

Abstract
Malaria is a parasitic illness caused by the genus Plasmodium from the apicomplexan phylum. Five plasmodial species of P. falciparum (Pf), P. knowlesi, P. malariae, P. ovale, and P. vivax (Pv) are responsible for causing malaria in humans. According to the World Malaria Report 2020, there were 229 million cases and ~ 0.04 million deaths of which 67% were in children below 5 years of age. While more than 3 billion people are at risk of malaria infection globally, antimalarial drugs are their only option for treatment. Antimalarial drug resistance keeps arising periodically and thus threatens the main line of malaria treatment, emphasizing the need to find new alternatives. The availability of whole genomes of P. falciparum and P. vivax has allowed targeting their unexplored plasmodial enzymes for inhibitor development with a focus on multistage targets that are crucial for parasite viability in both the blood and liver stages. Over the past decades, aminoacyl‐tRNA synthetases (aaRSs) have been explored as anti‐bacterial and anti‐fungal drug targets, and more recently (since 2009) aaRSs are also the focus of antimalarial drug targeting. Here, we dissect the structure‐based knowledge of the most advanced three aaRSs—lysyl‐ (KRS), prolyl‐ (PRS), and phenylalanyl‐ (FRS) synthetases in terms of development of antimalarial drugs. These examples showcase the promising potential of this family of enzymes to provide druggable targets that stall protein synthesis upon inhibition and thereby kill malaria parasites selectively.
Keywords: aminoacyl‐tRNA synthetase, antimalarial, drug, malaria, plasmodia
1. INTRODUCTION
Malaria is caused by the plasmodial parasites of the Apicomplexan phylum and causes high mortality and morbidity in endemic regions.1 Antimalarial drugs remain the main backbone of the therapeutic options available against malaria. The current malaria treatment involves the use of artemisinin‐based combination (ACT) therapy, chloroquine or sulfadoxine‐pyrimethamine combination therapy. However, the recent surge in antimalarial drug resistance poses a global health threat and impending disease resurgence.1, 2, 3, 4 This threat shall hinder the efforts towards malaria elimination, and emphasizes the need to identify novel drug scaffolds and validated drug targets with newer mechanisms of actions.5, 6 However, a dynamic proteome of the plasmodial species makes it difficult to select for multistage targets.7, 8 In this context, housekeeping pathways are attractive for multistage targeting. Here, we focus on aminoacyl‐tRNA synthetases (aaRSs) as potent antimalarial drug targets owing to their pivotal role in protein synthesis. The inhibition of aaRSs results in decimation of protein synthesis and subsequent cell death. The aaRSs undergo a two‐step reaction—step 1 includes the formation of aminoacyl‐adenylate complex followed by step 2 that involves the handing over of the amino acid to either the 2′‐ or 3′‐hydroxyl group on the 3′‐terminal adenosine of tRNA.8, 9, 10, 11, 12, 13 Based on their mode of tRNA charging and catalytic site topology, aaRSs are divided into Class I or Class II (Figure 1). The aaRSs were first proposed as potent antimalarial drug targets in 2008.14 In 2008, our group performed genomic analysis on P. falciparum and identified 37 aaRS genes that were responsible for 36 aaRS enzymes. Furthermore, while aaRSs comprise a large fraction of the overall proteome, localization and bioinformatics studies revealed 16 cytoplasmic aaRSs and 15 nucleus‐encoded apicoplastic aaRSs.14 Among these, alanyl‐ (ARS), cysteinyl‐ (CRS), glycyl‐ (GRS), and threonyl‐ (TRS) tRNA synthetases are present as single genes and are shared between cytoplasm and apicoplast. Intriguingly, the P. falciparum mitochondrial compartment was found to contain an enzymatically active phenylalanyl‐tRNA synthetase (FRS). Over the last decade, aaRSs have been successfully exploited for drug targeting and to date several aaRS inhibitors have been developed against multiple pathogens as listed in Table 1. Several of these known inhibitors have been structurally and functionally characterised and provide hope for development of anti‐infectives.
FIGURE 1.

The characteristics of Class I and Class II aminoacyl‐tRNA synthetases
TABLE 1.
Aminoacyl‐tRNA synthetase inhibitors developed against various pathogens
| Inhibitor | Target | Comment | References |
|---|---|---|---|
| 4‐Thiaisoleucine | IRS | Structural analogue of isoleucine, targets the cytoplasmic IRS. | 54 |
| A3, A5 | ARS | In‐silico and docking identified active site inhibition with IC50 ~ 4 μM | 55 |
| AN2690a, AN2279 | LRS | Member of benzoxaborols family | 56 |
| Ascamycin | FRS | 57 | |
| Borrelidin and analogues | TRS | P. falciparum IC50 ~ 1 nM | 37, 39, 45, 54, 55, 58, 59 |
| Cispentacin | PRS | 60 | |
| DWN12088 | PRS | 61 | |
| Furanomycin | IRS | 62 | |
| Graniticin | LRS | 63 | |
| Indolmycin | WRS | 64 | |
| Lysyl‐adenylate analogues | KRS | 50 analogues with μM inhibition reported | 65 |
| Mupirocin | IRS |
Clinical inhibitor of bacterial infection by S. aureus. Targets the active site of apicoplast IRS IC50 ~ 90 nM |
66 |
| Ochratoxin A | FRS | 67 | |
| REP8839/REP3123 | MRS | Inhibit P. falciparum with an IC50 ~ 150 nM | 68 |
| Sulfomyl adenosine analogues | SRS ERS QRS NRS YRS | Mimics the intermediate aminoacyl‐AMP with an IC50 in nM range | 58 |
| T‐3833261 | PRS | 69 | |
|
TCMDC‐140398 TCMDC‐140498 TCMDC‐140522 TCMDC‐140563 TCMDC‐140564 TCMDC‐140734 TCMDC‐141485 |
FRSb | GlaxoSmithkline's library screened molecules | 70 |
| TCMDC‐131575 | IRSb | GlaxoSmithkline's library screened molecules | 70 |
|
TCMDC‐139450 TCMDC‐139627 TCMDC‐140014 |
MRSb | GlaxoSmithkline's library screened molecules reference is number 70 for this too | |
| TCMDC‐139627 TCMDC‐141232 | YRSb | GlaxoSmithkline's library screened molecules | 70 |
Approved drug.
Hypothesized target.
1.1. Structurally validated aminoacyl‐tRNA synthetases
Over 30 Pf‐aaRSs crystal structures for seven different aaRSs are available in the PDB with either their respective substrates or inhibitors. These include—lysyl‐tRNA synthetase (KRS), editing domain of the leucyl‐ (LRS), prolyl‐ (PRS), phenylalanyl‐ (FRS), arginyl‐ (RRS), tryptophanyl‐ (WRS), and tyrosyl‐ (YRS).15, 16, 17, 18, 19, 20 The presence of several inhibitor binding sites make aaRSs tantalizing drug targets. The key binding sites in aaRSs include: (1) editing domain, (2) catalytic domain, (3) allosteric sites, (4) anticodon‐binding domain, and (5) aaRSs specific domains (Figure 2). The availability of several plasmodial aaRS three‐dimensional crystal structures provides a platform for rational drug design. In the sections below, we focus on three of the advanced aaRSs (lysyl‐, prolyl‐, and phenylalanyl‐tRNA synthetases) that have been structurally and functionally validated as potent antimalarial drug targets in either P. falciparum or P. vivax.
FIGURE 2.

Various aaRS domains are illustrated: the editing domain; catalytic domain; anticodon‐ binding domain; and parasite‐specific domains. Numbers indicates possible sites of interaction within the: (1) editing domain; (2) catalytic domain; (3) allosteric sites; (4) anticodon‐binding domains; and (5) plasmodial‐specific domains
1.2. Cytoplasmic lysyl‐tRNA synthetase (KRS)
Unlike other aaRSs, lysyl‐tRNA synthetase (KRS) belongs to both Class I (in archaea and bacteria) and Class II (prokaryotes and eukaryotes) family of aaRSs.19, 21, 22, 23 In addition to protein synthesis, KRSs have several noncanonical functions. For instance, the Homo sapiens KRS (HsKRS) acts as a cytokine resulting in the activation of macrophages and peripheral blood mononuclear cells inducing the production of TNF‐α.24 Further, KRS seems to be bound to multi‐synthetase complex (MSC) in α2β2 form, where the tetrameric interface of HsKRS (α4) is used by the p38 dimer (β2) to assemble a α2β2 subcomplex.25, 26 KRSs are also crucial in the synthesis of signalling molecules such as diadenosine polyphosphate (Ap4a) that are involved in DNA replication, regulation of ion channels and gene expression.27, 28 To date, the structural and biochemical attributes of KRSs have been elucidated for the following organisms—Cryptosporidium parvum, Entamoeba histolytica, Homo sapiens, Loa loa, and Plasmodium falciparum in context of the inhibitor cladosporin (CLD) and its derivatives.29, 30 The fungi—Aspergillus flavus and Cladosporium cladosporioides ‐ synthesize CLD [IUPAC: isocumarin 3,4‐dihydro‐6,8‐dihydroxy‐3‐(6‐methyltetrahydro pyran‐2‐ylmethyl)] compound.27 It is a chiral molecule whose scaffold is formed by the 6,8‐dihydroxylisocoumarin and methyltetrahydropyrene (mTHP) groups. These groups mimic the adenosine moiety, thereby competing with ATP (Figure 3a). CLD and its derivatives are amongst the most well studied inhibitors for the KRSs. Nearly a decade ago, CLD was reported to inhibit P. falciparum in cell‐based assays.27 Subsequent structural and biochemical studies validated that the cytoplasmic P. falciparum KRS (PfKRScyto) was the CLD drug target.27, 30 Thus far 19 (1 apo and 18 holo) three‐dimensional crystal structures of the PfKRS have been solved and deposited in the Protein Data Bank (PDB).15
FIGURE 3.

(a) Chemical structure of cladosporin (CLD) with constituent chemical fragments. (b) The anticodon binding (in sea green) and aminoacylation (in caramel) domain of the PfKRScyto are shown. The bound CLD (in blue) and the three conserved motifs, characteristic of Class II aaRSs are marked (in black). (c) Structural comparison of the CLD active site of PfKRScyto (pink) and HsKRS (blue) is shown. In PfKRScyto‐CLD, Phe342, His338, and Arg559 undergo rotameric conformational change to accommodate CLD. Val328 and Thr344 near the ATP‐binding pocket provides a hydrophobic cavity for mTHP ring of CLD to fit. (d) Sequence alignment of CLD selectivity residues in studied KRS complexes from Cryptosporidium parvum, Homo sapiens, Loa loa, and P. falciparum are shown. The vital residues that bestow CLD selectivity are highlighted in red box. CpKRS: Asn and Ala; HsKRS: Gln and Thr; LlKRS: Val and Ser and PfKRS: Val and Ser
1.2.1. Three‐dimensional structures of PfKRSs
The PfKRS exists as a dimer comprising of 19 β strands and 17 α helices. Each monomer harbors the N‐terminal OB‐fold anticodon‐binding (ABD) (E77‐P222) and a C‐terminal catalytic (CD) (T230‐P581) (Figure 3b). The catalytic domain has three conserved motifs: motif 1 (276‐PMMNLI‐281), 2 (329‐FRNE‐332), and 3 (557‐ID‐559) (Figure 3b). Structural analysis of the CLD bound PfKRS enzyme shows that the residues His338, Phe342, and Arg559 undergo an alteration in their rotameric‐conformation to avoid steric clash once bound to CLD. These residues are likely responsible for both the entry and placement of the isocoumarin moiety (dihydroxybenzene ring) of CLD in a stacking format (Figure 3c). The side chains of Arg330 and Asn503 provide a hydrophobic environment for the mTHP ring‐binding cavity in order to fit and lie with Phe342 on the opposite side. Phe342 along with Arg330 and Asn503 in the closed state of PfKRS stack with the benzopyran of CLD. The two‐hydroxyl groups of CLD and the side chains of Glu332 and Asn339 form hydrogen bonds. The secondary structural changes as well as the rotameric alterations in the active site residues, in particular—His338, Phe342, Arg559 in CLD bound PfKRS when compared to holo PfKRS suggests either the conformational selectivity model for enzyme substrate interactions or the ligand‐induced fit model.31 CLD selectivity is attributed to two residues—Val328 and Ser344 that are proximate to the ATP‐binding pocket (Figure 3c,d). The two residues Val328 and Ser344 in PfKRS due to their small size of their side chains result in a larger hydrophobic cavity adjacent to the residue Phe342 where the mTHP ring of CLD fits more snugly.
CLD inhibits the PfKRScyto in nanomolar range with >100‐fold selectivity when compared to HsKRS.27, 28, 30 However, CLD has reported poor metabolic stability and low bioavailability and thus the pharmacological properties of CLD need to be improved.26 To address this, CLD's three chiral centres were used to synthesize all of the eight possible stereoisomers (Figure 3e). Complete libraries of CLD stereoisomers thus developed were evaluated for anti‐plasmodial activity, in addition to enzyme‐, structure‐ and parasite‐based assays.32 Among these, the two CLD stereoisomers (CLD‐12 and CLD‐16) were most effective due to criticality of chiral positions at C3, C10, and C14.32 The two chiral centres C3 and C10 were found to be crucial for drug potency.32 Following this lead, CLD analogues were developed based on chemical modifications and alterations within the CLD scaffold.33 These analogues helped to identify molecules with similar potency as CLD but improved drug‐like properties.33 The authors studied structural and functional variations in the mTHP moiety, the aromatic dihydroisocoumarin ring as well as in the methylene functionality of the linker (Figure 3e).33 The CLD‐2 analog bound to the PfKRS enzyme (PDB: 6M0T) similar to the parent CLD molecule with an exception in the linker region, where the hydroxyl group resulted in the formation of a hydrogen bond with the side chain Glu500.33 This emphasizes the need to explore stereo‐chemical or functional modifications and their role in potency of CLD. In summary, a wealth of information on the structural basis of inhibitor binding to PfKRScyto has been generated to date. The target has been validated using cell‐based assays and therefore the continuing pursuit of drug‐like inhibitors that may mimic CLD is worthwhile in order to develop antimalarial scaffolds that may one day lead to a drug.
1.3. Prolyl‐tRNA synthetase (PRS)
The plant Dichroa febrifuga has been part of an old‐style antimalarial therapy in China. The quinazolinone‐type alkaloid known as “febrifugine (FF)” and its halogenated form, 7‐bromo‐6‐chloro derivative called halofuginone (HF) are of interest due to their antiparasitic activity (Figure 4a).34, 35 HF binding to human glutamylprolyl‐tRNA synthetase (HsEPRS) and the inhibition of the human prolyl‐tRNA synthetase (HsPRS) established that HF inhibits the PRSs via its 4‐quinazolinone group that mimics the 3′ end of bound tRNA and the piperidine ring that mimics the bound proline.36, 37, 38 To date, PRS three‐dimensional structures are available from the following parasites—Cryptosporidium parvum (CpPRS), Eimeria tenella (EtPRS), Giardia lamblia (GlPRS), Homo sapiens (HsPRS), Leishmania major (LmPRS), Plasmodium falciparum (PfPRS), and Toxoplasma gondii (TgPRS).36, 38, 39, 40, 41
FIGURE 4.
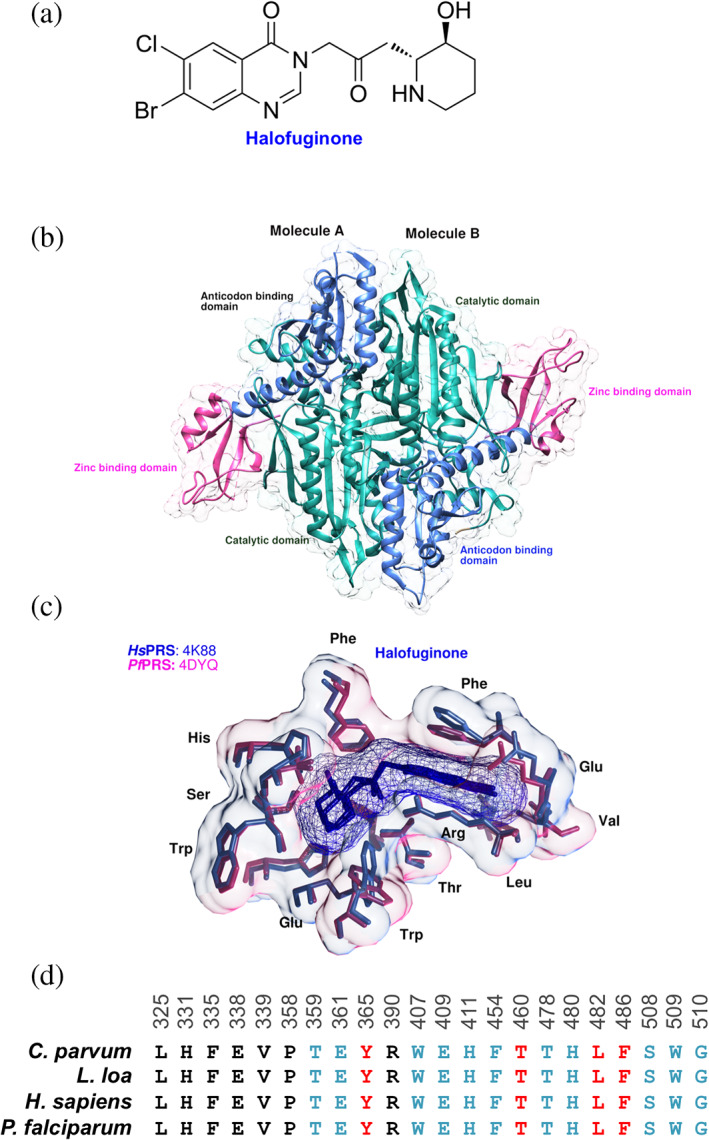
(a) Chemical structure of Halofuginone is shown. (b) The crystal structure of the PfPRScyto‐HF‐AMPPNP ternary complex. PfPRScyto dimer in the asymmetric unit of PfPRScyto‐HF‐AMPPNP crystals structure is shown with bound HF (yellow) and AMPPNP (pink). Catalytic (C‐domain, residues 255–532), anti‐codon binding (AB‐domain, residues 533–655), C‐terminal zinc binding like (Z‐domain, residues 656–746) domains as well as protein termini are marked. (c) Important HF binding pocket residues within 5 Å are shown. Protein residues are shown in stick representations with a transparent overlay of the corresponding Van der Waal's surface. (d) Multiple sequence alignment of likely HF binding site residues (blue) from Cryptosporidium parvum (Cp), Loa loa (Ll), Homo sapiens (Hs), and Plasmodium falciparum (Pf) are shown. The important secondary shell residues were coloured in red
1.3.1. Three‐dimensional structure of PfPRS with Halofuginone
PfPRS is a homo‐dimer containing three subdomains—catalytic (255–532), anticodon‐ (533–655), and a Z‐domain (C‐terminal‐zinc‐binding‐like) (656–746) (Figure 4b). PfPRS is a valuable anti‐malarial target.37 The dimeric interface (residues 316–351) is asymmetric in the PfPRS‐HF and has a “collapsed conformation” due to the presence of an empty active site with no substrates or inhibitors.41 This region forms the interface that makes the binding site for ATP, L‐Pro, and HF.42 Partly owing to the variable secondary structures and distinct orientations within the Chain A and Chain B of the dimeric interface, PfPRS adopts intrinsic active‐site asymmetry in the two chains.37, 38, 39, 40, 41 Structural analysis of the HF‐PfPRS showed that two key residues Phe405 and Trp407 are involved in the ATP binding and undergo a side‐chain rotameric changes.42 Upon binding of HF to PfPRS this rotameric change results in opening of the ATP binding motif.41 The residue Thr478 forms H‐bond with the hydroxylpiperidine ring and adopts alternate side chain conformation. Further, the residues Thr359 and Glu361 form H‐bonds with the piperidine ring via its N atom, and hydrophobic interactions result in the stabilization of the quinazolinone ring (Figure 4c).42 Analysis of the three‐dimensional crystal structure of the non‐hydrolysable ATP analog: adenosine 5′‐(β, γ‐imido) triphosphate (AMPPNP) bound PfPRS‐HF enabled the idea of further drug derivatization owing to the chemical spaces available within the structure (Figure 4c). The bromine of HF interacts with Phe335 via the main‐chain carbonyl O atom and is surrounded by hydrophobic stems of Glu338 and Val339 while the piperidine ring of HF is surrounded by Glu361, Trp407, Glu409, Thr478, His480, Ser508 thereby leaving no empty spaces for further additive modifications (Figure 4c). On the contrary, the chlorine atom is solvent exposed and forms hydrogen bonds with two water molecules.16, 43, 44, 45 Owing to these observations, crystallography and cell/enzyme‐based drug interaction assays were used to structurally and biochemically characterize the quinazolinone‐based compounds—HF, FF, 6‐fluoro febrifugine (6F‐FF), and tetrahydro quinazolinone febrifugine (Th‐FF). Among these, HF conferred higher thermal stability than other FF derivatives (FF and Th‐FF).43, 44, 45 Further to this, the multiple sequence alignment of HF active‐site residues and important shell residues suggested that HF can be used to target multiple parasitic pathogens (Figure 4d). In summary, a host of chemical information and structural data are available for PRS in complex with FF derivatives. The target has been validated using cell‐based assays and therefore the continuing pursuit of drug‐like inhibitors is worthwhile in order to develop antimalarial scaffolds that may one day lead to a drug.
1.4. Phenylalanyl‐tRNA synthetase (FRS)
The PfFRS genes code for three proteins that are localised to the subcellular compartments in the malaria parasite.14, 46 This FRS enzyme is unique as it has α‐ and β‐subunits, exists as a heterodimer and further dimerizes into a hetero‐tetrameric (αβ)2 assembly.47, 48 The FRS (αβ)2 heterotetramer binds to two molecules of tRNAPhe. The α‐subunit contains the active site that binds the substrates (L‐Phe, ATP, and 3′ end of tRNA). The (αβ)2 heterotetrameric arrangement is not a prerequisite for FRS activity. For instance, the human mitochondrial FRS (HsFRSmito) is monomeric, containing the amino acid binding site and the anti‐codon binding domain at its N‐ and C‐terminus, respectively, and tRNA binds in an open conformation. The aminoacylation reaction site is present on the α‐subunit, while the β‐subunit is involved in non‐cognate amino acids editing as well as tRNA recognition. The PfFRS α‐subunit is ~500 residue longer than the bacterial one and the β‐subunit is ~200 residues shorter than the bacterial counterpart. The α‐ and β‐subunit interface is occupied by a Mg2+ that may be critical for enzyme activity and stabilization of the complex.47 The plasmodial species contain two copies of tRNAPhe each for the nucleus and apicoplast. The mitochondria lack the tRNAPhe and it likely imports the Phe‐tRNAPhe complex from the cytoplasm.46 Inhibition of the FRScyto abrogates generation of Phe‐tRNAPhe that is required for protein synthesis in the cytoplasm and mitochondria.46
1.4.1. Crystal structure of cytosolic PvFRS
The (αβ)2 heterodimer arrangement of the PvFRScyto consists of eight domains.47 Among these eight domains, two are present in the α‐subunit (α1‐catalytic and α2) and six are within the β‐subunit (β1, β3‐β4 (constitute the editing site), β5, β6 (catalytic‐like) and β7).47 The α‐subunit is associated with β‐subunit via two‐subdomains of the β‐subunit, namely B1 and B2’ (Figure 5a). Bicyclic azetidine based small molecules were identified from a library of ~100,000 compounds (stereo‐chemically and structurally diverse compounds) synthesized by Broad Institute through Diversity Oriented Synthesis (DOS).49, 50, 51 The potent bicyclic azetidine BRD1389 compound is bound to the α‐subunit of the PvFRScyto enzyme.47 These bicyclic azetidine compounds exhibited killing activity against the different parasitic stages of Pf, that is, liver, blood and the transmission stages.49, 50 Initial phenotypic growth inhibition assays were conducted to target the blood stage in Pf drug resistant strain Dd2. Whole‐genome DNA‐sequencing from the BRD drug induced Pf drug resistant strain Dd2 (mutations on the α‐subunit—L550V, M316I, G512E, V545I) facilitated the identification of the BRD inhibitor target that is, PfcFRS. Further, in vivo studies showed low propensity for the mutations against BRD compounds, which specifically inhibit PfcFRS enzyme activity in nanomolar range.49 BRD1389 shows high selectivity for PfcFRS with an IC50 of 12 nM versus an IC50 of 1,200 nM for HscFRS.47, 49 Subsequently, BRD1389 was enzymatically characterized as a competitive inhibitor for L‐Phe, but noncompetitive inhibition was observed against ATP.47 This structural work established a unique inhibition mechanism where BRD1389 can bind and inhibit apo‐PfcFRS enzyme.
The homologous plasmodial cFRS enzyme—PvcFRS has been co‐crystallised with the BRD inhibitor—BRD1389 (PDB ID: 7BY6) (Figure 5a).47 BRD1389 contain core diazabicyclodecane ring, urea, 4‐cyclopropoxy phenyl, methoxymethyl and diarylacetylene moieties (Figure 5c). Structural characterization of the BRD1389‐PvcFRS revealed that the L‐Phe site was occupied by the diarylacetylene moiety, 4‐cyclopropoxy phenyl moieties occupy the auxiliary site and the bicyclic azetidine scaffold partly occupies the ATP site (Figure 5b). The structural analysis of the BRD1389‐PvFRS (PDB ID: 7BY6) identifies the following factors for strong affinity via the amino acid residues Ala541‐Leu544 (541‐AWGL‐544) that line the binding site for diarylacetylene moiety of BRD1389, while Asn519, Gln457, and Glu459 contribute to hydrophobic interactions (Figure 5c,d).47 The 4‐cyclopropoxy phenyl urea moiety is locked between the residues Val517, Ile483, and Pro549 and is buried in auxiliary site groove formed by the residues Gly506, His508, Glu510, Lys512, Lys513, Leu515, Arg548, Pro549, and Ile552 (Figure 5c,e). The bicyclic azetidine ring interacts with the Arg443, Glu445, His451, Phe455, and Ser545 near the ATP‐binding site (Figure 5c,f). A comparative analysis with the HsFRScyto suggests that the PvFRScyto accommodates the inhibitor via induced fit model as the protein undergoes conformational changes during ligand binding. These conformational changes are—(1) open conformation of the loop to accommodate the methoxymethyl group formed by the ATP binding pocket, (2) closed conformation of residues 508–513 (Loop 2) in the auxiliary pocket, and (3) the rotameric change of Arg548 in the auxiliary pocket accommodating of the urea moiety of BRD1389 compound (Figure 5g).47, 48 The residues Val458, Tyr480, and Ile483 within the auxiliary pocket of the PvFRScyto are not conserved between HsFRS and PvFRS, and possibly contribute to the selective binding of the BRD1389 (Figure 5h). BRD1389 has a unique mode of binding when compared to the single site inhibitor—CLD for KRS (mimics adenosine) or double site inhibitor—HF for PRS (mimics L‐Proline and 3′ end tRNA subsite). BRD1389 occupies the amino acid:phenylalanine pocket along with an auxiliary pocket without the engagement of ATP within the PvFRScyto.47 The structural characterization of the BRD‐bound PvFRScyto provides the opportunity for further modification of the BRD compound in order to enhance the drug properties and also utilize the ATP site. In summary, a host of chemical information and more recently structural data are available for FRS now. The target has been validated using cell‐based assays and therefore the continuing pursuit of FRS inhibitors is worthwhile.
FIGURE 5.

(a) The heterotetrameric assembly of PvFRScyto is shown, with different alpha‐ and beta‐subunits marked. The BRD1389 is shown in red surface. (b) The BRD1389 occupies the L‐Phe binding site (diarylacetylene moiety), ATP‐binding site (bicyclic azetidine scaffold) and the novel auxiliary site (4‐cyclopropoxy phenyl urea). (c) Chemical structure of the BRD1389 with its diarylacetylene core (in brown), 4‐cyclopropoxy phenyl urea (in blue) and bicyclic azetidine scaffold (in green). The active site residues of the BRD1389 in the alpha‐subunit of the PvFRScyto enzyme are shown. (d) Diarylacetylene core active site residues forming binding site (in blue) and residues with hydrophobic interactions (in pink). (e) Residues locking the 4‐cyclopropoxy phenyl urea within its active site (in blue) and the auxiliary site groove residues (in pink) are marked. (f) The residues interacting with the bicyclic azetidine ring that form the ATP‐binding site of the PvFRScyto are marked. (g) Superimposition of the PvFRScyto (plum) and HscFRS (seagreen) showing the showing the opening of Loop 1 (443–453) and closing of Loop 2 (508–513) upon BRD1389 binding is shown. (h) Sequence alignment of BRD1389 selectivity residues in Cryptosporidium parvum, Homo sapiens, Loa loa, and P. vivax are shown
2. SUMMARY
There is an urgent need for multistage antimalarials that target all or most stages of the malaria parasite. A few malaria parasite protein families have been exploited for antimalarial drug discovery. Being housekeeping enzymes, aminoacyl‐tRNA synthetases provide a unique opportunity for multistage drug targets of the plasmodial species. Several natural compounds and their derivatives are currently being explored to target malaria parasite aaRSs. We have reviewed the chemical and structural data that are available so far on three of the most advanced targets against malaria. These comprehensive studies have highlighted inhibitors that have been validated using cell‐based, enzyme‐based and structural binding assays. Their selectivity has been measured (vs. both human enzyme counterparts and against human cell lines) and their binding to target enzymes have been probed via high resolution crystal structures of enzyme‐drug complexes. However, the journey towards identification of the best drug‐like compounds is long and arduous, though very much ongoing. The global crisis of COVID‐19 may dent the dedication of funding allocation for malaria, as the wherewithal is required for pandemic management.52, 53 However, we feel that only sustained and enhanced funding of drug discovery projects in malaria will ensure that we are able to control and possibly eliminate this parasite in the coming decades.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Jyoti Chhibber‐Goel: Data curation; writing‐original draft; writing‐review & editing. Manickam Yogavel: Data curation; writing‐review & editing. Amit Sharma: Data curation; funding acquisition; writing‐review & editing.
Chhibber‐Goel J, Yogavel M, Sharma A. Structural analyses of the malaria parasite aminoacyl‐tRNA synthetases provide new avenues for antimalarial drug discovery. Protein Science. 2021;30:1793–1803. 10.1002/pro.4148
REFERENCES
- 1.World Heath Organization . World Malaria Report. 73, 1–4 (2020). [Google Scholar]
- 2.Wellems TE, Plowe CV. Chloroquine‐resistant malaria. J Infect Dis. 2001;184:770–776. [DOI] [PubMed] [Google Scholar]
- 3.Chhibber‐Goel J, Sharma A. Profiles of Kelch mutations in Plasmodium falciparum across South Asia and their implications for tracking drug resistance. Int J Parasitol Drugs Drug Resist. 2019;11:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaturvedi R et al. Geographical spread and structural basis of sulfadoxine‐pyrimethamine drug‐resistant malaria parasites. Int J Parasitol. 2021. 10.1016/j.ijpara.2020.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Pazhayam NM, Chhibber‐Goel J, Sharma A. New leads for drug repurposing against malaria. Drug Discov Today. 2019;24:263–271. [DOI] [PubMed] [Google Scholar]
- 6.Pushpakom S et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. [DOI] [PubMed] [Google Scholar]
- 7.Yang T et al. MalDA, Accelerating Malaria Drug Discovery. Trends Parasitol. 2021. 10.1016/j.pt.2021.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner MJ et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pham JS et al. Aminoacyl‐tRNA synthetases as drug targets in eukaryotic parasites. Int J Parasitol Drugs Drug Resist. 2014;4:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nyamai DW, Tastan Bishop Ö. Aminoacyl tRNA synthetases as malarial drug targets: a comparative bioinformatics study. Malar J. 2019;18:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson KE et al. Dual targeting of aminoacyl‐tRNA synthetases to the apicoplast and cytosol in Plasmodium falciparum. Int J Parasitol. 2012;42:177–186. [DOI] [PubMed] [Google Scholar]
- 12.Saint‐Léger A. Sinadinos, C. & Ribas de Pouplana, L. The growing pipeline of natural aminoacyl‐tRNA synthetase inhibitors for malaria treatment. Bioengineered. 2016;7:60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manickam Y et al. Drug targeting of one or more aminoacyl‐tRNA synthetase in the malaria parasite Plasmodium falciparum. Drug Discov Today. 2018;23:1233–1240. [DOI] [PubMed] [Google Scholar]
- 14.Bhatt TK et al. A genomic glimpse of aminoacyl‐tRNA synthetases in malaria parasite Plasmodium falciparum. BMC Genomics. 2009;10:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berman HM et al. The Protein Data Bank. Acta Crystallogr. D Biol Crystallogr. 2002;58:899–907. [DOI] [PubMed] [Google Scholar]
- 16.Jain V, Yogavel M, Sharma A. Dimerization of Arginyl‐tRNA Synthetase by Free Heme Drives Its Inactivation in Plasmodium falciparum. Structure. 2016;24:1476–1487. [DOI] [PubMed] [Google Scholar]
- 17.Khan S et al. An appended domain results in an unusual architecture for malaria parasite tryptophanyl‐tRNA synthetase. PloS One. 2013;8:e66224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonoiki E et al. Antimalarial Benzoxaboroles Target Plasmodium falciparum Leucyl‐tRNA Synthetase. Antimicrob Agents Chemother. 2016;60:4886–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh CY et al. Crystal structures of Plasmodium falciparum cytosolic tryptophanyl‐tRNA synthetase and its potential as a target for structure‐guided drug design. Mol Biochem Parasitol. 2013;189:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nachiappan M, Jain V, Sharma A, Manickam Y, Jeyakanthan J. Conformational changes in glutaminyl‐tRNA synthetases upon binding of the substrates and analogs using molecular docking and molecular dynamics approaches. J Biomol Struct Dyn. 2020;38:1575–1589. [DOI] [PubMed] [Google Scholar]
- 21.Ibba M et al. A euryarchaeal lysyl‐tRNA synthetase: resemblance to class I synthetases. Science. 1997;278:1119–1122. [DOI] [PubMed] [Google Scholar]
- 22.Ibba M et al. Substrate recognition by class I lysyl‐tRNA synthetases: a molecular basis for gene displacement. Proc Natl Acad Sci USA. 1999;96:418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Donoghue P, Luthey‐Schulten Z. On the evolution of structure in aminoacyl‐tRNA synthetases. Microbiol Mol Biol Rev MMBR. 2003;67:550–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park SG et al. Human lysyl‐tRNA synthetase is secreted to trigger proinflammatory response. Proc Natl Acad Sci USA. 2005;102:6356–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo M, Ignatov M, Musier‐Forsyth K, Schimmel P, Yang X‐L. Crystal structure of tetrameric form of human lysyl‐tRNA synthetase: Implications for multisynthetase complex formation. Proc Natl Acad Sci USA. 2008;105:2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang P et al. Structural context for mobilization of a human tRNA synthetase from its cytoplasmic complex. Proc Natl Acad Sci USA. 2011;108:8239–8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoepfner D et al. Selective and specific inhibition of the plasmodium falciparum lysyl‐tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma A, Yogavel M, Sharma A. Structural and functional attributes of malaria parasite diadenosine tetraphosphate hydrolase. Sci Rep. 2016;6:19981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan S et al. Structural analysis of malaria‐parasite lysyl‐tRNA synthetase provides a platform for drug development. Acta Crystallogr D Biol Crystallogr. 2013;69:785–795. [DOI] [PubMed] [Google Scholar]
- 30.Khan S, Sharma A, Belrhali H, Yogavel M, Sharma A. Structural basis of malaria parasite lysyl‐tRNA synthetase inhibition by cladosporin. J Struct Funct Genomics. 2014;15:63–71. [DOI] [PubMed] [Google Scholar]
- 31.Changeux, J.‐P. & Edelstein, S. Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol. Rep. 2011;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das P et al. Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite. J Med Chem. 2018;61:5664–5678. [DOI] [PubMed] [Google Scholar]
- 33.Babbar P et al. Design, Synthesis, and Structural Analysis of Cladosporin‐Based Inhibitors of Malaria Parasites. ACS Infect Dis. 2021. 10.1021/acsinfecdis.1c00092. [DOI] [PubMed] [Google Scholar]
- 34.Coatney GR, Cooper WC, Culwell WB. White, W. C. & Imboden, C. A. Studies in human malaria. XXV. Trial of febrifugine, an alkaloid obtained from Dichroa febrifuga lour., against the Chesson strain of Plasmodium vivax. J Natl Malar Soc US. 1950;9:183–186. [PubMed] [Google Scholar]
- 35.Derbyshire ER, Mazitschek R, Clardy J. Characterization of Plasmodium liver stage inhibition by halofuginone. ChemMedChem. 2012;7:844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller TL et al. Halofuginone and other febrifugine derivatives inhibit prolyl‐tRNA synthetase. Nat Chem Biol. 2012;8:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Son J et al. Conformational changes in human prolyl‐tRNA synthetase upon binding of the substrates proline and ATP and the inhibitor halofuginone. Acta Crystallogr. D Biol Crystallogr. 2013;69:2136–2145. [DOI] [PubMed] [Google Scholar]
- 38.Zhou H, Sun L, Yang X‐L, Schimmel P. ATP‐directed capture of bioactive herbal‐based medicine on human tRNA synthetase. Nature. 2013;494:121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larson ET et al. Structure of the prolyl‐tRNA synthetase from the eukaryotic pathogen Giardia lamblia. Acta Crystallogr. D Biol Crystallogr. 2012;68:1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linder MR et al. (2R,3S)‐(+)‐ and (2S,3R)‐(‐)‐Halofuginone lactate: synthesis, absolute configuration, and activity against Cryptosporidium parvum. Bioorg Med Chem Lett. 2007;17:4140–4143. [DOI] [PubMed] [Google Scholar]
- 41.Jain V, Kikuchi H, Oshima Y, Sharma A, Yogavel M. Structural and functional analysis of the anti‐malarial drug target prolyl‐tRNA synthetase. J Struct Funct Genomics. 2014;15:181–190. [DOI] [PubMed] [Google Scholar]
- 42.Jain V et al. Structure of Prolyl‐tRNA Synthetase‐Halofuginone Complex Provides Basis for Development of Drugs against Malaria and Toxoplasmosis. Structure. 2015;23:819–829. [DOI] [PubMed] [Google Scholar]
- 43.Kikuchi H et al. Exploration of a new type of antimalarial compounds based on febrifugine. J Med Chem. 2006;49:4698–4706. [DOI] [PubMed] [Google Scholar]
- 44.Kikuchi H et al. Synthesis of febrifugine derivatives and development of an effective and safe tetrahydroquinazoline‐type antimalarial. Eur J Med Chem. 2014;76:10–19. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi S et al. Catalytic Asymmetric Synthesis of Antimalarial Alkaloids Febrifugine and Isofebrifugine and Their Biological Activity. J Org Chem. 1999;64:6833–6841. [DOI] [PubMed] [Google Scholar]
- 46.Sharma A, Sharma A. Plasmodium falciparum mitochondria import tRNAs along with an active phenylalanyl‐tRNA synthetase. Biochem J. 2015;465:459–469. [DOI] [PubMed] [Google Scholar]
- 47.Sharma M et al. Structural basis of malaria parasite phenylalanine tRNA‐synthetase inhibition by bicyclic azetidines. Nat Commun. 2021;12:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finarov I, Moor N, Kessler N, Klipcan L, Safro MG. Structure of human cytosolic phenylalanyl‐tRNA synthetase: evidence for kingdom‐specific design of the active sites and tRNA binding patterns. Struct Lond Engl. 2010;1993(18):343–353. [DOI] [PubMed] [Google Scholar]
- 49.Kato N et al. Diversity‐oriented synthesis yields novel multistage antimalarial inhibitors. Nature. 2016;538:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maetani M et al. Synthesis of a Bicyclic Azetidine with In Vivo Antimalarial Activity Enabled by Stereospecific, Directed C(sp3)‐H Arylation. J Am Chem Soc. 2017;139:11300–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nielsen TE, Schreiber SL. Towards the optimal screening collection: a synthesis strategy. Angew Chem Int Ed Engl. 2008;47:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahi M, Baharia RK, Das P, Chhibber‐Goel J, Sharma A. Overlaying COVID‐19 mitigation plans on malaria control infrastructures. Trans R Soc Trop Med Hyg. 2021;115:6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahi M, Das P, Sharma A. Malaria elimination in India requires additional surveillance mechanisms. J Public Health Oxf Engl. 2021. 10.1093/pubmed/fdab106. [DOI] [PubMed] [Google Scholar]
- 54.Istvan ES et al. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc Natl Acad Sci USA. 2011;108:1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan S et al. Uneven spread of cis‐ and trans‐editing aminoacyl‐tRNA synthetase domains within translational compartments of P. falciparum. Sci Rep. 2011;1:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rock FL et al. An antifungal agent inhibits an aminoacyl‐tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. [DOI] [PubMed] [Google Scholar]
- 57.Isono K et al. Ascamycin and dealanylascamycin, nucleoside antibiotics from Streptomyces sp. J Antibiot (Tokyo). 1984;37(670–672). [DOI] [PubMed] [Google Scholar]
- 58.Novoa EM et al. Analogs of natural aminoacyl‐tRNA synthetase inhibitors clear malaria in vivo. Proc Natl Acad Sci USA. 2014;111:E5508–E5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Azcárate IG et al. Insights into the preclinical treatment of blood‐stage malaria by the antibiotic borrelidin. Br J.Pharmacol. 2013;169:645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konishi M et al. Cispentacin, a new antifungal antibiotic. I. Production, isolation, physico‐chemical properties and structure. J Antibiot (Tokyo). 1989;42:1749–1755. [DOI] [PubMed]
- 61.Park J. S. & Lee. Inhibition Of ProlyltRNA Synthetase As A Novel Mediator Of Cardiac Fibrosis. Circulation. 2017;136:A24036–A24036. [Google Scholar]
- 62.Katagiri, K.et al. A new antibiotic. Furanomycin, an isoleucine antagonist. J Med Chem. 10, 1149–1154 (1967). [DOI] [PubMed]
- 63.Ogilvie A, Wiebauer K, Kersten W. Inhibition of leucyl‐transfer ribonucleic acid synthetasymol. Biochem J. 1975;152:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner RG, Thorpe LF, Reuter W, Nierhaus KH. Indolmycin inhibits prokaryotic tryptophanyl‐tRNA ligase. Eur J Biochem. 1976;68:1–3. [DOI] [PubMed] [Google Scholar]
- 65.Hoen R et al. Selective inhibition of an apicoplastic aminoacyl‐tRNA synthetase from Plasmodium falciparum. Chembiochem Eur J Chem Biol. 2013;14:499–509. [DOI] [PubMed] [Google Scholar]
- 66.Nakama T, Nureki O, Yokoyama S. Structural basis for the recognition of isoleucyl‐adenylate and an antibiotic, mupirocin, by isoleucyl‐tRNA synthetase. J Biol Chem. 2001;276:47387–47393. [DOI] [PubMed] [Google Scholar]
- 67.Konrad I, Röschenthaler R. Inhibition of phenylalanine tRNA synthetase from Bacillus subtilis by ochratoxin A. FEBS Lett. 1977;83:341–347. [DOI] [PubMed] [Google Scholar]
- 68.Ochsner UA, Sun X, Jarvis T, Critchley I, Janjic N. Aminoacyl‐tRNA synthetases: essential and still promising targets for new anti‐infective agents. Expert Opin Investig Drugs. 2007;16:573–593. [DOI] [PubMed] [Google Scholar]
- 69.Shibata A et al. Discovery and pharmacological characterization of a new class of prolyl‐tRNA synthetase inhibitor for anti‐fibrosis therapy. PloS One. 2017;12:e0186587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gamo F‐J et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. [DOI] [PubMed] [Google Scholar]