

Abstract
Pharmacogenomic studies have successfully identified variants – typically with large effect sizes in drug target and metabolism enzymes – that predict drug outcome phenotypes. However, these variants may account for a limited proportion of phenotype variability attributable to the genome. Using genome-wide common variation, we measured the narrow-sense heritability [] of 7 pharmacodynamic and 5 pharmacokinetic phenotypes across 3 cardiovascular drugs, 2 antibiotics, and 3 immunosuppressants. We used a Bayesian Hierarchical Mixed Model, BayesR, to model the distribution of genome-wide variant effect sizes for each drug phenotype as a mixture of 4 normal distributions of fixed variance (0, 0.01%, 0.1% and 1% of the total additive genetic variance). This model allowed us to parse into bins representing contributions of no-, small-, moderate- and large-effect size variants respectively. For the 12 phenotypes, a median of 969 (range 235-6,304) unique individuals of European ancestry and a median of 1,201,626 (range 777,427-1,514,275) variants were included in our analyses. The number of variants contributing to ranged from 2,791 to 5,356 (median 3,347). Estimates for ranged from 0.05 (ACE-inhibitor induced cough) to 0.59 (gentamicin concentration). Small- and moderate-effect variants contributed a majority to for every phenotype (range 61-95%). We conclude that drug outcome phenotypes are highly polygenic. Thus, larger genome-wide association studies of drug phenotypes are needed both to discover novel variants and to determine how genome-wide approaches may improve clinical prediction of drug outcomes.
Keywords: heritability, Bayesian modeling, genome-wide variation
Introduction
An early and successful translation of precision medicine to clinical care is the use of genetic variant data to predict drug outcome phenotypes. For example, the HLA-B*57:01 allele predicts risk of abacavir hypersensitivity,1–4 and individuals with functional CYP3A5 (more common in individuals of African ancestry) require higher doses of tacrolimus.5,6 However, genetic potential for predicting drug outcomes has yet to be fully realized; the Food and Drug Administration table of Pharmacogenomic Biomarkers in Drug Labeling lists nearly 300 unique drugs,7 but only 115 have sufficient evidence to recommend a prescribing action.8
Drug-gene interactions identified to date include large-effect genetic variants in proteins critical for drug metabolism or response.9–11 For example, the antiplatelet drug clopidogrel requires activation by cytochrome P450 2C19; thus, genetic variants affecting CYP2C19 function strongly influence clopidogrel efficacy.12,13 However, these large-effect variants do not fully explain the variability of drug outcome phenotypes attributed to variation in the genome; while estimates of heritability for on-clopidogrel platelet reactivity range from 16 to 70%, common variants in CYP2C19 only explain 12% of the variation in clopidogrel response.13,14 Furthermore, for many drugs with significant interindividual variability, candidate-gene and genome-wide association studies (GWAS) have either failed to identify significant associations15,16 or accounted for only a small proportion of the overall phenotype variation.17,18
For non-pharmacologic phenotypes such as height, genome-wide variation contributes more to phenotypic variation than the relatively small number of statistically significant single nucleotide polymorphisms (SNPs) identified by GWAS.19 Using genome-wide approaches to combine many smaller effect size variants may explain increased variation in drug outcome phenotypes and enable pharmacogenomic prediction. Development of such pharmacogenomic predictors remains constrained by the sample size of pharmacogenomic studies; these studies depend on assembling a cohort with exposure to the drug of interest as well as documentation of clinically significant outcomes, many of which are rare or difficult to ascertain. Thus, comprehensive assessments of genomic architectures of drug outcome phenotypes are lacking.
Polygenic approaches, such as generalized linear mixed modeling (GLMM) or Bayesian non-linear models, calculate the proportion of phenotype variance explained by common SNPs with a minor allele frequency of greater than 1% (known as the narrow-sense heritability, ). For non-pharmacologic phenotypes, both GLMM and Bayesian models have demonstrated that the majority of the expected is accounted for when considering genome-wide variation, including SNPs that might otherwise fall well below the conventional Bonferroni corrected genome-wide significance threshold of 5x10−8.19–21 Since GLMM models assume that all SNPs have a non-zero effect on the phenotype, they account only for the influence of allele frequency on SNP effects. Bayesian models, however, have the added advantage of accounting for linkage disequilibrium (LD) by assuming that some SNPs will have no effect on the phenotype. While GLMM has been applied to a very limited number of pharmacogenomic phenotypes,22,23 no studies have explored pharmacogenomic outcomes using Bayesian models, limiting the polygenic exploration of pharmacogenomic phenotypes.
We hypothesized that Bayesian hierarchical models would demonstrate that common SNPs contribute more substantially to drug outcome variability than the small numbers of large-effect variants that have to date been associated to drug outcomes. We used an established method, BayesR,24 to calculate the and to estimate the extent to which is accounted for by SNPs of large, moderate and small effect sizes for drug outcomes. Our analyses were limited to individuals of White European ancestry due to the high sensitivity of Bayesian modeling to LD structure and the need to maximize sample sizes. We investigated drug outcome phenotypes for 8 different drugs, including 7 pharmacodynamic phenotypes, or those that affect drug action, and 5 pharmacokinetic phenotypes, or those that affect drug availability or concentration. Since these methods were initially developed for non-pharmacogenomic complex phenotypes, we included an analysis of clopidogrel outcomes for which heritability and genomic architecture have been previously assessed using other methods.
Methods
Study Population, Genotyping and Phenotyping
This study included multiple datasets: one from the International Clopidogrel Pharmacogenomics Consortium (ICPC, on-clopidogrel platelet reactivity);25,26 one from electronic MEdical Records and GEnomics (eMERGE) Network phase I and II (ACE-inhibitor associated cough);18 one previously analyzed cohort (major adverse cardiac events (MACE) while on statins)27 from Vanderbilt’s de-identified biobank and electronic health record system, BioVU;28 one from Children’s Oncology Group (COG) multi-institutional trials P9904 and P9905 (methotrexate clearance);29 and 4 datasets (8 phenotypes) newly assembled from BioVU (drug concentrations and nephrotoxicity with vancomycin, tacrolimus, gentamicin and cyclosporine). All datasets underwent standard quality control (QC) and were analyzed for relevant drug outcome phenotypes as described below. All phenotypes were adjusted for age, sex, and first 20 principal components (PCs) to account for structural features of the genome as done previously.21 The residuals were used in the final analyses, as BayesR does not have the capability to incorporate a covariate file. For methotrexate clearance, the adjusted clearance phenotype provided by the primary cohort was used instead.
This study was reviewed by the Institutional Review Board at Vanderbilt University Medical Center (VUMC) and determined to constitute non-human subject research.
On-clopidogrel platelet reactivity
The clopidogrel dataset was obtained from the ICPC,25,26 a combination of 17 studies from 13 different sites, retrospectively invited to be part of this consortium. The outcome phenotype was platelet reactivity, which, because of different assays across sites, was standardized by the Phenotype Subcommittee of the ICPC. Subjects with on-clopidogrel platelet reactivity data for whom DNA samples were available were genotyped on the Illumina Human Omni express exome chip at Rikagaku Kenkyūjyo (RIKEN) Center for Genomic Medicine (Japan).
ACE-inhibitor induced cough
The ACE-inhibitor dataset, as described previously,18 composed individuals from 6 sites of the eMERGE Network (VUMC, Marshfield Clinic, Northwestern University, Mayo Clinic and Group Health Research Institute, Geisinger Health System and Mount Sinai) and from Vanderbilt Electronic Systems for Pharmacogenomic Assessment (VESPA) study. The primary phenotype was cough induced by ACE-inhibitor use, as recorded by a health care provider. Subjects were genotyped on site-specific genotyping platforms, and SNPs common to all platforms were used for imputation and QC.
Major adverse cardiac events during statin therapy
The statin dataset was extracted from BioVU, as described previously.27 Cases of MACE included an acute myocardial infarction or the need for revascularization while on statins that occurred at least 180 days after the earliest recorded date of statin use. Controls were subjects who received statins and did not develop MACE. The dataset used a combination of natural language processing, ICD-9 and CPT codes, and lab values to determine cases, and age and sex matched controls in a 1:2 ratio. Genotyping was performed at RIKEN. The dataset was supplied by the authors of the published manuscript.
Methotrexate clearance in patients with acute lymphocytic leukemia
For methotrexate, the MTX clearance 9900 dataset used for analyses was obtained from dbGaP. The data were collected from pediatric patients with acute lymphoblastic leukemia from COG multi-institutional trials P9904 and P9905 as described previously.29 DNA from peripheral blood obtained at the time of patient remission was extracted and genotyped on the Affymetrix Genome-Wide Human SNP Array 6.0. Methotrexate clearance, adjusted for protocol, treatment arm, infusion, gender ancestry, was log2 transformed, and used as the final phenotype for our analyses.29
Nephrotoxicity on vancomycin, gentamicin, tacrolimus and cyclosporine
The datasets for vancomycin, gentamicin, cyclosporine and tacrolimus were extracted from BioVU subjects previously genotyped on the Illumina MEGAEX platform as part of a large institutional effort.28 Subjects were selected for a mention of the drug name of interest in their electronic health record at age 18 or older, and at least one measurement of both drug concentration and serum creatinine in their laboratory results, enabling study of both pharmacodynamic (nephrotoxicity, as indicated by peak serum creatinine) and pharmacokinetic (drug concentration) phenotypes. Peak creatinine was defined as the highest serum creatinine value between 1 and 14 days after the first drug concentration measurement. Peak creatinine values were positively skewed, and log10 transformed to follow a normal distribution. Outliers were defined as values less than 3 times the interquartile range below the 25th percentile or more than 3 times the interquartile range above the 75th percentile for the log-transformed variables. A review of a random subset of outliers found that they represented biologically implausible values; thus, the decision was made to exclude all outliers.
Vancomycin, gentamicin, tacrolimus, and cyclosporine drug concentration phenotypes
For BioVU subjects selected as discussed above, we extracted the first drug concentration available from laboratory values in electronic health record data. Drug dose and schedule was defined as that in the electronic health record closest in time to the drug concentration measurement, from 30 days beforehand to 3 days afterward. Dose data were highly variable for tacrolimus and cyclosporine, but not for vancomycin or gentamicin (where standard dosing is followed in clinical practice); thus, ratios of drug concentration to dose adjusted for 24 hour-dosing were used for tacrolimus and cyclosporine. Drug concentrations and the concentration to dose ratios were log10 transformed, correcting skew to a normal distribution. Outliers for drug concentration and concentration to dose ratios, defined above, were removed from final analyses.
Height
We used height, a phenotype known to have high heritability attributable to common SNPs,19 to benchmark the performance of the BayesR method. Height data were available for the clopidogrel, statin, vancomycin, tacrolimus, gentamicin and cyclosporine datasets. For the datasets extracted from BioVU, the height values used for analyses were the medians of all measurements in each subject’s electronic health record.
Quality Control and Processing
Due to the sensitivity of Bayesian models to variations in LD, these analyses were restricted to individuals of White European ancestry. To assess for European ancestry within each cohort, PC analyses in conjunction with the CEU, CHB, JPT, ASW, LWK, MKK and YRI HapMap populations, were used. Any subject with PC1 and PC2 more than 4 standard deviations away from the mean PC1 and PC2 of self-reported white subjects was removed from the final analyses.
Genotype data were phased and imputed using EAGLE2/MINIMAC4 on the Michigan Imputation server, with data from 1000 Genome Project Phase 3 Version 5 used as the reference. PLINK2 was used to convert imputed gene dosages to hard calls, which were then filtered by info scores greater than 0.8. QC was performed on each imputed dataset to include variants meeting the following criteria: biallelic SNPs only, minor allele frequency greater than 1% within the cohort, Hardy-Weinberg Equilibrium greater than 10−6, SNP genotyping rate of greater than 98%. The analyses did not include sex chromosomes, the HLA region on chromosome 6 (25500000-33500000), or the inversions on chromosomes 8 (8135000-12000000) and 17 (40900000-45000000, coordinates on GRCh37) as these regions are highly polymorphic or comprise large structural variation that may lead to aberrant results.
Any samples with genotype missing rate of >2% were removed from the final analyses. To account for cryptic relatedness, in each pair identified to have pi_hat greater than 0.2, the subject with a greater percentage of missing genetic data was removed. In each dataset, SNPs in high LD (r2> 0.9) were removed. The QC pipeline is shown in Figure S1. For European ancestry individuals who passed QC, the first 20 PCs were calculated and used to adjust the phenotypes.
BayesR
For each cohort and outcome of interest, BayesR (version 1), a hierarchical Bayesian Mixture Modeling method, was used to fit all the SNPs simultaneously to the adjusted outcome variables to give unbiased estimates of the SNPs effect sizes.24 This method assumes that the prior distribution of SNP effects can be modeled by K different normal distributions, where sum of the mixture proportions of each component is constrained to unity. We set K=4, as used previously,21 where each component was modeled as a normal distribution with a mean of 0 and a variance of 0, 0.01%, 0.1% and 1% of the additive genetic variance respectively, as shown below:
where is the additive genetic variance of the drug outcome phenotype for each of the datasets and are the mixture proportions of each component. To account for sparseness in the model, the mean and variance of the first component is set to zero. The components are referred to as no-, small-, moderate-, and large-effect SNPs. We also estimated the additive genetic variance, , as a hyperparameter from the model. The algorithm uses a Gibbs scheme to sample values from each unknown parameter’s posterior distribution.21,24 was calculated as:
where and were estimated by BayesR. Default prior distribution parameters were used, with the exception of the number of iterations (60,000), which were doubled from the default to allow for chain convergence given the smaller sample sizes of the datasets used. Conventional 89% high density credible intervals were calculated as described previously.30 To further test the robustness of the model, 3 pharmacodynamic phenotypes and 3 pharmacokinetic phenotypes representing the range of sample sizes were tested with prior distributions modeled as a mixture of 6 normal distributions of mean zero and a variance of 0.001%, 0.01%, 0.1%, 1% and 10% of the additive genetic variance.
Established, clinically tested, high-effect SNPs (rs4244285, CYP2C19*2, for clopidogrel and rs4149056, SLCO1B1*5, for methotrexate) were regressed on their respective phenotypes using the lm() function in R to assess their contribution to phenotype variability.
The results were processed using custom R scripts. All figures were annotated using Adobe Illustrator.
Results
Height heritability estimates and genomic architecture
Height measurements, available for 6 of the datasets (Table 1), were used to benchmark the performance of BayesR. After restricting analyses to individuals of White European ancestry who passed QC (Figure S1 and S2), the number of individuals available for height analyses ranged from 254 to 5,227. Height outcome data were normally distributed after adjusting for sex, age, and 20 PCs (Figure S3). Genotypes for a median of 1,217,676 (range 778,986-1,151,824) SNPs were input to the final models.
Table 1:
Height analyses data and results.
| Dataset | Clopidogrel | Statins | Vancomycin | Gentamicin | Tacrolimus | Cyclosporine |
|---|---|---|---|---|---|---|
| Subjects (n) | 1,509 | 4,843 | 5,227 | 254 | 1,180 | 508 |
|
| ||||||
| SNPs post-QC (n) | 778,986 | 1,515,824 | 1,050,868 | 1,248,133 | 1,187,219 | 1,248,265 |
|
| ||||||
| Female (n, (%)) | 328 (21.7) | 1,788 (36.9) | 2,293 (43.9) | 143 (56.3) | 449 (38.1) | 208 (40.9) |
|
| ||||||
| Age (mean, (SD), years) | 63.0 (11.1) | NA | 53.0 (13.6) | 43.5 (15.7) | 52.3 (12.0) | 49.2 (14.2) |
|
| ||||||
| Height (mean, (SD), cm) | 170.7 (8.8) | 172.3 (10.5) | 171.7 (10.7) | 169.4 (12.2) | 172.5 (10.2) | 171.5 (10.4) |
|
| ||||||
| 18.6 | 8.6 | 13.4 | 33.7 | 20.0 | 25.0 | |
|
| ||||||
| 0.43 [0.00, 0.85] | 0.19 [0.00, 0.42] | 0.24 [0.00,0.46] | 0.46 [0.00, 0.94] | 0.41 [0.00, 0.85] | 0.48 [0.00, 0.92] | |
|
| ||||||
| Large effect variant (prop., (# SNPs)) | 0.06 (19) | 0.05 (19) | 0.04 (17) | 0.32 (47) | 0.10 (26) | 0.21 (42) |
|
| ||||||
| Moderate-effect variant (prop., (# SNPs)) | 0.21 (215) | 0.39 (363) | 0.38 (377) | 0.34 (302) | 0.45 (400) | 0.33 (322) |
|
| ||||||
| Small-effect variant (prop., (# SNPs)) | 0.74 (6,468) | 0.55 (4,976) | 0.57 (5,079) | 0.34 (3,145) | 0.46 (4,027) | 0.45 (3,620) |
SD – Standard Deviation; – Additive Genetic Variance; - Narrow-sense Heritability, with conventionally calculated high density credible interval shown in brackets. Prop.: Proportion contributed to total . NA indicates data not available.
The estimates of for height ranged from 0.19 for the statin dataset to 0.48 for the cyclosporine dataset (Table 1 and Figure 1A). Credible intervals for each dataset were wide and included the expected value of ~0.40 based on prior studies of other datasets.20 BayesR also allowed us to describe the genomic architecture by parsing the into proportions accounted for by no-, small-, moderate- and large-effect SNPs. The contribution of large-effect SNPs ranged from 0.04 for vancomycin to 0.32 for gentamicin; thus, across all datasets, small- and moderate-effect SNPs accounted for the majority of height (Figure 1A).
Figure 1: Narrow-sense heritability () estimates of drug outcome phenotypes, divided into contributions from large-, moderate- and small-effect size variants.
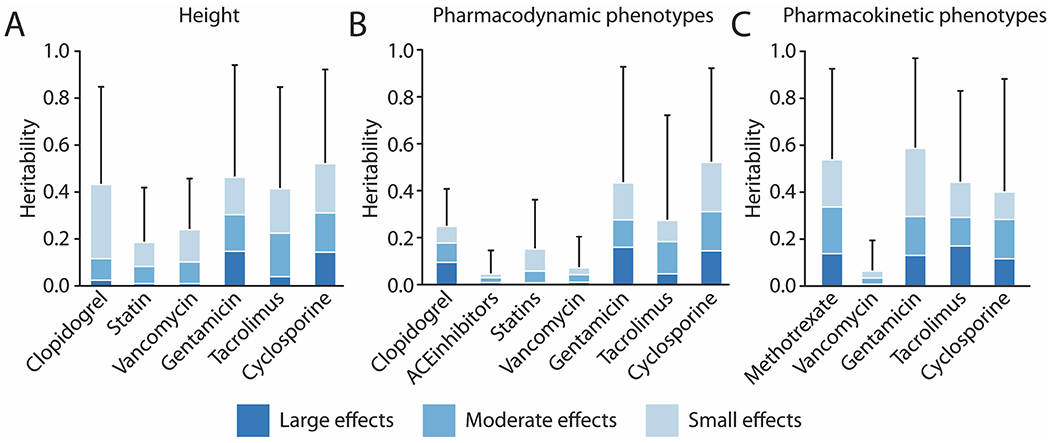
The horizontal axes represent the different datasets. A) Heritability of height as a positive control for 6 datasets. B) Heritability of 7 pharmacodynamic phenotypes (Clopidogrel: Platelet reactivity; ACE-inhibitor: Cough; Statins: Major Adverse Cardiac Events (MACE); Vancomycin, Gentamicin, Tacrolimus, Cyclosporine: Peak Creatinine). Clopidogrel () is a positive control. C) Heritability of 5 pharmacokinetic phenotypes (Methotrexate: Adjusted Drug Clearance; Vancomycin, Gentamicin: Drug trough; Tacrolimus, Cyclosporine: Plasma Concentration to Drug Ratio). Error bars represent conventional high density credible intervals for .
Drug outcome phenotype study populations
The 12 drug outcome phenotypes are shown in Table 2 (pharmacodynamic) and Table 3 (pharmacokinetic). The number of individuals of White European ancestry in the datasets ranged from 235 for gentamicin peak creatinine to 6,304 for vancomycin concentration. Demographic data for the individuals included in the final models are shown in Tables 2 and 3. Genotypes for a median of 1,201,626 (range 777,427-1,514,275) SNPs were available for the final models (Tables 2 and 3). Drug outcome phenotypes, adjusted for age or decade of birth (where available), sex and 20 PCs, used in the final analyses were normally distributed (Figures S4 and S5).
Table 2:
Pharmacodynamic phenotype analyses data and results.
| Clopidogrel | ACE inhibitors | Statins | Vancomycin | Gentamicin | Tacrolimus | Cyclosporine | |
|---|---|---|---|---|---|---|---|
| Subjects (n) | 2,518 | 5,925 | 5,834 | 5,776 | 235 | 1,033 | 273 |
|
| |||||||
| SNPs post-QC (n) | 777,427 | 1,024,789 | 1,514,275 | 1,052,294 | 1,246,692 | 1,188,309 | 1,263,625 |
|
| |||||||
| Female (n, (%)) | 583 (23.2) | 2,685 (45.3) | 2,083 (35.7) | 2,461 (42.6) | 115 (48.9) | 383 (37.0) | 121 (44.3) |
|
| |||||||
| Age (mean, (SD), years) | 64.8 (11.2) | NA | NA | 57.1 (15.5) | 49.2 (17.9) | 53.1 (11.8) | 53.1 (12.8) |
|
| |||||||
| Phenotype studied | Standardized Platelet Reactivity | Cough | Major Adverse Cardiac Events | Peak Creatinine | |||
|
| |||||||
| Cases (n, (%)) | - | 1,326 (22.3) | 1,708 (29.2) | - | - | - | - |
|
| |||||||
| Phenotype (median) | 0.006 | - | - | 1.10 | 1.10 | 1.69 | 1.50 |
|
| |||||||
| 0.24 | 0.01 | 0.03 | 0.02 | 0.28 | 0.09 | 0.15 | |
|
| |||||||
| 0.25 [0.00, 0.41] | 0.05 [0.00, 0.15] | 0.15 [0.00, 0.36] | 0.07 [0.00,0.20] | 0.43 [0.00, 0.93] | 0.27 [0.00, 0.72] | 0.52 [0.00, 0.92] | |
|
| |||||||
| Large effect variant (prop., (# SNPs)) | 0.38 (46) | 0.19 (50) | 0.05 (22) | 0.13 (36) | 0.37 (53) | 0.17 (44) | 0.28 (41) |
|
| |||||||
| Moderate-effect variant (prop., (# SNPs)) | 0.33 (293) | 0.46 (341) | 0.33 (342) | 0.46 (401) | 0.27 (282) | 0.50 (425) | 0.32 (314) |
|
| |||||||
| Small-effect variant (prop., (# SNPs)) | 0.29 (2,453) | 0.36 (2,902) | 0.62 (4,992) | 0.41 (3,211) | 0.36 (2,930) | 0.33 (2,467) | 0.40 (3,528) |
SD – Standard Deviation; C:D Ratio – Concentration to Dose Ratio; – Additive Genetic Variance; - Narrow-sense Heritability, with conventionally calculated high density credible interval shown in brackets. Prop.: Proportion contributed to total . NA indicates data not available.
Table 3:
Pharmacokinetic phenotype analyses data and results.
| Dataset | Methotrexate | Vancomycin | Gentamicin | Tacrolimus | Cyclosporine |
|---|---|---|---|---|---|
| Subjects (n) | 782 | 6,304 | 294 | 905 | 359 |
|
| |||||
| SNPs post-QC (n) | 1,212,948 | 1,052,259 | 1,247,598 | 1,190,303 | 1,256,128 |
|
| |||||
| Female (n, (%)) | 378 (48.3) | 2,726 (43.2) | 160 (54.4) | 329 (36.3) | 136 (37.9) |
|
| |||||
| Age (mean, (SD), years) | NA | 56.9 (15.6) | 47.0 (18.5) | 53.4 (11.6) | 52.7 (13.4) |
|
| |||||
| Phenotype studied | Clearance | Trough | C:D Ratio | ||
|
| |||||
| Phenotype (median) | 138.01 | 12 | 0.7 | 0.75 | 0.55 |
|
| |||||
| 0.06 | 0.02 | 0.53 | 0.26 | 0.27 | |
|
| |||||
| 0.54 [0.00, 0.93] | 0.06 [0.00,0.19] | 0.59 [0.00,0.97] | 0.44 [0.00,0.83] | 0.40 [0.00,0.88] | |
|
| |||||
| Large effect variant (prop., (# SNPs)) | 0.26 (41) | 0.10 (44) | 0.22 (37) | 0.38 (55) | 0.29 (49) |
|
| |||||
| Moderate-effect variant (prop., (# SNPs)) | 0.37 (338) | 0.42 (389) | 0.28 (265) | 0.28 (267) | 0.42 (359) |
|
| |||||
| Small-effect variant (prop., (# SNPs)) | 0.37 (3,276) | 0.47 (2,969) | 0.49 (4,455) | 0.34 (2,872) | 0.29 (2,634) |
Heritability estimates and genomic architecture of drug outcome phenotypes
The 7 pharmacodynamic phenotypes studied were on-clopidogrel platelet reactivity, angiotensin converting enzyme (ACE)-inhibitor associated cough, MACE during statin therapy, and peak creatinine during therapy with vancomycin, gentamicin, tacrolimus and cyclosporine. Estimates of for these phenotypes ranged from 0.05 for ACE-inhibitor cough to 0.52 for cyclosporine peak creatinine (Table 2, Figure 1B).
The 5 pharmacokinetic phenotypes studied were methotrexate clearance, vancomycin and gentamicin drug concentrations, and tacrolimus and cyclosporine concentration to dose ratios. Of these, the estimate was lowest for vancomycin concentration (0.06), and for the remaining 4 drugs ranged from 0.40 to 0.59 (Table 3, Figure 1C). Heritability estimates for the 6 phenotypes modeled as a mixture of 6 components were consistently higher than with 4 components (Tables S1 and S2).
Results of analysis of the genomic architecture for pharmacodynamic and pharmacokinetic phenotypes are shown in Figure 1 (panels B and C respectively). On-clopidogrel platelet reactivity resulted in of 0.25, with 46 large-effect SNPs contributing 0.09 (38% of ), and moderate- and small-effect SNPs contributing 33% and 29% of , respectively. For the remaining pharmacodynamic phenotypes, a range of 22 to 53 large-effect SNPs captured an average of 20% of , with the remainder captured by fewer than 5,500 moderate- and small-effect SNPs (Table 2 and S3, and Figure 1B). For pharmacokinetic phenotypes, a range of 37 to 55 large-effect SNPs captured an average of 25% of . The remainder of was equally divided between fewer than 5000 moderate- and small-effect SNPs (Table 3 and S4, and Fig 1C). The 6 phenotypes modeled using 6 components also demonstrated substantial contributions to from moderate-, small-, and very small-effect SNPs (Tables S1 and S2).
Thus, small- and moderate-effect SNPs represented over 99% of the SNPs contributing to drug outcome phenotype variability and were responsible for 61-95% of the total . Small- and moderate-effect SNPs contributed the greatest proportion of for MACE during statin therapy (95% of 0.15).
Based on conventional linear models, the contribution of CYP2C19*2 and SLCO1B1*5 in our datasets was found to be less than 6% and 5% for clopidogrel and methotrexate respectively (Table S5).
Discussion
In this study, we used a Bayesian hierarchical modeling method to estimate the variation in drug outcomes attributable to common variation in the genome, or , for 8 drugs across 12 different pharmacodynamic and pharmacokinetic phenotypes. Estimates of using these methods have not been previously pursued for drug outcome phenotypes. We found a majority of drug outcome phenotypes to have a substantial heritable component. We also showed that all 12 phenotypes are highly polygenic and that limiting to large-effect SNPs, especially those that are currently tested clinically, drastically underestimates the amount of drug outcome variation attributed to the genome. Our data indicate that larger GWAS are needed to explore the full genomic architecture of drug outcomes, and that SNP-based discovery may identify novel drivers of drug response.
Half of the drug outcome phenotypes studied here have estimates >40%, and an additional 2/12 have estimates >25%. These highly heritable phenotypes included pharmacodynamic phenotypes of on-clopidogrel platelet reactivity, peak creatinine while on gentamicin, tacrolimus or cyclosporine, and pharmacokinetic phenotypes for gentamicin, tacrolimus, cyclosporine, and methotrexate. Given that such a large portion of variability in the phenotype can be explained by common genomic variation, these drug outcomes represent candidates for pharmacogenomic study and implementation. Of these 8 traits, there is currently clinically actionable pharmacogenomic evidence for only 2 drug-gene pairs: clopidogrel-CYP2C19 and tacrolimus-CYP3A5.12,31 Although genetic mechanisms for variability in response or disposition of some of these drugs have been investigated and, in some cases, identified,27,32,33 clinical implementation of genotype-guided dosing is not yet recommended for other drug outcomes, as robust, valid predictors have not been defined. The high estimates support the hypothesis that additional, larger pharmacogenomic association studies could discover novel variants and enable the generation of valid and robust polygenic predictive models.
The heritability of ADP-stimulated platelet aggregation in response to clopidogrel, previously described using other methods, is estimated to be 70% in the genetically homogenous Amish population.13 Our estimate for the standardized platelet reactivity (25%) is lower, possibly due to the heterogeneity of the phenotype used by the ICPC, a standardized score of different platelet activity assays from different sites. Nonetheless, we found that our estimate was consistent with other estimates of heritability of the change in platelet aggregation due to clopidogrel.14
In comparing types of drug outcome phenotypes, we found that pharmacodynamic phenotypes showed a high variability in across different drugs, reflecting the highly disparate nature of drug response, influenced by genetic, clinical, and environmental factors. Pharmacokinetic phenotypes, on the other hand, had more consistent estimates across different drugs. However, the known mechanisms for drug absorption, distribution, metabolism, and excretion of the studied drugs are disparate (e.g. the 2 antibiotics are largely excreted unchanged by the kidneys, but tacrolimus and cyclosporine are metabolized by the cytochrome P450 3A family of enzymes). Thus, the SNPs driving variation of these traits are likely quite different.
The models allowed us to parse into proportions contributed by small-, moderate- and large-effect SNPs, enabling estimation of the additional variance to be explained by variants with smaller effect size. On-clopidogrel platelet reactivity provides comparison data for our genomic architecture results, since the heritability of this phenotype has been studied with multiple methods. Our methods estimated that large-effect SNPs contribute 29% of total , or 9% of the overall variation in phenotype, while previous studies indicate that the specific CYPC219*2 allele contributes 12% of the overall variation in a comparable phenotype in an Amish population.13
Our results indicate that drug outcome phenotypes follow a polygenic pattern, with small- and moderate-effect SNPs accounting for a majority of . For highly heritable phenotypes, genotype-guided approaches that are limited to large-effect SNPs will fail to incorporate variants accounting for 61% (for tacrolimus concentration to dose ratio) to 88% (for gentamicin drug concentration) of . Our analysis using single well-established common variants (CYP2C19*2 and SLCO1B1*5 for clopidogrel and methotrexate, respectively) found <10% of variability was explained by these single variants, further motivating a polygenic approach. Estimates of heritability were increased with prior distributions modeled as a mixture of 6 rather than 4 normal distributions, with most heritability from moderate- and small-effect variants. Multiple approaches for prior distributions in Bayesian modeling have been proposed.34 However, the ideal prior distribution for pharmacogenomic phenotypes to maximize heritability calculation while balancing computational burden is dependent on the genomic architecture of each trait,20 which can only be determined by larger pharmacogenomic studies.
For phenotypes with a relatively small , small- and moderate-effect SNPs explained up to 95% of the . Genome-wide predictors for some non-pharmacologic phenotypes with relatively small 35 enable the identification of individuals at the tails of the phenotype distribution with individuals’ risk equivalent to monogenic mutations,36 an approach that may be effective for drug outcomes as well. Thus, in clinical practice, polygenic risk scores for drug outcome phenotypes, especially those with high , are warranted to realize the full potential of pharmacogenomics. A first step in implementing such a vision will be to generate improved predictors, an exercise currently limited by the current sample sizes of pharmacogenomic GWAS. This study serves as an impetus to generate larger datasets with well-characterized exposures, drug responses, and SNP array data to generate improved predictors.
Our call for larger pharmacogenomic studies is extended to genetically diverse ancestries to fully realize the potential of pharmacogenomics. Examining the same phenotype in different genetic ancestry populations, and therefore different genomic linkage structures, will assist with fine mapping of pharmacogenomic variants for functional follow-up. Moreover, polygenic approaches devised in European ancestry populations, such as those suggested by our analyses, may have decreased predictive performance in other populations, further contributing to health disparities.37,38 Once in hand, such genomic predictors should be prospectively assessed, for instance in randomized or pragmatic trials, to determine their role in clinical practice. Alternately, strategies from health economics, quality improvement, and/or implementation science may contribute to the evidence supporting clinical pharmacogenomic prediction.
One limitation of our study is the use of smaller datasets than those previously explored via BayesR. We used both height and a previously studied drug outcome (on-clopidogrel platelet reactivity) to assess the potential accuracy of our results. We found our estimates of for height were generally consistent with previously published results.39 While the raw height estimates for the statin and vancomycin datasets were lower than the estimates for other datasets, credible intervals of these estimates included the expected value of ~40%. This finding may in part be due to the decreased precision of Bayesian methods with limited sample sizes; previous studies of this method to calculate height have used a dataset of approximately 700,000 people.20 Even for on-clopidogrel platelet reactivity, we found that the small sample size reduced the precision of our estimate of and resulted in wide credible intervals. Since Bayesian approaches are highly sensitive to ancestry-based genomic structure, we could not increase sample sizes by including people of non-European ancestries. The HLA and chromosome 8 and 17 inversion regions were excluded from these analyses, which could lead to an underestimation of the overall heritability.
Our study was also limited to previously constructed and available datasets. Many other drug-phenotype combinations may likewise benefit highly from genomic prediction. Such drug-phenotype combinations would include those requiring trial-and-error practices in the clinic, such as glycemic control from oral diabetes medications and depressive symptom relief from psychiatric medications, or the highly dangerous side-effects of commonly used drugs such as angioedema with ACE-inhibitors. We advocate for future studies to focus on curating datasets for drugs and outcomes, such as those mentioned above, to determine the heritability, genomic architecture, and polygenic predictors of these pharmacogenomic phenotypes.
In summary, our results demonstrate that generally, genome-wide variation significantly contributes to variability in drug outcomes. These phenotypes are polygenic with the majority of heritability attributed to moderate- and small-effect variants and may require a polygenic approach to predict drug response. Such an undertaking would involve larger GWAS aimed at identifying and validating additional variants to develop polygenic predictors with the potential to improve clinical care.
Supplementary Material
Study Highlights:
What is the current knowledge on the topic?
Interindividual variability in pharmacodynamic/pharmacokinetics phenotypes is a well-established phenomenon. Pharmacogenomic predictors for this variability often depend on one (or a few) large-effect variants in genes associated with drug metabolism.
What question did this study address?
Our study determined if leveraging genome-wide common variations would explain more variability in PD/PK phenotypes than current oligogenic approaches, and identify drugs that will benefit most from such polygenic techniques.
What does this study add to our knowledge?
We found that most of the PD/PK phenotypes we studied are highly heritable, but large-effect variants explain a small proportion of the heritability. The majority of the heritability was explained by small- and moderate-effect size variants.
How might this change clinical pharmacology or translational science?
This study shows the potential for polygenic approaches in the clinic to improve prediction of PD/PK phenotypes to fulfill the promise of precision medicine, and motivates the cultivation of large datasets to further define the impact of genomic variation on PD/PK phenotypes.
Acknowledgements:
The authors would like to thank Christian M. Shaffer for help with data extraction and coding resources, and the International Clopidogrel Pharmacogenomics Consortium (ICPC) for contributing data. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville, TN.
Funding information:
A.M. is supported by a grant from the American Heart Association (20PRE35180088) and from the Vanderbilt Medical Scientist Training Program (T32GM007347) . This work was supported by the National Institutes of Health (R01GM132204) to S.L.V. The ICPC research reported in this publication was supported by the National Heart, Lung, and Blood Institute U01HL105198, National Institute of General Medical Sciences R24GM61374 and NIH Genome Research Institute U24HG010615. Genome-wide SNP genotyping was supported by the Pharmacogenomics Research Network & CGM Global Alliance. Other support provided by the Deutsche Forschungsgemeinschaft (DFG), Germany grant numbers SCHW858/1-2, 374031971 – TRR 240, Klinische Forschungsgruppe-KFO-274 and in part, by the EU Horizon 2020 UPGx grant number 668353, and the Robert Bosch Stiftung, Stuttgart, Germany. The ACE-inhibitor dataset from electronic MEdical Records and GEnomics (eMERGE) Phase II data was supported by U01HG04603 (Vanderbilt), 1U02HG004608-01, 1U01HG006389 and NCATS/NIH grant UL1TR000427 (Marshfield/EIRH/Penn State), U01HG006375 and U01AG06781 (Group Health and University of Washington), U01HG04599 (Mayo Clinic), U01HG004609 (Northwestern University), U01HG006382 (Geisinger), an ARRA grant RC2GM092618, a Vanderbilt PGRN grant U19HL065962 and the Vanderbilt CTSA grant UL1TR000445 from NCATS/NIH. At Geisinger, the sample collection was supported by NIH (P30DK072488, R01DK088231 and R01DK091601), Pennsylvania Commonwealth Universal Research Enhancement Program, the Ben Franklin Technology Development Fund of PA, the Geisinger Clinical Research Fund and a Grant-In-Aid from the American Heart Association. This work used datasets from Vanderbilt University Medical Center’s BioVU, which is supported by institutional funding and by UL1TR000445 from NCATS. The tacrolimus dataset was supported by National Institutes of Health, NIGMS, K23GM100183. The MTX clearance 9900 dataset was generated at St. Jude Children’s Research Hospital and by the Children’s Oncology Group, and supported by grants CA36401, GM92666, CA98453, CA98413, CA114766, and CA21765 from the National Institutes of Health, ALSAC, the Jeffrey Pride Foundation and the National Childhood Cancer Foundation. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest: The authors declared no competing interests for this work.
Publisher's Disclaimer: Disclaimer: As an Associate Editor of Clinical Pharmacology & Therapeutics, Sara L. Van Driest was not involved in the review or decision process for this paper.
Data and Code Availability:
The ACE-inhibitor induced cough (phs000992.v1.p1), MACE while on statins (phs000963.v1.p1), and methotrexate clearance (phs000637.v1.p1) datasets are available at dbGaP (https://www.ncbi.nlm.nih.gov/gap/) at accession numbers as given. The ICPC dataset may be requested from International Clopidogrel Pharmacogenomics Consortium. The vancomycin, gentamicin, tacrolimus and cyclosporine datasets are available for use upon request from the authors with appropriate data use agreements in place. The code for BayesR is available online (https://github.com/syntheke/bayesR).
Supplemental File:
References:
- 1.Martin MA, Klein TE, Dong BJ, Pirmohamed M, Haas DW, Kroetz DL. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther. 2012;91(4):734–738. doi: 10.1038/clpt.2011.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mallal S et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727–732. doi: 10.1016/S0140-6736(02)07873-X [DOI] [PubMed] [Google Scholar]
- 3.Hetherington S et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359(9312):1121–1122. doi: 10.1016/S0140-6736(02)08158-8 [DOI] [PubMed] [Google Scholar]
- 4.Mallal S et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. N Engl J Med. 2008;358(6):568–579. doi: 10.1056/nejmoa0706135 [DOI] [PubMed] [Google Scholar]
- 5.Oetting WS et al. Genome-wide association study identifies the common variants in CYP3A4 and CYP3A5 responsible for variation in tacrolimus trough concentration in Caucasian kidney transplant recipients. Pharmacogenomics J. 2018;18(3):501–505. doi: 10.1038/tpj.2017.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oetting WS et al. Genetic variants associated with immunosuppressant pharmacokinetics and adverse effects in the DeKAF genomics genome-wide association studies. Transplantation. 2019;103(6):1131–1139. doi: 10.1097/TP.0000000000002625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Table of Pharmacogenomic Biomarkers in Drug Labeling | FDA. Accessed January 8, 2021. https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling
- 8.Clinical Pharmacogenetics Implementation Consortium. Accessed January 5, 2021. https://cpicpgx.org/genes-drugs/
- 9.Cooper GM et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112(4):1022–1027. doi: 10.1182/blood-2008-01-134247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi F et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5(3). doi: 10.1371/journal.pgen.1000433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormack M et al. HLA-A*3101 and Carbamazepine-Induced Hypersensitivity Reactions in Europeans. N Engl J Med. 2011;364(12):1134–1143. doi: 10.1056/nejmoa1013297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott SA et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94(3):317–323. doi: 10.1038/clpt.2013.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuldiner AR et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA - J Am Med Assoc. 2009;302(8):849–858. doi: 10.1001/jama.2009.1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bozzi L M et al. The Pharmacogenomics of Anti-Platelet Intervention (PAPI) Study: Variation in Platelet Response to Clopidogrel and Aspirin. Curr Vasc Pharmacol. 2015;14(1):116–124. doi: 10.2174/1570161113666150916094829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarasquete ME et al. Bisphosphonate-related osteonecrosis of the jaw is associated with polymorphisms of the cytoehrome P450 CYP2C8 in multiple myeloma: A genome-wide single nucleotide polymorphism analysis. Blood. 2008;112(7):2709–2712. doi: 10.1182/blood-2008-04-147884 [DOI] [PubMed] [Google Scholar]
- 16.Nicoletti P, Cartsos VM, Palaska PK, Shen Y, Floratos A, Zavras AI. Genomewide Pharmacogenetics of Bisphosphonate-Induced Osteonecrosis of the Jaw: The Role of RBMS3 . Oncologist. 2012;17(2):279–287. doi: 10.1634/theoncologist.2011-0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massey J et al. Genome-wide association study of response to tumour necrosis factor inhibitor therapy in rheumatoid arthritis. Pharmacogenomics J. 2018;18(5):657–664. doi: 10.1038/s41397-018-0040-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosley JD et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenomics J. 2016;16(3):231–237. doi: 10.1038/tpj.2015.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–569. doi: 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd-Jones LR et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat Commun. 2019;10(1). doi: 10.1038/s41467-019-12653-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moser G, Hong Lee S, Hayes BJ, Goddard ME, Wray NR, Visscher PM. Simultaneous Discovery, Estimation and Prediction Analysis of Complex Traits Using a Bayesian Mixture Model. PLoS Genet. 2015;11(4):1004969. doi: 10.1371/journal.pgen.1004969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mcgeachie MJ et al. Polygenic heritability estimates in pharmacogenetics: Focus on asthma and related phenotypes. Pharmacogenet Genomics. 2013;23(6):324–328. doi: 10.1097/FPC.0b013e3283607acf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chhibber A et al. Polygenic inheritance of paclitaxel-induced sensory peripheral neuropathy driven by axon outgrowth gene sets in CALGB 40101 (Alliance). Pharmacogenomics J. 2014;14(4):336–342. doi: 10.1038/tpj.2014.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erbe M et al. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J Dairy Sci. 2012;95(7):4114–4129. doi: 10.3168/jds.2011-5019 [DOI] [PubMed] [Google Scholar]
- 25.Bergmeijer TO et al. Genome-wide and candidate gene approaches of clopidogrel efficacy using pharmacodynamic and clinical end points—Rationale and design of the International Clopidogrel Pharmacogenomics Consortium (ICPC). Am Heart J. 2018;198:152–159. doi: 10.1016/j.ahj.2017.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma SS et al. Genomewide Association Study of Platelet Reactivity and Cardiovascular Response in Patients Treated With Clopidogrel: A Study by the International Clopidogrel Pharmacogenomics Consortium. Clin Pharmacol Ther. Published online July 9, 2020:cpt.1911. doi: 10.1002/cpt.1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei WQ et al. LPA variants are associated with residual cardiovascular risk in patients receiving statins. Circulation. 2018;138(17):1839–1849. doi: 10.1161/CIRCULATIONAHA.117.031356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roden D et al. Development of a Large-Scale De-Identified DNA Biobank to Enable Personalized Medicine. Clin Pharmacol Ther. 2008;84(3):362–369. doi: 10.1038/clpt.2008.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramsey LB et al. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood. 2013;121(6):898–904. doi: 10.1182/blood-2012-08-452839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makowski D, Ben-Shachar M, Lüdecke D. bayestestR: Describing Effects and their Uncertainty, Existence and Significance within the Bayesian Framework. J Open Source Softw. 2019;4(40):1541. doi: 10.21105/joss.01541 [DOI] [Google Scholar]
- 31.Birdwell KA et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther. 2015;98(1):19–24. doi: 10.1002/cpt.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Treviño LR et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27(35):5972–5978. doi: 10.1200/JCO.2008.20.4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosley J et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenomics J. 2016;16:231–237. doi: 10.1038/tpj.2015.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Carbonetto P, Stephens M. Polygenic Modeling with Bayesian Sparse Linear Mixed Models. Visscher PM, ed. PLoS Genet. 2013;9(2):e1003264. doi: 10.1371/journal.pgen.1003264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nielsen JB et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50(9):1234–1239. doi: 10.1038/s41588-018-0171-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Márquez-Luna C et al. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet Epidemiol. 2017;41(8):811–823. doi: 10.1002/gepi.22083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duncan L et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun. 2019;10(1):1–9. doi: 10.1038/s41467-019-11112-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wainschtein P et al. Recovery of trait heritability from whole genome sequence data. bioRxiv. Published online March 25, 2019:588020. doi: 10.1101/588020 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.