4°C with the mouse anti-human PML protein antibody (Abcam) diluted in blocking buffer (1:200). The primary antibody was removed, and cells were incubated for 1 h with diluted secondary antibody (1:500) donkey anti-mouse IgG Alexa Fluor 594 (Abcam). The secondary antibody was removed, proceeding then to telomere fluorescence in situ hybridization (telomere FISH). Cells were fixed again with 2% paraformaldehyde for 2 min, and dehydrated with cold ethanol series (70%, 85%, and 100%) for 2 min each step. Coverslips containing cells were pre-warmed at 85°C for 5 min in the dark. Hybridization buffer (70% formamide, 3 mM Tris, and 0.01% BSA in 1×PBS) was added to 200 nM PNA telomere FISH probe (TelG-FITC, PNA Bio). The mix was applied to the coverslips, then incubated for 15 min at 85°C. For hybridization, the coverslips were incubated in a humid chamber at room temperature for 2 h in the dark. Then, coverslips were washed (2×SSC plus 0.1% Tween-20) at 56°C for 15 min. Nuclei were counterstained with 1.5 μg/mL DAPI solution for 15 min at room temperature. The coverslips were mounted on slides and visualized in a fluorescence microscope (AxioImager.M2, Carl Zeiss). Images were acquired in the Applied Spectral Imaging software (Applied Spectral Imaging Ltd). Telomerase-deficient WI 38 VA13 subline 2RA cells (VA13, from ATCC) were used as the positive control.
C-circle assay:
The C-circle assay was performed according to methods described by Henson et al. (16). The final products of the C-circle reactions were blotted to a positively charged nylon membrane using a 96-well holder attached to a vacuum instrument. The membrane was hybridized and washed according to methods of the TeloTAGGG Telomere Length Assay Kit (Roche), using solutions from the kit. The human osteosarcoma cell lines U2-OS and Saos-2 (both from ATCC) were used as positive controls.
Hematopoietic differentiation:
Cells cultured in Matrigel (BD) and mTeSR medium (Stem Cell Technologies) were incubated in medium supplemented with 10 μM ROCK inhibitor (Millipore) for 1 h at 37°C. The cells were harvested with EDTA/PBS treatment (0.5 mM EDTA, Sigma-Aldrich) for 4 minutes at room temperature. Cell clumps were rinsed with mTeSR medium containing polyvinyl alcohol (Sigma-Aldrich) and transferred to ultra-low attachment plates (Corning Costar) to form embryoid bodies (EBs). On the following day, the medium was changed for STEMdiff APEL basal medium (Stem Cell Technologies) supplemented with 30 ng/mL VEGF, 30 ng/mL BMP4, 40 ng/mL SCF, and 50 ng/mL Activin A (all from R&D) for four days. Then, EBs were exposed for nine days to basal medium containing 300 ng/mL SCF, 300 ng/mL FLT3L, 10 ng/mL IL-3, 10 ng/mL IL-6, 50 ng/mL G-CSF, and 25 ng/mL BMP4 (all from R&D). At the end of day 13, EBs were collected and dissociated in single cells using 0.05% trypsin. Fifty thousand cells were seeded in methylcellulose medium with recombinant cytokines (MethoCult H4435) into 35 mm dishes. Fourteen days later, the grown colony-forming units (CFUs) were identified according to the standard morphological criteria and counted. The differentiation protocol was adapted from Ng et al. (2008) and Chadwick et al. (2003).
Statistical analyses:
The Prism software (GraphPad), version 5, was used for statistics and graph plotting. Linear regression was applied to qPCR and Southern blot results. One-way ANOVA was applied to compare telomerase activity among fibroblasts, iPSC clones and H1 ESC line at different passages.
Results
Derivation of DKC1[A353V] iPSCs
Induced pluripotent stem cells were derived from dermal fibroblasts of a 3-year-old male patient with severe DC (Fig. S1-A). The patient presented with short telomeres in his peripheral blood leukocytes, below the 10th percentile (7 kilobases) when compared to age-matched controls; mutational screening revealed a DKC1 mutation (c.1058 C>T, p.A353V). Patient’s clinical characteristics are available on Table 1. Reprogramming factors (OCT4, KLF4, SOX2, and MYC) were delivered using a lentiviral polycistronic vector under normoxic cell culture condition (21% O2). Twenty-seven independent iPSC clones were isolated and expanded, representing a reprogramming efficiency of 0.01%. The number of lentiviral integration sites was assessed by qPCR, and three clones with less than three viral integrations were selected for further experiments (referred to as c1, c2, and c3; Fig. S1-B). In order to reduce the potential effects of the inserted transgenes in some specific experiments, we derived three transgene-free clones by transient expression of Cre-recombinase, and the excision was confirmed by qPCR (Fig. S1-B).
Table 1.
Patient’s clinical characteristics.
| Patient (mutation) | Clinical manifestation | Family history |
|---|---|---|
| 3 years old, male DKC1 c.1058 C>T, p.A353V |
|
None (mother clinically healthy and wild-type for the DKC1 mutation) |
Transgene-free iPSC clones (c1-tf, c2-tf, and c3-tf) were further characterized for pluripotency. All clones retained the patient’s DKC1 mutation (Fig. 1-A), and were positive for the pluripotency markers SSEA4, TRA-1–81, NANOG, TRA-1–60, and OCT4 (Fig. 1-B). Spontaneous undirected in vitro differentiation (Fig. S2-A) showed downregulation of pluripotency marker genes (Fig. S2-B) and upregulation of endoderm, mesoderm, and ectoderm markers (Fig. S2-C). The three clones displayed differentiation ability and expression levels similar to the H1 hESC line.
Figure 1 – Characterization of the DKC1[A353V] iPSCs.
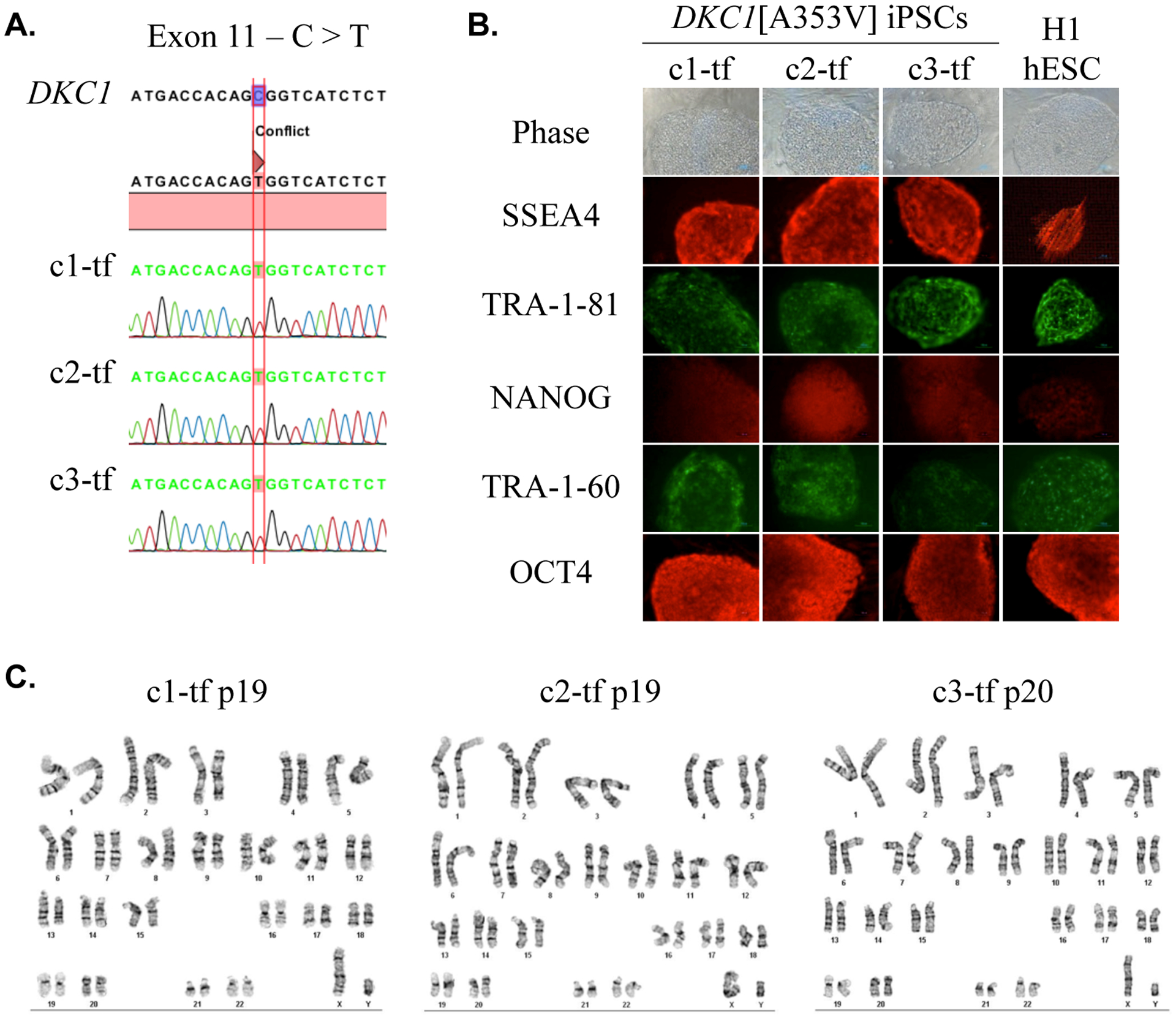
(A)DKC1 sequencing of the three iPSC clones (1-tf, 2-tf, and 3-tf) confirmed the nucleotide substitution C>T at position 1058 (exon 11) as found in the parental fibroblasts, which results in the missense mutation A353V. (B) Clones stained positive for the pluripotency markers SSEA4, TRA-1-81, NANOG, TRA-1-60, and OCT4. H1 hESC line is shown as positive control. Scale bar = 100 μm. (C) The three iPSC clones presented 46, XY normal karyotype in the band resolution (BR) analyzed: c1-tf, BR = 450–475; c2-tf and c3-tf, BR = 425–450.
The iPSC clones had normal karyotype in standard cytogenetic analysis (Fig. 1-C). A deeper investigation to detect DNA CNVs was performed at late passages (c1 p42, c2 p34, and c3 p40) using a single nucleotide polymorphism (SNP) array technology. No significant gain or loss of CNVs was detected. However, a deletion of 3 exons (17 to 19) of the DLG2 gene (chromosome 11) was detected in clone 2 after applying a more restrictive filter, which is not used in regular analyses for pathological lesions according to previous studies (Fig. S3). DLG2 has been implicated in cancer cell invasion (Chen et al., 2014) and, as it was not clear whether this alteration might play a role in the hematopoietic differentiation, the transgene-free version of the clone (c2-tf) was excluded from the differentiation experiments.
Telomere length shortens but stabilizes at late passages in the DKC1[A353V] iPSCs
In order to evaluate the telomere dynamics in the DKC1[A353V] iPSCs, we assessed telomere length, telomerase activity, and expression of genes encoding telomerase components (TERT, TERC, and DKC1) in three clones before transgene excision (c1, c2, and c3). Not cre-excised clones were used in telomere and telomerase dynamics experiments since Winkler et al. (2013) demonstrated that the presence of the reprogramming transgenes did not influence the expression of telomerase. Silencing of transgenes expressed by lentiviral vectors is a common phenomenon after reprogramming, thus retaining transgenes may not influence the expression of the endogenous telomerase genes (Winkler et al., 2013).
Consecutive telomere length measurements were performed by quantitative PCR (represented as a T/S ratio) along several passages (from 0 to 42). Compared to parental fibroblasts (time point 0), telomeres significantly eroded during the first passages. However, starting at passage 20, telomere length plateaued and did not significantly change upon prolonged culture time (Fig. 2-A). Clone 3 exhibited modest telomere elongation between passages 30 and 40. To validate the results, we also performed Southern blot analysis of seven samples previously analyzed by qPCR (Fig. 2-B). The correlation coefficient (r2 value) between results of both techniques was 0.95. In addition, telomere length was assessed in clones 1 and 3 after transgene removal at some passages (Fig. S4 and S5). We found that telomere length was similar to that observed in the clones at same passage before excision, supporting that the transgenes do not influence the telomere length dynamics.
Figure 2 – Telomere length over time and telomerase activity in DKC1[A353V] iPSCs.
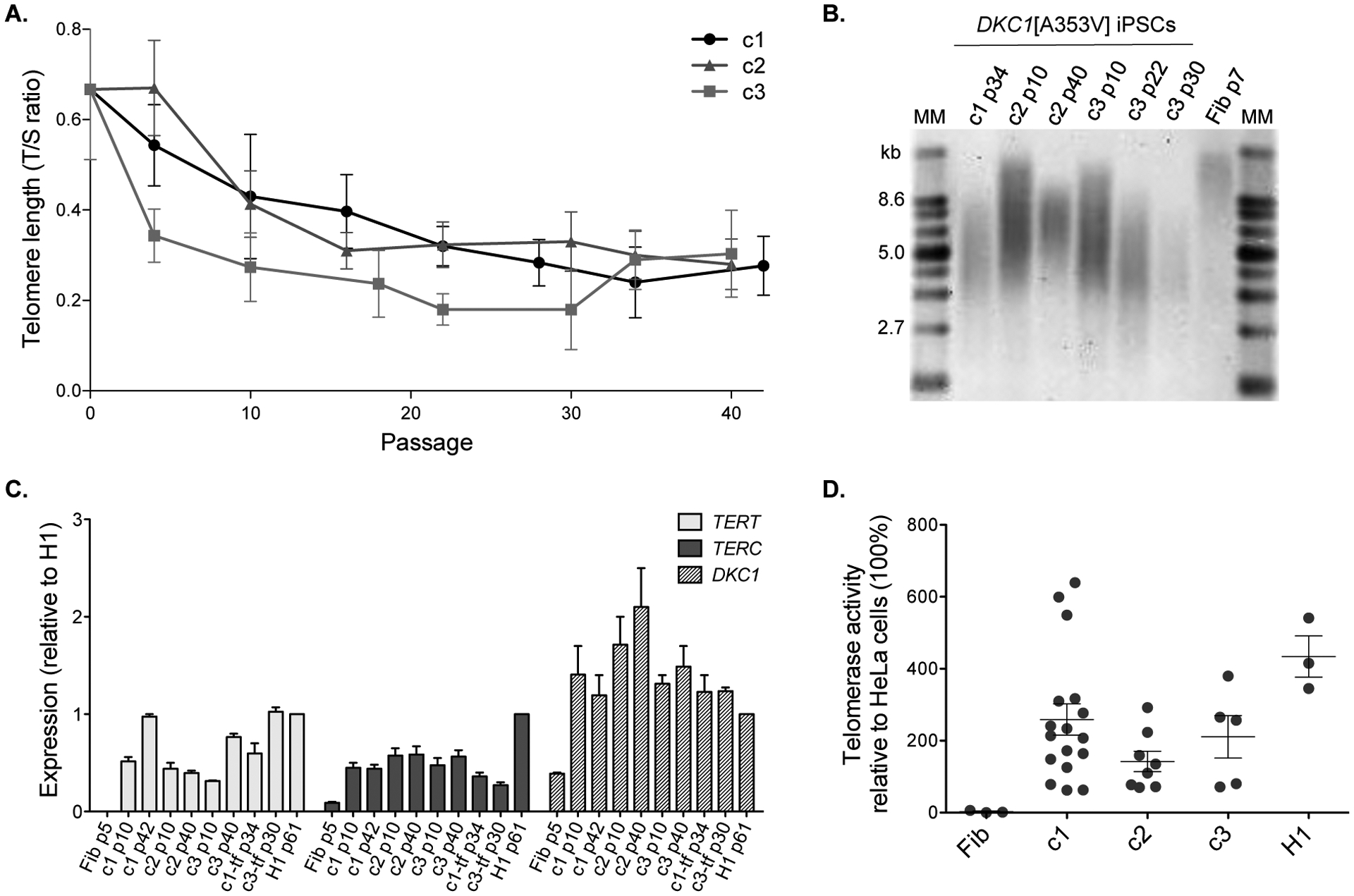
(A) Telomere lengths are represented as a T/S ratio. Passage 0 corresponds to the measurement of the parental fibroblasts (p7) immediately before reprogramming. (B) Telomere lengths by Southern blot from DKC1[A353V] fibroblasts (Fib p7) and iPSC clones 1, 2, and 3 at indicated passages (p). MM: molecular marker; kb: kilobases. (C) Expression levels of TERT, TERC, and DKC1 were assessed by RT-qPCR at different passages (p) in the clones (c) 1, 2, and 3, in the transgene-free clones 1-tf and 3-tf, and in the parental fibroblasts (Fib). The expression levels were normalized to GAPDH and then normalized to expression of the corresponding gene in H1 hESCs (control). (D) The telomerase activity of mutant iPSCs and H1 was measured using the TRAP assay and normalized to the activity of HeLa cells (set as 100%). Each circle represents a measurement of activity at a specific passage (p): fibroblasts at p7, iPSCs ranging from p8 to p116, and H1 at p35. Data on graph represent average and standard error of measurements at different passages for each clone or control. Activity was not statistically different (p > 0.05) between c1 and H1, and c3 and H1 (one-way ANOVA). Although the comparisons between fibroblasts and iPSCs were not statistically different, there is a clear trend of increased telomerase activity in the iPSC clones.
All clones were cultured long term and showed no sign of exhaustion (clone 1 up to passage 140), and maintained the ESC morphology without tendency of increased spontaneous differentiation in later passages (Fig. S6). These findings suggest that the DKC1[A353V] iPSCs were able to maintain telomere length stable at a sufficient level to guarantee self-renewal.
This telomere length behavior suggests functional telomere maintenance machinery in these pluripotent stem cells. Human pluripotent stem cells bypass telomere attrition and display unlimited capacity of division maintaining high levels of telomerase activity (Thomson et al., 1998). During the reprogramming process of somatic cells, TERT and TERC are up-regulated to sustain the pluripotent phenotype (Takahashi et al., 2007; Agarwal et al., 2010). Here we observed up-regulation of TERT, TERC, and also DKC1 in the three iPSC clones compared to the parental fibroblasts (Fig. 2-C). Telomerase activity was undetectable in the parental fibroblasts before reprogramming, but was similar to control ESCs at distinct passages (Fig. 2-D; telomerase activity was not statistically different between c1 and ESCs, and c3 and ESCs; p > 0.05, one-way ANOVA). There was no significant difference between activity measured in clones before and after excision of the reprogramming transgenes (Fig. S7; p > 0.05, one-way ANOVA).
Alternatively to telomerase, some cells maintain telomere length through telomerase-independent ALT mechanisms. In ALT-positive cells, telomeres are frequently colocalized with promyelocytic leukemia (PML) nuclear bodies, the ALT-associated PML bodies (APBs) (Yeager et al., 1999), which are the putative platforms for the recombination events. In our work, no colocalization of PML and telomeres was observed in the DKC1-mutant iPSCs at early, late, or very late passages (Fig. 3-A). Furthermore, recombination events in ALT-positive cells produce telomeres with heterogeneous lengths (Bryan et al., 1995), and lead to abundant extrachromosomal telomeric DNA that may be linear or circular (Tokutake et al., 1998; Cesare and Griffith, 2004; Henson et al., 2009). The accumulation of partially single-stranded telomeric (CCCTAA)n DNA circles (C-circles) represents a specific and quantifiable marker for ALT activity (Henson et al., 2009). DNA C-circles were not detected in the DKC1[A353V] iPSC clones (Fig. 3-B).
Figure 3 – Absence of alternative lengthening of telomeres in DKC1[A353V] iPSCs.
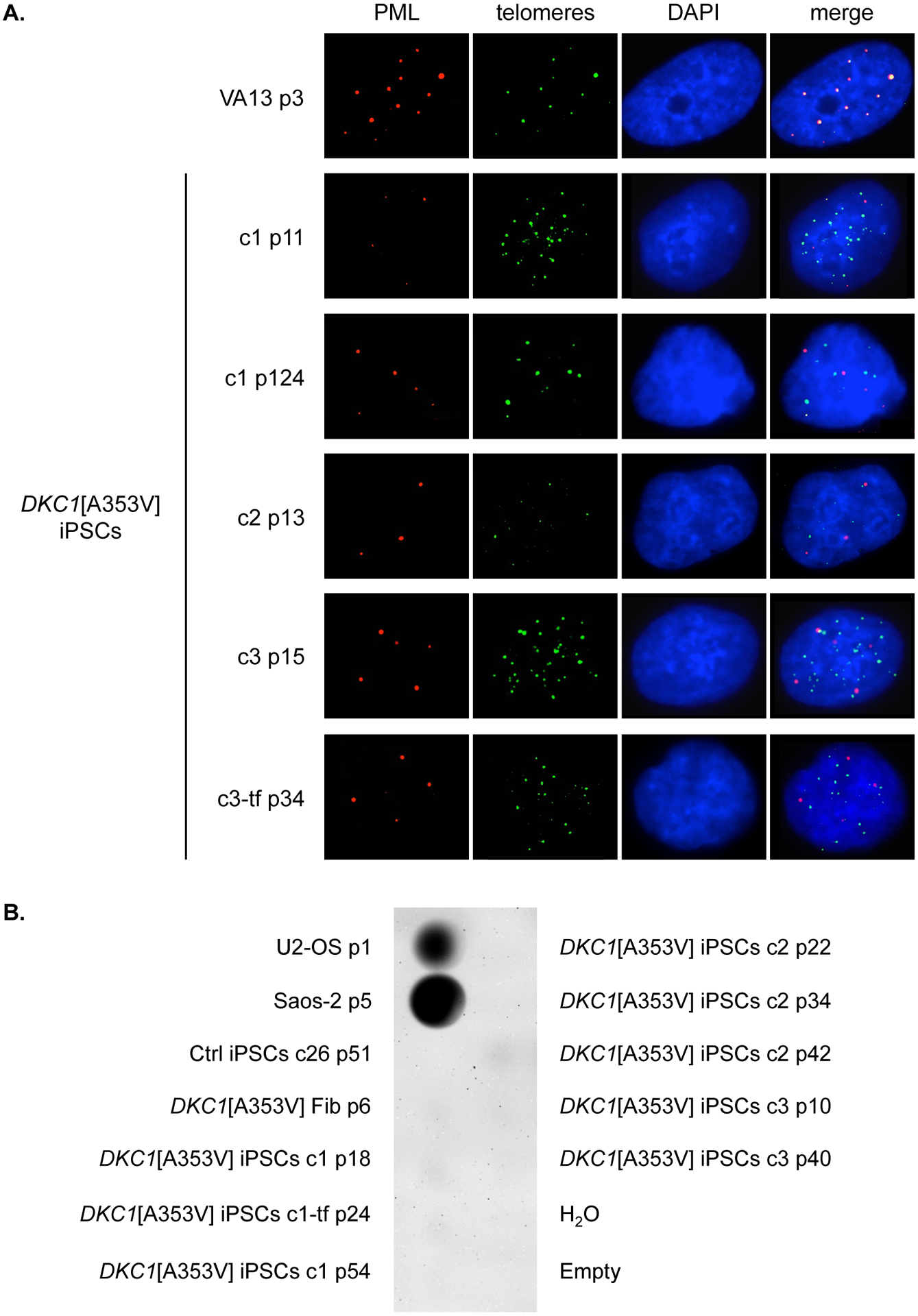
(A) Immunofluorescence of DKC1[A353V] fibroblasts (Fib p5) and iPSCs clones 1, 2, 3, and 3-tf at indicated passages (p). PML bodies were stained with an anti-PML antibody (red), and telomeres were labeled with FITC-conjugated PNA probe (green). Interphase nuclei were counterstained with DAPI (blue). Colocalization of PML bodies and telomeres was detected only in VA13 cells (positive control). (B) Dot blot of C-circle assay performed on genomic DNA from DKC1[A353V] fibroblasts (Fib p6) and iPSCs clones 1, 1-tf, 2, and 3 at indicated passages (p), as well as iPSCs from control (Ctrl iPSCs clone 26). Genomic DNA from the ALT-positive cell lines U2-OS and Saos-2 was set as positive control.
Collectively, these findings suggest that the mechanism of telomere length stabilization in the DKC1-mutant iPSC clones is likely to be mediated by telomerase, even in cells with a DKC1 mutation. However, we do not exclude that other unknown mechanisms may play a part in telomere elongation in these cells.
Increased hematopoietic differentiation capacity of DKC1[A353V] iPSCs
Since bone marrow failure is a hallmark of telomeropathies, hematopoietic differentiation was carried out in the DKC1-mutant clones to assess their capacity to derive hematopoietic lineages. The following experiments were performed in the clones after excision of the transgenes as, from our knowledge, no previous study has addressed the influence of exogenous reprogramming genes in the hematopoietic differentiation capacity of iPSCs. DKC1[A353V] clones 1-tf (p28) and 3-tf (p29 and p35) as well as H1 hESC line and previously described iPSC from healthy individuals we used as comparative controls (c26 p47, c26–3 p56, from Winkler et al., 2013). Differentiation was induced by exposing the iPSCs-derived EBs to a determined set of hematopoietic cytokines over a period of 13 days. Then, EBs were dissociated, and single cells were seeded on semi-solid methylcellulose-based medium containing cytokines for 14 days (Fig. 4-A). No difference in the formation and morphology of EBs was observed between control and DKC1-mutant iPSCs during the first 13 days of differentiation (Fig. 4-B). After 14 days of semi-solid culture, the hematopoietic colony-forming units (CFUs) were classified and counted according to their morphology (Fig. 4-C and D). The DKC1-mutant iPSCs c1-tf p28 and c3-tf p29 displayed significantly increased capacity of hematopoietic differentiation in comparison to control iPSCs, as evidenced by the total number of CFUs (Fig. 4-C). Of note, the differentiation capacity of clone c3-tf at later passage (p35) was reduced in comparison to earlier passage (c3-tf; p29) (Fig. 4-C). Also, the CFUs appeared bigger in size in the DKC1-mutant when compared to the controls (Fig. 4-D).
Figure 4 – Hematopoietic differentiation of the DKC1[A353V] iPSCs.
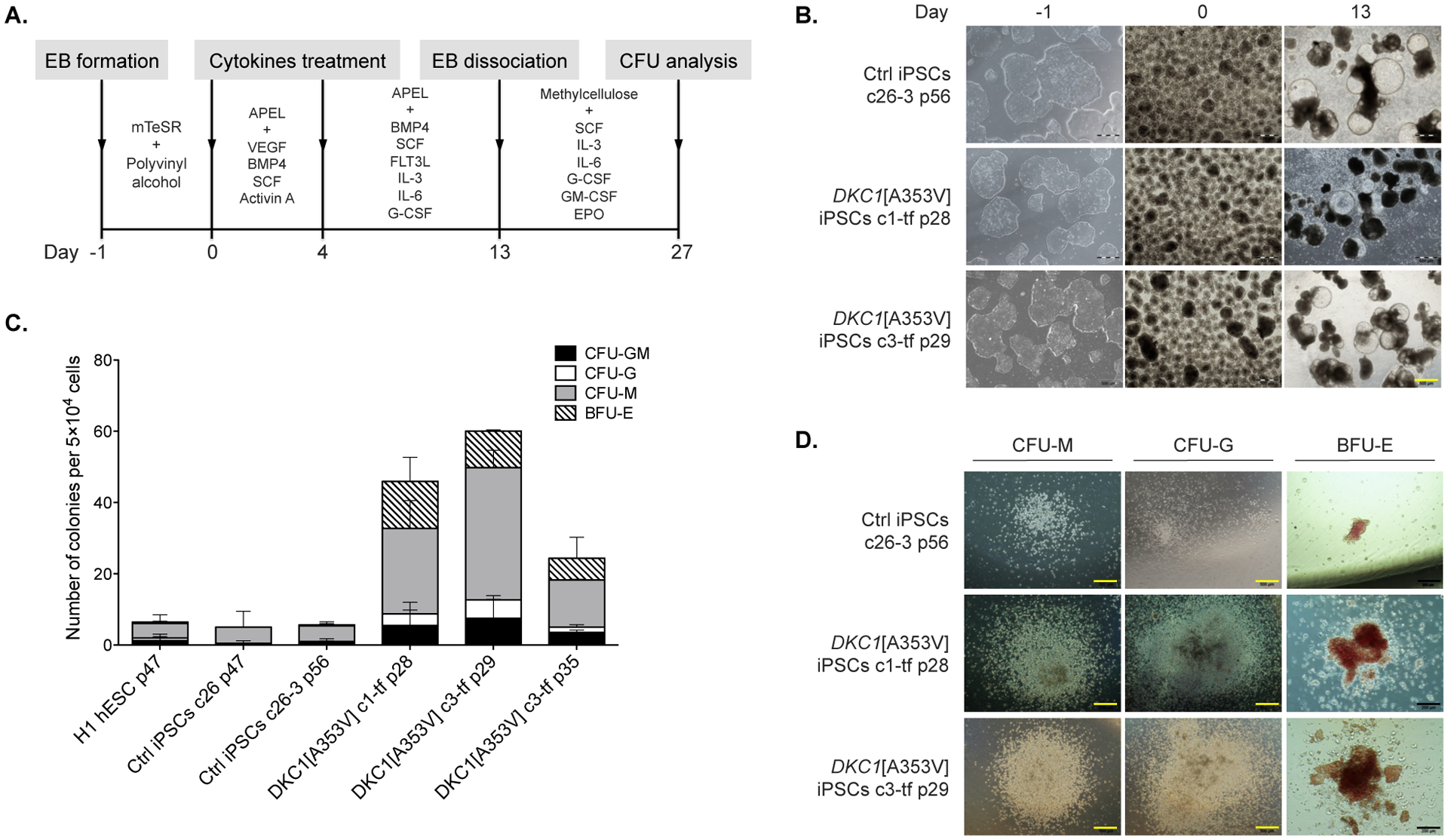
(A) Overview of differentiation: embryoid bodies (EBs) were derived from the iPSCs and guided to mesodermal differentiation with specific cytokines for three days (day 0 to 3). By day 4, the medium was replaced with a different cocktail of cytokines that promote maturation of the hematopoietic stem and progenitor cells. On day 13, EBs were harvested and dissociated, and single cells were seeded in methylcellulose containing cytokines. CFUs formed after 14 days in methylcellulose were classified and counted. (B) The iPSCs (control and DKC1-mutants) are shown during the differentiation process, from iPSC colonies in the undifferentiated state (on day −1), EBs on day 0, to differentiated EBs with cystic structures on day 13. Both iPSC mutants and control exhibited no morphological differences that could be detected under the inverted microscope during the differentiation. Yellow scale bar = 500 μm. (C) Qualitative and quantitative analysis of hematopoietic colonies derived from controls (H1 hESC, c26 and c26-3 iPSCs) as well as clones (c) of DKC1-mutant iPSCs at distinct passages (p). After CFU analysis, the DKC1-mutant iPSCs were found to be more prone to differentiate into myeloid lineages (c1-tf p28 and c3-tf p29). Counts are represented as the average and standard error of three independent experiments. CFU-GM, CFU-granulocyte, macrophage; CFU-G, CFU-granulocyte; CFU-M, CFU-macrophage; BFU-E, burst forming unit-erythroid. (D) Morphology of CFUs generated from iPSC control and DKC1-mutant clones. Black scale bars = 200 μm.
Discussion
In the present study, skin fibroblasts from a DC patient carrying a DKC1 mutation (p.A353V) were successfully reprogramed into the pluripotent state, retained the mutation from the parental cells, and did not acquire cytogenetic abnormalities or CNVs due to either the reprogramming process or the time in culture. The reprogramming efficiency (0.01%) was five-fold higher than that previously described for the iPSC derivation from DC-patient somatic cells (Agarwal et al., 2010; Batista et al., 2011). Our reprogramming experiments were conducted in normoxia (21% O2), whereas Batista et al. (2011) were able to reprogram DKC1-mutant fibroblasts with higher efficiency (0.04%) only after multiple rounds of viral infection and under hypoxia (5% O2), which is a condition that enhances the iPSC derivation (Yoshida et al., 2009).
The iPSCs are not a homogeneous clonal population in culture, and factors as culture conditions or stochastic events may yield variation in the experimental results. Telomere dynamics in iPSCs is heterogeneous even among multiple clones from the same starting cell population, but discrepancies are more evident in clones that acquire chromosomal abnormalities (Winkler et al., 2013). Thus, using multiple clones that were carefully screened for detection of cytogenetic abnormalities is crucial in assays with iPSCs (Chang et al., 2008; Jung et al., 2015). Here, three independent and cytogenetically normal DKC1-mutant clones were used for telomere dynamics experiments.
The missense change A353V, carried by the patient studied here, is the most common mutation in humans related to X-linked DC, found in approximately 40% of cases (Knight et al., 1999; Vulliamy et al., 2006). The A353V substitution affects the PUA RNA-binding domain of dyskerin, the putative site for interaction with TERC. Mouse ESCs carrying this genetic lesion display severe destabilization of Terc, reduction in telomerase activity, and progressive telomere erosion in vitro (Mochizuki et al., 2004). However, this mutation does not appear to completely abolish dyskerin function, and some residual activity may persist (Vulliamy et al., 2001).
In our DKC1-mutant iPSCs, telomeres showed severe erosion during the first passages after reprogramming of patient’s fibroblasts but reached stable short length after successive passaging, as opposed to clear telomere elongation that occurs in wild-type iPSCs (Winkler et al., 2013). The initial attrition observed in patient-derived iPSCs cannot be entirely attributed to the short telomeres of starting cells, as fibroblasts derived from older subjects display telomere elongation after reprogramming (Suhr et al., 2009). Thus, the reprogramming promotes elongation independent of the initial telomere length of somatic cells from healthy individuals. On the other hand, the telomere length of parental fibroblasts with impaired telomerase may influence later telomere dynamics. In a previous study, telomere erosion in iPSCs derived from patients with DKC1 mutations correlated with initial parental fibroblast telomere length (Gu et al., 2015). Of note, our patient had peripheral blood leukocytes’ telomere length between the first and 10th percentiles.
In addition, the initial erosion in telomeres of DKC1-mutant iPSCs may be explained, at least in part, by the late reactivation of telomerase during reprogramming. Telomerase is reactivated late along with endogenous Sox2 and Oct4 in a period when reprogrammed cells are already independent of exogenous transcription factors (Stadtfeld et al., 2008). In aggregate, deficient telomerase function of cells harboring a pathogenic DKC1 mutation along with delayed telomerase reactivation may explain the telomere attrition observed in the first passages of our iPSCs.
After the erosion in the first passages, telomere length stabilized and was sufficiently maintained to guarantee self-renewal. This may be explained by several reasons. First, the telomerase activity is the major mechanism responsible for telomere elongation in the induction of pluripotency in mice (Marion et al., 2009) and telomeropathy patient-derived iPSCs (TERT and TERC mutants) (Winkler et al., 2013). Although telomerase is potentially defective in our mutant cells, we observed DKC1 overexpression, which may compensate the deficient dyskerin function. As future approaches to provide insights into the mechanism behind the telomere maintenance in these iPSCs, knockdown-based experiments would be useful to address the effect of reduction in the DKC1 expression.
Second, ALT and telomerase pathways may coexist in the pluripotent state, as evidenced in murine (Wang et al., 2012) and porcine iPSCs (Ji et al., 2013) and may stabilize telomeres at later passages. ALT is activated in Terc−/− ESCs after crisis driven by extensive cell divisions (Niida et al., 2000). However, we failed to identify any evidence for active ALT mechanisms in DKC1-mutant clones while telomere lengths stabilize, suggesting that telomeres were maintained by a telomerase-dependent mechanism.
To our knowledge, no previous study addressed the hematopoietic differentiation potential of the DKC1-mutant pluripotent cells derived from patients’ somatic cells. The hematopoiesis in mammals is divided into two temporally and spatially distinct programs: the transient primitive, mainly in the fetus’ yolk sac, and the hematopoietic stem cell-driven adult-type, the definitive hematopoiesis, in the bone marrow (Orkin and Zon, 2008). The EB-based protocols for hematopoietic differentiation of iPSCs (the strategy we used) mostly recapitulate the primitive program, as the hematopoietic progenitors are not able to promote long-term reconstitution of all blood cell lineages after transplantation into mice (Vo and Daley, 2015).
Hematopoietic differentiation was carried out in two DKC1-mutant transgene-free iPSC clones, resulting in increased hematopoietic differentiation capacity compared to the controls. This observation is compatible with the phenotype of DC since patients usually do not present hematopoietic defects during fetal development (mostly driven by the primitive hematopoietic program); instead, hematopoietic defects manifest after birth, when the definitive hematopoietic program is responsible for giving rise to all blood cell lineages in the individual. In line with our findings, Fok et al. demonstrated that DKC1[A353V] hESCs with short telomeres produced significantly enhanced primitive erythroid colony-forming cells when differentiated into exclusively primitive hematopoietic progenitors.
The authors describe that the telomerase is tightly regulated throughout the hematopoietic specification and that short telomeres do not abrogate the differentiation capacity. The stable although short telomeres of our DKC1-mutant iPSCs represent a similar model compared to Fok’s model, and our findings corroborate their observations, although the mechanisms that favor the primitive hematopoietic program are not completely elucidated. Further investigation may be necessary, as it seems consistent that short telomeres favor primitive hematopoiesis. Understanding the mechanisms behind these in vitro observations might reflect the mechanisms that drive the apparent intact primitive hematopoiesis in the DC patients.
On the other hand, Fok et al. (2017) found that, in the definitive differentiation, the endothelial-to-hematopoietic transition was impaired in the DKC1[A353V] hESCs due to DNA damage accumulation and p53 signaling. As a future strategy to investigate whether the behavior of our patient-derived iPSCs is compatible with the CRISPR-derived DKC1-mutant hESCs, differentiation towards the definitive hematopoiesis may be tested in our iPSCs.
In a study modeling another bone marrow failure syndrome (the GATA2 deficiency) using patient-derived iPSCs, the differentiation towards mesoderm, hemogenic endothelial precursors (HEP), hematopoietic stem and progenitor cells, and natural killer cells was not significantly impaired. However, the maturation of HEP to hematopoietic lineages was defective in the GATA2-mutant iPSCs compared to isogenic controls, suggesting that the patient’s iPSCs were able only to reproduce the earlier steps of the hematopoiesis (Jung et al., 2018).
In conclusion, our findings suggest that the effect of the DKC1 mutation in the cells is context-dependent: in the pluripotent stage, telomere maintenance provided by telomerase with mutant dyskerin is reduced but sufficient to guarantee self-renewal. However, when cells undergo differentiation, critically short telomeres favor primitive hematopoietic potential.
Supplementary Material
Acknowledgments
This work was supported by the São Paulo Research Foundation (FAPESP) grant 13/08135-2 (FSD, RMAP, FGR, BAS, RTC), grants 12/18434-4 and 12/08119-4 (FSD doctoral scholarship).
Footnotes
The authors have declared that no conflict of interest exists.
References
- Agarwal S, and Daley GQ, 2011, Telomere dynamics in dyskeratosis congenita: the long and the short of iPS.: Cell Res, v. 21, p. 1157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD, Huo H, Okuka M, Dos Reis RM, Loewer S, Ng HH, Keefe DL, Goldman FD, Klingelhutz AJ, Liu L, and Daley GQ, 2010, Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients.: Nature, v. 464, p. 292–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, Giri N, Wernig M, Alter BP, Cech TR, Savage SA, Reijo Pera RA, and Artandi SE, 2011, Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells.: Nature, v. 474, p. 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA, 2007, Telomere length, stem cells and aging: Nat Chem Biol, v. 3, p. 640–9. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, and Reddel RR, 1997, Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines: Nat Med, v. 3, p. 1271–4. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Gupta J, Bacchetti S, and Reddel RR, 1995, Telomere elongation in immortal human cells without detectable telomerase activity: EMBO J, v. 14, p. 4240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado RT, and Young NS, 2009, Telomere diseases: N Engl J Med, v. 361, p. 2353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM, 2009, Telomere length measurement by a novel monochrome multiplex quantitative PCR method.: Nucleic Acids Res, v. 37, p. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, and Griffith JD, 2004, Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops: Mol Cell Biol, v. 24, p. 9948–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick K, Wang L, Li L, Menendez P, Murdoch B, Rouleau A, and Bhatia M, 2003, Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells: Blood, v. 102, p. 906–15. [DOI] [PubMed] [Google Scholar]
- Chang KH, Nelson AM, Fields PA, Hesson JL, Ulyanova T, Cao H, Nakamoto B, Ware CB, and Papayannopoulou T, 2008, Diverse hematopoietic potentials of five human embryonic stem cell lines: Exp Cell Res, v. 314, p. 2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, Parker M, Rusch M, Nagahawatte P, Wu J, Mao S, Boggs K, Mulder H, Yergeau D, Lu C, Ding L, Edmonson M, Qu C, Wang J, Li Y, Navid F, Daw NC, Mardis ER, Wilson RK, Downing JR, Zhang J, Dyer MA, and S. J. C. s. R. H. W. U. P. C. G. Project, 2014, Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma: Cell Rep, v. 7, p. 104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong YS, Wright WE, and Shay JW, 2002, Human telomerase and its regulation: Microbiol Mol Biol Rev, v. 66, p. 407–25, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokal I, 1999, Dyskeratosis congenita.: Br J Haematol, v. 105Suppl 1, p. 11–5. [PubMed] [Google Scholar]
- Dunham MA, Neumann AA, Fasching CL, and Reddel RR, 2000, Telomere maintenance by recombination in human cells: Nat Genet, v. 26, p. 447–50. [DOI] [PubMed] [Google Scholar]
- Fok WC, Niero ELO, Dege C, Brenner KA, Sturgeon CM, and Batista LFZ, 2017, p53 Mediates Failure of Human Definitive Hematopoiesis in Dyskeratosis Congenita: Stem Cell Reports, v. 9, p. 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu BW, Apicella M, Mills J, Fan JM, Reeves DA, French D, Podsakoff GM, Bessler M, and Mason PJ, 2015, Impaired Telomere Maintenance and Decreased Canonical WNT Signaling but Normal Ribosome Biogenesis in Induced Pluripotent Stem Cells from X-Linked Dyskeratosis Congenita Patients: PLoS One, v. 10, p. e0127414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Rodrigues F, Santana-Lemos BA, Scheucher PS, Alves-Paiva RM, and Calado RT, 2014, Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans: PLoS One, v. 9, p. e113747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, and Greider CW, 1990, Telomeres shorten during ageing of human fibroblasts.: Nature, v. 345, p. 458–60. [DOI] [PubMed] [Google Scholar]
- Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, and Dokal I, 1998, X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions: Nat Genet, v. 19, p. 32–8. [DOI] [PubMed] [Google Scholar]
- Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, and Reddel RR, 2009, DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity: Nat Biotechnol, v. 27, p. 1181–5. [DOI] [PubMed] [Google Scholar]
- Ji G, Ruan W, Liu K, Wang F, Sakellariou D, Chen J, Yang Y, Okuka M, Han J, Liu Z, Lai L, Gagos S, Xiao L, Deng H, Li N, and Liu L, 2013, Telomere reprogramming and maintenance in porcine iPS cells: PLoS One, v. 8, p. e74202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Cordes S, Zou J, Yu SJ, Guitart X, Hong SG, Dang V, Kang E, Donaires FS, Hassan SA, Albitar M, Hsu AP, Holland SM, Hickstein DD, Townsley D, Dunbar CE, and Winkler T, 2018, GATA2 deficiency and human hematopoietic development modeled using induced pluripotent stem cells: Blood Adv, v. 2, p. 3553–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Dunbar CE, and Winkler T, 2015, Modeling Human Bone Marrow Failure Syndromes Using Pluripotent Stem Cells and Genome Engineering: Mol Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan M, and Dokal I, 2008, Dyskeratosis congenita: a genetic disorder of many faces: Clin Genet, v. 73, p. 103–12. [DOI] [PubMed] [Google Scholar]
- Kirwan M, and Dokal I, 2009, Dyskeratosis congenita, stem cells and telomeres: Biochim Biophys Acta, v. 1792, p. 371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SW, Heiss NS, Vulliamy TJ, Greschner S, Stavrides G, Pai GS, Lestringant G, Varma N, Mason PJ, Dokal I, and Poustka A, 1999, X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene: Am J Hum Genet, v. 65, p. 50–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, Serrano M, and Blasco MA, 2009, Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells: Cell Stem Cell, v. 4, p. 141–54. [DOI] [PubMed] [Google Scholar]
- Mitchell JR, Wood E, and Collins K, 1999, A telomerase component is defective in the human disease dyskeratosis congenita.: Nature, v. 402, p. 551–5. [DOI] [PubMed] [Google Scholar]
- Ng ES, Davis R, Stanley EG, and Elefanty AG, 2008, A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies: Nat Protoc, v. 3, p. 768–76. [DOI] [PubMed] [Google Scholar]
- Niida H, Shinkai Y, Hande MP, Matsumoto T, Takehara S, Tachibana M, Oshimura M, Lansdorp PM, and Furuichi Y, 2000, Telomere maintenance in telomerase-deficient mouse embryonic stem cells: characterization of an amplified telomeric DNA: Mol Cell Biol, v. 20, p. 4115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI, 2008, Hematopoiesis: an evolving paradigm for stem cell biology: Cell, v. 132, p. 631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, and Cech TR, 2015, Human telomerase: biogenesis, trafficking, recruitment, and activation: Genes Dev, v. 29, p. 1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers A, Jean JC, Sommer CA, Omari A, Ford CC, Mills JA, Ying L, Sommer AG, Jean JM, Smith BW, Lafyatis R, Demierre MF, Weiss DJ, French DL, Gadue P, Murphy GJ, Mostoslavsky G, and Kotton DN, 2010, Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette: Stem Cells, v. 28, p. 1728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, and Mostoslavsky G, 2009, Induced pluripotent stem cell generation using a single lentiviral stem cell cassette: Stem Cells, v. 27, p. 543–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT, and Hochedlinger K, 2008, Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse: Cell Stem Cell, v. 2, p. 230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhr ST, Chang EA, Rodriguez RM, Wang K, Ross PJ, Beyhan Z, Murthy S, and Cibelli JB, 2009, Telomere dynamics in human cells reprogrammed to pluripotency: PLoS One, v. 4, p. e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S, 2007, Induction of pluripotent stem cells from adult human fibroblasts by defined factors.: Cell, v. 131, p. 861–72. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, and Jones JM, 1998, Embryonic stem cell lines derived from human blastocysts: Science, v. 282, p. 1145–7. [DOI] [PubMed] [Google Scholar]
- Tokutake Y, Matsumoto T, Watanabe T, Maeda S, Tahara H, Sakamoto S, Niida H, Sugimoto M, Ide T, and Furuichi Y, 1998, Extra-chromosomal telomere repeat DNA in telomerase-negative immortalized cell lines: Biochem Biophys Res Commun, v. 247, p. 765–72. [DOI] [PubMed] [Google Scholar]
- Vo LT, and Daley GQ, 2015, De novo generation of HSCs from somatic and pluripotent stem cell sources: Blood, v. 125, p. 2641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy TJ, Knight SW, Mason PJ, and Dokal I, 2001, Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita: Blood Cells Mol Dis, v. 27, p. 353–7. [DOI] [PubMed] [Google Scholar]
- Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, and Dokal I, 2006, Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation: Blood, v. 107, p. 2680–5. [DOI] [PubMed] [Google Scholar]
- Wang F, Yin Y, Ye X, Liu K, Zhu H, Wang L, Chiourea M, Okuka M, Ji G, Dan J, Zuo B, Li M, Zhang Q, Liu N, Chen L, Pan X, Gagos S, Keefe DL, and Liu L, 2012, Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells: Cell Res, v. 22, p. 757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler T, Hong SG, Decker JE, Morgan MJ, Wu C, Hughes WM, Yang Y, Wangsa D, Padilla-Nash HM, Ried T, Young NS, Dunbar CE, and Calado RT, 2013, Defective telomere elongation and hematopoiesis from telomerase-mutant aplastic anemia iPSCs: J Clin Invest, v. 123, p. 1952–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JM, and Collins K, 2006, Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita: Genes Dev, v. 20, p. 2848–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, and Reddel RR, 1999, Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body: Cancer Res, v. 59, p. 4175–9. [PubMed] [Google Scholar]
- Yoshida Y, Takahashi K, Okita K, Ichisaka T, and Yamanaka S, 2009, Hypoxia enhances the generation of induced pluripotent stem cells: Cell Stem Cell, v. 5, p. 237–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.