

Abstract
Biomolecular interactions between proteins regulate and control almost every biological process in the cell. Understanding these interactions is therefore a crucial step in the investigation of biological systems and in drug design. Many efforts have been devoted to unraveling principles of protein-protein interactions. Recently, we introduced a simple but robust descriptor of binding affinity based only on structural properties of a protein-protein complex. In Vangone and Bonvin (2015), we demonstrated that the number of interfacial contacts at the interface of a protein-protein complex correlates with the experimental binding affinity. Our findings have led one of the best performing predictor so far reported (Pearson’s Correlation r = 0.73; RMSE = 1.89 kcal mol-1). Despite the importance of the topic, there is surprisingly only a limited number of online tools for fast and easy prediction of binding affinity. For this reason, we implemented our predictor into the user-friendly PRODIGY web-server. In this protocol, we explain the use of the PRODIGY web-server to predict the affinity of a protein-protein complex from its three-dimensional structure. The PRODIGY server is freely available at: http://milou.science.uu.nl/services/PRODIGY.
Keywords: Protein contacts, Buried surface area, Web-server, Prediction, Protein interface, Kd, Protein-protein interactions, PPIs
Background
Interaction between biomolecules regulate and control almost every biological process in the cell. Studying and understanding these interactions is therefore a crucial step in the investigation of biological systems and in drug design. Many efforts have been devoted to unraveling principles of protein-protein interactions. For this purpose, we introduced a simple but robust descriptor of binding affinity based only on structural properties, mainly intermolecular contacts, of a protein-protein complex (Vangone and Bonvin, 2015). This approach led to the best predictor so far reported. Recently, we implemented our method in the PRODIGY web-server ( Xue et al., 2016 ) (http://milou.science.uu.nl/services/PRODIGY), an online tool to predict the binding affinity of a protein-protein complex given its three-dimensional structure. PRODIGY reports the binding affinity either as Gibbs free energy (ΔG, kcal mol-1) or dissociation constant (Kd, M). PRODIGY predicts the binding affinity using the formula reported in Vangone and Bonvin (2015): It counts the number of Interatomic Contacts (ICs) made at the interface of a protein-protein complex within a 5.5 Å distance threshold, and classifies them according to the polar/apolar/charged character of the interacting amino acids. This information is then combined with properties on the Non-Interacting Surface (NIS), which we have previously shown to influence the binding affinity ( Kastritis et al., 2011 ). For training and testing, we used the binding affinity benchmark of protein-protein complexes published in Kastritis and Bonvin (2010). A recent updated version of this benchmark can be found at: http://bmm.crick.ac.uk/~bmmadmin/Affinity ( Vreven et al., 2015 ).
Further information about the benchmark, the prediction model and its accuracy can be found online on the ‘Dataset’ and ‘Method’ pages of the PRODIGY web-server, respectively.
Equipment
A computer with internet access
Software
-
A web browser (the PRODIGY server has been tested successfully on Chrome, Firefox and Safari)
PRODIGY web server address: http://milou.science.uu.nl/services/PRODIGY
Software repositories for running a local version (not described in this protocol) under a Linux or MacOSX operating system:
PRODIGY repository (https://github.com/haddocking/binding_affinity)
freeSASA (http://freesasa.github.io)
Procedure
-
The software
-
Technical description
PRODIGY is made freely available to the scientific community either as standalone software (https://github.com/haddocking/binding_affinity), which can be used locally on a desktop computer, or more conveniently as an online web-server, for which the usage is explained in this protocol. The PRODIGY software consists of a collection of Python scripts, a few Perl scripts to handle the online submission and the open-source tool freeSASA (Mitternacht, 2016) used to calculate the solvent accessible surface area, using default NACCESS (Hubbard and Thornton, 1993) parameters for atomic radii (http://freesasa.github.io).
-
Data requirement
-
Input file – mandatory
The main input required to perform binding affinity prediction is a text file containing the atomic coordinates describing the 3D structure of the protein-protein complex (or ensemble of complexes). They can either be experimental structures solved e.g., by X-ray crystallography or NMR spectroscopy, which can be obtained from the worldwide Protein Data Bank (wwPDB) (http://www.wwpdb.org) ( Berman et al., 2003 ), or structures modeled through computational approaches, e.g., by homology modelling or docking approaches. The 3D coordinates should be provided to the server in PDB or mmCIF format.
The input structure/structures can be provided in different ways:
By uploading a PDB or mmCIF file.
By providing a PDB code for automatic retrieval from the wwPDB (http://www.wwpdb.org).
By uploading an archive file (.tar, .tgz, .zip, .bz2 or .tar.gz) containing multiple PDB/mmCIF files. This option allows the submission of a unique file when many structures have to be analyzed (e.g., models derived from docking simulations).
-
Chains – mandatory
It is necessary to specify chains identifiers for the molecules involved in the interaction. If one (or both) interacting molecule is made of multiple chains at the interface, they all have to be provided separated by comma.
-
Temperature – optional
The user can specify at which temperature to perform the calculation of the dissociation constant (Kd). If nothing is specified, PRODIGY will use 25 °C by default.
-
Job name – optional
If provided, the job name will be used to identify your run. Otherwise a random label will be assigned.
-
Email – optional
If an email is provided, a link to the results will be sent when the job has completed.
-
-
-
How to use PRODIGY web-server
-
Submitting a prediction
Here we describe the process of submitting a prediction run to the PRODIGY web-server (http://milou.science.uu.nl/services/PRODIGY). As example, we will use the protein-protein complex between an antibody (FAB) and HIV-1 capsid protein p24, that is present in the Protein Data Bank (PDB) with the access code ‘1E6J’.
Open an internet browser and go to http://milou.science.uu.nl/services/PRODIGY.
-
Fill in the PRODIGY input page (Figure 1):
Insert the PDB code ‘1E6J’ into the ‘Structure’ box for automatic retrieval from the wwPDB.
-
The complex 1E6J is made of 3 chains: P (corresponding to HIV-1 capsid protein p24) and L and H (corresponding to the FAB). Considering that in this protocol we want to investigate the binding affinity at the interface between the antibody (chains L + H) and the antigen (chain P), you will need to insert:
Interactor 1 ID_chain(s): P
Interactor 2 IC_chain(s): L, H
-
Personalize your job by defining some (optional) parameters if needed:
In the box related to the temperature, change the 25 °C default if needed (note that this only affects the calculation of the dissociation constant and not the binding affinity ΔG).
Give a name to your prediction run. No spaces or special characters other then ‘-‘ or ‘_’ are allowed. For this example we will name our run ‘1E6J_prediction’.
Add your email address to be notified when your job is done and receive the link to the results page.
We are now ready to send the prediction to PRODIGY: Click on the Submit button at the bottom of the page.
A prediction usually does not take more than a few minutes. After this time, you will be redirected to the result page. If an email has been provided (see the above step B1c.iii), you will be notified when the prediction is complete and receive a link to the results page. Please note that the results are only stored for 2 weeks.
-
The result page
The result page is organized in three sections, reporting different information:
-
Binding affinity and Kd prediction
The name identifiers of your complex, which contains the PDB code of the retrieved file (or the name of the input you upload) is reported, together with the predicted ΔG (in kcal mol-1) and Kd (in M) values at the given temperature. In this example, -9.1 kcal mol-1 has been predicted for ΔG, corresponding to a Kd of 2.1e-07 M at 25 °C.
-
Prediction details
Number of ICs calculated within a threshold of 5.5 Å and % NIS classified according to the charged/polar/apolar character of the amino acids are reported. In this case, for example, there are 7 ICs between charged and polar residues and the % NIS charged atoms is 20.48.
-
Further, the full table (format .txt) of ICs is provided and can be viewed by clicking on the link reported under ‘Table of the ICs at the interface’. The format of the table is the following:
#chain1 #aa1 #res_num1 #chain2 #aa2 #res_num2
H → THR → 33 P THR 210
In which chain ID, residue type and residue number are reported for both residues interacting in Protein 1 and Protein 2.
-
Download outputs
In this foldable menu, it is possible to download a ready-to-run Pymol script (.pml) (http://www.pymol.org) that will highlight the interaction interface by displaying and coloring the interacting residues, see Figure 2. Further, it is possible to download a compressed file (.tgz) with all the result files.
-
-
-
Useful information
Make sure to check and input the correct chain_IDs for the PDB file that you are uploading/retrieving: chain_IDs have to be present in the file, and correspond to the chains that are interacting. In this example, the FAB has two chains labeled as L and H, and both of them are interacting with the HIV1 capsid protein, which is labeled as chain P.
PRODIGY can deal with files consisting of an ensemble of structures (e.g., as is typical for NMR structures). In the current implementation, only the first model will be considered for prediction. If you wish to analyze every model present in such an ensemble you should split the PDB file into single-model PDB files and submit them all as an archive file. A collection of useful Python scripts for the manipulation of PDB files, such as splitting of ensemble file, residue renumbering, changing chain ID and so on, can be found in our freely available pdb-tools GitHub repository available online at https://github.com/haddocking/pdb-tools.
The PRODIGY web-server currently only supports the 20 canonical amino acids.
Information about the web-server input/output, the prediction method and its performance, and the dataset used for training/testing the method can be found online under the Manual/Method/Dataset PROGIDY pages respectively. These are reachable through the corresponding tabs located at the beginning of each page.
-
Distribution/Software download
The PRODIGY web-server is made freely available to the scientific community at: http://milou.science.uu.nl/services/PRODIGY. The prediction scripts are also available from our GitHub repository for local setup and usage at: https://github.com/haddocking/binding_affinity.
The collection of software developed by the HADDOCK group can be found at: http://www.bonvinlab.org/software.
The freeSASA software (Mitternacht, 2016) used to calculate the solvent accessible surface area can be downloaded from http://freesasa.github.io.
Figure 1. Example view of an input page of the PRODIGY web-server.

Figure 2. A three-dimensional representation of the complex 1E6J with the color-coding of the PRODIGY script (.pml).
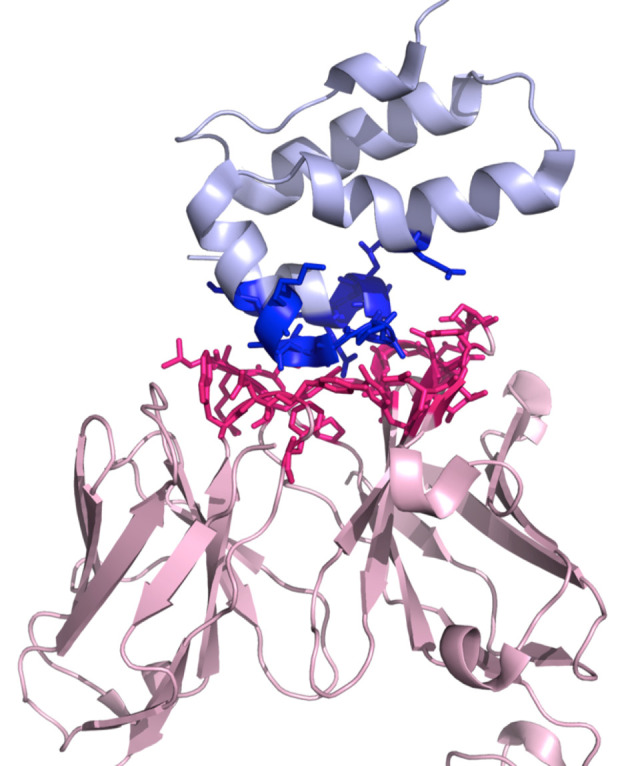
This script can be downloaded from the PRODIGY output page. Interactor 1 is shown in light pink (chains L and H in this example) and Interactor 2 in light blue (chain P), respectively. The interacting residues are represented in sticks in blue and dark pink for Interactor 1 and Interactor 2, respectively.
Notes
To run the ready-to-run Pymol script (.pml) provided by PRODIGY (see step B2c), open a Pymol session with the PDB code that you submitted to PRODIGY and follow one of the possible options:
From the bar menu of Pymol, choose File → Run and navigate in the directory where the PRODIGY Pymol script has been saved. Then select the .pml file clicking on ‘Open’.
In the Pymol terminal bar, type @ followed by the .pml file. Please note, if the Pymol session is not open in that folder, the user will need to type the full path. For example: @home/my_path/prodigy_pymol_script.pml
Acknowledgments
This protocol has been adapted from: Vangone and Bonvin (2015) and Xue et al. (2016). Anna Vangone was supported by H2020 Marie-Skłodowska-Curie Individual Fellowship MCSA-IF-2015 [BAP-659025].
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Berman H., Henrick K. and Nakamura H.(2003). Announcing the worldwide Protein Data Bank. Nat Struct Biol 10(12): 980. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard S. J. and Thornton J. M.(1993). Naccess. Computer Program. [Google Scholar]
- 3.Kastritis P. L. and Bonvin A. M.(2010). Are scoring functions in protein-protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J Proteome Res 9(5): 2216-2225. [DOI] [PubMed] [Google Scholar]
- 4.Kastritis P. L., Moal I. H., Hwang H., Weng Z., Bates P. A., Bonvin A. M. and Janin J.(2011). A structure-based benchmark for protein-protein binding affinity. Protein Sci 20(3): 482-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitternacht S.(2016). FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Res 5: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vangone A., and Bonvin A. M. J. J.(2015). Contacts-based prediction of binding affinity in protein-protein complexes. eLife 4: 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vreven T., Moal I. H., Vangone A., Pierce B. G., Kastritis P. L., Torchala M., Chaleil R., Jimenez-Garcia B., Bates P. A., Fernandez-Recio J., Bonvin A. M. and Weng Z.(2015). Updates to the integrated protein-protein interaction benchmarks: Docking benchmark version 5 and affinity benchmark version 2. J Mol Biol 427(19): 3031-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xue L. C., Rodrigues J. P., Kastritis P. L., Bonvin A. M. and Vangone A.(2016). PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics. [DOI] [PubMed] [Google Scholar]