

Abstract
Rationale
Arginine vasopressin (AVP) is a neuropeptide that modulates both physiological and emotional responses to threat. Until recently, drugs that target vasopressin receptors (V1a) in the human central nervous system were unavailable. The development of a novel V1a receptor antagonist, SRX246, permits the experimental validation of vasopressin’s role in the regulation of anxiety and fear in humans.
Objectives
Here, we examined the effects of SRX246 in a proof-of-concept translational paradigm of fear (phasic response to imminent threat) and anxiety (prolonged response to potential threat).
Methods
Healthy volunteers received both SRX246 and placebo in a randomized, double-blind, counter-balanced order separated by a 5–7-day wash-out period. Threat consisted of unpleasant electric shocks. The “NPU” threat test probed startle reactivity during predictable threat (i.e., fear-potentiated startle) and unpredictable threat (i.e., anxiety-potentiated startle).
Results
As predicted, SRX246 decreased anxiety-potentiated startle independent of fear-potentiated startle.
Conclusions
As anxiety-potentiated startle is elevated in anxiety and trauma-associated disorders and decreased by traditional anxiolytics such as SSRIs and benzodiazepines, the V1a receptor is a promising novel treatment target.
Keywords: Fear-potentiated startle, Anxiety-potentiated startle, NPU threat test, Threat of shock, V1a receptor
Introduction
Psychiatric drug development is quite challenging, and many companies have abandoned the search for new therapies (Griebel and Holmes 2013; Kesselheim et al. 2015). Current treatment response rates for anxiety and trauma-based disorders rarely exceed 60% (Berger et al. 2009), and no new agents for the treatment of these disorders have been approved by the Food and Drug Administration (FDA) or European authorities in the last decade (Sartori and Singewald 2019). In response to less investment in drug development at a time of great need, the National Institutes of Mental Health (NIMH) has encouraged researchers to (1) engage in early-phase clinical trials to explore novel treatment mechanisms and (2) develop translational biomarkers for use in proof-of-concept studies (Anderzhanova et al. 2017; Grillon et al. 2019; Krystal et al. 2019).
One promising target is the arginine vasopressin (AVP) receptor V1a. AVP is a neuropeptide that modulates both amygdaloid activation to threatening stimuli (Brunnlieb et al. 2013) and anxious mood in humans (Thompson et al. 2006), and clinical studies have suggested that AVP may promote anxiety disorders (Peskind et al. 1998) and post-traumatic stress disorder (Pitman et al. 1993; de Kloet et al. 2008; Feldman et al. 2014). Consistent with this, levels of AVP in cerebral spinal fluid decrease following treatment with anxiolytics (i.e., 6 weeks of fluoxetine) (De Bellis et al. 1993).
AVP has three receptors: V1a, V1b (primarily in the anterior pituitary), and V2 (primarily in the kidney) (Caldwell et al. 2008; Carter 2017). V1a receptor (V1aR) levels are high in the lateral septum, bed nucleus of the stria terminalis (BNST), and central amygdala (CeA) and moderate in the anterior/lateral hypothalamus, medial preoptic area, and hippocampus. The receptors are not detectable in the basolateral amygdala (Ross et al. 2019). Since many V1AR-expressing regions modulate fear and anxiety (Davis et al. 2010), rodent studies have focused on the effect of V1aR on anxious behavior (Caldwell et al. 2008). V1aR knock-out mice exhibit attenuated anxious behavior in paradigms such as the elevated plus maze (Bielsky et al. 2004; Egashira et al. 2007), the open field, and the light/dark box without changes in muscle tone or coordination (Bielsky et al. 2004). V1aR antagonists also decrease anxious behavior in the elevated plus maze (Wigger et al. 2004; Bleickardt et al. 2009; Bayerl et al. 2016), the elevated zero maze, conditioned lick expression, and rat-pup separation-induced ultrasonic vocalization (Bleickardt et al. 2009). Thus, pre-clinical studies indicate that the V1aR modulates anxiety in animal models.
Clinical studies have tested V1aR’s role on neural activation in response to emotional faces. Meyer-Lindenberg and colleagues have demonstrated an association between a V1aR variant gene (AVPR1A) and increased amygdaloid activation during the presentation of angry and fearful faces (Meyer-Lindenberg et al. 2009), and Lee et al. found that a V1aR antagonist suppressed the increase in amygdaloid activation to angry faces produced by intranasal AVP (Lee et al. 2013). Here, we describe the effect of a novel V1aR antagonist (SRX246; Azevan Pharmaceuticals Inc.) on non-social fear and anxiety in humans.
Etiological theories have postulated that distressing feelings of apprehension about potential danger are central to pathologic anxiety (Zinbarg and Barlow 1996; Barlow 2000; Grupe and Nitschke 2013). Underlying such feelings is a perceived unpredictability over negative events, leading to an exaggerated and prolonged anticipatory response to uncertain threats (i.e., anxiety) (Barlow 2000; Grupe and Nitschke 2013). Some patients, however, also suffer from excessive reactivity to imminent threat (i.e., fear) (Barlow 2000). These responses are characterized by distinct symptoms: anxiety is a state of hypervigilance and hyperarousal, and fear is a fight-or-flight response. Indeed, responses to acute threat (“fear”) and potential threat (“anxiety”) are separate constructs of the negative valence systems according to the NIMH Research Domain Criteria (RDoC). While the circuits underlying anxiety and fear are actively studied, results in both human and animal literature support that these two constructs are mediated by partially overlapping, but nevertheless distinct, brain regions (e.g., BNST and CeA respectively) (Davis et al. 2010, but see Shackman and Fox 2016). Taken together, these features suggest that anxiety and fear may respond to different treatments.
We developed the Neutral-Predictable-Unpredictable (“NPU”) threat test to experimentally induce anxiety and fear in humans. The NPU threat test is a translational paradigm consisting of a neutral condition (N) and two threat conditions that involve administration of predictable (P) and unpredictable (U) electric shocks (Schmitz and Grillon 2012). During the task, loud white noise bursts (“startle probe”) elicit the startle reflex, a highly reliable cross-species measure of aversive state (Grillon and Baas 2003). Increased startle during unpredictable threat operationally defines anxiety (i.e., anxiety-potentiated startle), while increased startle during predictable threats operationally defines fear (i.e., fear-potentiated startle) (Schmitz and Grillon 2012). The NPU threat test generates highly robust and replicable responses (Kaye et al. 2016) and is therefore well-suited for repeated-measure study designs (Grillon et al. 2006, 2007, 2011, 2015; Lago et al. 2018; Kaye et al. 2019).
The distinction between anxiety-potentiated startle and fear-potentiated startle has clinical implications. Individuals with panic, social anxiety, specific phobias, and posttraumatic stress disorders have enhanced anxiety-potentiated startle, but not fear-potentiated startle, compared to healthy controls (Grillon et al. 1998, 2008, 2009b; Gorka et al. 2017b). Further, in healthy participants, conventional pharmaceutical treatments such as selective-serotonin receptor inhibitors (SSRIs) (Grillon et al. 2009a) and benzodiazepines (Grillon et al. 2006) selectively reduce anxiety-potentiated startle without affecting fear-potentiated startle. These findings support that at least some patients have exaggerated responses to unpredictable threats (i.e., anxiety) that can be preferentially reduced with traditional anxiolytics. Therefore, the NPU threat test is a promising tool to test the effect of novel therapeutics on biomarkers of clinically relevant threat circuitry (Avery et al. 2016; Grillon et al. 2019; Sartori and Singewald 2019).
This proof-of-concept study was designed to assess whether a novel V1aR antagonist, SRX246, demonstrates anxiolytic properties in the NPU threat test. Since in animal models, V1aRs are found in the BNST (Ring 2005; Caldwell 2017) and V1aR antagonists decrease anxious behavior (Caldwell et al. 2008), we hypothesized that SRX246 would decrease anxiety-potentiated startle compared to placebo. We did not make a prediction about the effect of SRX246 on fear-potentiated startle since, to our knowledge, there are no published studies examining the effect of V1aR on fear (e.g., fear conditioning).
Materials and methods
Participants
Power calculations were guided by our previous study in which we used a similar design to test the effects of a corticotropin-releasing hormone antagonist on anxiety-potentiated startle (Grillon et al. 2015). In that study, the effect of the control treatment on anxiety-potentiated startle had a Cohen’s d effect size of 0.53. Given the possibility that the effect of SRX246 may be weaker than the control treatment, we used a conservative dz of 0.46. Assuming 80% power and a two-tailed test of 0.05, we estimated that 36 subjects were required to test the effect of SRX246 on anxiety-potentiated startle.
All procedures were in accordance with the ethical standards of the Helsinki Declaration of 1975 (as revised in 1983). Healthy men and women were recruited from the Washington DC metropolitan area via flyers, notecards, and advertisements. All participants gave written informed consent approved by the NIMH Institutional Review Board and were compensated for their participation. Criteria for eligibility included (1) age 21–50, (2) body mass index 18.5–34.0 kg/m2, (3) body weight < 50 kg, (4) English-speaking, (5) no past or present active suicidal ideation as assessed by Columbia-Suicide Severity Rating Scale (C-SSRS; Posner et al. 2011), (6) no past or present Axis I psychiatric disorder (including substance dependence) as assessed by SCID-I/NP (First 2002), (7) no past or present significant medical/brain illness that may interfere with drug action/metabolism/excretion or psychophysiology measures as determined by principal investigators (Supplementary Materials and Methods), and (8) startle response 3 times greater than baseline EMG activity for 5 to 9 startle probes during the Startle Reactivity Screening Visit (see below). Further, all participants agreed to (1) no medication and herbal remedies within 7 days of the first drug dose (SRX246 or placebo) until 7 days of the last drug dose (with the exception of birth control); (2) no exposure to other study drugs or devices for 14 days prior to the first drug dose and throughout the study; (3) use of at least two forms of birth control including hormonal contraceptives, intrauterine device, barrier plus spermicide, surgical sterilization, and refrainment from heterosexual intercourse; and (4) abstinence from illicit drugs (including marijuana).
Thirty-seven participants (n = 37) completed the protocol (Figure S1 ). One participant was excluded from analysis due to poor EMG electrode contact. The final sample included 36 subjects (16 males, 20 females) with a mean age of 28.94 years (SD 6.82) and weight of 53.80 kg (SD 14.06).
Drug (SRX246, placebo)
We used a double-blind, crossover trial design (Fig. 1). Each participant received a total of SRX246 300 mg (180 mg morning, 120 mg evening) by mouth daily for 10–14 doses over 5–7 days (SRX), followed or preceded by matching placebo (PLC). The half-life of SRX246 (120 mg twice daily after 7 days) is 3.96 ± 0.57 h, and maximum concentration (Cmax) is reached at 1.5 h (Clinical Investigator’s Brochure). Drug order was randomized by the National Institute of Health (NIH) pharmacy prior to data collection. Safety was monitored by the NIH Data and Safety Monitoring Board. Participants took their first and last dose of drug/placebo at the NIH. They were given a standard 7-day AM/PM pill box pre-filled with their individualized regimen (i.e., dependent on visit dates), an instruction sheet, and a drug log to be completed at home. Drug compliance was assessed by drug log and serum plasma level of SRX246 (Supplementary Materials and Methods). Participants and investigators were asked at each experimental visit to guess which treatment the participant had received throughout the week.
Fig. 1.
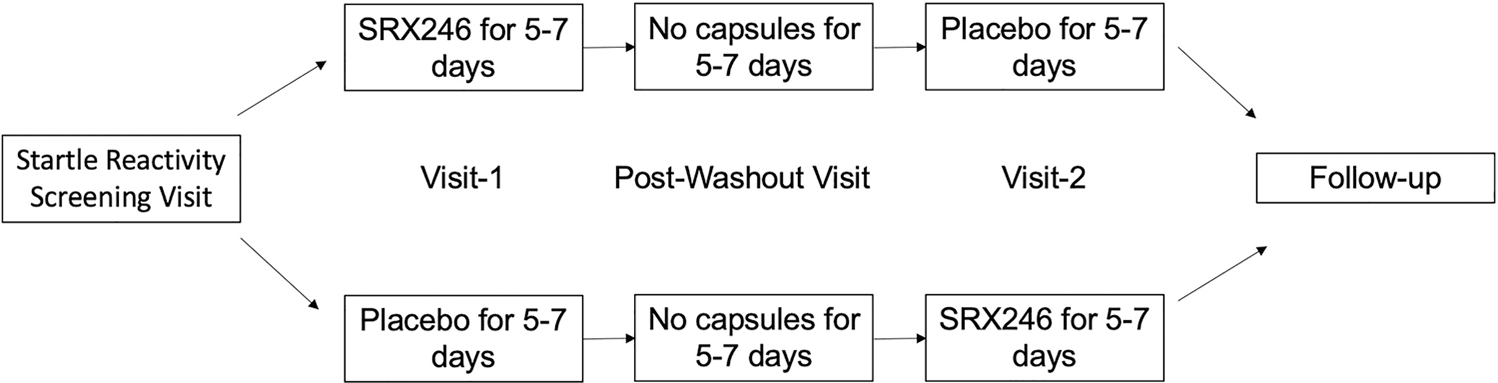
Study design. After startle reactivity screening test, participants were randomized to receive either SRX246 or matching placebo for 5–7 days. Then, participants underwent the NPU threat test during Visit-1. After a washout period, participants received either placebo or SRX246, whichever they did not receive previously, and repeated the NPU threat test during Visit-2. This was followed by a clinician interaction (either phone call or visit)
Startle
Eye-blink startle was elicited with 40-ms bursts of 103-dB white noise (probe) delivered over headphones and recorded with EMG electrodes under the left eye (BIOPAC Systems Inc., CA). EMG data were digitized (1000 Hz) and filtered (30–500 Hz) (MATLAB; Mathworks, MA). Peak blink amplitude was determined in the 20- to 120-ms window following probe onset and compared to the 50-ms pre-probe baseline. Stimuli were delivered via Presentation (Neurobehavioral Systems, Inc., CA).
Study design
Each participant completed four visits, including a Startle Reactivity Screening Visit, Visit-1 for experimental testing, Post-Washout Visit and Visit-2 for experimental testing at the NIH Clinical Center (Fig. 1). At each visit, clinicians confirmed protocol compliance by participant report, medical record review, medical safety assessments, and toxicology screens (Supplementary Materials and Methods). Alcohol was not confirmed beyond participant report; however, clinicians could administer a breathalyzer test upon suspicion of acute alcohol intoxication (not indicated in any participants). Clinicians assessed for adverse events by participant report, targeted physical exam if indicated, and medical safety assessments at every visit (Supplementary Materials and Methods).
During the Startle Screening Reactivity Visit, participants underwent nine startle probes (“startle habituation”) to determine the overall startle reactivity. Participants who did not have an observable startle response to at least five of these startle probes were compensated for the visit and discontinued from the study. Otherwise, participants proceeded to take the first dose of SRX246 or placebo.
During Visit-1, participants took their last dose of either SRX246 or placebo immediately after blood and urine collection (medical safety assessments, toxicology screens, plasma SRX246 level; Supplementary Materials and Methods). After about 70 min, participants underwent startle habituation. Investigators performed a shock work-up about 10 min prior to the NPU threat test by administering shocks on participants’ left forearms at increasing intensities until the participants rated the shock as highly uncomfortable but not painful. This shock intensity was used throughout the NPU threat test, which began about 90 min after drug ingestion (corresponding with Cmax). Finally, participants completed an emotion recognition task (not under threat of shock). All measures, conditions, data exclusions, and power analyses have been reported here for the NPU threat test task; results of the emotion recognition task will be reported elsewhere.
Participants had a washout period of 5–7 days and then returned to the clinic. At the Post-Washout Visit, participants took the first dose of either SRX246 or placebo (whichever they did not receive during the Startle Screening Reactivity Visit).
Visit-2 was the same as Visit-1. Seven to 14 days after Visit-2, a clinician contacted each participant to assess for adverse events or concerns. Both SRX246 and placebo were safe and well-tolerated. Throughout the entire study (no-drug, SRX, PLC), there were a total of 130 adverse events. The most frequent adverse events included headache (13% of events: 6 no-drug, 6 SRX, 6 PLC), elevated creatine kinase (7%: 3 no-drug, 3 SRX, 3 PLC), and abdominal pain (5%: 1 no-drug, 4 SRX, 2 PLC).
NPU threat test
The NPU threat test is a well-validated experimental tool (Kaye et al. 2016) that examines phasic fear and prolonged anxiety by delivering predictable and unpredictable shock, respectively (Schmitz and Grillon 2012). Participants were exposed to three distinct shock conditions (Neutral, Predictable, Unpredictable; NPU) each lasting 120 s (Fig. 2a). During each shock condition, three 8-s duration geometric shapes (“cue”) were presented, separated by inter-trial intervals (“no-cue”), on a computer screen (Fig. 2b). During the neutral condition, a shock was never administered. During the predictable condition, a shock could be administered only if a cue was on the screen. A shock was not administered during every cue in order to avoid habituation to the shock. During the unpredictable condition, a shock could be administered at any time (regardless of cue). Participants were informed of each condition’s potential for shock prior to the test, and conditions were signaled during the test with words on top of the computer screen, i.e., “No Shocks at Anytime,” “Shock Only During Cue,” or “Shock at Anytime.” Participants were asked to continuously rate their subjective anxiety on a scale of 0 (not at all anxious) to 10 (extremely anxious) throughout the test by pressing the left and right arrow keys on the keyboard.
Fig. 2.
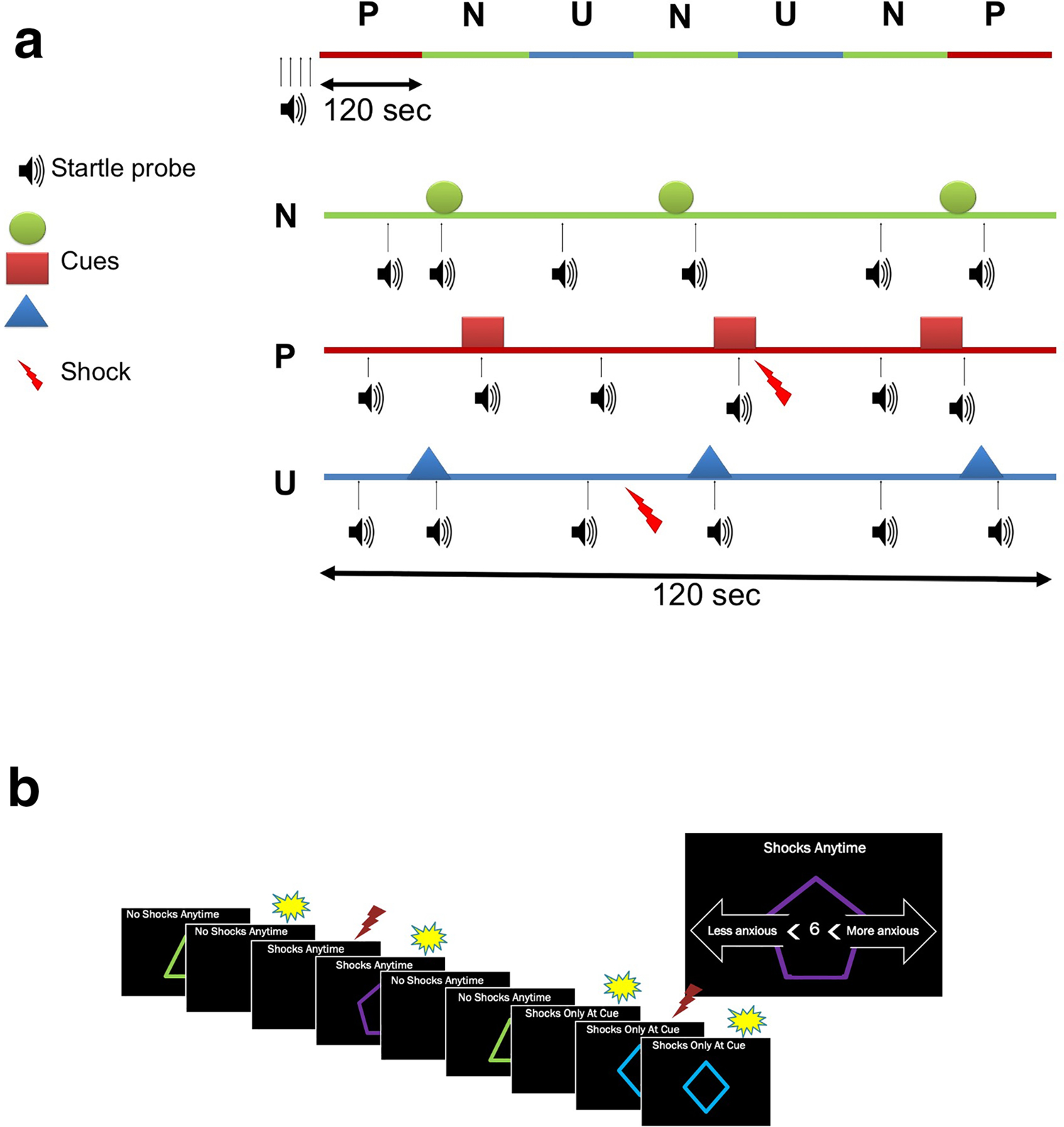
NPU threat test. a Schematic representation of stimulus presentation and shock delivery during each condition in the NPU threat test. Prior to each run, subjects were exposed to 4 startle probes to reduce initial startle reactivity. The upper line represents one of two potential orders of P (predictable), N (neutral), U (unpredictable) threat conditions. The lower lines are examples of each threat condition, including startle probes, visual threat cues, and shock delivery. Each condition had three visual threat cues (8 s) separated by inter-trial intervals. No shock was delivered in the N condition. In the P condition, shock was delivered only if a cue was on the computer screen. In the U condition, shock was delivered independently of cue. There were two shocks per P and U conditions (total of eight) and six startle probes per N, P, and U conditions ( adapted from Schmitz and Grillon 2012). b Participants were asked to rate their online subjective anxiety on a scale of 1–10 by pressing arrows on the keyboard throughout the NPU threat test. During the N condition, the display read “No Shocks at Anytime” at the top of the screen; during the P condition, the display read “Shocks Only During Cue”; and during the U condition, the display read “Shocks at Anytime.” The visual threat cues consisted of colored shapes that were displayed intermittently throughout each condition. Startle probes are represented by yellow bursts, and shocks are represented by red lightning
Participants completed two NPU runs during every experimental visit (Visit-1 and Visit-2). Prior to each run, subjects were exposed to 4 startle probes to reduce initial startle reactivity. Shock conditions were then presented in one of two orders: P N U N U N P or U N P N P N U. The order was counterbalanced across participants (i.e., first order for first run or second order for first run). Startle probes were separated by at least 20 s and occurred at least 8 s after a shock. Each run lasted 14 min (120 s × 7) and consisted of 8 shocks (2 per predictable and unpredictable condition) and 42 startle probes (3 during cue, 3 during no-cue during each condition).
Data analysis
All statistics were conducted with SPSS Statistics Subscription (IBM, NY).
Drug
The effect of drug order was tested on number of SRX doses, number of PLC doses, and SRX plasma levels with univariate ANOVAs.
Baseline startle
To assess effects of SRX246 on baseline startle, raw startle magnitudes were averaged within each startle habituation session and were analyzed with a drug (no-drug, SRX, PLC) × drug-order rANOVA.
NPU startle t-scores
Raw startle magnitudes were (1) standardized into t-scores ([z-scores × 10] + 50) within sessions for each participant as we have done in the past (Robinson et al. 2012; Lago et al. 2018) and (2) averaged within each condition (Neutral, Predictable, Unpredictable) and stimulus type (cue, no-cue) for each subject and experimental visit. Anxiety-potentiated startle was operationally defined as the change in startle during no-cue from the neutral to unpredictable condition and fear-potentiated startle as the change in startle from the no-cue to cue during the predictable condition (Grillon et al. 2009a, 2011; Ballard et al. 2014; Lago et al. 2018). Startle t-scores were analyzed first with a drug (SRX, PLC) × response (anxiety-potentiated startle, fear-potentiated startle) rANOVA and then by a drug (SRX, PLC) × response (anxiety-potentiated startle, fear-potentiated startle) × drug-order (SRX first, PLC first) rANOVA.
NPU online subjective anxiety ratings
Online ratings were recorded continuously. To match the startle measures, only ratings recorded immediately prior to the startle probe (within approximately 2 ms) were analyzed. Ratings were averaged within each condition and stimulus type for each subject and experimental visit. Results were analyzed with a drug (SRX, PLC) × response (anxiety-potentiated startle, fear-potentiated startle) × drug-order (SRX first, PLC first) rANOVA.
Questionnaires
Shock level and retrospective shock discomfort were analyzed with drug (SRX, PLC) × drug-order (SRX first, PLC first) rANOVAs. Drug blinding questionnaires were analyzed with a rater (participant, investigator) × drug (SRX, PLC) × drug-order (SRX first, PLC first) rANOVA.
Results
Drug
Number of SRX doses (mean 13.11, SEM 0.23), PLC doses (mean 13.02, SEM 0.26), and SRX plasma level (mean 10.48, SEM 1.38) did not differ between drug-order groups (SRX first, PLC first) (all p > 0.813, η2 < 0.003).
Baseline startle
Startle habituation did not differ from no-drug, SRX, or PLC (f[2,60] = 1.21, p = 0.306, η2 = 0.039), regardless of drug order.
NPU startle t-scores
Startle t-scores were first analyzed with a drug (SRX, PLC) × response (anxiety-potentiated, fear-potentiated) rANOVA. There was a trend for a drug × response interaction (f[1,35] = 3.06, p = 0.089, η2 = 0.080) on startle t-scores. SRX had lower anxiety-potentiated startle than PLC (mean − 1.09, SEM 0.52; t[35] = − 2.09, p = 0.044; 95% CI [− 2.14, − 0.03]). Drug did not affect fear-potentiated startle (mean 0.95, SEM 1.10; t[35] = 0.86, p = 0.396; 95% CI [− 1.29, 3.19]).
The drug (SRX, PLC) × response (anxiety-potentiated startle, fear-potentiated startle) × drug-order (SRX first, PLC first) rANOVA on startle t-scores revealed a 3-way interaction between drug × response × drug order (f[1,34] = 4.34, p = 0.045, η2 = 0.113; Tables 1 and 2). To better understand the nature of this interaction, it was decomposed by response (i.e., drug × drug order separately for anxiety-potentiated startle and fear-potentiated startle). For anxiety-potentiated startle, there was a significant drug main effect, which reflected reduced anxiety-potentiated startle with SRX compared to PLC (f[1,34] = 4.36, p = 0.044, η2 = 0.114). The drug × drug-order interaction was not significant (f[1,34] = 1.00, p = 0.324, η2 = 0.029) (Fig. 3).
Table 1.
Startle scores. Mean (SEM). T-scores
| SRX first |
PLC first |
Total sample |
|||||
|---|---|---|---|---|---|---|---|
| PLC | SRX | PLC | SRX | PLC | SRX | ||
| Neutral | No-cue | 47.2 (0.9) | 48.0 (0.8) | 46.5 (0.8) | 47.6 (0.9) | 46.9 (0.6) | 47.8 (0.6) |
| Cue | 48.0 (0.9) | 48.0 (0.8) | 47.5 (0.8) | 48.3 (0.9) | 47.7 (0.6) | 48.1 (0.6) | |
| Predictable | No-cue | 49.4 (1.0) | 49.3 (0.9) | 48.2 (0.8) | 48.8 (0.8) | 48.8 (0.6) | 49.0 (0.6) |
| Cue | 50.4 (1.0) | 53.6 (1.4) | 54.0 (1.3) | 52.6 (1.0) | 52.2 (0.8) | 53.1 (0.8) | |
| Unpredictable | No-cue | 49.7 (0.7) | 50.0 (1.1) | 50.8 (1.0) | 50.3 (1.0) | 50.3 (0.6) | 50.1 (0.7) |
| Cue | 49.6 (0.8) | 52.6 (1.3) | 53.8 (1.2) | 51.2 (1.1) | 51.7 (0.8) | 51.9 (0.8) | |
Table 2.
Startle scores. Mean (SEM). Threat-potentiated t-scores
| SRX first |
PLC first |
Total sample |
||||
|---|---|---|---|---|---|---|
| PLC | SRX | PLC | SRX | PLC | SRX | |
| APS | 2.5 (0.8)* | 1.9 (0.8)* | 4.3 (1.0)* | 2.7 (0.7)* | 3.4 (0.7) | 2.3 (0.5) |
| FPS | 1.0 (0.8)^ | 4.8 (1.2)^ | 5.7 (1.3) | 3.9 (0.8) | 3.4 (0.9) | 4.3 (0.7) |
Fig. 3.
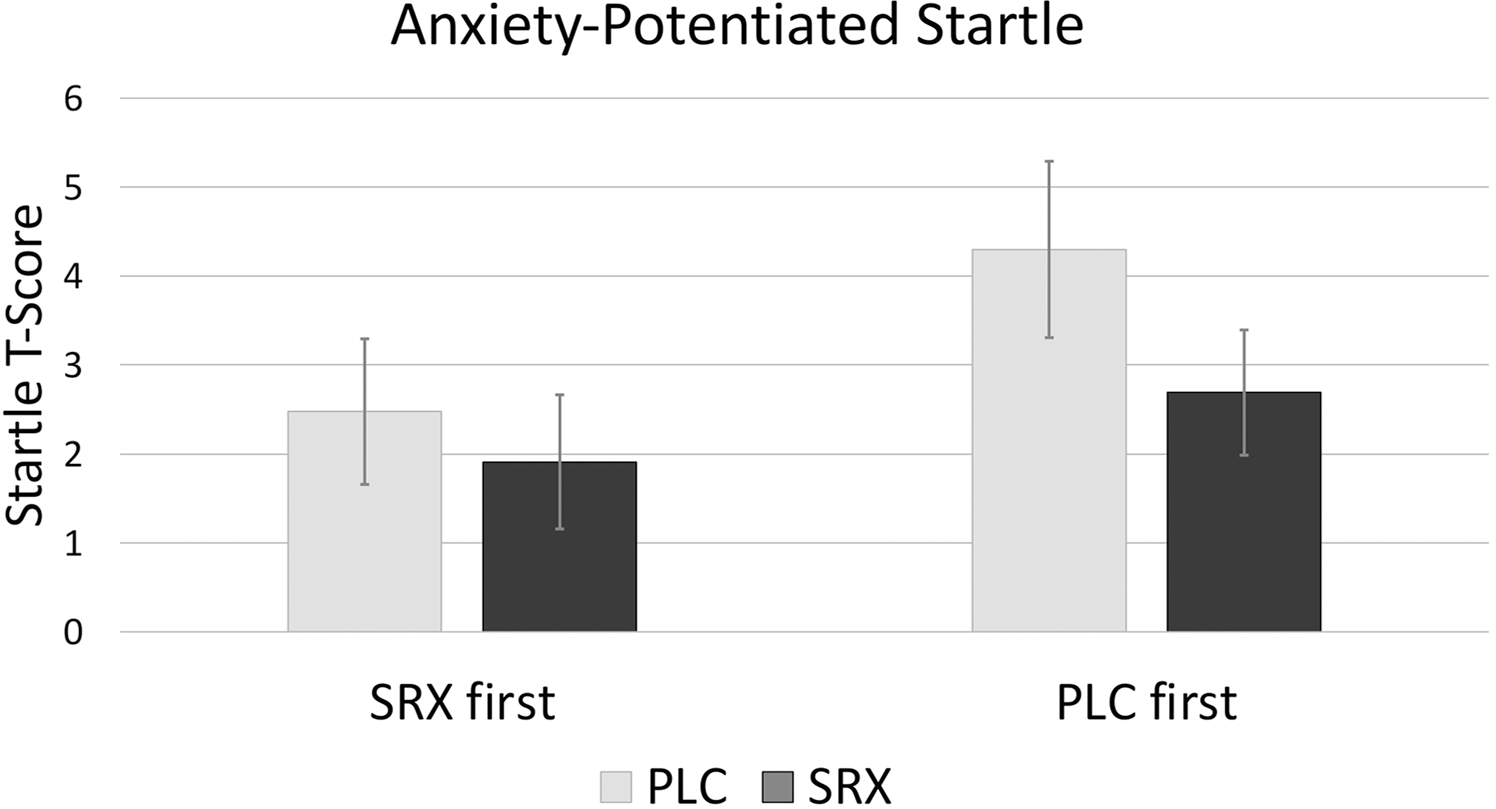
Anxiety-potentiated startle. Mean (SEM). Anxiety-potentiated startle was reduced with SRX compared to PLC (p < 0.05) regardless of drug order
For fear-potentiated startle, there was a significant drug × drug-order interaction (f[1,34], p = 0.008, η2 = 0.189). Participants who received SRX first had higher fear-potentiated startle with SRX than PLC (mean 3.79, SEM 1.34; t[17] = 2.83, p = 0.012, 95% CI [0.96, 6.61]). Participants who received PLC first had larger fear-potentiated startle with PLC than SRX, but this effect did not reach significance (mean 1.89, SEM 1.51; t[17] = 1.25, p = 0.227, 95% CI [− 1.29, 5.07]). For NPU raw startle results, see Supplemental Information.
NPU online subjective anxiety ratings
The drug (SRX, PLC) × ratings (anxiety-potentiated rating, fear-potentiated rating) × drug-order (SRX first, PLC first) rANOVA for online anxiety revealed a drug × drug-order interaction (f[1,34] = 4.66, p = 0.038, η2 = 0.121). When SRX was given first, participants reported lower (non-significant) online anxiety ratings during SRX than PLC (mean − 0.35, SEM 0.23; t[17] = − 1.49, p = 0.155, 95% CI [− 0.84, 0.14]). When PLC was given first, participants reported lower (also non-significant) online anxiety ratings during PLC than SRX (mean − 0.46, SEM 0.29; t[17] = − 1.58, p = 0.134, 95% CI [− 1.08, 0.16]). There was a main effect of condition (f[1,34] = 57.59, p < 0.001, η2 = 0.629), with overall higher anxiety-potentiated ratings than fear-potentiated ratings (mean 1.38, SEM 0.18; t[35] = 7.56, p < 0.001, 95% CI [1.00, 1.75]). The drug × condition interaction was not significant (f[1,34] = 0.09, p = 0.762, η2 = 0.003). Drug order did not affect rating effects.
Shock
There was a drug × drug-order interaction on shock level (f[1,34] = 5.07; p = 0.031, η2 = 0.130). When it was given first, PLC tended to have a lower shock level (mA) than SRX (mean − 1.70, SEM 0.86; t[17] = − 1.99, p = 0.063, 95% CI [− 3.50, 0.10]). Shock level did not differ by drug when SRX was given first (mean 1.45, SEM 1.11; t[17] = 1.31, p = 0.208, 95% CI [− 0.89, 3.79]). Drug did not modulate retrospective shock discomfort (f[1,34] = 0.41, p = 0.526, η2 = 0.012), regardless of drug order.
Blinding
The rater (participant, investigator) × drug (SRX, PLC) × drug-order (SRX first, PLC first) rANOVA did not reveal any effects (e.g., main effects: rater f[1,34] = 0.11; p = 0.744, η2 = 0.003; drug f[1,34] = 2.21; p = 0.146, η2 = 0.061; drug order f[1,34] = 0.22; p = 0.642, η2 = 0.006), indicating that the double-blind was successfully implemented.
Discussion
The objective of this study was to determine the effect of the novel V1aR antagonist SRX246 on an experimental model of fear and anxiety in humans. The main finding supports the hypothesis that SRX246 has anxiolytic properties. Specifically, SRX246 decreased anxiety-potentiated startle. The effects of SRX246 on fear-potentiated startle, however, were difficult to interpret due to a drug-order interaction. While results suggest that SRX246 may act as an anxiolytic in humans, this would need to be demonstrated in a clinical trial.
Since many compounds have affected “anxiety” in animals but not humans, drug developers must make crucial “go/no go” decision when selecting an agent for a clinical trial. This decision may be assisted by the use of experimental models of anxiety in healthy human subjects, based on the assumption that patients are more likely to benefit from a drug with anti-anxiety effects in the model than a drug with no anti-anxiety effects. Preliminary evidence for this assumption comes from studies that show (1) conventional anxiolytics (e.g., benzodiazepines and SSRIs) that are used to treat anxiety patients and also reduce the magnitude of anxiety-potentiated startle (Baas et al. 2002; Grillon et al. 2006, 2009a, 2015) and (2) candidate anxiolytics with no clinical efficacy (e.g., CRH antagonist) that do not affect anxiety-potentiated startle (Grillon et al. 2015). The predictive validity of this approach remains to be demonstrated, however, by showing that a candidate anxiolytic that reduces anxiety-potentiated startle in healthy humans can decrease symptoms in patients.
Previous studies showed that alprazolam (1 mg), diazepam (15 mg), and citalopram (20 mg daily for 2 weeks but not × 1) decreased anxiety-potentiated startle without affecting fear-potentiated startle. In addition, alprazolam and diazepam, but not citalopram, also reduced baseline startle (Baas et al. 2002; Grillon et al. 2006, 2007, 2009a, 2015). Baseline startle reduction reflects the sedative effect of benzodiazepines. In the present study, SRX246 did not affect baseline startle. This is consistent with previous studies that have shown that SRX246 does not have sedative effects in animals or humans (SRX246 Investigational Drug Brochure). Given that SRX246, SSRIs, and benzodiazepines have different mechanisms of action, it is notable that all these agents successfully decreased anxiety. These results suggest that SRX246 could offer relief for patients who are resistant to other treatments and may be associated with improved tolerability and compliance compared to benzodiazepines and SSRIs. It should be noted, however, that different classes of anxiolytics have not been tested using the NPU threat test paradigm.
The effect of SRX246 on fear-potentiated startle was inconclusive due to a significant drug-order interaction. Participants who received SRX first (and PLC second) had higher startle response to predictable threat with SRX than PLC, while participants who received PLC first (and SRX second) had larger startle response to predictable threat with PLC than SRX (non-significant). In other words, the magnitude of fear-potentiated startle was larger during the first session compared to the second session in both groups (SRX first, PLC first). Although we have found no drug-order effect in most of our past studies (Grillon et al. 2006, 2007, 2011, 2015), it has happened occasionally (Grillon et al. 2007). One explanation may be that results differed by number of SRX and PLC doses; however, analyses were run while covarying for number of SRX and PLC doses, and findings did not change (see Supplemental Information). Another factor is the reporting of subjective anxiety during the task. Indeed, one key difference between the present study and our past psychophysiological studies is the use of continuous online subjective ratings of fear and anxiety. We now believe that such ratings affect threat-potentiated startle. For example, reporting online subjective anxiety may reduce the magnitude of FPS and/or APS. As such, the process of reporting online subjective ratings may affect threat processing to imminent threat during the predictable condition, resulting in reduced FPS from session 1 to session 2. Finally, the power analyses and sample size calculations for this study were promised on detecting a medium-sized effect for APS. Thus, we may have been underpowered to detect a smaller effect of SRX246 on FPS. We can conclude that the effect of SRX246 on fear-potentiated startle, if any, may have been weaker than the effect of repeat testing and thus difficult to detect with the sample size used in this study.
Although we cannot conclude definitively that SRX246 does not affect fear, it is clear that this compound affects anxiety in our experimental model. Why would SRX246 decrease anxiety but not fear, and what does this finding tell us about its potential symptom targets? While the V1aR is found in both the BNST and CeA (Dumais and Veenema 2016), which broadly modulate anxiety and fear respectively (Davis et al. 2010; Avery et al. 2016; Shackman and Fox 2016), AVP is synthesized in the BNST but not the CeA (Godino and Renard 2018). Indeed, animal studies support AVP innervation of the dorsal raphe nucleus from the BNST (Rood and Beck 2014) (which may explain the comparable anxiolytic results of SRX246 and SSRIs). Thus, the BNST may play a stronger role in V1aR activity than the CeA. As discussed previously, a sustained state of sensitization characterized by hyperarousal and hypervigilance is a characteristic of many anxiety and stress disorders (Barlow 2000; Grillon 2002; Mineka and Oehlberg 2008; Grupe and Nitschke 2013). We believe that anxiety-potentiated startle is a “read-out” of this behavioral sensitization. Consistent with this view is increasing evidence of exaggerated responses to unpredictable threat in anxiety and stress disorders across a wide range of measures (startle, skin conductance, brain activation), procedures (context conditioning, verbal threat, shocks, unpleasant pictures), and laboratories (Grillon et al. 1994, 2008, 2009b; Morgan et al. 1995; Pole et al. 2003, 2009; Gorka et al. 2013a, 2017a, b; Grupe et al. 2016; Brinkmann et al. 2017a, b; Lieberman et al. 2020). Consequently, we propose that these symptoms (hypervigilance and hyperarousal) are potential indications for SRX246.
The differentiation between objective and subjective measures of experimental anxiety has been discussed at length (LeDoux and Pine 2016). In brief, these measures are thought to reflect different subcortical and cortical neural processes. It should also be noted that, based on the results of this study, we cannot differentiate between the neural effects of AVP and OT. Although OT has much less affinity (34.9 nmol/L) for V1aR than AVP (1.4 nmol/L) (Åkerlund et al. 1999), both peptides interact with the receptor (Carter 2017). While AVP and OT were historically thought to antagonize one another in response to threat, with OT “registering” safety and AVP “registering” danger, this model is currently viewed as too simplistic (Carter 2017). Therefore, our current results clearly support the anxiolytic effects of V1aR antagonism, but not necessarily the anxiogenic effects of only AVP.
Our study has a number of strengths, including the use of a well-validated experimental model of unpredictable and predictable threat to obtain objective measures of defensive responses (NPU threat test) (Grillon et al. 1998, 2008, 2009b; Gorka et al. 2013b, 2015, 2017b; Ballard et al. 2014; Klahn et al. 2016, 2017). Additionally, we used a within-subject, placebo-controlled, cross-over design to minimize individual threat sensitivity confounds. Subjects were not on other medications or herbal supplements and were free from illicit substances. Thus, the effects we saw were likely to have been caused by SRX246. Finally, both investigators and subjects were successfully blinded to drug treatment, and compliance was ensured by measuring SRX246 plasma levels (spot checks).
One limitation of the study was the sample size, which, as discussed previously, may have resulted in too little power to detect a potential effect of drug on fear-potentiated startle (power analyses were conducted based upon our previous drug studies; Grillon et al. 2015). Another limitation was lack of control for reproductive state. As SRX246 is an investigational drug, participants were required to be on two forms of birth control to participate in the study, but female hormonal fluctuations were not standardized. Clinical studies have not shown an effect of gender on AVP’s modulation of anxiety however (Thompson et al. 2006; Colloca et al. 2016; Lu et al. 2019). The study design also does not discriminate between the effects of single or multiple doses. Subjects were instructed to take the drug (SRX246 and PLC) for 5–7 days due to pharmacokinetic studies showing that peak and sustained levels are stable after this amount of exposure (Clinical Investigator’s Brochure). Nevertheless, we wanted to limit the confound of drug levels by incorporating a witnessed dose ingestion in the clinic with NPU threat testing coinciding with Cmax.
In conclusion, our results suggest that SRX246, a novel V1aR antagonist, decreases anxiety-potentiated startle in healthy humans. Since anxiety-potentiated startle is elevated in anxiety and trauma disorders (Grillon et al. 2008, 2009b; Gorka et al. 2017b) and decreased by traditional anxiolytics (Grillon et al. 2006, 2009a, 2015), the V1aR appears to be a promising target for drug developers who are interested in these disorders. Future studies should determine if results are replicable, and if so, whether SRX246 modulates anxiety-potentiated startle in a patient population.
Supplementary Material
Acknowledgements
We would like to thank Dr. Carlos Zarate, Jr. and Dr. Peixiong Yuan for assistance with plasma sample storage and shipping. We would like to thank NIMH nurses for their assistance with clinical coverage. We would like to thank Dr. Deborah Roberts for her assistance with protocol management.
Funding
This study was supported by the Intramural Research Program of the National Institute of Mental Health (grant number ZIAMH002798), Protocol 17-M-0046, NCT03036397 (clinicaltrials.gov). Azevan Pharmaceuticals Inc. provided SRX246 and placebo without charge and funded analysis of plasma samples for drug content. No NIH investigator involved in this study received any payment or other benefits from Azevan Pharmaceuticals Inc. T.L., E.P., E.B., A.M., A.B., C.R., N.B., E.D., S.P., M.E., and C.G. have nothing to disclose. M.B. and N.S. serve as officers at Azevan Pharmaceuticals and hold equity in the Company.
Footnotes
Declaration
Conflict of interest T.L., E.P., E.B., A.M., A.B., C.R., N.B., E.D., S.P., M.E., and C.G. have nothing to disclose. M.B. and N.S. serve as officers at Azevan Pharmaceuticals Inc. and hold equity in the Company. Azevan Pharmaceuticals Inc. provided SRX246 and placebo without charge and funded analysis of plasma samples for drug content. T.L., E.P., E.B., A.M., A.B., C.R., N.B., S.P., M.E., and C.G. did not receive any payment or other benefits from Azevan Pharmaceuticals Inc.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00213-021-05861-4.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Åkerlund M, Bossmar T, Brouard R et al. (1999) Receptor binding of oxytocin and vasopressin antagonists and inhibitory effects on isolated myometrium from preterm and term pregnant women. Br J Ostet Gynaecol 106:1047–1053 [DOI] [PubMed] [Google Scholar]
- Anderzhanova E, Kirmeier T, Wotjak CT (2017) Animal models in psychiatric research: the RDoC system as a new framework for endophenotype-oriented translational neuroscience. Neurobiol Stress 7:47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery SN, Clauss JA, Blackford JU (2016) The human BNST: functional role in anxiety and addiction. Neuropsychopharmacology 41:126–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas JMP, Grillon C, Böcker KBE et al. (2002) Benzodiazepines have no effect on fear-potentiated startle in humans. Psychopharmacology 161:233–247 [DOI] [PubMed] [Google Scholar]
- Ballard ED, Ionescu DF, Vande VJL et al. (2014) Increased fear-potentiated startle in major depressive disorder patients with lifetime history of suicide attempt. J Affect Disord 162:34–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow DH (2000) Unraveling the mysteries of anxiety and its disorders from the perspective of emotion theory. Am Psychol 55:1247–1263 [DOI] [PubMed] [Google Scholar]
- Bayerl DS, Hönig JN, Bosch OJ (2016) Vasopressin V1a, but not V1b, receptors within the PVN of lactating rats mediate maternal care and anxiety-related behaviour. Behav Brain Res 305:18–22 [DOI] [PubMed] [Google Scholar]
- Berger W, Mendlowicz MV, Marques-Portella C et al. (2009) Pharmacologic alternatives to antidepressants in posttraumatic stress disorder: a systematic review. Prog Neuropsychopharmacol Biol Psychiatry 33:169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielsky IF, Hu S-B, Szegda KL et al. (2004) Profound impairment in social recognition and reduction in anxiety-like behavior in vasopressin V1a receptor knockout mice. Neuropsychopharmacology 29:483–493 [DOI] [PubMed] [Google Scholar]
- Bleickardt CJ, Mullins DE, MacSweeney CP et al. (2009) Characterization of the V1a antagonist, JNJ-17308616, in rodent models of anxiety-like behavior. Psychopharmacology 202:711–718 [DOI] [PubMed] [Google Scholar]
- Brinkmann L, Buff C, Feldker K et al. (2017a) Distinct phasic and sustained brain responses and connectivity of amygdala and bed nucleus of the stria terminalis during threat anticipation in panic disorder. Psychol Med 47:2675–2688 [DOI] [PubMed] [Google Scholar]
- Brinkmann L, Buff C, Neumeister P et al. (2017b) Dissociation between amygdala and bed nucleus of the stria terminalis during threat anticipation in female post-traumatic stress disorder patients. Hum Brain Mapp 38:2190–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunnlieb C, Münte TF, Tempelmann C, Heldmann M (2013) Vasopressin modulates neural responses related to emotional stimuli in the right amygdala. Brain Res 1499:29–42 [DOI] [PubMed] [Google Scholar]
- Caldwell HK (2017) Oxytocin and vasopressin: powerful regulators of social behavior. Neurosci 23:517–528 [DOI] [PubMed] [Google Scholar]
- Caldwell HK, Lee HJ, Macbeth AH, Young WS III (2008) Vasopressin: behavioral roles of an “original” neuropeptide. Prog Neurobiol 84:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS (2017) The oxytocin-vasopressin pathway in the context of love and fear. Front Endocrinol (Lausanne) 8:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colloca L, Pine DS, Ernst M et al. (2016) Vasopressin boosts placebo analgesic effects in women: a randomized trial. Biol Psychiatry 79:794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C (2010) Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35:105–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bellis MD, Gold PW, Geracioti TD Jr et al. (1993) Association of fluoxetine treatment with reductions in CSF concentrations of corticotropin-releasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry 150:656–657 [DOI] [PubMed] [Google Scholar]
- de Kloet C, Vermetten E, Geuze E et al. (2008) Elevated plasma arginine vasopressin levels in veterans with posttraumatic stress disorder. J Psychiatr Res 42:192–198 [DOI] [PubMed] [Google Scholar]
- Dumais KM, Veenema AH (2016) Vasopressin and oxytocin receptor systems in the brain: sex differences and sex-specific regulation of social behavior. Front Neuroendocrinol 40:1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egashira N, Tanoue A, Matsuda T et al. (2007) Impaired social interaction and reduced anxiety-related behavior in vasopressin V1a receptor knockout mice. Behav Brain Res 178:123–127 [DOI] [PubMed] [Google Scholar]
- Feldman R, Vengrober A, Ebstein R (2014) Affiliation buffers stress: cumulative genetic risk in oxytocin-vasopressin genes combines with early caregiving to predict PTSD in war-exposed young children. Transl Psychiatry 4:e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB (2002) The DSM series and experience with DSM-IV. Psychopathology 35:67–71 [DOI] [PubMed] [Google Scholar]
- Godino A, Renard GMM (2018) Effects of alcohol and psychostimulants on the vasopressin system: behavioural implications. J Neuroendocrinol 30:e12611. [DOI] [PubMed] [Google Scholar]
- Gorka SM, Nelson BD, Sarapas C et al. (2013a) Relation between respiratory sinus arrythymia and startle response during predictable and unpredictable threat. J Psychophysiol 27:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorka SM, Nelson BD, Shankman SA (2013b) Startle response to unpredictable threat in comorbid panic disorder and alcohol dependence. Drug Alcohol Depend 132:216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorka SM, Liu H, Sarapas C, Shankman SA (2015) Time course of threat responding in panic disorder and depression. Int J Psychophysiol 98:87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorka SM, Lieberman L, Klumpp H et al. (2017a) Reactivity to unpredictable threat as a treatment target for fear-based anxiety disorders. Psychol Med 47:2450–2460 [DOI] [PubMed] [Google Scholar]
- Gorka SM, Lieberman L, Shankman SA, Phan KL (2017b) Startle potentiation to uncertain threat as a psychophysiological indicator of fear-based psychopathology: an examination across multiple internalizing disorders. J Abnorm Psychol 126:8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel G, Holmes A (2013) 50 years of hurdles and hope in anxiolytic drug discovery. Nat Rev Drug Discov 12:667–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C (2002) Associative learning deficits increase symptoms of anxiety in humans. Biol Psychiatry 51:851–858 [DOI] [PubMed] [Google Scholar]
- Grillon C, Baas J (2003) A review of the modulation of the startle reflex by affective states and its application in psychiatry. Clin Neurophysiol 114:1557–1579 [DOI] [PubMed] [Google Scholar]
- Grillon C, Ameli R, Goddard A et al. (1994) Baseline and fear-potentiated startle in panic disorder patients. Biol Psychiatry 35:431–439 [DOI] [PubMed] [Google Scholar]
- Grillon C, Morgan CA III, Davis M, Southwick SM (1998) Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biol Psychiatry 44:1027–1036 [DOI] [PubMed] [Google Scholar]
- Grillon C, Baas JMP, Pine DS et al. (2006) The benzodiazepine alprazolam dissociates contextual fear from cued fear in humans as assessed by fear-potentiated startle. Biol Psychiatry 60:760–766 [DOI] [PubMed] [Google Scholar]
- Grillon C, Levenson J, Pine DS (2007) A single dose of the selective serotonin reuptake inhibitor citalopram exacerbates anxiety in humans: a fear-potentiated startle study. Neuropsychopharmacology 32:225–231 [DOI] [PubMed] [Google Scholar]
- Grillon C, Lissek S, Rabin S et al. (2008) Increased anxiety during anticipation of unpredictable but not predictable aversive stimuli as a psychophysiologic marker of panic disorder. Am J Psychiatry 165:898–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Chavis C, Covington MF, Pine DS (2009a) Two-week treatment with the selective serotonin reuptake inhibitor citalopram reduces contextual anxiety but not cued fear in healthy volunteers: a fear-potentiated startle study. Neuropsychopharmacology 34:964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Pine DS, Lissek S et al. (2009b) Increased anxiety during anticipation of unpredictable aversive stimuli in posttraumatic stress disorder but not in generalized anxiety disorder. Biol Psychiatry 66:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Heller R, Hirschhorn E et al. (2011) Acute hydrocortisone treatment increases anxiety but not fear in healthy volunteers: a fear-potentiated startle study. Biol Psychiatry 69:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Hale E, Lieberman L et al. (2015) The CRH1 antagonist GSK561679 increases human fear but not anxiety as assessed by startle. Neuropsychopharmacology 40:1064–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Robinson OJ, Cornwell B, Ernst M (2019) Modeling anxiety in healthy humans: a key intermediate bridge between basic and clinical sciences. Neuropsychopharmacology 44:1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupe DW, Nitschke JB (2013) Uncertainty and anticipatino in anxiety: an integrated neurobiological and psychological perspective. Nat Rev Neurosci 14:488–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupe DW, Wielgosz J, Davidson RJ, Nitschke JB (2016) Neurobiological correlates of distinct post-traumatic stress disorder symptom profiles during threat anticipation in combat veterans. Psychol Med 46:1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JT, Bradford DE, Curtin JJ (2016) Psychometric properties of startle and corrugator response in NPU, affective picture viewing, and resting state tasks. Psychophysiology 53:1241–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JT, Fronk GE, Zgierska AE et al. (2019) Acute prazosin administration does not reduce stressor reactivity in healthy adults. Psychopharmacology 236:3371–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesselheim AS, Hwang TJ, Franklin JM (2015) Two decades of new drug development for central nervous system disorders. Nat Rev Drug Discov 14:815–816 [DOI] [PubMed] [Google Scholar]
- Klahn AL, Klinkenberg IAG, Notzon S et al. (2016) Prepare for scare-impact of threat predictability on affective visual processing in spider phobia. Behav Brain Res 307:84–91 [DOI] [PubMed] [Google Scholar]
- Klahn AL, Klinkenberg IA, Lueken U et al. (2017) Commonalities and differences in the neural substrates of threat predictability in panic disorder and specific phobia. NeuroImage Clin 14:530–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal AD, Pizzagalli DA, Mathew SJ et al. (2019) The first implementation of the NIMH FAST-FAIL approach to psychiatric drug development. Nat Rev Drug Discov 18:82–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lago TR, Hsiung A, Leitner BP et al. (2018) Exercise decreases defensive responses to unpredictable, but not predictable, threat. Depress Anxiety 35:868–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Pine DS (2016) Using neuroscience to help understand fear and anxiety: a two-system framework. Am J Psychiatry 173:1083–1093 [DOI] [PubMed] [Google Scholar]
- Lee RJ, Coccaro EF, Cremers H et al. (2013) A novel V1a receptor antagonist blocks vasopressin-induced changes in the CNS response to emotional stimuli: an fMRI study. Front Syst Neurosci 7:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman L, Funkhouser CJ, Gorka SM et al. (2020) The relation between posttraumatic stress symptom severity and startle potentiation to predictable and unpredictable threat. J Nerv Ment Dis 208(5):397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Lai J, Du Y et al. (2019) Sexual dimorphism of oxytocin and vasopressin in social cognition and behavior. Psychol Res Behav Manag 12:337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Kolachana B, Gold B et al. (2009) Genetic variants in AVPR1A linked to autism predict amygdala activation and personality traits in healthy humans. Mol Psychiatry 14:968–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineka S, Oehlberg K (2008) The relevance of recent developments in classical conditioning to understanding the etiology and maintenance of anxiety disorders. Acta Psychol (Amst) 127:567–580 [DOI] [PubMed] [Google Scholar]
- Morgan CA III, Grillon C, Southwick SM et al. (1995) Fear-potentiated startle in posttraumatic stress disorder. Biol Psychiatry 38:378–385 [DOI] [PubMed] [Google Scholar]
- Peskind ER, Jensen CF, Pascualy M et al. (1998) Sodium lactate and hypertonic sodium chloride induce equivalent panic incidence, panic symptoms, and hypernatremia in panic disorder. Biol Psychiatry 44:1007–1016 [DOI] [PubMed] [Google Scholar]
- Pitman RK, Orr SP, Lasko NB (1993) Effects of intranasal vasopressin and oxytocin on physiologic responding during personal combat imagery in Vietnam veterans with posttraumatic stress disorder. Psychiatry Res 48:107–117 [DOI] [PubMed] [Google Scholar]
- Pole N, Neylan TC, Best SR et al. (2003) Fear-potentiated startle and posttraumatic stress symptoms in urban police officers. J Trauma Stress 16:471–479 [DOI] [PubMed] [Google Scholar]
- Pole N, Neylan TC, Otte C et al. (2009) Prospective prediction of post-traumatic stress disorder symptoms using fear potentiated auditory startle responses. Biol Psychiatry 65:235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner K, Brown GK, Stanley B et al. (2011) The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168:1266–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring RH (2005) The central vasopressinergic system: examining the opportunities for psychiatric drug development. Curr Pharm Des 11:205–225 [DOI] [PubMed] [Google Scholar]
- Robinson OJ, Overstreet C, Allen PS et al. (2012) Acute tryptophan depletion increases translational indices of anxiety but not fear: serotonergic modulation of the bed nucleus of the stria terminalis? Neuropsychopharmacology 37:1963–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rood BD, Beck SG (2014) Vasopressin indirectly excites dorsal raphe serotonin neurons through activation of the vasopressin1A receptor. Neuroscience 260:205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AP, McCann KE, Larkin TE et al. (2019) Sex-dependent effects of social isolation on the regulation of arginine-vasopressin (AVP) V1a, oxytocin (OT) and serotonin (5HT) 1a receptor binding and aggression. Horm Behav 116:1045678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori SB, Singewald N (2019) Novel pharmacological targets in drug development for the treatment of anxiety and anxiety-related disorders. Pharmacol Ther 204:1–33 [DOI] [PubMed] [Google Scholar]
- Schmitz A, Grillon C (2012) Assessing fear and anxiety in humans using the threat of predictable and unpredictable aversive events (the NPU-threat test). Nat Protoc 7:527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackman AJ, Fox AS (2016) Contributions of the central extended amygdala to fear and anxiety. J Neurosci 36:8050–8063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RR, George K, Walton JC et al. (2006) Sex-specific influences of vasopressin on human social communication. PNAS 103:7889–7894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigger A, Sánchez MM, Mathys KC et al. (2004) Alterations in central neuropeptide expression, release, and receptor binding in rats bred for high anxiety: critical role of vasopressin. Neuropsychopharmacology 29:1–14 [DOI] [PubMed] [Google Scholar]
- Zinbarg RE, Barlow DH (1996) Structure of anxiety and the anxiety disorders: a hierarchical model. J Abnorm Psychol 105:181–193 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.