

Abstract
Structure–activity relationships of 2-phenyl-imidazo[2,1-i]purin-5-ones as ligands for human A3 adenosine receptors (ARs) were investigated. An ethyl group in the 8-position of the imidazoline ring of 4-methyl-2-phenyl-imidazopurinone leading to chiral compounds was found to increase affinity for human A3 ARs by several thousand-fold. Propyl substitution instead of methyl at N4 decreased A3 affinity but increased A1 affinity leading to potent A1-selective AR antagonists. The most potent A1 antagonist of the present series was (S)-8-ethyl-2-phenyl-4-propyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (S-3) exhibiting a Ki value of 7.4 nM at rat A1 ARs and greater than 100-fold selectivity versus rat A2A and human A3 ARs. At human A1 ARs 2-phenyl-imidazo[2,1-i]purin-5-ones were generally less potent and therefore less A1-selective (S-3: Ki=98 nM). 2-, 3-, or 4-Mono-chlorination of the 2-phenyl ring reduced A3 affinity but led to an increase in affinity for A1 ARs, whereas di- (3,4-dichloro) or polychlorination (2,3,5-trichloro) increased A3 affinity. The most potent and selective A3 antagonist of the present series was the trichlorophenyl derivative (R)-8-ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (R-8) exhibiting a subnanomolar Ki value at human A3 ARs and greater than 800-fold selectivity versus the other AR subtypes. Methylation of 4-alkyl-2-phenyl-substituted imidazo[2,1-i]purin-5-ones led exclusively to the N9-methyl derivatives, which exhibited largely reduced AR affinities as compared to the unmethylated compounds. [35S]GTPγS binding studies of the most potent 2-phenyl-imidazo[2,1-i] purin-5-ones at membranes of Chinese hamster ovary cells expressing the human A3 AR revealed that the compounds were inverse agonists at A3 receptors under standard test conditions. Due to their high A3 affinity, selectivity, and relatively high water-solubility, 2-phenyl-imidazo[2,1-i]purin-5-ones may become useful research tools.
Introduction
A3 adenosine receptors (A3 ARs) are the youngest member of the AR family of G-protein-coupled receptors, which consists of four different subtypes, A1, A2A, A2B and A3.1 A number of selective antagonists for human A3 ARs have been developed during the past years.2 However, all of the potent and selective A3 antagonists developed so far, including triazoloquin-azolines (e.g., MRS-1177, MRS-1220),3,4 pyrazolotri-azolopyrimidines (e.g., MRE-3005F20, MRE-3008F20),5,6 isoquinolines (e.g., VUF-8504),7-9 and dihydropyridines (e.g., MRS-1334),10,11 are highly lipophilic and display a very low degree of water-solubility. More water-soluble A3 antagonists are required as pharmacological tools for in vitro and in vivo studies. A3 antagonists have potential as novel drugs; postulated therapeutic applications include inflammatory diseases, glaucoma, and stroke.2
Imidazo[2,1-i]purinones and related tricyclic purine derivatives derived from xanthines have been developed as water-soluble AR antagonists with selectivity for A1 or A2A ARs, respectively, depending on their substitution pattern.12-14 Recently, we discovered that a 2-phenyl-substituted imidazopurinone, namely (R)-8-ethyl-4-methyl-2-phenyl-imidazo[2,1-i]purin-5-one (R-1, PSB-11, Table 3) possesses high affinity for human A3 ARs (Ki = 2.3 nM) and is highly selective versus all other AR subtypes.13 The present study was aimed at investigating the properties and the structure–activity relationships of 2-phenyl-imidazopurinones as adenosine receptor ligands, particularly with regard to substituents at the 2-phenyl group, and at the nitrogen atoms N4 and N9.
Table 3.
Adenosine receptor affinities and selectivities of 2-phenylimidazo[2,1-i]purin-5-one derivatives

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | R2 | R4 | R8 |
Ki (nM)±SEM or % displacement at concentration indicated (in brackets) |
||||
| A1 Affinity rat brain cortical membranes [3H]CCPA |
A1 Affinity human brain cortical membranes [3H]CCPA |
A2A Affinity rat brain striatal membranes [3H]MSX-2 |
A2A Affinity human brain caudate-putamen membranes [3H]CGS-21680 |
A3 Affinity human recombinant receptors [125I]AB-MECA |
||||
| N1/N9-unsubstituted imidazopurinones | ||||||||
| R-1 (PSB-11) | Phenyl | Methyl | (R)-Ethyl | 44013a | 1640±50 | 210013b | 1280±430 | 2.313 3.5±0.39c |
| S-1 | Phenyl | Methyl | (S)-Ethyl | 11513a | 305±25 | 330013b | 440±80 | 9.813 |
| 2 | Phenyl | Methyl | H | 2230±200 | n.d.d | 14,100±4,460 | n.d. | 18,900±3200 |
| S-3 | Phenyl | Propyl | (S)-Ethyl | 7.4±1.4 | 98±12 | 863±203 | 3400±2400 | 860±640 |
| S-4 | 3,4-Dichlorophenyl | Propyl | (S)-Ethyl | 46±1 | n.d. | 26%e (2.5 μM) | n.d. | 116±17 |
| S-5 | 2-chlorophenyl | Methyl | (S)-Ethyl | 17±1 | 90±11 | 4170±350 | 2500±1200 | 420±340 |
| S-6 | 3-Chlorophenyl | Methyl | (S)-Ethyl | 30±2 | n.d. | 2530±240 | n.d. | 184±129 |
| S-7 | 4-Chlorophenyl | Methyl | (S)-Ethyl | 63±20 | n.d. | 2630±320 | n.d. | 12±1 |
| R-8 (PSB-10) | 2,3,5-Trichlorophenyl | Methyl | (R)-Ethyl | 805±55 | 1700±200 | 6040±260 | 2700±500 | 0.433±0.045c 0.997±0.311f |
| N9-methylated derivatives | ||||||||
| S-9 | 3,4-Dichlorophenyl | Propyl | (S)-Ethyl | 2800±1,100g | 36,000±16,500g | 13,000±2,000g | 6600±600g | 2350±1150f,g |
| R-10 | 2,3,5-Trichlorophenyl | Methyl | (R)-Ethyl | 12%±1e (10 μM) | 35,500±18,500g | 17%±1e (10 μM) | 0%e (10 μM) | 395±55f |
Determined versus [3H]CHA.
Determined versus [3H]CGS21680.
Determined versus [3H]NECA.
n.d. = not determined.
Percentage inhibition at concentration indicated.
Determined versus [3H]PSB-11.
Estimated value; full curve could not be determined due to limited water solubility.
Results and Discussion
Chemistry
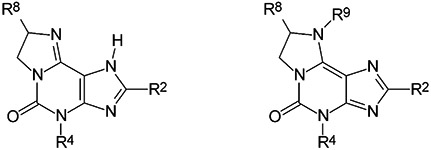
2-Phenylimidazo[2,1-i]purin-5-one derivatives were synthesized from the corresponding 3-methyl- or 3-propyl-substituted 8-phenylxanthine derivatives, respectively, in analogy to described procedures.13-16 Imidazopurinones S-3 and R-8 were methylated using methyl iodide and sodium hydride in dry dimethylformamide to yield N9-methylated derivatives S-9 and R-10 (Fig. 1). Methylation of a 2-unsubstituted imidazo[2,1-i]purin-5-one had been reported to yield the N1-methylated product.15 For the 2-phenyl-substituted imidazo[2,1-i]purinones, we found methylation to occur predominantly or exclusively at N9. If the methylation was performed in dimethylformamide using methyl iodide in the presence of potassium carbonate N9- and N1-methylated products were formed in 85 and 15% yield, respectively.14 The use of sodium hydride instead of potassium carbonate led to the exclusive formation of N9-methylated products S-9 and R-10 (Fig. 1).
Figure 1.

Tautomeric structures and alkylation of imidazo[2,1-i]purin-5-ones.
The position of the methyl group could unambiguously be assigned by NMR spectroscopy. While a methyl group at N1 of 2-phenyl-substituted imidazo[2,1-i]purinones was found at ca. 4.05 ppm (in DMSO-d6),13,14 the signal for a methyl group at N9 was at 3.54 (S-9), or 3.60 ppm (R-10), respectively. Hetero-multiple quantum coherence (HMQC) experiments with compound R-10 showed a correlation between the methyl group and C8 (at 62.0 ppm). However, neither a correlation with C2 (at 160.6 ppm) nor with C9b (at 109.6 ppm), the C atom neighboring N1, could be observed. Therefore, the methyl group is clearly attached to N9 rather than N1.
Yields, melting points and analytical data of the new compounds are collected in Table 1. 1H NMR spectral data were in accordance with the proposed structures (see Experimental).
Table 1.
Yields, melting points and analytical data of synthesized 2-phenylimidazo[2,1-i]purin-5-one derivatives
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd | R2 | R4 | R8 | R9 | Yield (%) |
mp (° C) |
a | Concentration (mg/mL) |
Formula | Analyses |
| R-1 | Phenyl | Methyl | Ethyl | H | 51 | > 250 | 4.5b | 10.0 | C16H17N5O | 295.1423c |
| 2 | Phenyl | Methyl | H | H | 60 | 303 | — | — | C14H13N5O*H2O | Cd, H, Nd |
| S-3 | Phenyl | Propyl | Ethyl | H | 79 | 232 | −27.3 | 5.1 | C18H21N5O*2H2O | C, He, N |
| S-4 | 3,4-Dichlorophenyl | Propyl | Ethyl | H | 53 | 283 | −11.8 | 5.1 | C18H19N5OCl2 | C, H, N |
| S-5 | 2-Chlorophenyl | Methyl | Ethyl | H | 42 | 125 | −15.3 | 7.7 | C16H16N5OCl*H2O | C, H, N |
| S-6 | 3-Chlorophenyl | Methyl | Ethyl | H | 60 | 283 | −10.5 | 4.4 | C16H16N5OCl | C, H, N |
| S-7 | 4-Chlorophenyl | Methyl | Ethyl | H | 42 | 262 | −19.5 | 5.7 | C16H16N5OCl*2H2O | C, Hf, N |
| R-8 | 2,3,5-Trichlorophenyl | Methyl | Ethyl | H | 70 | 160 | 12.3 | 3.7 | C16H14N5O Cl3 | 397.0259c |
| S-9 | 3,4-Dichlorophenyl | Propyl | Ethyl | Methyl | 76 | 215 | 3.9 | 5.7 | C19H21N5OCl2 | 405.1123c |
| R-10 | 2,3,5-Trichlorophenyl | Methyl | Ethyl | Methyl | 58 | 214 | −10.7 | 3.6 | C17H16N5OCl3*0.5H2O | C, H, N |
Determined in DMSO.
Determined for the hydrochloride in methanol.13
Determined by high resolution mass spectroscopy.
C: calcd, 58.94; found, 58.48; N: calcd, 24.55; found, 23.86.
H: calcd, 7.01; found, 6.40.
H: calcd, 5.51; found, 4.94.
Analysis of enantiomeric purity
A recently developed capillary electrophoresis (CE) method13 was used to determine the enantiomeric purity of compounds 3–8 (see Table 2). As chiral discriminators cyclodextrins were added to the inlet buffer (β-cyclodextrin or 2-hydroxypropyl-β-cyclodextrin plus sulfated β-cyclodextrin), and sulfated β-cyclodextrin was added to the outlet buffer. The separations were performed at a pH value of 4.5 (phosphate buffer), at which the compounds were protonated. The compounds were detected and identified by their UV spectra using a diode array detector. Conditions were optimized for each compound (Table 2). Typical separations are shown in Figure 2 for the racemate RS-8 (A) and the enantiomer S-3 (B). All compounds investigated exhibited a high degree of enantiomeric purity; contamination by the other enantiomer was lower than 1.5% in all cases.
Table 2.
Analysis of enantiomeric purity of chiral compounds by capillary electrophoresis
| Enantiomer | Cyclodextrin(s) in inlet buffera | Cyclodextrin in outlet buffera |
Capillary effective length |
Determined purity (%) ±R.S.D.b (%) |
Determined impurityc (%) ±R.S.D.b (%) |
|---|---|---|---|---|---|
| S-3 | HPCD (6 mM) + s-b-CD (1 mg/mL) | s-β-CD (1 mg/mL) | 50 cm | 98.90±0.06 (n = 4) | 1.10±0.06 |
| S-4 | HPCD (6 mM) + s-β-CD (1 mg/mL) | s-β-CD (0.5 mg/mL) | 50 cm | 98.80±0.08 (n = 3) | 1.20±0.08 |
| S-5 | HPCD (6 mM) + s-β-CD (1 mg/mL) | s-β-CD (1 mg/mL) | 50 cm | 99.32±0.05 (n = 4) | 0.68±0.05 |
| S-6 | HPCD (6 mM) + s-β-CD (0.5 mg/mL) | s-β-CD (0.5 mg/mL) | 50 cm | 99.80±0.04 (n = 3) | 0.20±0.04 |
| S-7 | HPCD (6 mM) + s-β-CD (0.5 mg/mL) | s-β-CD (0.5 mg/mL) | 50 cm | 99.29±0.10 (n = 4) | 0.71±0.10 |
| R-8 | β-CD (6 mM) | s-β-CD (2.5 mg/mL) | 40 cm | 98.75±0.22 (n = 8) | 1.25±0.22 |
Phosphate buffer, 50 mM, pH 4.5; β-CD = β-cyclodextrin; HPCD = 2-hydroxypropyl-β-cyclodextrin; s-β-CD = sulfated β-cyclodextrin.
R.S.D. = relative standard deviation.
Contamination by the other enantiomer.
Figure 2.

Typical electropherograms of chiral separation of two chiral compounds. A. Racemate RS-8. Inlet buffer: phosphate 50 mM, β-cyclodextrin 6 mM, pH 4.5. Outlet buffer: phosphate 50 mM, sulfated β-cyclodextrin 2.5 mg/mL, pH 4.5. Fused silica capillary, 40 cm effective length×75 μm I.D. ×375 O.D.; constant current: 90 μA; injection time: 5 s with 0.5 p.s.i.; B. Enantiomer S-3. Inlet buffer: phosphate 50 mM, hydroxypropyl-β-cyclodextrin 6 mM, sulfated β-cyclodextrin 1 mg/mL, pH 4.5. Outlet buffer: phosphate 50 mM, sulfated β-cyclodextrin 1 mg/mL, pH 4.5. Fused silica capillary, 50 cm effective length×75 μm I.D.×375 O.D.; constant current: 90 μA; injection time: 20 s with 0.5 p.s.i.
Biological activity
The new compounds were investigated in radioligand binding assays at A1 ARs of rat brain cortical membranes using the A1-selective radioligand [3H]2-chloro-N6-cyclopentyladenosine ([3H]CCPA], and at A2A ARs of rat brain striatal membranes using the A2A-selective radiolig and [3H]3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine ([3H]MSX-2).
The compounds were additionally investigated at native human A1 and A2A ARs using membrane preparations of post-mortem human brain. [3H]CCPA was used as a radioligand for the human A1 and [3H]-2-[[4-(carboxyethyl)phenyl]ethyl]amino] - 5′ - N - (ethylcarbonyl)amino] adenosine ([3H]CGS21680) for the human A2A ARs. All compounds were investigated at recombinant human A3 ARs expressed in CHO or HEK cells using [125I]AB-MECA as an A3 radioligand. In a few cases, [3H]NECA, or the novel radioligand [3H]PSB-11, was used for A3 binding assays. Ki values determined with different A3 radioligands were very similar (see Table 3, compounds R-1 and R-8).
Structure–activity relationships
2-Phenylimidazopurinone R-1 (PSB-11) had been found in a previous study to possess high affinity for human A3 ARs (Ki = 2.3 nM).13 The compound had been shown to be 190-fold selective versus rat A1 ARs, and exhibited even higher selectivity (> 900-fold) versus rat A2A and mouse A2B ARs. A similar Ki value at human recombinant A3 ARs was found using a different radioligand, [3H]NECA (Ki = 3.5 nM, Table 3). R-1 has now been shown to possess similarly high affinity versus the human A1 and A2A receptor subtypes (Table 3). Chiral 2-phenyl-substituted imidazopurinones, such as compounds R-1 and S-1 had exhibited only a low degree of enantioselectivity at ARs.13 For the analysis of the structure–activity relationships of compounds 3–10 only one single enantiomer was synthesized. The ethyl residue in the 8-position at the imidazoline ring of 4-methyl-2-phenyl-imidazopurinone proved to be essential for high affinity at A3 ARs. Imidazopurinone 2, lacking the ethyl group, was several thousand-fold less potent at A3 ARs than the analogous ethyl derivatives R-1 and S-1. At A1 and A2A ARs the decrease in potency by removal of the ethyl group was much less pronounced (only 4–19-fold). An isomer of 2, in which the 4-methyl group was (formally) moved to the N1-position (1-methyl-2-phenyl-imidazo[2.1-i]purin-5-one) had previously been shown to exhibit high A3 affinity (Ki = 47 nM, 400-fold as potent as 2) but similar A1 and A2A affinity as 2. The corresponding 1,4-dimethyl derivative, however, had shown reduced A3 affinity.13 These results indicate that imidazopurinones can exhibit different binding modes at A3 ARs depending on the substitution pattern. This conclusion is corroborated by recently published data on another imidazopurinone derivative unsubstituted in the 8-position, KF26777 (2-(p-bromophenyl)-4-propyl-substituted imidazo[2,1-i]purin-5-one), which has been reported to be a potent and selective antagonist at human A3 ARs.16,17
In xanthine derivatives the exchange of a 3-methyl group (corresponding to the 4-position in imidazo[2,1-i] purinones) by a propyl group generally results in a large increase in affinity for human A3 ARs.2,18 However, the same modification in imidazopurinones, such as the 4-propyl derivatives S-3 and S-4, appeared to decrease A3 affinity to a large extent. The propyl derivative S-3, for example, exhibited 88-fold lower affinity for A3 ARs than its methyl homologue S-1. Conversely, A1 (16-fold, rat; 3-fold, human receptors) and A2A affinity (4-fold, rat; 8-fold, human receptor) were increased by the exchange of methyl (S-1) for propyl (S-3). As a matter of fact, compound S-3 is a very potent and selective A1 AR antagonist with a Ki value of 7.4 nM at rat A1 ARs and over 100-fold selectivity versus rat A2A ARs. The compound is less potent at human A1 ARs (Ki = 98 nM) and less selective (36-fold versus human A2A, 9-fold versus human A3 ARs).
Chloro-substitution of the 2-phenyl group was explored. Chlorine atoms at different positions of the phenyl ring did not have much influence on A2A affinity, which was generally found to be rather low with all of the 2-phenylimidazopurinones, typically in the micromolar concentration range. At A1 ARs, chlorination in the 2-, 3- or 4-position of the phenyl ring caused a moderate (2–7-fold) increase in affinity, with the following rank order of potency: 2-chlorophenyl (S-5) > 3-chlorophenyl (S-6) > 4-chlorophenyl (S-7) > phenyl (S-1). However, the introduction of two or three chlorine atoms as in compounds S-4 (3,4-dichlorophenyl) and R-8 (2,3,5-trichlorophenyl) resulted in a moderate decrease or no change in A1 affinity (compare S-4/S-3 and R-1/R-8). At A3 ARs the effect of chlorination was different (opposite to the effects at A1 ARs) and much more pronounced. The affinity decreased within the following sequence: phenyl (S-1) ≥ 4-chlorophenyl (S-7) >> 3-chlorophenyl (S-6) > 2-chlorophenyl (S-5). However, polychlorination led to an increase in A3 affinity. Thus, the 3,4-dichlorophenyl derivative S-4 was 7-fold more potent than the corresponding phenyl derivative S-3, and the 2,3,5-trichlorophenyl derivative R-8 exhibited 8-fold higher A3 affinity as compared to the corresponding unsubstituted phenyl derivative R-1. Meanwhile, the trichlorophenyl derivative R-8 has successfully been also used as a precursor for the preparation of a novel A3 antagonist radioligand ([3H]PSB-11), in which the chlorine atoms were replaced by tritium.19 Compound R-8 is the most potent A3 antagonist of the present series. It exhibits a subnanomolar Ki value (Ki = 0.433 nM versus [3H]NECA) and therefore belongs to the most potent A3 antagonists described to date. In addition, R-8 is highly selective (> 800-fold) versus the other AR subtypes.
Methylation at N9 of 2-phenylimidazopurinones S-4 and R-8 led to a large decrease in affinity at A1 and A3 ARs (compare S-4/S-9 and R-8/R-10). The affinity decrease was most pronounced in compound R-10 as compared to R-8 with respect to the A3 AR (almost 400-fold). This result confirms the important role of a hydrogen bond donor/hydrogen bond acceptor motif (N1-H/N9, or N9-H/N1, respectively, in imidazo[2,1-i] purinones) for A1 and also for A3 receptor ligands.20 By methylation this structure is destroyed.
8-Ethyl-2-phenylimidazopurinones were generally weaker (2–13-fold) at human as compared to rat A1 ARs. In contrast, the compounds appeared to be somewhat more potent at human than at rat A2A receptors. Compounds, such as R-1 and R-8, were highly A3-selective in a comparison at the human receptor subtypes.
pH-Dependence of AR binding
The pKa value of imidazo[2,1-i]purinones had been determined to be around 7, close to physiological pH value.13,14 At physiological pH the compounds are expected to be partly protonated, which is believed to be the reason for their improved water-solubility in comparison with xanthine derivatives. The cation is stabilized by delocalization of the positive charge. In order to investigate whether the neutral molecule or the cation binds preferably to the receptors, and which species will bind at pH 7.4 (the physiologic pH value used in all of our experiments), we performed pH-dependent radioligand binding experiments at A1 ARs using compound S-3, a potent, selective A1 antagonist. The results are shown in Figure 3. At an alkaline pH of 8.0, at which the compound is unprotonated, the same low Ki value is obtained as at physiologic pH of 7.4. If the pH value is reduced to 6 in order to produce the protonated compound, the Ki value is statistically significantly reduced by about 2-fold. This may indicate that the neutral molecule binds preferably to the receptor and that it is probably the species binding to the receptors under physiological conditions. An alternative explanation would be that our results reflect different ionization states of the receptor protein at pH 8.0 and 7.4 versus pH 6.0.
Figure 3.

pH-Dependence of A1 adenosine receptor affinity of imidazopurinone derivative S-3; Ki values at pH 7.4 and 8.0 were not statistically significantly different from each other; both Ki values were statistically significantly different from Ki value at pH 6.0 (unpaired t-test).
[35S]GTPγS binding studies
Selected imidazo[2,1-i]purin-5-one derivatives have earlier been shown to act as antagonists at A1-, A2A-, and A2B ARs in adenylate cyclase assays.13 We have now investigated two potent A3-selective imidazopurinones, R-1 and R-8, in [35S]GTPγS binding studies using recombinant human A3 ARs expressed in Chinese hamster ovary (CHO) cells. The results are shown in Figure 4. The AR agonist NECA and the A3-selective AR antagonist MRS-1523, a pyridine derivative (Ki value at human A3 ARs=19 nM)21 were included as standard compounds for comparison. The binding of agonists to certain G-protein-coupled receptors (GPCR) may be modulated by GTP, which binds to the G-protein. Thus, the addition of GTP has been shown to cause a rightward shift of the competition curve for agonists but not for antagonists at A1 and A3 ARs, both of which are Gi-coupled receptors.6,22 While GTP modulates agonist binding, conversely, agonists can also modulate GTP binding to the G-protein. Thus, agonists increase the binding of [35S]GTPγS, a radiolabelled stable analogue of GTP.23 This is shown in Figure 4 at the A3 AR for the agonist NECA, which caused a concentration-dependent increase in [35S]GTPγS binding with an EC50 value of 77 nM. This corresponds well with the Ki value of NECA obtained in radioligand binding experiments at human A3 ARs (Ki = 35 nM).6 Neutral antagonists would not have any influence on [35S]GTPγS binding, while inverse agonists lead to a reduction in [35S]GTPγS binding.24 While agonists are believed to bind preferentially to the active receptor conformation, neutral antagonists are thought to have no preference for either conformation, and inverse agonists to bind preferentially to an inactive conformation.25 In the present [35S]GTPγS binding study we have found that the new A3-selective imidazopurinones R-1 and R-8 caused a concentration-dependent reduction in [35S]GTPγS binding to 84 and 81%, respectively, of control binding. The EC50 values of this effect corresponded with the Ki values of the compounds at A3 ARs, compound R-8 being several-fold more potent than R-1 (see Fig. 4 and Table 3). Based on these results, the new A3 antagonists could be characterized as inverse agonists under the applied test conditions. In contrast, the standard A3 antagonist MRS-152321 did not have any effect on [35S]GTPγS binding and thus behaved as a neutral antagonist in this test system. Also, the recently described imidazopurinone derivative KF26777 (2-(p-bromophenyl)-7,8-dihydro-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one dihydrochloride), structurally related to R-1 (PSB-11) and R-8 (PSB-10), had no effect on [35S]GTPγS binding and behaved as a neutral, competitive antagonist at human A3 adenosine receptors expressed in human embryonic kidney (HEK) cells.17
Figure 4.

Stimulation (by NECA) and inhibition (by 2-phenylimidazo[2,1-i]purin-5-one derivatives R-1 and R-8) of [35S]GTPγS binding to membranes prepared from Chinese hamster ovary cells expressing the human A3 adenosine receptor; NECA (agonist): EC50 = 77 ± 10 nM, max. stimulation (control=100%): 147%; compound R-1: IC50 = 36 ± 3 nM, max. inhibition: to 81%; compound R-8: IC50 = 4.0 ± 0.3 nM, max. inhibition: to 84% of control [35S]GTPγS binding. MRS 1523 caused no significant inhibition of [35S]GTPγS binding.
Conclusion
In summary, structure–activity relationships of 2-phenylimidazo[2,1-i]purinones as AR ligands have been explored with respect to the 8-phenyl ring (chlorine substitution), the N4-alkyl residue (methyl versus propyl), 8-ethyl substitution versus hydrogen, and N9-methylation. 8-Ethyl-4-methyl - 2 - (2,3,5-trichlorophenyl) - (8R) - 4,5,7,8-tetra-hydro-1H-imidazo[2,1-i]purin-5-one (R-8) was developed as a very potent and selective ligand for human A3 ARs. The compounds R-1 and R-8 were shown to exhibit inverse agonistic activity in [35S]GTPγS binding studies at human recombinant A3 ARs expressed in Chinese hamster ovary cells under standard conditions. Due to their assumed relatively good water-solubility 2-phenylimi-dazopurinones may become useful research tools for studying A3 adenosine receptors and their functions.
Experimental
Synthetic procedures.
NMR spectra were performed on a Varian XL-300 (1H: 300 MHz, 13C: 75 MHz), a Bruker AMX 500 (1H: 500 MHz, 13C: 125 MHz), and a Bruker DRX 500 (1H: 500 MHz, 13C: 125 MHz). The chemical shifts of the deuterated solvent served as internal standard: δ (ppm) DMSO: 1H: 2.50; 13C: 39.1; CHCl3:1H: 7.24; 13C: 77.0; CH2Cl2: 1H: 5.32; 13C: 53.5. Coupling constants (J) are given in Hertz (Hz). All compounds were checked for purity by TLC using aluminium sheets with silica gel 60 F254 (Merck). Preparative HPLC was performed with a Knauer HPLC pump 64 using a Eurospher column (type 100-C18: 7μm, 250*20 mm). The melting points were determined by a Büchi 545 melting point apparatus and are uncorrected. Optical rotation was determined on a Perkin–Elmer 241 polarimeter using solutions in DMSO.
Elemental analyses were performed by the Institute of Pharmaceutical Chemistry, University of Bonn. The mass spectra were collected on an MS-50 A.E.I. mass spectrometer. Only selected NMR data for one representative member of each class of compounds is given.
Additional NMR data of the tricyclic purinone derivatives are available as Supporting Information. Analysis of enantiomeric purity was performed as described.13
(R)- and (S)-aminobutanol of high optical purity was obtained from Merck; (R)-aminobutanol: , (S)-aminobutanol: .
As an example, the preparation of 8-ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (R-8) is described in detail. The other compounds were synthesized analogously.
8-Ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (R-8). 6-Amino-1-methyl-5-[(2,3,5-trichlorobenzylidene)amino]-1H-pyrimidin-2,4-dione.
5,6-Diamino-1-methyluracil (1.88 g, 12 mmol) and 2,3,5-trichlorobenzaldehyde (2.72 g, 13 mmol) were suspended in 50 mL of ethanol and refluxed for 3 h. The precipitate was collected by filtration, washed with ethanol and dried in vacuo. Yield: 87%. The yields for analogous compounds ranged from 87–89%. 1H NMR (DMSO-d6): δ (ppm) 3.34 (s, 3H, CH3), 7.57 (s, 2H, NH2), 7.68 (d, 4JHH = 2.5 Hz, ar), 8.46 (d, 4JHH = 2.6 Hz, ar), 10.04 (s, 1H, CH), 10.74 (s, 1H, NH).
3-Methyl-8-(2,3,5-trichlorophenyl)-3,7-dihydro-1H-purin-2,6-dione.
6-Amino-1-methyl-5-[(2,3,5-trichlorobenzylidene)amino]-1H-pyrimidin-2,4-dione (12.2 g, 35 mmol) was dissolved in 200 mL of thionyl chloride. The solution was stirred for 8 h at rt. Excess SOCl2 was removed by distillation. To the residue saturated aq NaHCO3 solution was added until a pH value of 7 was obtained. The product was collected by filtration. Yield: 93%. The yields for analogous compounds ranged from 91–93%. 1H NMR (DMSO-d6): δ (ppm) 3.40 (s, 3H, CH3), 7.80 (d, 4JHH = 2.4 Hz, 1H, ar), 8.01 (d, 4JHH = 2.5 Hz, 1H, ar), 11.17 (s, 1H, NH).
3-Methyl-6-thioxo-8-(2,3,5-trichlorophenyl)-2,3,6,7-tetrahydro-1H-purin-2-one.
A mixture of 3-methyl-8-(2,3,5-trichlorophenyl)-3,7-dihydro-1H-purin-2,6-dione (1.3 g, 3.9 mmol) and phosphorus pentasulfide (1.4 g, 6.3 mmol) in 200 mL of dry pyridine was refluxed for 8 h. After cooling the mixture to rt, 400 mL of H2O was added and the solution was left standing for 2 h. Then it was reduced in vacuo to 1/3 of its volume. The formed precipitate was collected by filtration and suspended in 100 mL of 2N NaOH. The solution was filtered and the product was precipitated by adding dilute HCl solution to achieve a pH value of 4, and the precipitate was collected by filtration. Yield: 86%. 1H NMR (DMSO-d6): δ (ppm) 3.42 (s, 3H, CH3), 7.83 (d, 4JHH = 2.5 Hz, 1H, ar), 8.03 (d, 4JHH = 2.4 Hz, 1H, ar), 12.36 (s, 1H, NH).
3-Methyl-6-methylthio-8-(2,3,5-trichlorophenyl)-3,7-dihydro-2H-purin-2-one.
3-Methyl-6-thioxo-8-(2,3,5-trichlorophenyl)-2,3,6,7-tetrahydro-1H-purin-2-one (1.0 g, 2.8 mmol) was suspended in 16 mL of 0.5 N NaOH. EtOH (ca. 5 mL) was added until a clear solution was obtained. At rt, 4.2 mmol (0.25 mL) of CH3I was added slowly. After stirring for 1 h, the product was collected by filtration and washed with H2O (10 mL). Yield: 86%. 1H NMR (DMSO-d6): δ (ppm) 2.65 (s, 3H, S-CH3), 3.50 (s, 3H, N-CH3), 7.88 (d, 4JHH = 2.5 Hz, 1H, ar), 8.03 (d, 4JHH = 2.4 Hz, 1H, ar).
6-(1-Hydroxymethyl-(1R)-propylamino)-3-methyl-8-(2,3,5-trichlorophenyl)-3,7-dihydro-2H-purin-2-one.
A mixture of 3-methyl-6-methylthio-8-(2,3,5-trichlorophenyl)-3,7-dihydro-2H-purin-2-one (0.37 g, 1 mmol) and 0.44 g (5 mmol) of (R)-2-aminobutanol in 1 mL of DMSO was heated for 1 h at 150 °C. The solvent and the excess amount of alcohol were removed in vacuo. The residue was purified by silica gel column chromatography using a linear gradient CH2Cl2→CH2Cl2/MeOH (93/7, v/v) followed by recrystallization from a mixture of H2O:EtOH (20:80). Yield: 43%. 1H NMR (DMSO-d6): δ (ppm) 0.93 (m, 3H, CH3), 1.40–1.82 (m, 2H, CH2), 3.43 (s, 3H, CH3), 3.48–3.73 (m, 2H, CH2), 4.10 (br, 1H, OH), 4.97 (m, 1H, CH), 7.51 (d, 3JHH = 7.9 Hz, NH), 7.73, (s, 1H, ar), 7.99 (s, 1H, ar).
8-Ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (R-8).
To 30 mL of thionyl chloride (cooled to 0 °C), 0.3 mmol of 6-(1-hydroxymethyl-1R-propylamino)-3-methyl-8-(2,3,5-trichlorophenyl)-3,7-dihydro-2H-purin-2-one was added. The mixture was refluxed for 60 min. Excess thionyl chloride was removed by vacuum distillation. The residue was purified by silica gel chromatography using a linear gradient CH2Cl2→CH2Cl2/MeOH (93/7, v/v) followed by recrystallization from a mixture of H2O:EtOH (10:90). Yield: 43%. 1H NMR (DMSO-d6): δ (ppm) 0.93 (t, 3JHH = 7.3 Hz, 3H, CH3), 1.67 (m, 2H, CH2), 3.46 (s, 3H, CH3), 3.78 (dd, 2JHH = 9.7 Hz, 3JHH = 5.4 Hz, 1H, CH2); 4.28–4.15 (m, 2H, CH, CH2); 7.77 (d, 4JHH = 2.6H, 1H, ar); 7.94 (d, 4JHH = 2.6 Hz, 1H, ar).
2-(3,4-Dichlorophenyl)-8-ethyl-9-methyl-4-propyl-(8S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (S-9), 4,9-dimethyl-8-ethyl-2-(2,3,5-trichlorophenyl)-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (R-10).
To a solution of the imidazo[2,1-i]purin-5-one (0.2 mmol) S-4, or R-8, respectively, in 5 mL of DMF sodium hydride (20–30 mg) was added portionwise. After stirring for 15 min at 0 °C, 93 μL (1.5 mmol) of methyl iodide was added, and the mixture was stirred for 45 min at rt. The excess sodium hydride was filtered off and the product was extracted with CHCl3, washed with water and brine and subsequently concentrated by rotary evaporation. The product was purified by silica gel chromatography using a linear gradient: CH2Cl2→CH2Cl2/MeOH (95:5, v/v). Further purification of compound R-10 could be achieved by preparative HPLC using a linear gradient: MeOH/H2O (60:40)→MeOH/H2O (90:10) over 30 min.
S-9:1H NMR (CDCl3): δ (ppm) 0.90 (m, 6H, 2*CH3), 1.73 (m, 1H, CH2), 1.85 (m, 2H, CH2), 1.95 (m, 1H, CH2);3.57 (s, 3H, NCH3), 3.87 (dd, 2JHH=10.5 Hz, 3JHH = 7.25 Hz, 1H, CH2), 4.00 (m, 1H, CH); 4.12 (t, 3JHH = 7.6 Hz, 2H, CH2); 4.28 (dd, 2JHH = 10.5 Hz, 1H, 3JHH=10.5 Hz, CH2); 7.45 (d, 3JHH = 8.3 Hz, 1H, ar); 8.07 (dd, 3JHH = 8.3 Hz, 3JHH = 1.9 Hz, 1H, ar); 8.34 (d, 3JHH=1.9 Hz, 1H, ar). 13C NMR (CDCl3): δ (ppm) 7.9 (CH3); 11.1 (CH3); 21.2 (CH2); 24.3 (CH2); 31.8 (N9CH3); 45.4 (N4CH2); 48.6 (C7); 62.0 (C8); 110.2 (C9b); 125.9 (Car); 128.2 (Car); 130.2 (Car); 131.3 (Car); 132.1 (Car); 135.8 (Car); 147.4 (C3a); 147.5 (C9a); 157.9 (C5); 160.9 (C2).
R-10:1H NMR (CDCl3): δ (ppm) 1.00 (t, 3JHH = 7.5 Hz, 3H, CH3); 1.74 (ddq, 2JHH = 14.8 Hz, 3JHH = 7.5 Hz, 1H, CH2); 1.96 (ddq, 2JHH = 14.8 Hz, 3JHH = 7.5 Hz, 3JHH = 3.2 Hz, 1H, CH2); 3.60 (s, 3H, CH3); 3.66 (s, 3H, CH3); 3.94 (dd, 2JHH = 11.4 Hz, 3JHH = 7.2 Hz, 1H, CH2); 4.07 (m, 1H, CH); 4.37 (dd, 2JHH = 11.34 Hz, 3JHH = 9.8 Hz, CH2); 7.43 (d, 4JHH = 2.5 Hz, 1H, ar); 7.85 (d, 4JHH = 2.5 Hz, 1H, ar). 13C NMR (CDCl3): δ (ppm) 8.0 (CH3); 24.4 (CH2); 30.3 (N4CH3); 32.1 (N9CH3); 48.6 (C7); 62.0 (C8); 109.6 (C9b); 129.4 (Car); 129.9 (Car); 130.0 (Car); 132.2 (Car); 134.7 (Car); 137.1 (Car); 147.7 (C3a); 147.8 (C9a); 157.4 (C5); 160.4 (C2).
Receptor binding studies
Radioligand competition experiments were performed as previously described using rat brain cortical membrane preparations as a source of A1 ARs, and rat brain striatal membrane preparation for A2A AR assays.13,26,27 Frozen rat brains (unstripped) were obtained from Pel-Freez, Rogers, Arkansas, USA. For assays at human A3 ARs, CHO or HEK cell membranes recombinantly expressing the human A3 receptor subtype were used as described.13,28 Human post-mortem brain membrane preparations were used for radioligand binding assays at human A1 and A2A ARs; human frontal cortex was used as A1 AR source, caudate-putamen as A2A AR-containing tissue. The latter was prepared as previously described.29 Human frontal cortex membranes were prepared analogously. Frontal cortex was pooled from three male subjects (36, 40 and 44 years old), caudate-putamen was from five subjects (1 female, 50 years, 4 male, 23, 35, 35, and 40 years old) with no history of brain diseases (‘control brains’). The brains were dissected no longer than 24–48 h post mortem.
[3H]2-Chloro-N6-cyclopentyladenosine ([3H]CCPA) was used as A1 radioligand, [3H]3-(3-hydroxypropyl)-7-methyl - 8 - (m - methoxystyryl) - 1 - propargylxanthine ([3H]MSX-2) as A2A ligand at rat receptors, and [3H]-2- [[4-(carboxyethyl)phenyl]ethyl]amino] - 5′ - N - (ethylcarbonyl)amino]adenosine ([3H]CGS21680) as A2A radioligand at human A2A ARs.26-29 [125I]N2-methyl-3,4-dihydroxy-5-[6-[3-iodobenzyl)amino]-9H-9-purinyl)-3-tetrahydro-2-furancarboxamide ([125I]AB-MECA), [3H]N2-ethyl-3,4-dihydroxy-5-[6-amino]-9H-9-purinyl)-3- tetrahydro-2-furancarboxamide ([3H]NECA), or [3H]2-phenyl - 8 - ethyl - 4 - methyl - (8R)-4,5,7,8 - tetrahydro-1H-imidazo[2,1-i]purin-5-one ([3H]PSB-11), respectively, were employed as radioligands for the human recombinant A3 ARs.13,18,19,28
Curves were determined using 6–7 different concentrations of test compounds spanning 3 orders of magnitude. At least two to three separate experiments were performed each in duplicate or triplicate.
Data were analyzed using Graph Pad PRISM version 3.0 (San Diego, CA, USA). For statistical comparison between groups, data were subjected to analysis of variance followed by unpaired t-test.
Radioligand binding assays at human post-mortem brain cortical A1 adenosine receptors
The assays were performed essentially as the assays at rat A1 ARs using 0.5 nM of [3H]CCPA (KD = 3.8 nM), 2-chloroadenosine (400 μM) to define nonspecific binding in 50 mM Tris–HCl buffer (pH 7.4) in a final volume of 1 mL. Membrane suspensions were washed with the buffer before starting the assay by centrifugation at 35,000g for 20 min (4 °C) and subsequently preincubated with 0.12 I.U./mL of adenosine deaminase for 30 min. Protein concentration in the assay was ca. 75 μg/mL. Incubation was at 23 °C for 90 min in a shaking water-bath and was terminated by rapid filtration through glass fiber GF/B filters.
[35S]GTPγS-binding studies
The incubation mixture contained in a total volume of 200 μL, 50 mM Tris–HCl pH 7.4, 1 mM EDTA, 100 mM NaCl, 0.2 I.U./ mL adenosine deaminase, 5 mM MgCl2, 1 mM DTT, 10 μM GDP, 0.5% (w/v) bovine serum albumin, 0.1–0.5 nM [35S]GTPγS (about 30,000 cpm) and test compound diluted in DMSO. To start the incubation, 7.5 μg of membrane protein were added to the glass tubes and kept for 45 min at 25 °C on a shaking water bath. Incubation was terminated by adding 4 °C cold rinse buffer (50 mM Tris–HCl pH 7.4, 5 mM MgCl2) and rapid filtration through glass fiber filters (Whatman GF/B) using a Brandell cell harvester. Nonspecific binding was determined in the presence of 10 μM of unlabelled GTPγS and was less than 0.5% of total [35S]GTPγS binding. Filters were punched out and transferred to minivials and 3 mL of Ultima Gold scintillation cocktail (Canberra Packard) was added. After 180 min of incubation, the samples were counted in an LSC Tricarb 2100TR (Canberra Packard). EC50 and IC50 values were calculated from dose–response curves using the program Graph Pad Prism 3.00, and the inbuilt equations for sigmoidal dose response with variable slope. Experiments were carried out in triplicates in at least two independent experiments.
1H and 13C NMR spectral data of intermediate and final products
Additional NMR spectral data are provided for most intermediate and all final products.
Acknowledgements
We thank Stefanie Weyler, Ulrike Reith and Sonja Hinz for performing some of the radioligand binding assays. V. Ozola was on leave from the Latvian Institute of Organic Synthesis in Riga, Latvia and was supported by a Humboldt fellowship (Roman Herzog program). This work was supported by the Deutsche Forschungs-gemeinschaft within the Graduiertenkolleg GRK 677 (scholarship for B. Schumacher). C. E. Müller is grateful for support by the Fonds der Chemischen Industrie and the Bundesministerium für Bildung und Forschung (BMBF). The post mortem human brain material was kindly provided by Professor Dr. P. Falkai, University of Bonn, Clinic for Psychiatry and Psychotherapy.
Footnotes
Supporting information available. Additional 1H and 13C NMR data of synthesized compounds.
References and Notes
- 1.Ralevic V; Burnstock G Pharmacol. Rev 1998, 50, 413. [PubMed] [Google Scholar]
- 2.Müller CE Mini-Reviews Med. Chem 2001, 1, 433. [DOI] [PubMed] [Google Scholar]
- 3.Kim Y-C; Ji X; Jacobson KA J. Med. Chem 1996, 39, 4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y-C; de Zwart M; Chang L; Moro S; von Frijtag Drabbe Künzel JK; Melman N; IJzerman AP; Jacobson KA J. Med. Chem 1998, 41, 2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baraldi PG; Cacciari B; Romagnoli R; Spalluto G; Klotz K-N; Leung E; Varani K; Gessi S; Merighi S; Borea PA J. Med. Chem 1999, 42, 4473. [DOI] [PubMed] [Google Scholar]
- 6.Varani K; Merighi S; Gessi S; Klotz K-N; Leung E; Baraldi PG; Cacciari B; Romagnoli R; Spalluto G; Borea PA Mol. Pharmacol 2000, 57, 968. [PubMed] [Google Scholar]
- 7.Van Muijlwijk-Koezen JE; Timmerman H; Link R; van der Goot H; IJzerman AP J. Med. Chem 1998, 41, 3987. [DOI] [PubMed] [Google Scholar]
- 8.Van Muijlwijk-Koezen JE; Timmerman H; Link R; van der Goot H; IJzerman AP J. Med. Chem 1998, 41, 3994. [DOI] [PubMed] [Google Scholar]
- 9.Van Muijlwijk-Koezen JE; Timmerman H; van der Goot H; Menge WM; Frijtag von Drabbe Künzel J; de Groote M; IJzerman AP J. Med. Chem 2000, 43, 2227. [DOI] [PubMed] [Google Scholar]
- 10.Jiang J; van Rhee AM; Melman N; Ji X; Jacobson KA J. Med. Chem 1996, 39, 4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang J; van Rhee AM; Chang L; Patchornik A; Ji X; Evans P; Melman N; Jacobson KA J. Med. Chem 1997, 40, 2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki F; Shimada J; Nonaka H; Ishii A; Shiozaki S; Ichikawa S; Ono E J. Med. Chem 1992, 35, 3578. [DOI] [PubMed] [Google Scholar]
- 13.Müller CE; Thorand M; Qurishi R; Dieckmann M; Jacobson KA; Padgett WL; Daly JW J. Med. Chem 2002, 45, 3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hipp J Dissertation, University of Würzburg, 1997. [Google Scholar]
- 15.Shimada J; Kuroda T; Suzuki FJ Heterocyclic Chem. 1993, 30, 241. [Google Scholar]
- 16.Tsumuki H; Saki M; Nonaka H; Ichimura M; Shimada J; Suzuki F; Ichikawa S; Kosaka N Kyowa Hakko Kogyo Co., Ltd., Japan, World Patent WO98/15555, 1998.
- 17.Saki M; Tsumuki H; Nonaka H; Shimada J; Ichimura M Eur. J. Pharmacol 2002, 444, 133. [DOI] [PubMed] [Google Scholar]
- 18.Hayallah AM; Sandoval-Ramirez J; Reith U; Schobert U; Preiss B; Schumacher B; Daly JW; Müller CE J. Med. Chem 2002, 45, 1500. [DOI] [PubMed] [Google Scholar]
- 19.Müller CE; Diekmann M; Thorand M; Ozola V Bioorg. Med. Chem. Lett 2002, 12, 501. [DOI] [PubMed] [Google Scholar]
- 20.Müller CE Exp. Opin. Ther. Patents 1997, 7, 419. [Google Scholar]
- 21.Van Rhee AM; Jiang J-L; Melman N; Olah ME; Stiles GL; Jacobson KA J. Med. Chem 1998, 41, 3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geis U; Grahner B; Pawłowski M; Drabczynska A; Gorczyca M; Müller CE Pharmazie 1995, 50, 333. [PubMed] [Google Scholar]
- 23.Lorenzen A; Fuss M; Vogt H; Schwabe U Mol. Pharmacol 1993, 44, 115. [PubMed] [Google Scholar]
- 24.Shryock JC; Ozeck MJ; Belardinelli L Mol. Pharmacol 1998, 53, 886. [PubMed] [Google Scholar]
- 25.De Ligt RAF; Kourounakis AP; IJzerman AP Br. J. Pharmacol 2000, 130, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess S; Müller CE; Frobenius W; Reith U; Klotz K-N; Eger K J. Med. Chem 2000, 43, 4636. [DOI] [PubMed] [Google Scholar]
- 27.Müller CE; Maurinsh J; Sauer R Eur. J. Pharm. Sci 2000, 10, 259. [DOI] [PubMed] [Google Scholar]
- 28.Klotz K-N; Hessling J; Hegler J; Owman C; Kull B; Fredholm BB; Lohse MJ Naunyn-Schmiedberg’s Arch. Pharmacol 1998, 357, 1. [DOI] [PubMed] [Google Scholar]
- 29.Sauer R; Maurinsh J; Reith U; Fülle F; Klotz K-N; Müller CE J. Med. Chem, 2000, 43, 440. [DOI] [PubMed] [Google Scholar]