

Abstract
Cannabinoid receptors are a potential target for anti-inflammatory and pain therapeutics. There are two subtypes, CB1 and CB2, and Δ9-tetrahydrocannabinol activates both of them, providing an analgesic effect but also psychoactive side effects. The psychoactive side effects are considered to be caused by activation of CB1, but not CB2. ABK5 is a CB2 subtype selective agonist that has a very different structure from known cannabinoid receptor agonists. Here, we report anti-inflammatory effects of ABK5 using the T-cell line Jurkat cells, and antinociceptive effect in an inflammatory pain model in rats. Production of the cytokines IL-2 and TNF-α was measured in stimulated Jurkat cells and MOLT-4 cells, and CXCL12-mediated chemotaxis of Jurkat cells was evaluated by a transwell migration assay. Anti-inflammatory and antinociceptive effects of ABK5 were also evaluated in a hindpaw CFA model in rats. ABK5 significantly decreased production of IL-2 and TNF-α measured as both mRNA and protein levels, and reduced chemotaxis towards CXCL12. It also attenuated edema and increased mechanical threshold in the hindpaw of CFA-treated rats. These results suggest that ABK5 is a good lead compound for the development of potential anti-inflammatory and analgesic agents.
Keywords: Cannabinoid receptor CB2 agonist, Pro-inflammatory cytokines, Cell migration, Anti-inflammation, Inflammatory pain
1. Introduction
Cannabinoids – including drugs based on Δ9-tetrahydrocannabinol (THC), one of the most well studied cannabinoids – have various beneficial therapeutic effects. For example, cannabinoids are used to treat diseases or symptoms such as neuropathic pain and cancer-related pain, and chemotherapy-induced nausea and vomiting.1 Despite their beneficial effects, the use of THC-related drugs is limited by unwanted side effects, including psychoactive effects such as dizziness, disorientation, and impairment of memory.2 The main binding targets of THC and related drugs are cannabinoid receptors, which have two subtypes, CB1 and CB2. Cannabinoid receptors are G protein-coupled receptors (GPCRs), which typically couple to Gi protein and decrease cAMP production through inhibition of adenylate cyclase.3 CB1 is abundant in the central nervous system (CNS) and peripheral neurons, and is involved in the control of neurotransmitter release.4,5 Given this widespread physiological role, activation of CB1 via the orthosteric site is considered to cause unwanted psychoactive effects.5 On the other hand, CB2 is mainly localized on immune cells and involved in regulating release of chemicals such as inflammatory mediators.6,7 Although multiple studies suggest that expression of CB2 includes CNS neurons,6,8–11 activation of CB2 is not considered to cause psychoactive effects.12–15
Chronic pain, which is pain that lasts for more than 3 months, is a major public health problem in the United States. Approximately 20% of American adults have chronic pain and 8% suffer from severe pain that affects their daily life.16 Although various analgesic agents are available, those drugs can cause severe side effects or may not manage pain adequately. Therefore, efficacious analgesic agents with fewer side effects are needed. CB2 is a good target for developing new analgesic agents.13,14 Activation of CB2 in immune cells by endocannabinoids such as 2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamine (AEA) or by exogenous agonists such as THC, CP55940 and some other synthetic compounds reduced inflammation.17–22 Prolonged inflammation after injury causes chronic pain through excessive production of inflammatory mediators that sensitize pain-sensing neurons.23 Therefore, reduction of inflammation by CB2 activation is believed to contribute to alleviation of associated pain. CB2-mediated antinociception has been demonstrated in various pain models including inflammatory pain models, while no CB1-mediated psychoactive effects were observed.14,24–28
ABK5 is a CB2 subtype selective agonist with a distinct structure from known cannabinoid receptor ligands such as THC and CP55940 (Fig. 1). In a high throughput screening assay ABK5 was found to inhibit cAMP production through activation of CB2 and Gi coupling in cells expressing CB2.29 Previously, we reported that ABK5 bound CB2 with high affinity (Ki = 16 nM), with no CB1 binding, and caused G protein coupling and downstream signaling effects such as phosphorylation of ERK1/2 and MEK.30 In addition, ABK5 inhibited proliferation of Jurkat cells, which are a human T cell line,30 suggesting that ABK5 would have anti-inflammatory effects.
Fig. 1.
Chemical structures of a CB2 subtype selective agonist (ABK5) and some compounds that bind both CB1 and CB2 (THC and CP55940) as agonists.
In the present study, we further evaluated the pharmacological effects of ABK5 in anti-inflammation and antinociception assays. Cannabinoids are reported to inhibit the function of T cells21,31 which are important players in inflammation. For example, activation of CB2 in T cells reduced interleukin (IL)-2 production through inhibition of T cell receptor signaling,32 and a CB2 agonist also inhibited CXCL12-mediated chemotaxis of T cells.21 Therefore, Jurkat cells were selected to evaluate whether ABK5 also reduces pro-inflammatory cytokine production and decreases chemotaxis by CXCL12. Cytokine production was confirmed in a second T-cell line. In addition, we examined the effect of ABK5 in a Complete Freund’s adjuvant (CFA) model of inflammatory pain in rats.
2. Materials and methods
2.1. Drugs and reagents
ABK5 was purchased from ChemBridge Corporation (San Diego, CA USA). CP55940 and CLCX12 were obtained from R&D Systems (Minneapolis, MN USA). Complete Freund’s adjuvant (CFA; 1 mg/mL killed M. tuberculosis suspended in mineral oil), phytohemagglutinin (PHA) and phorbol 12-myristate 13-acetate (PMA) were purchased from Millipore Sigma (St. Louis, MO USA).
2.2. Cell culture and drug treatment
Jurkat cells and MOLT-4 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS and 1% antibiotic-antimycotic (Life Technologies, Carlsbad, CA USA) and were maintained at 37 °C and 5% CO2 saturation. Jurkat cells between passage four to seventeen were used in experiments.
For cytokine analysis, Jurkat cells and Molt-4 cells were seeded at 0.75 × 106 cells/mL and incubated with dimethyl sulfoxide (DMSO, <0.01%) or 0.1–10 μM CP55940 or ABK5 for 30 min. Then cells were stimulated by 1 μg/mL PHA and 12.5 ng/mL PMA for 3 and 24 h. Cells were harvested for RNA analysis and the culture supernatant was used for measurement of IL-2 and TNF-α concentrations by human IL-2 and TNF-α ELISA kits (Thermo Fisher Scientific; Waltham, MA USA) according to manufacturer’s instructions.
2.3. Quantitative real-time PCR
0.75 × 106 Jurkat cells were lysed and total RNA was extracted using TRIzol Reagent (Life Technologies, Carlsbad, CA USA) followed by reverse transcription by a High-Capacity cDNA Reverse Transcription kit (Life Technologies, Carlsbad, CA USA) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using an Applied Biosystems 7500 Fast Real-Time PCR system in a 10 μL reaction volume containing 2 μL diluted cDNA and 0.5 μM each of forward and reverse primers, and PowerUp SYBR Green Master Mix (Life Technologies, Carlsbad, CA USA). Primers used in the reaction are as follows: human 36B4 forward, 5′-GAAGTGCTTGATATCACAGAGGAA-3′, and human 36B4 reverse, 5′-TGATGCAACAGTTGGGTAGC-3′, and human IL-2 forward, 5′-AAGTTTTACATGCCCAAGAAGG-3′, and human IL-2 reverse, 5′-AGTCCCTGGGTCTTAAGTGAAA-3′, and human TNF-α forward, 5′-CAGCCTCTTCTCCTTCCTGAT′, human TNF-α reverse, 5′-GCCAGAGGGCTGATTAGAGA-3’. The PCR cycles are as follows: 50 °C for 2 min, 95 °C for 2 min, 40 cycles of 95 °C for 3 s and 60 °C for 30 s.
2.4. Migration assay
A Transwell Multiple Well Plate with permeable polycarbonate membrane inserts (0.5 μm, 24-well, Corning, Corning, NY USA) were used in migration assays. Jurkat cells were seeded at 2.5 × 106 cells/mL in RPMI medium containing 2.5% FBS and treated with DMSO or ABK5 for 2 h. Then 150 μL of cells were transferred to the top well. 400 μL of RPMI medium containing 0–1000 ng/mL CXCL12 and appropriate concentration of ABK5 was loaded to the bottom chamber. Cells were allowed to migrate for 2 h and cells migrated from the top well to the bottom chamber were counted or measured by CellTiter 96 AQueous One Cell Proliferation Assay (Promega; Madison, WI USA) following the manufacturer’s instructions. For evaluation of cell viability, the same procedure used for the migration assay was performed without the transwells and the viable cells were counted or measured by CellTiter 96 AQueous One Cell Proliferation Assay.
2.5. Animals
Adult female Sprague–Dawley rats were used (60–90 days old, weighing 175–275 g) for the antinociception assay. Rats were bred in-house from Envigo stock (Livermore, CA USA), and housed under a 12:12 h light:dark cycle (lights on at approximately 6 a.m.) in a vivarium room maintained at 21 ± 2 °C. Rats were housed in pairs or triplets, and had ad libitum access to food and water except during testing; cages contained TekFresh® bedding (Envigo, Denver, CO USA). Rats were tested during the light part of the cycle, at approximately 8 a.m. – 1 p.m. ABK5 was dissolved in DMSO to which 1:1:8 ethanol:cremaphor:saline was added, for a final DMSO concentration of 10%; this DMSO solution was used as the vehicle for testing control rats. ABK5 was administered i.p. in a volume of 2 mL/kg. All procedures were approved by the Institutional Animal Care and Use Committee, Washington State University, and rats were treated in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals (2011).
2.6. Apparatus for rat behavioral experiment
An electronic von Frey aesthesiometer (IITC Life Science, Woodland Hills, CA USA) was used to measure mechanical threshold. To assess hindpaw weight-bearing, an incapacitance meter (Stoelting, Wood Dale, IL USA) was used. Horizontal locomotor activity was measured using a photobeam apparatus (Optovarimex, Columbus Instruments, Columbus, OH USA), in which 15 photobeams cross the width of a 20 × 40 × 23-cm clear Plexiglas rodent cage, with photobeams spaced 2.5 cm apart and 6 cm above the cage floor. To assess edema, maximum dorsal-ventral hindpaw thickness was measured with calipers.
2.7. Behavioral procedure
Rats were randomly assigned to treatment groups (n 8–9/group). The experimenter was blinded to treatment group assignment (to ABK5 dose). On Day 1, rats were weighed, and then each was placed into a hanging wire cage that allowed access to the plantar surface of the hindpaws, to habituate for approximately 15 min. Then baseline measurements were taken on several tests. First, the threshold at which the rat responded when the von Frey probe was applied to the plantar surface of the right hindpaw was recorded in g, with three assessments made over approximately 2 min with 30 s between assessments. Subsequently, rats were placed into the incapacitance apparatus and weight-bearing in g on each hindpaw was recorded for 15 s, three times over approximately 1 min. Then rats were placed into the locomotor apparatus, and the number of photobeams broken in 10 min was recorded. Finally, right hindpaw thickness was measured in mm with calipers. Rats were then lightly anesthetized with isoflurane, and paw inflammation/pain was induced with a 0.1-mL injection of CFA into the plantar surface of the right hindpaw. Three h later (when pain/inflammation was near-maximal), vehicle or a single dose of ABK5 (5, 10 or 20 mg/kg) was injected i.p. The same dose was administered again at approximately 6 p.m., and again at approximately 8 a.m. and 6 p.m. on Days 2 and 3; rats were weighed each morning and injection volumes were adjusted accordingly. On Day 4 at approximately 8 a.m., the same dose was again injected, and rats were re-tested on all behavioral tests at 30, 60, 120, 180 and 240 min post-injection, to determine mechanical hypersensitivity (allodynia), biased weight-bearing (weight placed on the inflamed hindpaw), and locomotor activity (10-min periods). After all behavioral tests were completed at the 240-min time point, right hindpaw thickness was measured.
2.8. Data analysis
Results are presented as the mean ± one standard error of the mean (S.E.M.). For evaluation of CB2 mRNA levels in Jurkat cells, Student’s t-test was performed to determine significant differences between vehicle- and PHA/PMA-treated cells at each time point. For cytokine analysis (mRNA and protein concentration) and the migration assay, one-way ANOVA followed by Dunnett’s post hoc test was used to determine significant differences from the vehicle-treated group.
For the animal study, because there are individual differences in some measures (e.g., weight-bearing and paw thickness differ by rat body weight), each rat’s mechanical threshold, weight-bearing, and locomotor data after vehicle or drug treatment were transformed to percent of its own baseline scores (obtained on Day 1 before CFA was given). For example, for the von Frey test, at each time point after vehicle or drug, data were transformed as follows: (mechanical threshold in g/baseline mechanical threshold in g) × 100. Percent baseline data for the von Frey, weight-bearing, and locomotor tests were then analyzed by ANOVA, with variables of drug dose (4 levels) and time post-injection (5 levels, repeated measure). Paw thickness data were analyzed by one-way ANOVA, with the independent variable of drug dose (4 levels). Dunnett’s post hoc test was used to determine which doses differed from vehicle. For analyses of both biochemical and behavioral data, significance level was set at p < 0.05.
3. Results
3.1. Expression of cannabinoid receptors in jurkat cells and changes by PHA/PMA stimulation
In general, CB2 is expressed abundantly and endogenously in immune cells, but the level of expression may vary depending on the type of immune cells and the activation state of the receptor.33 Although we confirmed that Jurkat cells express CB2 when cells are not stimulated,30 stimulation of Jurkat cells may alter CB2 expression. Therefore, we first tested CB2 mRNA levels at various time points after stimulation by PHA/PMA using the conditions of a typical experiment. At 1.5 and 3 h of stimulation, CB2 mRNA decreased; in contrast, CB2 mRNA significantly increased at 24 h after stimulation (Fig. 2). At all time points, the Ct values were between 23 and 25, suggesting enough CB2 expression for testing ABK5 on these cells even at the lowest expression level. CB1 expression was also tested, but it was very low (Ct > 33) or not detected for all the time points used in our current experimental conditions (data not shown).
Fig. 2.
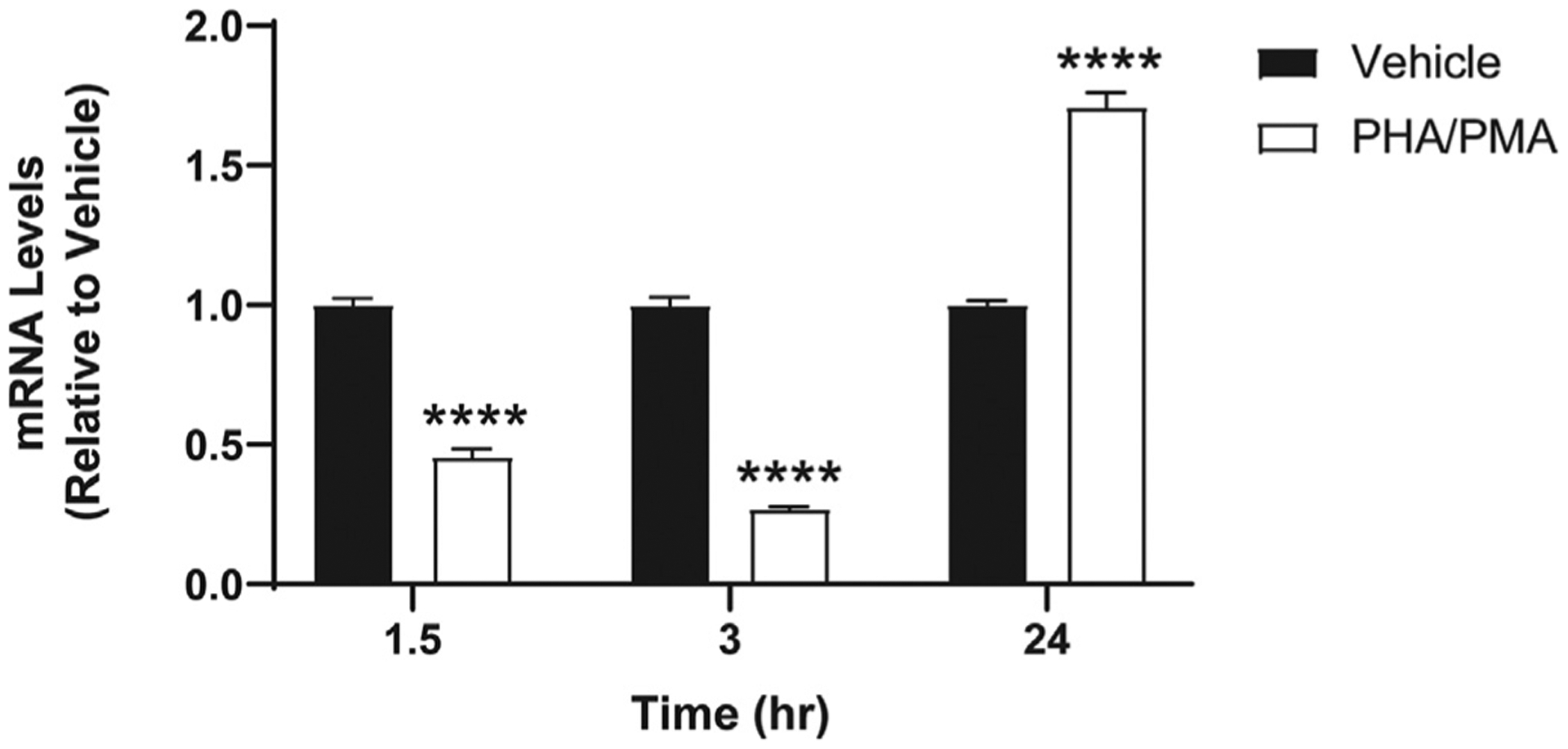
CB2 mRNA levels in Jurkat cells after PHA/PMA stimulation. Cells were treated with vehicle (equivalent volume of DMSO and water as the volumes of PHA and PMA used for treatment) or 1 μg/mL PHA and 12.5 ng/mL PMA for 1.5–24 h as described in Materials and Methods. Results are indicated as the mean ± S.E.M. of three independent assays performed in triplicate for each concentration. Student’s t-test was used and ****p < 0.05 versus vehicle control group.
3.2. ABK5 treatment inhibits PHA/PMA-induced IL-2 and TNF-α production in T cell lines
To test whether ABK5 has anti-inflammatory effects, we examined whether ABK5 can inhibit cytokine production in T cells. Jurkat cells are a human leukemic T cell line that are known to produce pro-inflammatory cytokines such as IL-2 and TNF-α after stimulation by PHA and PMA.34 First, to verify that activation of CB2 by an agonist inhibits cytokine production, we treated Jurkat cells with the non-subtype selective cannabinoid receptor agonist CP55940 and stimulated the cells by PHA/PMA. We evaluated the change in cytokine production by mRNA levels (Fig. 3A) and protein levels (Fig. 3B) released to the culture medium. CP55940 dose-dependently decreased IL-2 (F(3,32) = 33.18, p < 0.0001) and TNF-α (F(3,32) = 20.92, p < 0.0001) mRNA in cells. The highest concentration of CP55940, 10 μM, produced an 84% decrease in IL-2 mRNA relative to vehicle-treated cells, as well as a 41% decrease in TNF-α mRNA (Fig. 3A). Consistent with mRNA results, the IL-2 (F(3,32) = 25.84, p < 0.0001; Fig. 3B) and the TNF-α (F(3,32) = 46.47, p < 0.0001; Fig. 3C) concentration in the cell culture supernatant was also significantly decreased by the 10 μM CP55040 treatment. Similar to CP55940, ABK5 reduced IL-2 (F(3,30) = 61.00, p < 0.0001) mRNA levels in a dose-dependent manner, with the largest decrease at 10 μM (66% decrease compared to vehicle-treated cells) (Fig. 3D). The IL-2 protein released to the culture medium showed the same effect: it was significantly decreased at 10 μM ABK5 (F(3,32) = 11.86, p < 0.0001; Fig. 3E). ABK5 (10 μM) also reduced TNF-α mRNA (F(3,32) = 21.44, p < 0.0001; Fig. 3D) as well as protein (F(3,32) = 20.62, p < 0.0001; Fig. 3F). The stimulatory effect of PHA/PMA is confirmed in Fig. 3G, which shows about 1000-fold increase of IL-2 and 130-fold increase of TNF-α mRNA levels. The protein level of these cytokines was under the detection limit without PHA/PMA stimulation.
Fig. 3.
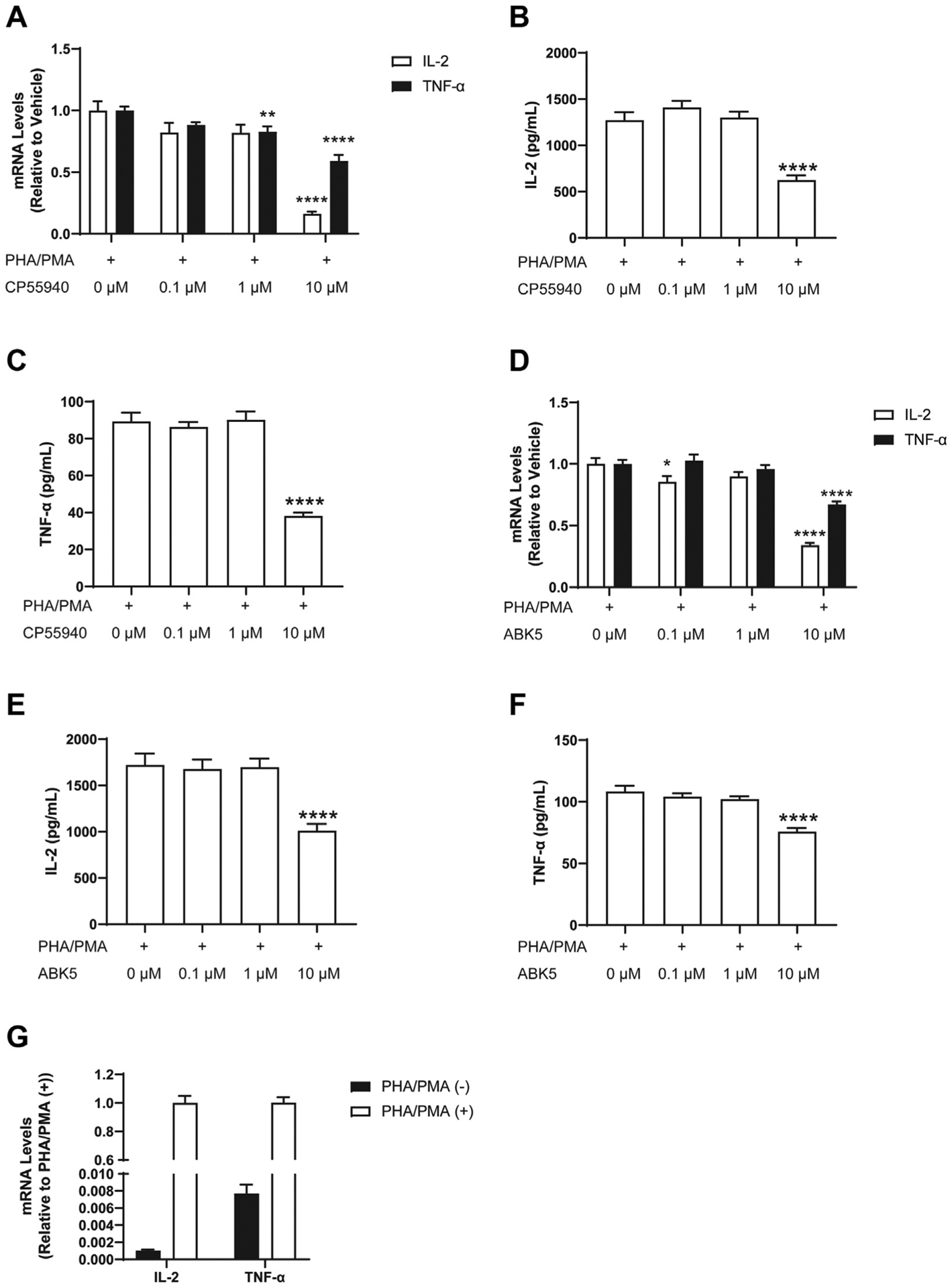
Effect of CP55940 and ABK5 on cytokine production. Jurkat cells were pretreated with DMSO (<0.01%) or various concentrations of CP55940 (A, B, C) or ABK5 (D, E, F) for 30 min and stimulated by PHA/PMA for 24 h. IL-2 and TNF-α mRNA levels were measured by RT-PCR in cells treated with (A) CP55940 and (D) ABK5. The concentrations of IL-2 and TNF-α protein released to the culture medium were tested by ELISA for cells treated with (B, C) CP55940 and (E, F) ABK5. The 0 μM CP55940 or ABK5 in frames A-F is just PHA/PMA stimulation alone. (G) Effect of PHA/PMA stimulation on IL-2 and TNF-α mRNA levels. Results are indicated as the mean ± S.E.M. of three independent assays performed in duplicate or triplicate for each concentration. One-way ANOVA plus Dunnett’s post-hoc test were used and *p < 0.05, **p < 0.01, ****p < 0.0001 versus vehicle control group.
The inhibitory effect of ABK5 was also tested in another human T cell line, MOLT-4 cells. PHA/PMA treatment significantly increased TNF-α mRNA levels in MOLT-4 cells, and pretreatment with 10 μM ABK5 decreased TNF-α mRNA levels (F(2,21) = 11, p < 0.0001; Fig. 4). This is consistent with the observation in Jurkat cells.
Fig. 4.
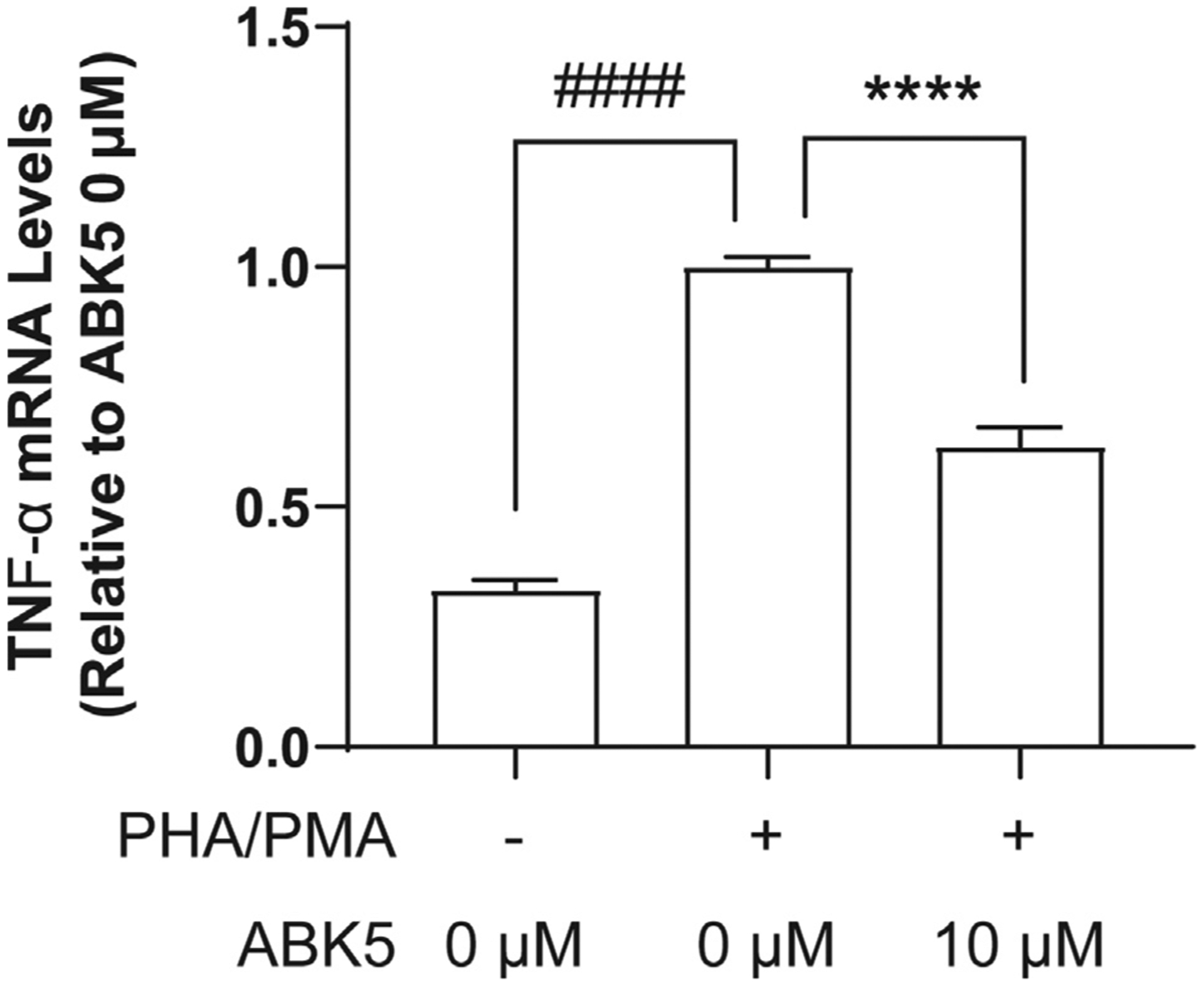
Effect of ABK5 on TNF-α mRNA levels in MOLT-4 cells. MOLT-4 cells were pretreated with DMSO (<0.01%) or 10 μM ABK5 and stimulated with PHA/PMA for 24 h. Results are indicated as the mean ± S.E.M. of three independent assays performed in duplicate or triplicate for each concentration. One-way ANOVA plus Dunnett’s post-hoc test were used and ****p < 0.0001 is versus DMSO and PHA/PMA treated (control group), ####p < 0.0001 is also versus DMSO and PHA/PMA treated (control group).
3.3. ABK5 decreases CXCL12 mediated jurkat cell migration
Chemotaxis of T cells is also an important response during inflammation, causing accumulation of T cells in inflamed tissue. To examine whether ABK5 can inhibit chemotaxis, we used a transwell migration assay to evaluate CXCL12-mediated migration of Jurkat cells. First, we determined the best CXCL12 concentration to promote cell migration. Various CXCL12 concentrations between 0 and 1000 ng/mL were added to the bottom chamber and cell migration was evaluated after 2 h incubation. Migration of Jurkat cell peaked at 100 ng/mL (Fig. 5A). Therefore, 100 ng/mL CXCL12 was used for later studies. Migration of Jurkat cells was significantly decreased by 10 and 25 μM ABK5 (F(2,23) = 34.68, p < 0.0001; Fig. 5B). Since ABK5 can inhibit cell proliferation, 30 we also evaluated whether cell viability had changed. Under the conditions used, no difference in cell viability was observed in cells treated with ABK5 relative to vehicle, which suggests that decreased cell migration is not due to a change in viable cell numbers (F(2,6) = 3.82, p = 0.0851; Fig. 5C).
Fig. 5.
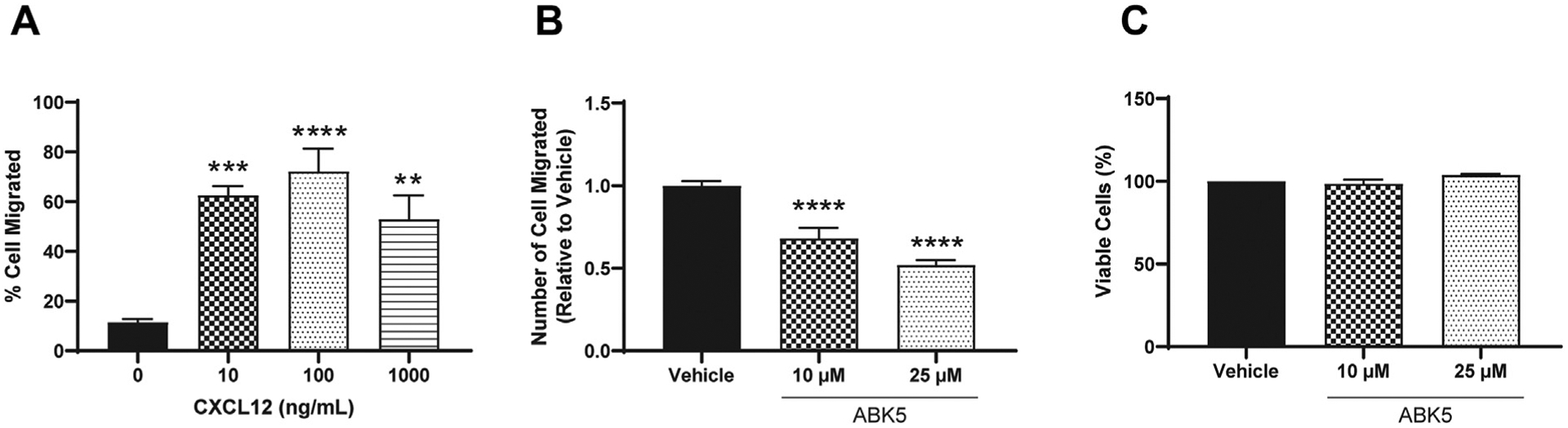
Effect of ABK5 on CXCL12 mediated Jurkat cell migration. (A) Jurkat cell migration in response to different concentrations of CXCL12. Various concentrations of CXCL12 were added to lower chamber and cells were allowed to migrate for 2 h. Cell numbers migrated to the lower chamber and are indicated as % cell migrated relative to cell numbers loaded on the top chamber. (B) Transwell migration of Jurkat cells. Cells were treated with vehicle or various concentrations of ABK5 for 2 h and then allowed to migrate through transwells for 2 h. Cell numbers migrated to the lower chamber are indicated as relative to vehicle (DMSO) alone. Results are indicated as the mean ± S.E.M. of three independent assays performed in triplicate for each concentration. (C) Cell viability after the migration assay. Cells were treated exactly the same but without transwells and viability was determined. Results are indicated as the mean ± S.E.M. of three independent assays performed for each concentration. One-way ANOVA plus Dunnett’s post-hoc test were used for both migration and viability assays. **p < 0.01, ***p < 0.001, ****p < 0.0001 versus vehicle control group.
3.4. ABK5 attenuates mechanical allodynia and hindpaw edema in rats
Since anti-inflammatory effects of ABK5 were observed in Jurkat cells, we evaluated its ability to decrease inflammation and pain in a rat model of inflammatory pain. Fig. 6 shows the effects of 3 days of twice-daily treatment with vehicle or ABK5 in CFA-treated rats. CFA-treated rats that received only vehicle for 3 days showed a reduction in mechanical threshold to approximately 60% of baseline, placed approximately 50% as much weight on the right hindpaw and locomoted approximately 50% as much after CFA compared to before CFA (see vehicle-treated group in each panel). Additionally, hindpaw thickness more than doubled (to approximately 215% of baseline) 3 days after CFA injection. ABK5 significantly increased mechanical threshold (F(3,31) = 5.75, p = 0.003; Fig. 6A) and reduced paw edema (F(3,31) = 3.04, p = 0.044; Fig. 6D) in CFA-treated rats, although it did not increase weight-bearing on the inflamed paw (Fig. 6B) or restore pain-suppressed locomotor activity (Fig. 6C).
Fig. 6.
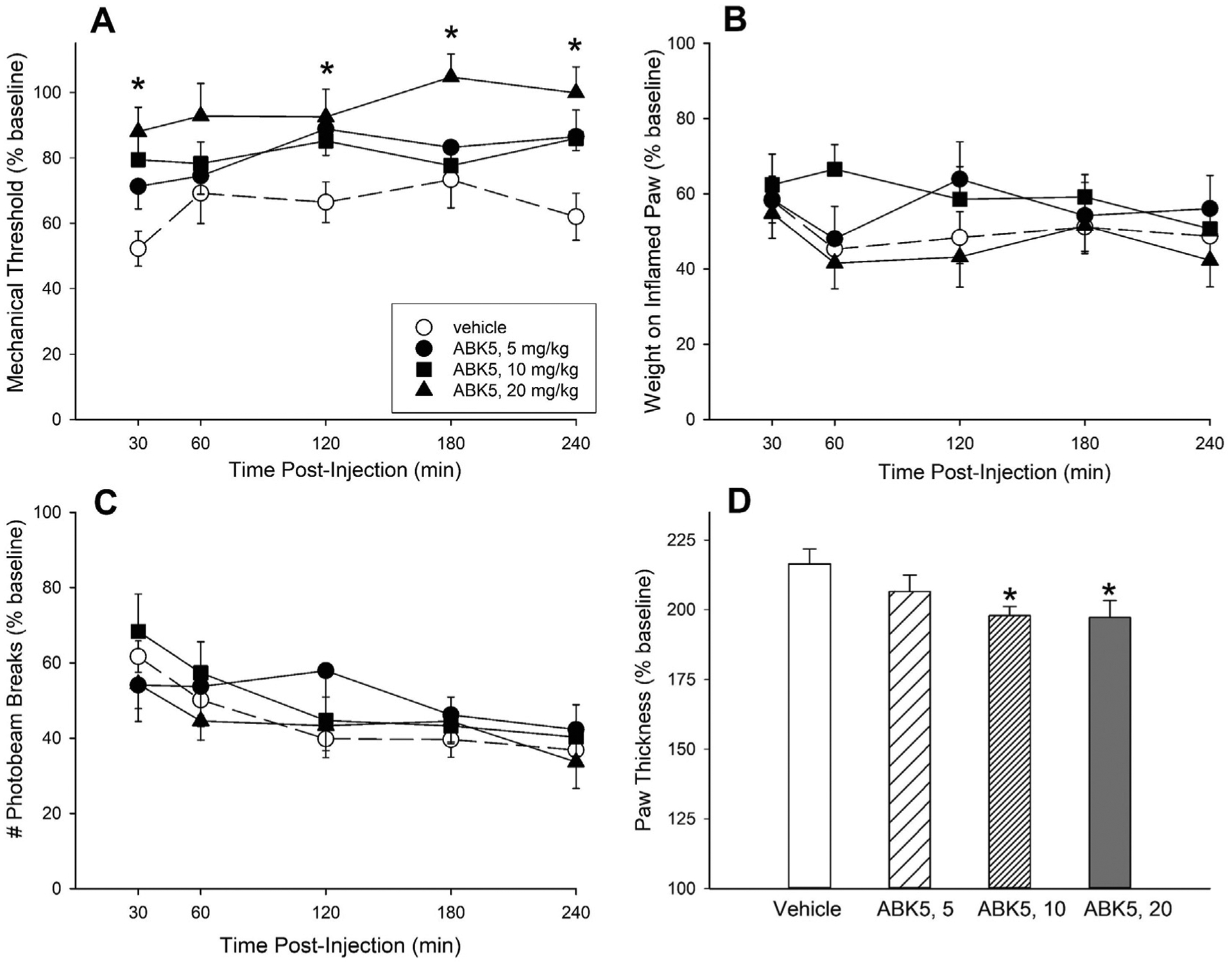
Effect of ABK5 on inflammatory pain and edema in female rats. CFA was injected into one hindpaw, and starting 3 h later, rats were treated twice-daily for 3 days with vehicle or ABK5 i.p. On Day 4, mechanical threshold (von Frey test; A), biased weight-bearing (B), and locomotor activity (C) were assessed from 30 to 240 min after the final injection. Paw thickness, an index of edema (D) was assessed after all behavioral tests were completed at the 240-min time point. Each point/bar is the mean ± 1 S E M. of 8–9 rats. *Significantly different from vehicle-treated controls, p < 0.05.
4. Discussion
Among the various therapeutic effects of cannabinoids, antinociceptive effects have been of particular interest. Due to the high prevalence of chronic pain in the United States16 and abuse of prescribed opioids,35 cannabinoids are considered a possible alternative class of analgesic agents. However, prototypical cannabinoids such as THC and its synthetic analogs can cause unwanted psychoactive side effects, likely caused by nonspecific activation of the two cannabinoid receptors CB1 and CB2.36 Although specifically activating CB2 (and not CB1) is considered to be a good strategy for developing new analgesic agents with fewer side effects, there is still no CB2 subtype selective agonist on the market.
Analgesic effects of cannabinoids are produced through blocking or modulating pain signals transmitted from the periphery to the brain and may also involve modulation of associated inflammation.37 Multiple studies have reported effects of endocannabinoids as well as plant-derived and synthetic cannabinoids on reducing cell proliferation, cytokine production, and chemotaxis in cell and animal models.17–21,31,32,38,39 The compound ABK5 is a CB2 selective agonist with CB2 Ki = 16 nM and no binding to CB1.30 We previously observed anti-proliferation effects of ABK5,30 however, it was not clear if ABK5 could inhibit cytokine production and migration. Using other compounds, CB2 activation has been reported to inhibit IL-2, TNF-α and IFN-γ production in Jurkat cells and activated peripheral blood mononuclear cells (PBMC)s.17,32 Our current results show that both the non-subtype selective agonist CP55940 and the subtype selective ABK5 inhibit IL-2 and TNF-α production in PHA/PMA-stimulated Jurkat cells, in a manner comparable to these prior studies. ABK5 also reduced TNF-α production in MOLT-4 cells, further evidence of the anti-inflammatory effect of the compound on T cells. Other CB2 agonists are also shown to inhibit CXCL12-mediated chemotaxis in Jurkat cells,21 which we also observed in cells treated with ABK5. These results suggest that ABK5 has anti-inflammatory effects through modulating multiple mechanisms of inflammation.
A 10 μM CP55940 treatment produced an 84% decrease of IL-2 mRNA and a 44% decrease of TNF-α mRNA relative to vehicle treatment, while ABK5 treatment produced 66% and 33% decreases of IL-2 mRNA and TNF-α mRNA, respectively (Fig. 3A, D). A decrease of these cytokines at the protein level by both CP55940 and ABK5 was also confirmed by ELISA (Fig. 3B, C, E, F), and the protein results showed the same trends as the mRNA results. Therefore, CP55940 had a slightly stronger anti-cytokine effect than ABK5 at the same dose; this was not surprising considering that CP55940 can activate both CB1 and CB2 receptors and has a higher binding affinity to these receptors than does ABK5. Although CB2 is known as the predominant cannabinoid receptor in immune cells, CB1 may also be expressed at a low level, and upregulation of CB1 in activated immune cells has been reported.40 However, CB1 expression was not detected or very low in the Jurkat cells used here with or without stimulation. Therefore, under our current experimental conditions, CB1 is less likely to be involved in the observed anti-inflammatory effects. This difference in CB1 expression can be due to the different stimulation method used (anti-CD3/CD28 antibody versus PHA/PMA), and it will be interesting to test the anti-inflammatory effects of ABK5 in different cells and under physiological conditions such as those offered by PBMCs activated by anti-CD3/CD28.40 However, the greater effect of CP55940 compared to ABK5 may simply reflect the somewhat higher affinity of CP55940 (Ki = 0.3 nM)30 than ABK5 (Ki = 16 nM)30 for CB2.
The anti-inflammatory effects of cannabinoid receptor agonists are suggested to be caused by inhibition of T-cell receptor activation, since cannabinoid receptor agonists inhibited anti-CD3 antibody-induced Jurkat cells and splenocyte proliferation but not PMA/ionomycin-induced splenocyte proliferation.41 However, inhibition of cytokine production including IFN-γ, TNF-α and IL-17 is reported in human PBMCs stimulated by PMA/ionomycin.15 In the present study, we stimulated Jurkat cells with PHA/PMA, which causes partial T-cell receptor activation but also non-T-cell receptor specific T-cell activation,42,43 and CP55940 and ABK5 inhibited IL-2 and TNF-α production. Taken together, these observations suggest that the anti-inflammatory effect of CB2 activation may not be only through inhibition of T-cell receptor activation but also through targeting T-cell receptor signal transduction.
A variety of endogenous and exogenous cannabinoid receptor agonists are reported to inhibit pain-related behaviors in rodent inflammatory pain models, in a CB1- and CB2-dependent manner.44 Since CB2 activation is not considered to cause psychoactive effects, some CB2 agonists have been tested on animals, and these reduce edema and pain-related behaviors.25,45 Our results showed that ABK5 significantly reduced paw edema, with efficacy similar to that of systemically administered THC,46 another indication that ABK5 is anti-inflammatory. ABK5 also significantly increased the mechanical threshold that had been decreased by CFA treatment, suggesting its capability of ameliorating pain. These results are consistent with prior studies using other putative CB2 selective agonists.25,45 Although ABK5 showed positive effects on these indices of inflammation and pain, CFA-induced biased weight-bearing and reduced locomotor activity were not reinstated by twice-daily treatment with ABK5. To the extent that weight-bearing and locomotion are considered more functional measures of pain than noxious stimulus-evoked paw withdrawal assays in rodents,47 these results suggest that selective CB2 activation alone may have limited analgesic efficacy, or that ABK5 lacks sufficient potency, efficacy, or duration of action in vivo to reinstate pain-suppressed activity. Since ABK5 is a lead compound found from a high throughput screening effort,29 further structure optimization needs be done to determine whether in vivo potency and/or efficacy can be increased.
In summary, our present study shows that the CB2 subtype selective agonist ABK5 reduces inflammation through inhibiting pro-inflammatory cytokine production and T-cell migration, and reduces mechanical allodynia and hindpaw edema in an inflammatory pain model in rats. These results suggest that ABK5 is a good lead compound as an analgesic agent for inflammatory pain. Developing drugs based on this compound may yield effective analgesic agents with fewer side effects than existing analgesics.
Acknowledgements
We thank Dr. Michael Lynes (University of Connecticut, Storrs, CT) for providing Jurkat cells and Dr. José Manautou (University of Connecticut, Storrs, CT) for his plate reader. This work was supported in part by National Institutes of Health, Grant DA040920.
List of nonstandard abbreviations
- ABK5
ethyl 2-(2-(N-(2,3-dimethylphenyl)phenylsulfonamido)acetamido)benzoate
- AEA
N-arachidonoylethanolamine
- 2-AG
2-arachidonoylglycerol
- CNS
central nervous system
- CFA
Complete Freund’s adjuvant; (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)-phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol
- DMSO
dimethyl sulfoxide
- GPCR
G protein-coupled receptor
- ERK
extracellular signal-regulated kinase
- IL
interleukin
- PMA
phorbol 12-myristate 13-acetate
- PHA
phytohemagglutinin
- PBMC
peripheral blood mononuclear cells
- PVDF
polyvinylidene fluoride
- RPMI
Roswell Park Memorial Institute
- THC
Δ9-tetrahydrocannabinol
- TNF
Tumor necrosis factor
Footnotes
Peer review under responsibility of Japanese Pharmacological Society.
Declaration of competing interest
The authors declare there are no conflicts of interest.
References
- 1.Pertwee RG. Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc Lond B Biol Sci. 2012;367(1607):3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol. 2009;156(3):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes. 2006;30(Suppl 1):S13–S18. [DOI] [PubMed] [Google Scholar]
- 4.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned DNA. Nature. 1990;346(6284):561–564. [DOI] [PubMed] [Google Scholar]
- 5.Busquets-Garcia A, Bains J, Marsicano G. CB(1) receptor signaling in the brain: extracting specificity from ubiquity. Neuropsychopharmacology. 2018;43(1):4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galiègue S, Mary S, Marchand J, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte sub-populations. Eur J Biochem. 1995;232(1):54–61. [DOI] [PubMed] [Google Scholar]
- 7.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. [DOI] [PubMed] [Google Scholar]
- 8.Liu Q-R, Pan C-H, Hishimoto A, et al. Species differences in cannabinoid receptor 2 (CNR2 gene): identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Gene Brain Behav. 2009;8(5):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stempel AV, Stumpf A, Zhang H-Y, et al. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the Hippocampus. Neuron. 2016;90(4):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashton JC, Friberg D, Darlington CL, Smith PF. Expression of the cannabinoid CB2 receptor in the rat cerebellum: an immunohistochemical study. Neurosci Lett. 2006;396(2):113–116. [DOI] [PubMed] [Google Scholar]
- 11.Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78(6):1192–1197. [DOI] [PubMed] [Google Scholar]
- 12.Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatr. 2015;77(5):475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malan TP Jr, Ibrahim MM, Deng H, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93(3):239–245. [DOI] [PubMed] [Google Scholar]
- 14.Malan TP, Ibrahim MM, Lai J, Vanderah TW, Makriyannis A, Porreca F. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol. 2003;3(1):62–67. [DOI] [PubMed] [Google Scholar]
- 15.Chen D-j, Gao M, Gao F-f, Su Q-x, Wu J. Brain cannabinoid receptor 2: expression, function and modulation. Acta Pharmacol Sin. 2017;38(3):312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlhamer J, Lucas J, Zelaya C, et al. Prevalence of chronic pain and high-impact chronic pain among adults - United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67(36):1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cencioni MT, Chiurchiu V, Catanzaro G, et al. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PloS One. 2010;5(1), e8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coopman K, Smith LD, Wright KL, Ward SG. Temporal variation in CB2R levels following T lymphocyte activation: evidence that cannabinoids modulate CXCL12-induced chemotaxis. Int Immunopharm. 2007;7(3):360–371. [DOI] [PubMed] [Google Scholar]
- 19.Joseph J, Niggemann B, Zaenker KS, Entschladen F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Canc Immunol Immunother. 2004;53(8):723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Do Y, McKallip RJ, Nagarkatti M, Nagarkatti PS. Activation through cannabinoid receptors 1 and 2 on dendritic cells triggers NF-kappaB-dependent apoptosis: novel role for endogenous and exogenous cannabinoids in immunoregulation. J Immunol (Baltimore, Md : 1950). 2004;173(4):2373–2382. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh S, Preet A, Groopman JE, Ganju RK. Cannabinoid receptor CB2 modulates the CXCL12/CXCR4-mediated chemotaxis of T lymphocytes. Mol Immunol. 2006;43(14):2169–2179. [DOI] [PubMed] [Google Scholar]
- 22.Yoshihara S, Morimoto H, Ohori M, Yamada Y, Abe T, Arisaka O. The cannabinoid receptor agonist WIN 55212–2 inhibits neurogenic inflammations in airway tissues. J Pharmacol Sci. 2005;98(1):77–82. [DOI] [PubMed] [Google Scholar]
- 23.Pinho-Ribeiro FA, Verri WA Jr, Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol. 2017;38(1):5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibrahim MM, Rude ML, Stagg NJ, et al. CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122(1–2):36–42. [DOI] [PubMed] [Google Scholar]
- 25.Quartilho ABS, Mata Heriberto PBS, Ibrahim Mohab MMS, et al. Inhibition of inflammatory hyperalgesia by activation of peripheral CB2Cannabinoid receptors. Anesthesiology: J Am Soc Anesthesiol. 2003;99(4):955–960. [DOI] [PubMed] [Google Scholar]
- 26.Rahn EJ, Thakur GA, Wood JAT, Zvonok AM, Makriyannis A, Hohmann AG. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: antinociception without central nervous system side-effects. Pharmacol Biochem Behav. 2011;98(4):493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kikuchi A, Ohashi K, Sugie Y, Sugimoto H, Omura H. Pharmacological evaluation of a novel cannabinoid 2 (CB2) ligand, PF-03550096, in vitro and in vivo by using a rat model of visceral hypersensitivity. J Pharmacol Sci. 2008;106(2):219–224. [DOI] [PubMed] [Google Scholar]
- 28.Masocha W, Thomas A. Indomethacin plus minocycline coadministration relieves chemotherapy and antiretroviral drug-induced neuropathic pain in a cannabinoid receptors-dependent manner. J Pharmacol Sci. 2019;139(4): 325–332. [DOI] [PubMed] [Google Scholar]
- 29.Ogawa LM, Burford NT, Liao Y-H, et al. Discovery of selective cannabinoid CB2 receptor agonists by high-throughput screening. Slas Discov: Advan Life Sci R&D. 2017;23(4):375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott CE, Tang Y, Alt A, et al. Identification and biochemical analyses of selective CB2 agonists. Eur J Pharmacol. 2019;854:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan M, Kiertscher SM, Cheng Q, Zoumalan R, Tashkin DP, Roth MD. Delta 9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. J Neuroimmunol. 2002;133(1–2):124–131. [DOI] [PubMed] [Google Scholar]
- 32.Borner C, Smida M, Hollt V, Schraven B, Kraus J. Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. J Biol Chem. 2009;284(51):35450–35460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basu S, Dittel BN. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res. 2011;51(1):26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manger B, Hardy KJ, Weiss A, Stobo JD. Differential effect of cyclosporin A on activation signaling in human T cell lines. J Clin Invest. 1986;77(5):1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G. Drug and opioid-involved overdose deaths - United States, 2013–2017. MMWR Morb Mortal Wkly Rep. 2018;67(5152):1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhopeshwarkar A, Mackie K. CB2 Cannabinoid receptors as a therapeutic target-what does the future hold? Mol Pharmacol. 2014;86(4):430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starowicz K, Finn DP. Cannabinoids and pain: sites and mechanisms of action. Adv Pharmacol. 2017;80:437–475. [DOI] [PubMed] [Google Scholar]
- 38.Lombard C, Nagarkatti M, Nagarkatti P. CB2 cannabinoid receptor agonist, JWH-015, triggers apoptosis in immune cells: potential role for CB2-selective ligands as immunosuppressive agents. Clin Immunol. 2007;122(3):259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tambaro S, Casu MA, Mastinu A, Lazzari P. Evaluation of selective cannabinoid CB1 and CB2 receptor agonists in a mouse model of lipopolysaccharide-induced interstitial cystitis. Eur J Pharmacol. 2014;729:67–74. [DOI] [PubMed] [Google Scholar]
- 40.Börner C, Höllt V, Kraus J. Activation of human T cells induces upregulation of cannabinoid receptor type 1 transcription. Neuroimmunomodulation. 2007;14(6):281–286. [DOI] [PubMed] [Google Scholar]
- 41.Lee M, Yang KH, Kaminski NE. Effects of putative cannabinoid receptor ligands, anandamide and 2-arachidonyl-glycerol, on immune function in B6C3F1 mouse splenocytes. J Pharmacol Exp Therapeut. 1995;275(2):529–536. [PubMed] [Google Scholar]
- 42.Altman A, Mally MI, Isakov N. Phorbol ester synergizes with Ca2+ ionophore in activation of protein kinase C (PKC)alpha and PKC beta isoenzymes in human T cells and in induction of related cellular functions. Immunology. 1992;76(3):465–471. [PMC free article] [PubMed] [Google Scholar]
- 43.Martin EPG, Arnaud J, Alibaud L, et al. Molecular mechanisms in the TCR (TCRαβ–CD3δε,γε) interaction with ζ2 homodimers: clues from a ‘phenotypic revertant’ clone. Int Immunol. 1999;11(7):1005–1015. [DOI] [PubMed] [Google Scholar]
- 44.Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153(2):319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elmes SJ, Winyard LA, Medhurst SJ, et al. Activation of CB1 and CB2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain. 2005;118(3):327–335. [DOI] [PubMed] [Google Scholar]
- 46.Craft RM, Kandasamy R, Davis SM. Sex differences in anti-allodynic, anti-hyperalgesic and anti-edema effects of Δ(9)-tetrahydrocannabinol in the rat. Pain. 2013;154(9):1709–1717. [DOI] [PubMed] [Google Scholar]
- 47.Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp Therapeut. 2006;319(2):507–514. [DOI] [PubMed] [Google Scholar]