

Abstract
The Rel proteins of the NF‐κB complex comprise one of the most investigated transcription factor families, forming a variety of hetero‐ or homodimers. Nevertheless, very little is known about the fundamental kinetics of NF‐κB complex assembly, or the inter‐conversion potential of dimerised Rel subunits. Here, we examined an unexplored aspect of NF‐κB dynamics, focusing on the dissociation and reassociation of the canonical p50 and p65 Rel subunits and their ability to form new hetero‐ or homodimers. We employed a soluble expression system to enable the facile production of NF‐κB Rel subunits, and verified these proteins display canonical NF‐κB nucleic acid binding properties. Using a combination of biophysical techniques, we demonstrated that, at physiological temperatures, homodimeric Rel complexes routinely exchange subunits with a half‐life of less than 10 min. In contrast, we found a dramatic preference for the formation of the p50/p65 heterodimer, which demonstrated a kinetic stability of at least an order of magnitude greater than either homodimer. These results suggest that specific DNA targets of either the p50 or p65 homodimers can only be targeted when these subunits are expressed exclusively, or with the intervention of additional post‐translational modifications. Together, this work implies a new model of how cells can modulate NF‐κB activity by fine‐tuning the relative proportions of the p50 and p65 proteins, as well as their time of expression. This work thus provides a new quantitative interpretation of Rel dimer distribution in the cell, particularly for those who are developing mathematical models of NF‐κB activity.
Keywords: DNA binding, native mass spectrometry, NF‐kappaB, NF‐κB, protein complex, protein dynamics, protein–protein interaction, Rel proteins, subunit exchange, transcription factor
Abbreviations
- DSF
differential scanning fluorimetry
- ESI
electrospray ionization
- GuHCl
guanidinium hydrochloride
- Ig
immunoglobulin
- IKK
inhibitory κB kinase
- MALS
multi‐angle light scattering
- MS
mass spectrometry
- NLS
nuclear localisation signal
- RHD
Rel homology domain
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEC
size‐exclusion chromatography
1. INTRODUCTION
Since its discovery in the mid‐1980s, the transcription factor NF‐κB has served as a key component in understanding gene regulation—particularly, given its pivotal role in what is now among the best‐understood eukaryotic signaling cascades.1, 2 NF‐κB was originally identified as a protein complex that bound to, and selectively activated, the transcription of a genomic enhancer region in the immunoglobulin (Ig) κ light‐chain gene in B cells.3, 4 Since then, NF‐κB has been found in almost all animal cell types, where it plays an essential role as a transcriptional regulator prominently associated with immunity and inflammatory responses triggered by stress (e.g., oxidative damage) or pathogens.5 NF‐κB also regulates other fundamental cellular processes, including growth and apoptosis.6 This protein complex is therefore central to a wide range of signaling pathways,7 and recent studies of mutations in humans and other species affecting the NF‐κB system have revealed its critical role in immune regulation.8 Altogether, NF‐κB's far‐reaching significance makes it an important target for continued biochemical and biophysical investigation.
The NF‐κB complex in mammals is canonically described as a dimer formed from members of a family of five related “Rel” proteins, so‐called due to their homology to the retroviral oncoprotein v‐Rel.9 In recent years, this class of proteins has grown to encompass a number of evolutionary‐related transcription factors that play critical roles in defining cellular patterning and signaling.10, 11, 12 The Rel family of proteins mediate the sequence‐specific activation of κB target genes through a common architecture containing a DNA‐binding motif and a dimerisation domain, which together comprise the highly conserved Rel homology domains (RHDs)13 (Figure 1a). The RHDs of these family members are comprised of a highly conserved set of DNA binding and dimerisation domains, both of which lie toward the N‐terminal portion of their respective proteins.14 These regions of the Rel proteins have served as the critical core components for biochemical and biophysical analyses of NF‐κB.
FIGURE 1.
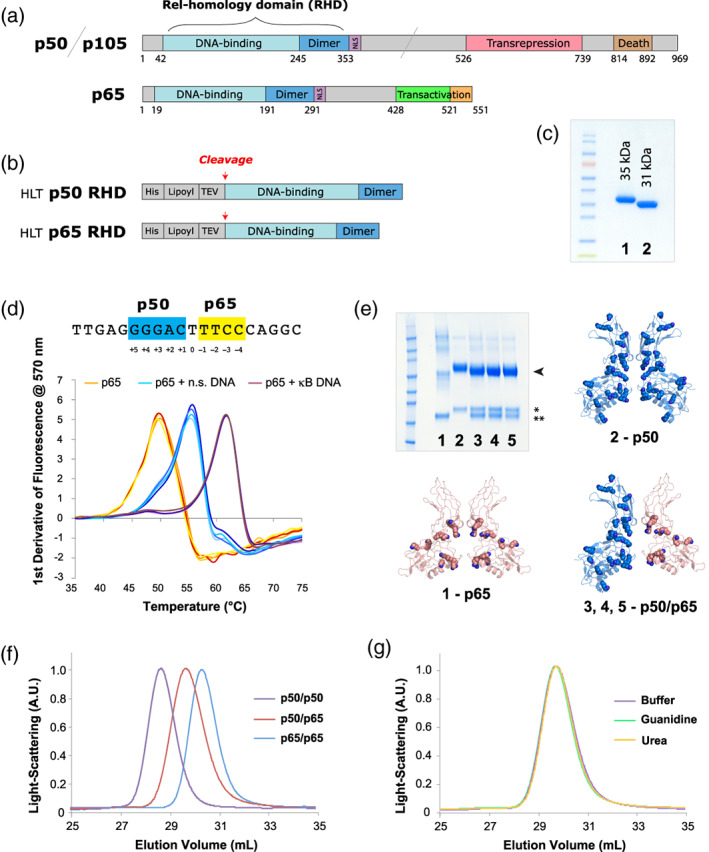
Recombinant proteins used in this work. (a) The full‐length variants of p50/p105 and p65. (b) The HLT fusion proteins used for expression and purification. (c) SDS‐PAGE gel of purified samples. (All samples used in this figure were the first‐generation constructs, for example, p50‐1 RHD and p65‐1 RHD, described here briefly as p50 RHD and p65 RHD.): 1–p50 RHD, 2–p65 RHD. (d) Nucleotide binding by NF‐κB. The sequence of the canonical p50/p65 heterodimer Ig‐like binding site is shown with the p50 half‐site in blue and the p65 half‐site in yellow. Below are shown DSF temperature melts of the p65 RHD subunits of NF‐κB (with each condition showing four repeats, in slight variations of the colors listed below). p65 RHD was heat‐denatured in 50 mM NaCl buffer in the absence of oligonucleotides (orange), presence of heparin or nonspecific DNA (cyan), and presence of specific κB site DNA (purple). Four repeats of each melt (varying shades of the colors above) are shown. (e) SDS‐PAGE of crosslinked samples of various RHD preparations, stained with Coomassie blue: 1–p65 RHD, 2–p50 RHD, 3–p50/p65 RHD mixed in buffer, 4–p50/p65 RHD refolded from urea denaturation, 5–p50/p65 RHD refolded from GuHCl denaturation. The bands corresponding to dimeric crosslinked species are indicated with an arrowhead, and the isolated p50 (*) and p65 (**) monomers are indicated with asterisks. The structure of the p50 (blue, PDB ID: 1SVC) and p65 (peach, PDB ID: 2RAM) homodimers, as well as the p50/p65 heterodimer (blue/peach, PDB ID: 1VKX)—all of which have had their corresponding double‐stranded DNA oligos omitted—are shown as cartoon representations, with lysine side chains depicted as spheres. (f) SEC analysis of p50 RHD (purple) and p65 RHD (blue) homodimers, as well as the p50/p65 RHD heterodimer (red). (g) Gel filtration of the heterodimer species generated either by directly mixing the p50 and p65 RHD homodimers (purple), or by denaturing the RHD homodimers in GuHCl (green) or urea (orange), followed by refolding via overnight dialysis into buffer. DSF, differential scanning fluorimetry; GuHCl, guanidinium hydrochloride; HLT, His‐lipoyl‐TEV; RHD, Rel homology domain; SDS‐PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; SEC, size‐exclusion chromatography
The Rel proteins are further subdivided based on their C‐terminal domains.14 The Class I precursor proteins p105 and p100 contain large C‐terminal ankyrin‐repeat regions, which function as transrepressors of nuclear localization and DNA binding, followed by a death domain. In contrast, the Class II Rel proteins, RelA (p65, used hereafter), RelB, and c‐Rel, contain C‐terminal transcription‐activation (transactivation) domains that serve as potent activators of gene expression. NF‐κB dimers bind to κB sites within the promoters/enhancers of target genes and regulate transcription through the recruitment of additional proteins.15 Because they lack transactivation domains, p50 and p52 may repress gene transcription, unless these proteins are associated with Rel Class II members capable of directing transcription. Constitutive binding of p50 or p52 homodimers to κB promoters may therefore dampen NF‐κB transactivation, a process that is balanced against the activating capabilities of alternate NF‐κB dimers.15
Dimerisation of the limited repertoire of Rel proteins allows targeting of the complex to ~10 bp genomic κB sites with the consensus sequence 5′‐GGGRNNYYCC‐3′, where R is a purine (A or T), N is any nucleotide, and Y is a pyrimidine (T or C).16 Among the most well‐known of these DNA binding sites is the sequence 5′‐GGGACTTTCC‐3′ identified by methylation interference experiments—this sequence comprises a portion of the Ig κ light‐chain enhancer region.3, 4, 17 κB DNA often possesses a deep major groove at the Rel binding site, providing a general architectural mode of recognition.18 Variability in NF‐κB binding sites, including nontraditional sequence motifs or the presence of isolated subsites, has further highlighted the inherent plasticity of NF‐κB dimers for recognizing cognate κB motifs.19, 20 Binding to κB DNA by NF‐κB is followed by recruitment of additional coactivators and RNA polymerase to initiate gene transcription.
A preponderance of the biochemical and structural work on the NF‐κB RHDs focuses on the two most common mammalian Rel proteins, p50, and p65. Structural characterization of the p50/p65 heterodimer demonstrate that κB DNA sites are typically pseudo‐symmetric, containing a 5 bp p50 subsite, a 4 bp p65 subsite, and a 1 bp spacer between them.16 Despite these general principles, there is some evidence of flexibility in κB site composition. For instance, Rel dimers presented with only one optimal ~5 bp subsite, can still bind to the overall κB site by making base‐specific contacts to the optimal subsite, and only phosphate backbone contacts at the other nucleotides.16, 21, 22, 23 Interestingly, an RNA aptamer with no sequence homology to κB sites has also been designed to bind Rel dimers with high affinity, demonstrating that the recognition modes of these proteins are plastic.14, 21, 24
Numerous X‐ray crystal structures of Rel hetero‐ and homodimers dimers bound to DNA have been solved, notably including structures of the p50/p50,25, 26 p65/p65,27, 28 and p50/p6529, 30 RHD dimers. Together, these structures have provided key atomic insight of the transcription factor core. Detailed biochemical interaction maps as well as structures of higher‐order NF‐κB complexes have additionally indicated that interactions between multiple transcription factors and their regulatory elements provide further degrees of transcriptional control.31, 32, 33, 34, 35
The activity of isolated (monomeric) Rel proteins is regulated through inhibitory interactions with the set of ankyrin‐rich Inhibitor of κB (IκB) proteins.21, 22, 23, 36, 37 These proteins each resemble the C‐terminal ankyrin‐rich transrepression domains of p105 and p100, and include the IκBα, IκBβ, IκBγ, IκBε (NEMO), IκBζ, and Bcl‐3 members.14, 38, 39, 40, 41, 42 Biochemical and structural data demonstrate that IκB members bind the DNA‐contacting regions and nuclear localisation signals (NLS) of the Rel proteins, blocking their gene targeting function.43, 44, 45 IκB members thus prevent nuclear shuttling of the Rel components of NF‐κB and obviate their ability to contact nuclear DNA.
NF‐κB activity is further modulated through a complex signal transduction cascade31, 33 regulating the inhibitory κB kinase (IKK) formed from IKKα, IKKβ, and IKKγ.21, 23 In the classical pathway, activation of the IKK complex leads to phosphorylation by IKKβ of two N‐terminal serine residues of IκBα.21 These modifications target IκBα for ubiquination and degradation by the 26S proteasome. In an alternate pathway, the p100/RelB complex is activated by IKK phosphorylation of the C‐terminal region of p100, leading to ubiquitination and subsequent 26S proteasomal degradation of the p100 C‐terminal ankyrin repeats. This process generates the p52/RelB NF‐κB transcriptional core.46 In a related capacity, p50 can be generated from p105 by cleavage of a glycine‐rich region between the Rel core and ankyrin‐repeat domain, the latter of which is then targeted for proteasomal degradation.47, 48 The death domains of p105 and p100 direct protein–protein interactions necessary for the recruitment of IKK, leading to site‐specific phosphorylation and subsequent proteolysis to form either p50 or p52, respectively.49 The p50 or p52 subunits of NF‐κB may also be produced directly from arrested translation. Removal of IκB components thus frees the Rel subunits from the inhibitory complex and allows nuclear translocation and targeting of cognate κB sites.
Infections, inflammatory cytokines, and engagement of antigen receptors all induce NF‐κB activation, highlighting the central role of this transcription factor in immunity. NF‐κB additionally functions in the regulation of cell differentiation, proliferation, and survival.50 Consequently, disruptions in the regulation of NF‐κB are linked to a wide range of disorders, spanning from autoimmunity, susceptibility to HIV, and the development of cancer.51, 52 Because of its central role in cell survival and growth, NF‐κB activity is therefore tightly controlled at multiple levels by positive and negative regulators.
Many of the gene targets of NF‐κB affect rapid responses to acute signs of cellular stress.39 Hence, once activated, the Rel subunits of NF‐κB continue to be tightly regulated in order to allow responses to new gene induction stimuli, as well as prevent deleterious long‐acting target gene upregulation. As is often observed for potent regulatory molecules, NF‐κB can activate expressions of its own repressor, specifically by binding to enhancers of the IκBα gene.14 Nascent IκBα can then bind nuclear NF‐κB and transport it back to the cytoplasm in an inhibited state. A series of studies using confocal microscopy on live, stimulated cells has provided dramatic, real‐time insight into this cyclical NF‐κB shuttling behavior.53 These remarkable, tunable properties of NF‐κB have allowed it to evolve into a leading paradigm for nuclear translocation as a means of transcriptional regulation.
Despite these advances, there is sparse information quantifying the relative kinetic stabilities of the p50 and p65 NF‐κB dimers, or the factors guiding the prevalence of particular NF‐κB species. We therefore investigated the biophysical properties that underlie the frequency of NF‐κB dimer populations to gain a better understanding of the assembly dynamic and equilibrium of these vital proteins in their cellular context. A better understanding of these biophysical properties is thereby essential to further understanding and probing NF‐κB activity.
2. MATERIALS AND METHODS
2.1. Protein expression and purification
p50 RHD and p65 RHD were cloned as fusions to a “His‐lipoyl‐TEV” (HLT) solubility and purification tag54 (Figure 1b). Unlike prior work,25, 26, 27, 28, 29, 30 we found that the Rel protein fusions could be expressed solubly and at high yield in Escherichia coli (~20 mg/L of culture) at 22°C. The proteins were initially purified by nickel‐affinity chromatography. After digestion with TEV protease, the cleaved RHDs were further purified by successive rounds of heparin and gel filtration chromatography, and were judged >95% pure by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) (Figure 1c).
The proteins used in these experiments were as follows: His6‐lipoyl‐(TEV)‐p65‐RHD‐1 (p65‐1) containing residues 19–291 of human p65, His6‐lipoyl‐(TEV)‐p65‐RHD‐2 (p65‐2) containing residues 19–325 of human p65, His6‐lipoyl‐(TEV)‐p50‐RHD‐1 (p50‐1) containing residues 42–353 of human p50, and His6‐lipoyl‐(TEV)‐p50‐RHD‐2 (p50‐2) containing residues 42–367 of human p50. These boundaries were established by comparison with those used in prior biophysical and biochemical analyses of the mouse homologs of p50 and p65.25, 26, 27, 28, 29, 30
The HLT‐Rel fusions were expressed as soluble proteins in E. coli. Briefly, cells were grown to an OD600 of 0.5–0.8 in 2xYT medium, before being induced with 1 mM IPTG and incubated at 22°C overnight. Cells were resuspended (50 mM phosphate buffer pH 8.0, 300 mM NaCl, protease inhibitors, and 10 mM β‐mercaptoethanol) and lysed using a combination of a cell‐cracker and lysozyme. Cleared lysate was loaded onto a nickel‐affinity column. Protein was eluted in buffer supplemented with 250 mM imidazole pH 8.0 and dialyzed overnight (25 mM Tris buffer pH 7.5, 300 mM NaCl, 10% glycerol, 5 mM DTT) in the presence of tobacco etch virus (TEV) protease. The dialyzed solution was diluted 10‐fold (25 mM Tris buffer pH 7.5, 10% glycerol, 5 mM DTT), and cleaved proteins were separated from the His‐tagged lipoyl domain using a heparin column. Protein was eluted from the heparin column with a gradient over 20 column volumes of elution buffer (25 mM Tris buffer pH 7.5, 1 M NaCl, 10% glycerol, and 5 mM DTT). Combined fractions were concentrated using Centricon concentrators (Millipore) and further purified by gel filtration. Proteins were purified with a HiLoad 26/60 Superdex 200 column (GE Healthcare) using phosphate buffer (25 mM phosphate buffer pH 7.2, 300 mM NaCl, 5 mM DTT, and 10% glycerol). All purification steps were carried out at 4°C. The samples were judged >95% pure by SDS‐PAGE.
2.2. Chemical unfolding and refolding
Purified recombinant p50 and p65 RHDs were denatured by rapid dilution of 200 μl of ~100 μM protein stock into 1.8 ml of either buffered 8 M urea, buffered 10 M guanidinium hydrochloride (GuHCl), or buffered alone (25 mM phosphate buffered pH 7.2, 300 mM NaCl, 5 mM DTT, and 10% glycerol). The proteins were then dialyzed overnight at 4°C against buffer lacking any denaturant and concentrated to 200 μl.
2.3. Thermal unfolding
Samples were prepared by mixing protein samples with SyproOrange (Invitrogen) just prior to the experiment, as follows: a 5,000x stock of SyproOrange (as supplied by the manufacturer) was diluted 10x in DMSO to yield a 500x stock, and 45 μl of the 500x stock solution were mixed with 95 μl of 10 μM protein. Four identical samples of 20 μl each were then dispensed into four separate optically clear reaction tubes. The samples were placed in the rotor and locked into place. The following settings were used for measuring protein thermal stability: ramp from 30 to 90°C, raising the temperature 0.6°C per step. The maximum value of the first derivative of the fluorescence curves (i.e., at the mid‐point of the unfolding reaction, at which the rate of change in fluorescence should be the largest) defined the melting temperature. The values presented are the averages and SD across measurements from two separate experiments.
2.4. BS3 protein–protein interaction crosslinking
BS3 is a water‐soluble amine‐reactive crosslinker. 10 mM stock solutions were freshly prepared just prior to a crosslinking reaction by dissolving ~1.1 mg of BS3 into 200 μl ddH2O. Crosslinking was performed by addition of 1 mM BS3 to 100 μl of 1 mg/ml of protein in PBS. Crosslinking reactions were performed for approximately 1 hr at 20°C and were quenched by addition of 1 M Tris pH 7.5 to a 20 mM final concentration.
2.5. Mass spectrometry
The protein storage buffer was exchanged to 500 mM ammonium acetate, pH 6.9 by passing through Micro Bio‐Spin columns (BioRad) prior to MS analysis. Previous results have demonstrated that this buffer does not adversely affect the exchange properties of protein–protein complexes, although dramatically improves the quality of the resulting spectra.55 Typically, 3 μl of protein solution was analyzed by quadropole ion‐mobility time‐of‐flight (Synapt, Waters, Milford MA) according to a previously described protocol.56 To preserve non‐covalent interactions, the following instrument parameters used on the Synapt: capillary voltage 1.2–1.8 kV, sample cone 150 V, trap and transfer collision energy 100 V, backing pressure 5 mbar, source pressure 6–7 × 10−2 mbar, trap and IMS pressure 5 × 10−2 and 5 × 10−1 mbar, respectively, ToF analyzer pressure 2 × 10−6 mbar. The following parameters were used on the LCT Premier: capillary voltage 1.1–2.0 kV, sample cone 200 V, ion guide 150 V, aperture 150 V, ion energy 55 V, source pressure 2.8 mbar, ToF analyzer pressure 4.5 × 10−7 mbar. All spectra were calibrated internally using a solution of cesium iodide (100 mg/ml). Data were processed with MassLynx 4.0 software (Waters, UK), and were typically smoothed +/− 50 data points over two rounds. Although most peaks could be clearly resolved, in instances with overlapping peaks, the relative contribution of each species was estimated using the trend reference peaks that could be distinguished. Kinetic data were subsequently analysed in KaleidaGraph (Synergy Software).
2.6. DNA oligos
Unlabelled oligos were purchased with standard desalting purification (IDT). The nonspecific DNA sequence employed was derived from the p53 B600 binding oligo, GTCAGGGGATCCCCTGAC (palindromic p53 binding site shown in italics).
3. RESULTS
3.1. DNA‐binding behavior of Rel subunits
Previous work has described the RHD DNA‐binding mechanism in detail using protein sequences derived from Mus musculus.57 Given our lab's broader interests in inherited immunological diseases in humans, we focused instead on the human variants of the NF‐κB p50 and p65 subunits (Figures S1–S3). We expressed and purified the Homo sapiens p50 and p65 RHDs (see Methods), and confirmed that these proteins possessed DNA‐binding properties consistent with previously described Rel family members. We probed the interaction of p50 and p65 RHD with nucleic acids using differential scanning fluorimetry (DSF). As expected, both p50 RHD and p65 RHD possess melting profiles that are heavily influenced by the presence and sequence of oligonucleotides, with canonical κB sites showing the strongest degree of stabilization (Figure 1d, Tables S1 and S2). The p50 and p65 RHDs bound specific κB sites under 50 mM NaCl conditions with a remarkable 10°C stabilization over the protein alone (Tables S1 and S2). In contrast, at 150 mM NaCl, the RHDs were stabilized equally well by both oligonucleotide mimics (heparin) and specific κB DNA sequences, with a modest increase in melting temperature of ~5°C (Tables S1 and S2). Nonspecific DNA showed considerably less stabilization; with a small ~1°C increase in melting temperature. Presumably, this difference between heparin and nonspecific DNA is due to the comparably greater charge per unit length of heparin.
Prior work supports our observation that the affinity of Rel proteins for oligonucleotides is inversely proportional to salt concentration (i.e., these proteins show decreasing affinity for nucleic acids in the presenting of increasing salt concentrations).57 The consistency of this effect across species indicates that the Rel proteins possess weaker or more transient interactions with oligonucleotides at greater ionic strength, likely due to the abundance of charge–charge interactions stabilizing DNA binding.25, 26, 27, 28, 29, 30 Together, these observations demonstrate the functional competency of the recombinant human p50 and p65 RHD, and their overarching consistency with the mouse proteins.
Together, these results suggest that NF‐κB employs a bimodal DNA‐binding strategy, in which the Rel proteins assume either a flexible nonspecific DNA‐bound conformer, or alternately an ordered and rigid conformer when bound to κB DNA. These modes of interaction facilitate tracking of the transcription factor along DNA as it performs a one‐dimensional search for high‐affinity sites—indeed a similar mechanism was shown for the tumor suppressor p53.58 In cases where κB sites are inaccessible due to binding by nucleosomes, chromatin remodeling is required to reveal these sites and allow binding of the NF‐κB Rel core.59 This tracking model is further supported by the presence of general DNA backbone contacts observed across a wide variety of Rel protein structures, in addition to site‐specific interactions mediated in the context of κB DNA sequences.1, 60
3.2. Crosslinking analysis of Rel dimer composition
Previous studies have generated the p50/p65 RHD heterodimer by either complete chemical denaturation of homodimeric species followed by subsequent refolding,29 incubation of p65 RHD with an excess of its p50 RHD partner,30 as well as co‐expression of the p50 and p65 RHDs with removal of any homodimeric populations.24 Importantly, given these prior descriptions, we presumed that some degree of Rel dimer purification would be required to produce a reasonably homogeneous heterodimer population. In addition, the relative rates of dimer subunit exchange were not discernible from existing literature.24, 30, 61, 62
We performed a series of experiments in which we compared the apparent composition of the dimeric species formed upon mixing p50 RHD and p65 RHD under denaturing conditions, followed by refolding. Using previously described protocols employed to establish many of the essential biochemical properties of the NF‐κB complex,28, 43, 44, 61, 62 we denatured the Rel proteins with either urea or GuHCl, and refolded them by rapid dilution and overnight dialysis in standard buffer. As a control, we mixed an aliquot of the native p50 and p65 RHDs and incubated them alongside the other samples. We visualized the composition of the NF‐κB dimers by subsequently chemical crosslinking at room temperature for ~1 hr. These refolding and/or crosslinking procedures generated an essentially quantitative yield of the p50/p65 RHD heterodimer, as judged by SDS‐PAGE and size‐exclusion chromatography multi‐angle light scattering (SEC MALS) (Figure 1e–g). These crosslinking experiments demonstrated that the heterodimer comprises the predominant species in solution and forms readily in buffers routinely used for biochemical experiments on NF‐κB.
These results clearly demonstrate dramatically different crosslinking behaviors between different kinds of Rel homo‐ and heterodimers. In particular, homodimeric p65 RHD showed a particularly poor capacity to crosslink to its binding partner, whereas both the p50 RHD homodimer and p50/p65 RHD heterodimer crosslinked very efficiently. Because the BS3 crosslinker is amine reactive, this difference in crosslinking behavior can likely be attributed to the abundance of lysines in p50 RHD (23 per monomer) as compared to p65 RHD (11 per monomer) (Figure 1e). The crosslinking competency of the heterodimer indicates that, although relatively few lysines in p65 are capable of crosslinking, this capacity can be substantially increased in the context of the p50/p65 heterodimer. This is presumably due to the large number of available moieties for crosslinking in p50 that are proximal to p65 RHD (i.e., along the inner faces of the dimerisation and DNA‐binding domains). In addition, inter‐RHD movement may either restrict the crosslinking capacity of p65 RHD or favor p50 RHD crosslinking to an exaggerated degree.
The crosslinking reactions were all performed using an equivalent overall concentration of Rel proteins. We thereby presume that chemical‐crosslinker‐induced multimerisation is responsible for the lowered intensity of the bands seen for p65 RHD in Figure 1e in effect, creating a series of distinct species that “spread out” the primary intensity of the p65 RHD band into numerous distinct multimerised species. For instance, a single crosslinking event between two RHDs of adjacent p65 units would likely occur between the DNA‐binding domains of p65, rather than between the dimerisation domains of p65. Consequently, the resulting crosslinked species is likely able to form additional protein–protein interactions through the un‐modified dimerization domains. These sequential crosslinking reactions would thereby form progressively higher‐weight molecular species (dimers, trimers, etc.). This behavior is not observed with the same frequency in p50 RHD due to the abundance of lysines in p50 RHD—this creates a higher probability for forming a “self‐sealing” crosslinked dimer interface. Given the above rationale explaining the lack of crosslinkable lysines within the dimerisation domains of p65 RHD, the diversity of crosslinked species are unlikely to be biologically relevant, and instead are artifacts of the crosslinking reaction.
Our results and interpretation support the structural models of the Rel protein dimers for both human p50 and p65 RHDs, featuring a crosslinkable dimer interface in the p50 homodimer and p50/65 heterodimer, but a generally unreactive interface in the p65 homodimer. Taken together, these experiments also indicate that the heterodimeric complex is particularly stable relative to either homodimer, and demonstrate an unexpectedly dynamic behavior of the homodimeric Rel proteins.
3.3. Subunit exchange between Rel homodimers
The observation that the p50 RHD and p65 RHD homodimers rapidly dissociate and stably reassociate to preferentially form the p50/p65 RHD heterodimer suggested a significant energetic preference for this mixed species. We investigated subunit exchange dynamics within the p65 and p50 RHD dimers by using two slightly different lengths of each of these polypeptides, varying in unstructured regions that extend away from the dimerisation interface (Figure 2a). These analogous domain boundaries have been used across several manuscripts describing the structures of the mouse homologs of p50 and p65, none of which attribute any significance of these regions to dimer formation. (These assumptions about the Rel domain boundaries' lack of influence on dimer formation are corroborated by our results, described below.) These variants of the p50 RHD, termed p50‐1 and p50‐2, as well as p65 RHD, termed p65‐1 and p65‐2 (Figure 2b), could then be combined to study the kinetics of subunit exchange and the resulting protein species populations (Figure 2c–e).
FIGURE 2.
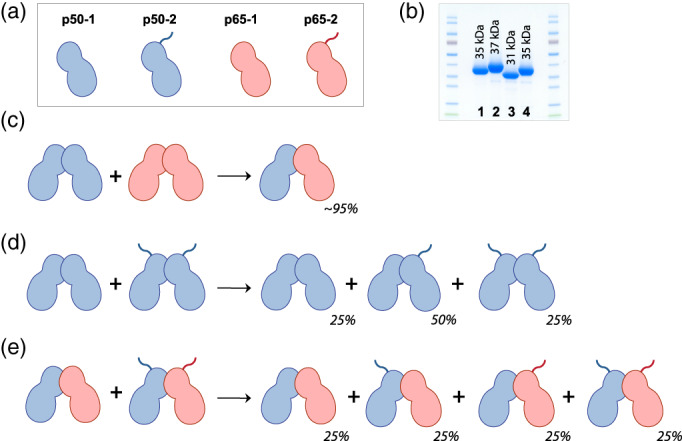
NF‐κB species populations resulting from subunit exchange between Rel dimers. (a) Schematic representation of subunit exchange. Proteins are annotated as either “−1” or “−2” variants based on subtle differences in their amino acid length, as demonstrated in the SDS‐PAGE gels at right: 1–p50‐1 RHD, 2–p50‐2 RHD, 3–p65‐1 RHD, 4–p65‐2 RHD. These polypeptide differences encompass regions outside of the dimerisation domains, and therefore do not affect the association properties of the Rel dimers. RHD, Rel homology domain; SDS‐PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis
Native electrospray ionization mass spectrometry (ESI‐MS) data were acquired for three sets of Rel proteins: (1) p50‐1 and p50‐2, (2) p65‐1 and p65‐2, and (3) p50‐1 and p65‐1, using techniques as previously described.55, 63 Experiments were conducted both at room temperature (Figures S4–S6), as well as 37°C (Figures 3 and 4). These data represent the convolution of several Gaussian distribution envelopes for each species, which changed in their relative proportions over time.
FIGURE 3.
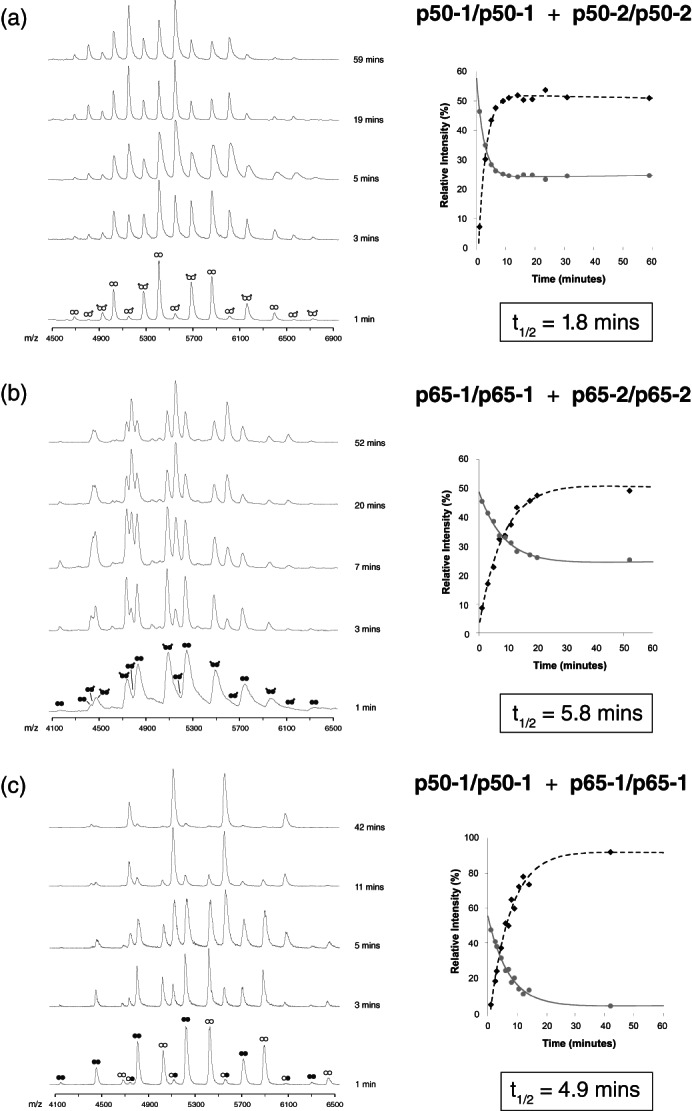
Subunit exchange between Rel dimers at 37 °C. (a) ESI‐MS time‐course of subunit exchange between p50‐1/p50‐1 (white circles) and p50‐2/p50‐2 (white circles with tails) RHD homodimers. The panel insert shows the relative populations of each species from 0‐60 minutes. (b) ESI‐MS time‐course of subunit exchange between p65‐1/p65‐1 (black circles) and p65‐2/p65‐2 (black circles with tails) RHD homodimers. The panel insert shows the relative populations of each species from 0‐60 minutes. (c) ESI‐MS time‐course of subunit exchange between p50‐1/p50‐1 (white circles) and p65‐1/p65‐1 (black circles) RHD homodimers. The panel insert shows the relative populations of each species from 0‐60 minutes. For Figure 3a‐c, note that values obtained for the homodimeric species have been averaged to generate the decay curve. ESI, electrospray ionization; MS, mass spectrometry; RHD, Rel homology domain
FIGURE 4.
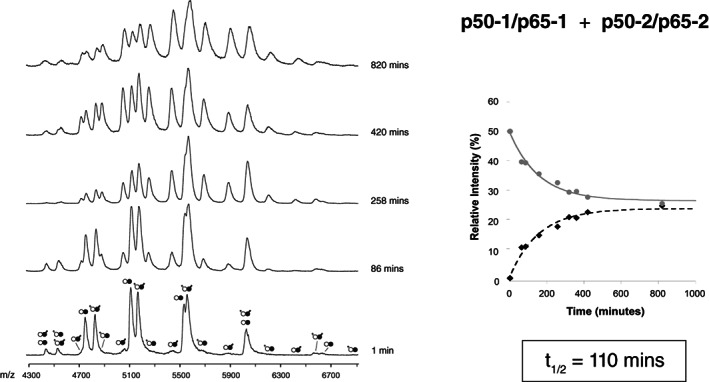
Subunit exchange between Rel dimers at 37 °C. ESI‐MS time‐course of subunit exchange between p50‐1/p65‐1 (white and black circles) and p50‐2/p65‐2 (white circles with tails and black circles with tails) RHD heterodimers. The panel insert shows the relative populations of each species from 0‐1000 minutes. Note that values obtained for the p50‐1/p65‐1 and p50‐2/p65‐2 heterodimeric species have been averaged to generate the decay curve, and that values obtained for the p50‐1/p65‐2 and p50‐2/p65‐1 heterodimeric species have been averaged to generate the corresponding growth curve. ESI, electrospray ionization; MS, mass spectrometry; RHD, Rel homology domain
For both the bi p50‐1/p50‐2 and p65‐1/p65‐2 homodimer shuffling experiments (i.e., p50‐1 shuffling with p50‐2 homodimers, and p65‐1 homodimers shuffling with p65‐2 homodimers), we anticipated that the relative proportion of the resulting dimeric species would be 25% p‐1/p‐1, 50% p‐1/p‐2, and 25% p‐2/p‐2 (Figure 2d), reflecting a purely probabilistic association. Our experimental results validated this assumption, demonstrating that the additional short polypeptide stretch distinguishing the p‐1 and p‐2 variants of both p50 RHD (Figure S4 and Figure 3b) and p65 RHD (Figure S5 and Figure 3c) did not perturb the overall equilibrium dimer populations. This result fits with the body of biophysical work conducted on the RHDs of NF‐κB family members, which demonstrate that these regions present no structural significance as it relates to dimerization—that is, these regions are either disordered in crystal structures of Rel dimers, or are spatially incapable of interacting in the context of the dimeric complexes. Therefore, as anticipated, an isolated p50‐1 subunit has equal probability for associating with either p50‐1 or p50‐2. Similarly, an isolated p65‐1 subunit has an equal probability for associating with either p65‐1 or p65‐2. The equivalence of the p‐1 and p‐2 variants in the context of p50 RHD and p65 RHD is an essential and verifiable component of all subsequent analyses.
Although isolated p50 RHD and p65 RHD populations exchanged subunits in a purely probabilistic manner, the combination of p50 RHD and p65 RHD dramatically favored the formation of the heterodimeric complex: at equilibrium, the reaction was compromised of ~95% heterodimer (Figure 3c, S5). Moreover, the kinetics of subunit exchange was distinct between these complexes. In room temperature conditions, the t 1/2 was 18 min for p50‐1/p50‐2 (Figure S4), 44 min for p65‐1/p65‐2 (Figure S5), and 38 min for p50‐1/p65‐1 (Figure S6). At 37°C, the trend was similar, although considerably faster owing to enhanced dynamics at higher temperatures, with a t 1/2 of 1.8 min for p50‐1/p50‐2 (Figure 3a), 5.8 min for p65‐1/p65‐2 (Figure 3b), and 4.9 min for p50‐1/p65‐1 (Figure 3c). Since the heterodimeric complex forms at a rate that is nearly that of the dissociation rate of the p65 homodimer, these results suggest that the slow dissociation rate of p65 dimers likely comprises the rate‐limiting step in heterodimer formation.
We additionally queried whether preformed heterodimers could exchange subunits, by mixing the p50‐1/p65‐1 heterodimer with the p50‐2/p65‐2 heterodimer. At room temperature, even after hours of incubation, no appreciable reaction could be observed (Figure S6). Similarly, the reaction proceeded slowly (although measurably) at 37°C, with a t 1/2 of 110 min. These results provide strong evidence for the kinetic stability of the p50/p65 heterodimer, and demonstrate why it often comprises the majority of NF‐κB species within the cell (see Section 4).
The basis for the distinct behavior of the p50 and p65 RHDs is clarified by examination of the subunit interfaces in dimer crystal structures62 (Figure 5b–e), with particular attention paid to the distinct interfaces as viewed from above and below the dimerisation domains. Across all dimers, the dimerisation domain interface has a remarkable degree of dyad symmetry. All interfaces, as seen from either above or below, feature pairs of positively and negatively charged residues that seal the edges of the interfaces, and create a stronger propensity for subunit association (Figure 5b–e). As seen from above, these residue pairs are D273‐R246 and R307‐D217 in p50/p65, D273‐R307 + R307‐D273 in the p50 homodimer, and D217‐R246 + R246‐D17 in the p65 homodimer. These electrostatic interactions form the fundamental basis of dimer stability.
FIGURE 5.
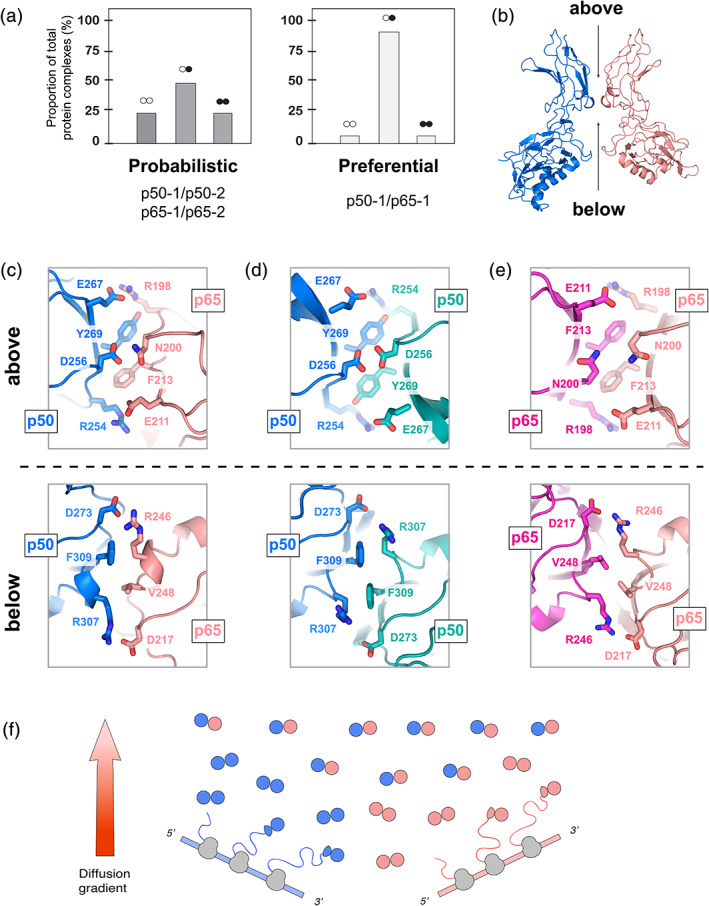
Subunit exchange between Rel species dramatically favors heterodimer formation. (a) The expected and observed population proportions are shown as a percentage of the aggregate populations. (b) Interface between the dimerisation domains of p50 (left, blue) and p65 (right, peach) in the structure of the p50/p65 heterodimer (PDB: 1VKX). The corresponding double‐stranded DNA oligo is omitted. Opposite faces of the dimerisation interface are shown from above (top) and from below (bottom). for the following: (c) p50/p65 (PDB ID: 1VKX), (d) p50/p50 (PDB ID: 1SVC), (e) p65/p65 (PDB ID: 2RAM). (f) Model of the apparent preference for the p50/p65 heterodimer, showing that (in the absence of further polypeptide modification or sequestration), the heterodimeric species should comprise the majority of cellular species after even modest diffusion away from sites of translation. p50 is represented in blue, and p65 is represented in peach
wAdditional residues in these interfaces direct dimer‐specific stability (Figure 5c–e). As seen from above, contacts between aromatic or hydrophobic residues (as well as the methylene chains of the flanking charged residues mentioned above) form the interface between adjacent Rel proteins. However, additional distinct residues between subunits help define dimer stability. Importantly, p50 projects D256 into the center of the dimer interface, resulting in two abutting negatively charged residues in the center of the p50 homodimer. In contrast, the analogous residue N200 in p65 results in two neutrally charged residues in the center of the p65 homodimer interface62 (Figure 5c). The juxtaposition of two negatively charged aspartates destabilizes the p50 homodimer,57 and is likely the most important factor in the rapid dissociation of the p50 homodimer. In contrast, the p65 homodimer accommodates the two neutral asparagines via formation of inter‐subunit water‐mediated hydrogen bonds.57 The p50/p65 heterodimer is likewise stabilized by hydrogen bonds between D256 of p50 and N200 of p65 (Figure 5c).
Viewed from above, there is an additional layer of aromatic residues, one presented by each dimerization domain (Figure 5c–e). In the p65 homodimer, the phenyl ring of F213 is almost completely buried within the dimer interface between adjacent subunits, and forms a well‐packed hydrophobic interface. In contrast, the p50 homodimer presents two Tyr in the heterodimer, the aromatic ring of Y269 is largely buried within a hydrophobic pocket formed by both by the p50 and p65 dimerisation domains. The Tyr hydroxyl, however, remains solvent accessible. The differences in chemistry between Phe and Tyr both appear to be well‐accommodated within the dimer interface, by counterbalancing the favorable hydrophobic surface burial of the phenyl ring against minimization of the burial of polar or hydrogen‐bonding competent moieties of the Tyr phenolic ring.
Examination of the alternate face of the dimerisation domain closest to the DNA‐binding domains (Figure 5b–e), that is, the dimerization interface as viewed from below, demonstrates an additional asymmetric pair of residues consisting of F309 in p50 and V248 in p65. In the case of the p50 homodimer, the Phe–Phe pair interact across the densely packed dimer interface,25, 26 although these are positioned in an orientation that is incapable of stabilizing the interaction via π‐stacking. The Phe rings pack against the long hydrophobic methylene chain of inter‐subunit R198 as well as laterally against its cognate Phe (Figure 5d). In contrast, the Val–Val pair in the p65 homodimer results in a “steric gap” in the center of the interface27, 28 (Figure 5e), likely contributing to this complex's considerably faster exchange rate relative to the p50/p65 heterodimer. The p50/p65 heterodimer strikes a balance between the two homodimeric interfaces, thereby forming an interface containing a Phe‐Val pair that fills the spatial void presented by a Val dyad alone.
Given that prior work has demonstrated a remarkable degree of similarity in the interactions and orientations of the Rel dimerisation domains in isolation as well as in the DNA‐bound RHD complexes,62 these results are likely to be inherent properties of the Rel architecture that are invariant with the presence of nucleic acids. With subtle side chain rearrangements relative to either homodimer, the dimerisation interface of p50/p65 ultimately forms a well‐packed interface29, 64, 65 that is dramatically stabilized over that in either homodimer. Together, these results thus rationalize the kinetically stable p50/p65 heterodimer.
4. CONCLUSIONS
Although it is one of the most investigated transcription factors, little is known about the fundamental kinetics of NF‐κB complex formation or the inter‐conversion potential of hetero‐ and homodimers, especially when considering NF‐κB's short nuclear translocation timescale. Here, we examined the dissociation and reassociation of subunits to form new NF‐κB hetero‐ or homodimers using a combination of gel filtration, chemical crosslinking, and native MS. Our results demonstrate that the core p50 and p65 homodimeric components of NF‐κB routinely exchange subunits with a half‐life of less than 10 min at physiological temperature, highlighting a dramatic preference for the formation of the p50/p65 heterodimer. The heterodimer was also remarkably kinetically stable, with a dissociation half‐life that is more than 15‐fold longer than that of either homodimer.
Together, our result suggests that the relative levels of Rel proteins present in a cell may be a means for modulating the populations of various Rel dimer species, in addition to well‐characterized mechanisms like phosphorylation or cellular localization (e.g., export from or import into the nucleus). Our studies of these Rel dimers thus provide new insight into the means by which NF‐κB complexes must be regulated by either expression levels or additional post‐translational modifications in order to maintain a variety of NF‐κB species (and functional outputs), as well as specifically target certain subsets of NF‐κB response elements. Likewise, our results underscore how essential these regulatory subunits or protein modifications are for maintaining NF‐κB species diversity in the cell.
Current literature suggests that the p50 homodimer is more abundant than the p65 homodimer, as based on gel shift analyses of cellular extracts.57 This is partly due to the sequestration of p65 homodimers in the cytoplasm by the inhibitory protein IκBα, which when dissociated from p65 by a mild detergent induces appreciable quantities of p65 homodimers.66 These observations nevertheless reinforce the overwhelming preponderance of the p50/p65 heterodimer, consistent with our observations using purified recombinant proteins. Our results indicate that protein expression levels are likely to play a key role in modulating the preponderance of certain types of NF‐κB activity. Additionally, the observed NF‐κB species in solution are likely to reflect their populations bound to DNA, as suggested by prior work demonstrating that subunit exchange within transcriptional complexes ceases once bound to DNA.55 These equilibrium analyses of NF‐κB dimer species in solution thereby further clarify the overall transcriptional activity of the NF‐κB complex.
It is intriguing to speculate that the relative association/dissociation rates of the Rel complexes influences NF‐κB function in the cell. For example, the comparatively rapid subunit dissociation of the p50 homodimer may be an evolved negative function to minimize the persistence of the transcription‐repressive p50 homodimer. In contrast, the slower dissociation rate of p65 homodimers would favor a transcriptionally active form of the NF‐κB complex.
There are several mechanisms that may explain the routinely observed NF‐κB species in vivo, which seemingly contradict our observation for a pronounced or nearly exclusive preference for the formation of the p50/p65 heterodimer. One explanation is an unequal expression of p50 or p65, leading to a favorable presence of either homodimer. For example, a higher expression of p50 in comparison to p65 would enable the p50 homodimer to persist alongside the p50/p65 heterodimer; and, in contrast, the higher expression of p65 in comparison to p50 would result in the presence of the p65 homodimer alongside the p50/p65 heterodimer. Our analysis therefore presents a distinct interpretation of how transcriptional control can be modulated by altering the relative abundance of either the p50 or p65 subunits to direct NF‐κB to a particular subset of κB sites. The relative proportion of the NF‐κB Rel subunits themselves, as opposed to the abundance of inhibitory subunits like IκBα, may thereby produce an inherent degree of DNA binding‐site selectivity.
Similarly, the simultaneous presence of the p50 and p65 homodimers in the cell is likely mediated by post‐translational modification that modulates Rel assembly to favor a particular form of NF‐κB.60 The fact that homodimers are more prevalent than expected in vivo highlights how other non‐Rel subunits of the NF‐κB complex,67 as well as factors arising from the unique environment of chromatin,68 could alter the stability of these transcriptional complexes under different conditions.
Based on our observations, p50 and p65 homodimer formation should be able to occur during or soon after translation in the high effective protein concentration of the translational milieu (Figure 5e). However, heterodimers are likely to form once the homodimers diffuse from the site of translation. Our work thus provides a new model for understanding NF‐κB transcriptional activity and offers the possibility of modulating NF‐κB DNA specificity using compounds that can selectively target the unique dimerisation interfaces of p50/p50, p65/p65, or p50/p65 complexes.
Despite the preference for formation of the p50/p65 heterodimer in vitro, chromatin inaccessibility likely occlude many heterodimer DNA‐binding sites, and thereby preferentially shifts the dimer equilibrium to favor binding of other forms of NF‐κB to particular targets. This possibility is further underscored when considering that transcription factor activity is actively regulated by the accessibility of κB sites, a property that is modulated by dynamic alterations chromatin structure.69, 70 Our results highlight that additional in vivo experimentation is needed to further elucidate the underlying mechanisms leading to the formation and transcriptional activity of the wide array of NF‐κB species.
Last, the preferential affinity of p50 and p65 for each other can be used as a working assumption for future experimental and theoretical investigations of these critical models of transcriptional activity. In particular, these considerations might be important for better understanding the interactions of these proteins with other members of the Rel transcription factor family. The conspicuous affinity of p50 and p65 for forming heterodimers should also be considered for the research of other Rel subunits, where it may underlie a number of general physiological behavior as well as courses of disease. More globally, such considerations may guide many other transcription factor families that depend on homo‐ or heterodimer formation.
CONFLICT OF INTEREST
The authors declare no competing or conflicting interests.
AUTHOR CONTRIBUTIONS
Matthew Biancalana: Conceptualization; data collection; data curation; formal analysis; investigation; methodology; validation; visualization; writing‐original draft; writing‐review & editing. Eviatar Natan: Data collection; data curation; formal analysis; investigation; methodology; writing‐original draft; validation; writing‐review & editing. Michael J. Lenardo: Funding acquisition; project administration; resources; supervision; writing‐original draft; writing‐review & editing. Alan R. Fersht: Funding acquisition; project administration; resources; supervision; writing‐original draft; writing‐review & editing.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENT
We thank Stephen Ambrose for assistance with MS data collection.
Biancalana M, Natan E, Lenardo MJ, Fersht AR. NF‐κB Rel subunit exchange on a physiological timescale. Protein Science. 2021;30:1818–1832. 10.1002/pro.4134
Michael J. Lenardo and Alan R. Fersht are regarded as co‐last authors and co‐supervisors
Funding information Medical Research Council Laboratory of Molecular Biology, Protein and Nucleic Acid Division; National Institutes of Health, National Institute of Allergy and Infectious Diseases
REFERENCES
- 1.Ghosh S. Handbook of transcription factor NF‐kappaB. Boca Raton: CRC Press; 2007. [Google Scholar]
- 2.Gilmore TD. Introduction to NF‐κB: Players, pathways, perspectives. Oncogene. 2006;25:6680–6684. [DOI] [PubMed] [Google Scholar]
- 3.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer‐binding protein Nf‐kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. [DOI] [PubMed] [Google Scholar]
- 4.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 5.Perkins ND. Integrating cell‐signalling pathways with NF‐κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin J, Albert S. Series introduction: The transcription factor NF‐κB and human disease. J Clin Invest. 2001;107:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baltimore D. NF‐κB is 25. Nature. 2011;12:683–685. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF‐kappaB: A blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmore TD. Multiple mutations contribute to the oncogenicity of the retroviral oncoprotein v‐Rel. Oncogene. 1999;18:6925–6937. [DOI] [PubMed] [Google Scholar]
- 10.Gilmore TD, Temin HM. Different localization of the product of the v‐rel oncogene in chicken fibroblasts and spleen cells correlates with transformation by REV‐T. Cell. 1986;44:791–800. [DOI] [PubMed] [Google Scholar]
- 11.Steward R, Zusman SB, Huang LH, Schedl P. The dorsal protein is distributed in a gradient in early drosophila embryos. Cell. 2003;55:487–495. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan JC, Kalaitzidis D, Gilmore TD, Finnerty JR. Rel homology domain‐containing transcription factors in the cnidarian Nematostella vectensis . Develop Genes Evol. 2006;217:63–72. [DOI] [PubMed] [Google Scholar]
- 13.Baeuerle PA, Baltimore D. NF‐kappa B: Ten years after. Cell. 1996;87:13–20. [DOI] [PubMed] [Google Scholar]
- 14.Gilmore TD. Introduction to NF‐kappaB: Players, pathways, perspectives. Oncogene. 2006;25:6680–6684. [DOI] [PubMed] [Google Scholar]
- 15.Hayden MS, Ghosh S. Shared principles in NF‐kappaB signaling. Cell. 2008;132:344–362. [DOI] [PubMed] [Google Scholar]
- 16.Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF‐kappaB transcription factors: Structural views. Oncogene. 1999;18:6845–6852. [DOI] [PubMed] [Google Scholar]
- 17.Lenardo M, Pierce JW, Baltimore D. Protein‐binding sites in Ig gene enhancers determine transcriptional activity and inducibility. Science. 1987;236:1573–1577. [DOI] [PubMed] [Google Scholar]
- 18.Tisné C, Hantz E, Hartmann B, Delepierre M. Solution structure of a non‐palindromic 16 base‐pair DNA related to the HIV‐1 kappa B site: Evidence for BI‐BII equilibrium inducing a global dynamic curvature of the duplex. J Mol Biol. 1998;279:127–142. [DOI] [PubMed] [Google Scholar]
- 19.Siggers T, Chang AB, Teixeira A, et al. Principles of dimer‐specific gene regulation revealed by a comprehensive characterization of NF‐κB family DNA binding. Nat Immunol. 2011;13:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong D, Teixeira A, Oikonomopoulos S, et al. Extensive characterization of NF‐κB binding uncovers non‐canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011;12:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine‐responsive IkappaB kinase that activates the transcription factor NF‐kappaB. Nature. 1997;388:548–554. [DOI] [PubMed] [Google Scholar]
- 22.Baeuerle PA, Baltimore D. I kappa B: A specific inhibitor of the NF‐kappa B transcription factor. Science. 1988;242:540–546. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh S, Karin M. Missing pieces in the NF‐kappaB puzzle. Cell. 2002;109 Suppl:S81–S96. [DOI] [PubMed] [Google Scholar]
- 24.Chen FE, Kempiak S, Huang DB, Phelps C, Ghosh G. Construction, expression, purification and functional analysis of recombinant NFkappaB p50/p65 heterodimer. Protein Eng. 1999;12:423–428. [DOI] [PubMed] [Google Scholar]
- 25.Müller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC. Structure of the NF‐kappa B p50 homodimer bound to DNA. Nature. 1995;373:311–317. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF‐kappa B p50 homodimer bound to a kappa B site. Nature. 1995;373:303–310. [DOI] [PubMed] [Google Scholar]
- 27.Chen YQ, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF‐kappa B p65 homodimer. Nat Struct Biol. 1998;5:67–73. [DOI] [PubMed] [Google Scholar]
- 28.Chen YQ, Sengchanthalangsy LL, Hackett A, Ghosh G. NF‐kappaB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Struct Fold Des. 2000;8:419–428. [DOI] [PubMed] [Google Scholar]
- 29.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF‐kappaB bound to DNA. Nature. 1998;391:410–413. [DOI] [PubMed] [Google Scholar]
- 30.Escalante CR, Shen L, Thanos D, Aggarwal AK. Structure of NF‐kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon‐beta promoter. Struct Fold Des. 2002;10:383–391. [DOI] [PubMed] [Google Scholar]
- 31.Perkins ND. Integrating cell‐signalling pathways with NF‐kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. [DOI] [PubMed] [Google Scholar]
- 32.Moorthy AK, Huang DB, Wang VY, Vu D, Ghosh G. X‐ray structure of a NF‐kappaB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem kappaB sites. J Mol Biol. 2007;373:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouwmeester T, Bauch A, Ruffner H, et al. A physical and functional map of the human TNF‐alpha/NF‐kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. [DOI] [PubMed] [Google Scholar]
- 34.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon‐beta enhanceosome. Cell. 2007;129:1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panne D. The enhanceosome. Curr Opin Struct Biol. 2008;18:236–242. [DOI] [PubMed] [Google Scholar]
- 36.Steward R, Zusman SB, Huang LH, Schedl P. The dorsal protein is distributed in a gradient in early drosophila embryos. Cell. 1988;55:487–495. [DOI] [PubMed] [Google Scholar]
- 37.Petersen AJ, Katzenberger RJ, Wassarman DA. The innate immune response transcription factor relish is necessary for neurodegeneration in a drosophila model of ataxia‐telangiectasia. Genetics. 2013;194:133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldwin AS Jr. Series introduction: The transcription factor NF‐kappaB and human disease. J Clin Invest. 2001;107:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor‐kappaB: Its role in health and disease. J Mol Med. 2004;82:434–448. [DOI] [PubMed] [Google Scholar]
- 40.Kim HJ, Hawke N, Baldwin AS. NF‐kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–747. [DOI] [PubMed] [Google Scholar]
- 41.Kawakami K, Scheidereit C, Roeder RG. Identification and purification of a human immunoglobulin‐enhancer‐binding protein (NF‐kappa B) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc Natl Acad Sci U S A. 1988;85:4700–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malek S, Huxford T, Ghosh G. Ikappa Balpha functions through direct contacts with the nuclear localization signals and the DNA binding sequences of NF‐kappaB. J Biol Chem. 1998;273:25427–25435. [DOI] [PubMed] [Google Scholar]
- 44.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF‐kappaB complex reveals mechanisms of NF‐kappaB inactivation. Cell. 1998;95:759–770. [DOI] [PubMed] [Google Scholar]
- 45.Wan F, Anderson DE, Barnitz RA, et al. Ribosomal protein S3: A KH domain subunit in NF‐kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. [DOI] [PubMed] [Google Scholar]
- 46.Chen ZJ. Ubiquitin signalling in the NF‐kappaB pathway. Nat Cell Biol. 2005;7:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin L, Ghosh S. A glycine‐rich region in NF‐kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol Cell Biol. 1996;16:2248–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rape M, Jentsch S. Productive RUPture: Activation of transcription factors by proteasomal processing. Biochim Biophys Acta. 2004;1695:209–213. [DOI] [PubMed] [Google Scholar]
- 49.Beinke S, Belich MP, Ley SC. The death domain of NF‐kappa B1 p105 is essential for signal‐induced p105 proteolysis. J Biol Chem. 2002;277:24162–24168. [DOI] [PubMed] [Google Scholar]
- 50.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF‐κB signaling pathways. Nat Immunol. 2011;12:695–708. [DOI] [PubMed] [Google Scholar]
- 51.Baker RG, Hayden MS, Ghosh S. NF‐κB, inflammation, and metabolic disease. Cell Metabol. 2011;13:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature. 2006;441:431–436. [DOI] [PubMed] [Google Scholar]
- 53.Ashall L, Horton CA, Nelson DE, et al. Pulsatile stimulation determines timing and specificity of NF‐kappaB‐dependent transcription. Science. 2009;324:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hipps DS, Packman LC, Allen MD, et al. The peripheral subunit‐binding domain of the dihydrolipoyl acetyltransferase component of the pyruvate dehydrogenase complex of Bacillus stearothermophilus: Preparation and characterization of its binding to the dihydrolipoyl dehydrogenase component. Biochem J. 1994;297:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Natan E, Hirschberg D, Morgner N, Robinson CV, Fersht AR. Ultraslow oligomerization equilibria of p53 and its implications. Proc Natl Acad Sci U S A. 2009;106:14327–14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Analyt Chem. 2002;74:1402–1407. [DOI] [PubMed] [Google Scholar]
- 57.Phelps CB. Mechanism of kappa B DNA binding by Rel/NF‐kappa B dimers. J Biol Chem. 2000;275:24392–24399. [DOI] [PubMed] [Google Scholar]
- 58.Melero R, Rajagopalan S, Lázaro M, et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc Natl Acad Sci U S A. 2011;108:557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lone IN, Shukla MS, Charles Richard JL, Peshev ZY, Dimitrov S, Angelov D. Binding of NF‐κB to nucleosomes: Effect of translational positioning, nucleosome remodeling and linker histone H1. PLoS Genet. 2013;9:e1003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huxford T, Ghosh G. A structural guide to proteins of the NF‐kappaB signaling module. Cold Spring Harb Perspect Biol. 2009;1:a000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huxford T. Preparation and crystallization of dynamic NF‐kappa B / Ikappa B complexes. J Biol Chem. 2000;275:32800–32806. [DOI] [PubMed] [Google Scholar]
- 62.Huang DB, Huxford T, Chen YQ, Ghosh G. The role of DNA in the mechanism of NFkappaB dimer formation: Crystal structures of the dimerization domains of the p50 and p65 subunits. Struct Fold Des. 1997;5:1427–1436. [DOI] [PubMed] [Google Scholar]
- 63.Natan E, Joerger AC. Structure and kinetic stability of the p63 tetramerization domain. J Mol Biol. 2012;415:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF‐kappaB complex. Cell. 1998;95:749–758. [DOI] [PubMed] [Google Scholar]
- 65.Berkowitz B. The X‐ray crystal structure of the NF‐kappa B p50/p65 heterodimer bound to the interferon beta ‐kappa B site. J Biol Chem. 2002;277:24694–24700. [DOI] [PubMed] [Google Scholar]
- 66.Ganchi PA, Sun SC, Greene WC, Ballard DW. A novel NF‐kappa B complex containing p65 homodimers: Implications for transcriptional control at the level of subunit dimerization. Mol Cell Biol. 1993;13:7826–7835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wan F, Anderson DE, Barnitz RA, et al. Ribosomal protein S3: A KH domain subunit in NF‐κB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. [DOI] [PubMed] [Google Scholar]
- 68.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF‐kappa B and cytokine‐inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 69.Smale ST. Transcriptional regulation in the immune system: A status report. Trends Immunol. 2014;35:190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hubner MR, Eckersley‐Maslin MA, Spector DL. Chromatin organization and transcriptional regulation. Curr Opin Genet Dev. 2013;23:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information