

SUMMARY
Neuropsychiatric disorders are often accompanied by cognitive impairments/intellectual disability (ID). It is not clear whether there are converging mechanisms underlying these debilitating impairments. We found that many autism and schizophrenia risk genes are expressed in the anterodorsal (AD) subdivision of anterior thalamic nuclei, which has reciprocal connectivity with learning and memory structures. CRISPR-Cas9 knockdown of multiple risk genes selectively in AD thalamus led to memory deficits. While AD is necessary for contextual memory encoding, the neighboring anteroventral (AV) subdivision regulates memory specificity. These distinct functions of AD and AV are mediated through their projections to retrosplenial cortex, using differential mechanisms. Furthermore, knockdown of autism and schizophrenia risk genes PTCHD1, YWHAG, or HERC1 from AD led to neuronal hyperexcitability, and normalization of hyperexcitability rescued memory deficits in these models. This study identifies converging cellular to circuit mechanisms underlying cognitive deficits in a subset of neuropsychiatric disease models.
IN BRIEF
Neuropsychiatric disorders are often accompanied by cognitive impairments. Roy et al. report that the knockdown of several autism and schizophrenia risk genes from anterodorsal thalamus leads to hyperexcitability and cognitive deficits. Normalization of hyperexcitability rescues cognitive deficits across models, revealing a converging mechanism underlying cognitive deficits.
Graphical Abstract
INTRODUCTION
Intellectual disability (ID)/cognitive impairment is characterized by significant limitations in cognitive functions, including reasoning, learning, memory, and adaptive behaviors, which co-occur with many neuropsychiatric disorders, including autism spectrum disorder (ASD) and schizophrenia (Morgan et al., 2008; Matson and Shoemaker, 2009). Cognitive impairments in these disorders have been commonly linked to dysfunction within hippocampal and cortical circuits (O’Tuathaigh et al., 2007; Kvajo et al., 2008; Golden et al., 2018), however whether converging neurobiological mechanisms underlie cognitive impairments across disorders has not been established. This issue has an important implication: if common mechanisms can be identified, therapeutic approaches capable of treating cognitive impairments in a subset of neuropsychiatric disorders may be developed.
PTCHD1 is mutated in some ASD patients with ID (Chaudhry et al., 2015). These patients have multiple symptoms including attention deficits, hyperactivity, sleep abnormality, and memory deficits. Our previous study in mice showed that the selective deletion of PTCHD1 from the thalamic reticular nucleus (TRN) was responsible for attention deficits, hyperactivity, and sleep abnormality, but not memory deficits (Wells et al., 2016). Interestingly, in addition to TRN, PTCHD1 exhibits strong expression in one other brain region, the anterodorsal (AD) thalamus, but not in well-known memory structures such as hippocampus, entorhinal cortex, or amygdala (Lein et al., 2007).
AD thalamus is part of the understudied anterior thalamic nuclei (ATN) complex, which also contains anteroventral (AV) and anteromedial (AM) subdivisions. ATN has reciprocal connectivity with frontal cortical areas, hippocampal subregions, and hypothalamic nuclei involved in memory functions (Jankowski et al., 2013). Lesion studies have suggested a potential role for ATN in spatial navigation (Winter et al., 2015) and cognitive tasks (Aggleton et al., 1991; Mitchell and Dalrymple-Alford, 2006; Savage et al., 2011; Warburton and Aggleton, 1999). Recent work has indicated that ATN are necessary for fear memory encoding and remote memory retrieval (Yamawaki et al., 2019; Vetere et al., 2021). Robust reductions in the number of ATN neurons was reported in tissue from patients (Young et al., 2000), suggesting a potential role for ATN dysfunction in schizophrenia. For these reasons, we hypothesized that AD thalamus dysfunction underlies memory deficits in PTCHD1 mutant mice (Wells et al., 2016), which may extend to other ASD and schizophrenia models.
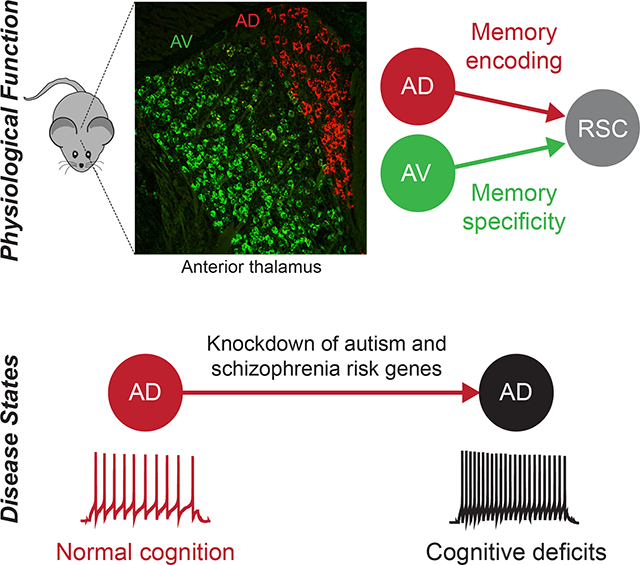
PTCHD1 led us to focus on AD thalamus, whose precise role in memory remains unclear. In this study, using wild type mice we showed that the AD→retrosplenial cortex (RSC) circuit is necessary for memory encoding, whereas the neighboring AV→RSC circuit regulates memory specificity. We observed that AD thalamus shows a high percentage of ASD and schizophrenia risk gene expression. The knockdown (KD) of different risk genes from AD leads to cognitive deficits. Several KD models had AD neuronal hyperexcitability that correlated with an impairment in learning-induced synaptic strengthening. We demonstrated that rescuing AD hyperexcitability in KD models is sufficient to restore multiple memory functions. Together, this study identifies cellular, circuit, and behavioral convergence underlying cognitive deficits in a subset of neuropsychiatric disease models.
RESULTS
Expression of ASD and schizophrenia risk genes in AD thalamus
Fluorescent in situ hybridization (FISH) revealed that the ASD/ID gene, PTCHD1, is selectively expressed in AD thalamus within ATN (Figure 1A). By examining gene expression in the Allen Brain Atlas (Lein et al., 2007) using 45 syndromic ASD risk genes from the SFARI database, we noticed that 10 risk genes had clear AD expression. Their expression pattern could be divided into three groups, namely expression only in AD thalamus within ATN, higher expression in AD thalamus relative to other ATN subdivisions, and high expression in the entire ATN. We focused on four of these risk genes, two of which had higher expression in AD vs. other ATN subdivisions (contactin associated protein 2 or CNTNAP2, ATPase Na+/K+ transporting subunit alpha 3 or ATP1A3), while the other two showed expression only in AD within ATN (mechanistic target of rapamycin kinase or MTOR, tyrosine 3- monooxygenase/tryptophan 5-monooxygenase activation protein gamma or YWHAG) (Figure 1A). Following a similar approach but this time using the top ten schizophrenia risk genes (Singh et al., 2020), we noticed that three risk genes exhibited high expression in AD thalamus, namely glutamate ionotropic receptor AMPA type subunit 3 (GRIA3), calcium voltage-gated channel subunit alpha 1G (CACNA1G), and HECT and RLD domain containing E3 ubiquitin protein ligase family member 1 (HERC1) (Figure 1B). Given that AD thalamus was the only ATN subdivision to exhibit expression of many risk genes, it is possible that AD thalamus-specific dysfunction contributes to disease phenotypes in a subset of different disorders.
Figure 1. Memory Impairments in AD Thalamus-Specific PTCHD1 Knockdown Mice.

(A-B) FISH staining of ASD (A), schizophrenia risk genes (B), in ATN. Anterodorsal (AD), anteroventral (AV).
(C) 11 excitatory neuron clusters in mouse thalamus from DropViz (89,027 cells, n = 6 mice) (left), top differentially expressed (DE) genes from the highlighted cluster (right). Rspo3 (R-spondin 3), Col27a1 (collagen type XXVII alpha 1 chain), Syndig1 (synapse differentiation inducing 1), Megf11 (multiple EGF like domains 11), Hs3st4 (heparan sulfate-glucosamine 3-sulfotransferase 4).
(D) FISH staining in ATN, parvalbumin (PV) neurons in TRN, DAPI staining (blue).
(E) Antibody staining in ATN.
(F) FISH staining in marmoset ATN.
(G) Retrograde CTB labeling from PreSub or RSC in ATN. Average of 296 CTB555+ and 271 CTB488+ cells were observed in AD. 84% of all PreSub-projecting neurons send collaterals to RSC (n = 3 mice).
(H) Circuit-based PTCHD1 knockdown (KD) strategy (left), FISH staining after KD (right). Ptchd1 expression is decreased by 96% (fluorescence intensity) in KD mice as compared to mCh controls in Figure S1H (n = 3 mice per group).
(I) CFC behavior. mCh control mice received an AAV expressing mCherry in AD in place of the AAV expressing sgRNAs. Long-term memory (LTM) recall test (mCh n = 9, KD n = 10 mice).
(J) T-maze behavior (mCh n = 9, KD n = 10 mice). Dashed line indicates chance level (50% correct).
Dashed line indicates the border between AD and AV. Two-tailed unpaired t test (I, J). For statistical comparisons, **p < 0.01; NS, not significant.
Data are presented as mean ± SEM.
Molecular marker and outputs of AD thalamus
To test our hypothesis, we needed to develop an approach to selectively manipulate risk genes in AD thalamus within ATN. We started by determining whether specific molecular markers could be identified within ATN. Taking advantage of the DropViz RNA-sequencing dataset (Saunders et al., 2018), we focused on 11 excitatory neuron clusters in mouse thalamus (Figure 1C). One of these clusters had the highest levels of complement C1q like 2 (C1QL2) gene expression. Staining experiments showed that C1QL2 is selectively expressed in AD thalamus within ATN (Figures 1D–1E) (Vertes et al., 2015). Similar to mice, C1QL2 mRNA was restricted to AD thalamus in the ATN of marmosets (Figure 1F), and we also observed C1QL2 expression in human tissue containing anterior thalamus (Figure S1A). Thus, C1QL2 is an AD thalamus-specific molecular marker conserved from rodents to primates.
C1QL2+ AD neurons are excitatory (Figure S1B). It is known that AD neurons primarily project to pre-subiculum (PreSub) and retrosplenial cortex (RSC) (Jankowski et al., 2013) (Figure 1G, and see Figures S1C–S1D). These retrograde tracing experiments also showed that AV thalamus projects to RSC but not PreSub, indicating that both AD and AV subdivisions converge on RSC. By examining the overlap of two different tracers in AD, we found that the majority of AD neurons send collaterals to both PreSub and RSC (Figure 1G). Further, using FISH we directly demonstrated that C1QL2+ AD neurons have a high degree of overlap with AD neurons projecting to either downstream target (Figure S1E). Using calcium/calmodulin-dependent protein kinase II (CAMKII)-Cre mice, glutamate decarboxylase 2 (GAD2)-Cre mice, and Cre-dependent, monosynaptic retrograde tracing (Wickersham et al., 2007), we found that most AD neurons project to excitatory CaMKII+ neurons in PreSub (Figure S1F). Applying a similar strategy to RSC, we observed that AD and AV project to both excitatory and inhibitory neurons (Figure S1G).
Memory impairments in multiple AD thalamus-specific risk gene knockdown mice
To test whether AD dysfunction plays a role in memory deficits in a PTCHD1 model, we took advantage of our finding that AD but not AV projects to PreSub. We optimized a circuit-based CRISPR-Cas9 viral approach, which included a retrograde rabies virus (RV)-expressing Cre (Chatterjee et al., 2018) injected in PreSub and a virus expressing target guide RNAs combined with a Cre-dependent SpCas9 virus (Xu et al., 2018) injected in AD, to knockdown (KD) PTCHD1 in AD (Figure 1H, and see Figure S1H). Using the contextual fear conditioning (CFC) memory paradigm, we found that PTCHD1 KD in AD did not alter foot shock-induced freezing during CFC training, but led to a long-term memory (LTM) recall deficit (Figure 1I). Using a spatial working memory paradigm, we found that PTCHD1 KD did not alter days to criterion during training or performance when the delay between sample and choice phases was short (10 s), but led to a working memory impairment when we used a more demanding long delay (60 s) (Figure 1J). These observations provide evidence linking an ASD/ID risk gene, PTCHD1, to behaviorally relevant AD circuit dysfunction. We performed KD of another ASD risk gene YWHAG (Figure 2A). YWHAG KD mice exhibited significant CFC memory deficits (Figure 2B, and see Figures S1I–S1M). Strikingly, AD thalamus-specific KD of schizophrenia risk genes GRIA3 (Figures 2D–2E), CACNA1G (Figures 2G–2H), or HERC1 (Figures 2J–2K) all led to CFC memory deficits. Furthermore, YWHAG, GRIA3, CACNA1G, and HERC1 KD mice were impaired in the long delay working memory test (Figures 2C, 2F, 2I, 2L), indicating that AD dysfunction induces cognitive impairments in a subset of different disease models.
Figure 2. Knockdown of Several ASD and Schizophrenia Risk Genes from AD Thalamus Leads to Memory Impairments.

(A-C) FISH staining (A), CFC behavior (B), T-maze behavior (C) (n = 9 mice per group). Ywhag expression is decreased by 94% (fluorescence intensity) in KD mice as compared to mCh controls (n = 3 mice per group).
(D-F) FISH staining (D), CFC behavior (E), T-maze behavior (F) (n = 9 mice per group). Gria3 expression is decreased by 92% (fluorescence intensity) in KD mice as compared to mCh controls (n = 3 mice per group).
(G-I) FISH staining (G), CFC behavior (H), T-maze behavior (I) (n = 9 mice per group). Cacna1g expression is decreased by 90% (fluorescence intensity) in KD mice as compared to mCh controls (n = 3 mice per group).
(J-L) FISH staining (J), CFC behavior (K), T-maze behavior (L) (n = 9 mice per group). Herc1 expression is decreased by 97% (fluorescence intensity) in KD mice as compared to mCh controls (n = 3 mice per group).
Dashed line indicates the border between AD and AV. Control FISH staining (A, D, G, J) from mCh mice. Dashed line in T-maze (C, F, I, L) indicates chance level (50% correct).
Two-tailed unpaired t test (B-C, E-F, H-I, K-L). For statistical comparisons, *p < 0.05, **p < 0.01; NS, not significant.
Data are presented as mean ± SEM.
Because many ASD and schizophrenia risk genes are not only highly expressed in AD thalamus but the KD of several risk genes selectively from AD lead to cognitive deficits, we wanted to know how this convergence compared to well-known cognitive brain regions. We examined the expression of 428 ASD (category S, 1, and 2 from the SFARI database) and schizophrenia (FDR < 5%) (Singh et al., 2020) risk genes using the Allen Brain Atlas (Lein et al., 2007) with a focus on AD thalamus and two other memory brain regions, hippocampal CA1 and mediodorsal thalamus (MD). While 21% of these risk genes were robustly expressed in MD, 48% and 57% were expressed in AD and CA1 respectively (Figure S1N). Among the risk genes that we functionally tested in AD thalamus (Figures 1A–1B), other than PTCHD1 all risk genes are also expressed in CA1 (Lein et al., 2007). To determine whether the KD of these risk genes from CA1 alters memory, we injected a virus expressing target guide RNAs combined with a constitutive SpCas9 virus (Figure S2A). While the KD of YWHAG, GRIA3, CACNA1G, and HERC1 from AD thalamus led to CFC memory deficits, in CA1 only GRIA3 and CACNA1G KD mice showed comparable memory deficits (Figures S2B–S2C). These experiments indicate that for a subset of risk genes expressed in both AD and CA1, risk gene KD shows a greater functional convergence in AD thalamus.
Inputs and electrophysiological properties of AD and AV thalamus
Because some risk genes are not only expressed in AD but also in neighboring AV thalamus (Figures 1A–1B), it is important to understand the cellular/circuit properties and behavioral contributions of these two ATN subdivisions in wild type mice. By examining highly expressed genes in other thalamic clusters (Figure 1C), we found that collagen type XXV alpha 1 chain (COL25A1) mRNA is selectively expressed in AV thalamus within ATN in mice (Figure 3A, and see Figure S2D for marmosets). We next wanted to map brain-wide inputs to AD and AV. By injecting a retrograde Cre virus (Tervo et al., 2016) in PreSub combined with Cre-dependent RV-mCherry injection in ATN, we characterized inputs to AD thalamus with high specificity (Figure 3B). For selective AV labeling, we injected the retrograde Cre virus in RSC combined with Cre-dependent RV-mCherry injection targeting AV (Figure 3C, and see Figures S2E–S2F). Given that the starter cells in AV are less dense than COL25A1+ AV neurons, it is likely that these experiments underestimate input cell numbers to this subdivision. Nevertheless, by normalizing inputs to each ATN subdivision to their respective starter cell counts, we found that most structures projected to both AD and AV (Figure 3D, and see Figures S2G–S2I), however prelimbic cortex input was observed for AV but not AD. Interestingly, most inputs had more neurons projecting to AV than AD. The granular division of RSC did not fit this pattern as it sent a larger input to AD.
Figure 3. Inputs and Electrophysiological Properties of AD and AV Thalamus.

(A) FISH staining in ATN.
(B-D) Mapping brain-wide inputs to AD or AV. RV starters (yellow) in AD (B) or AV (C), average RV-positive cell counts (D) (n = 3 mice for AD, n = 4 mice for AV, normalized starters across groups). PrL (prelimbic cortex), Cg1 (cingulate cortex area 1), Cg2 (cingulate cortex area 2), M2 (secondary motor cortex), S1BF (primary somatosensory cortex barrel field), RSA (retrosplenial agranular cortex), RSG (retrosplenial granular cortex). Dashed line in panel C indicates the border between AD and AV, see also Figures S2E–S2F.
(E-I) RV-GFP labeling (E) of AD neurons (green), recorded neurons (red) (F), after-depolarization potential (ADP) amplitude (G), Ih current-induced sag (H), excitability (I) (22 AD RV+, 17 AD RV−, 18 AV neurons, n = 3 mice).
(J-K) Terminals of ChR2-eYFP injected into PreSub (left) or ChR2-mCherry injected into RSC (right) (J), connectivity between AD, AV, PreSub, and RSC (K).
One-way ANOVA followed by Bonferroni post-hoc test (G-H), and two-way ANOVA with repeated measures followed by Bonferroni post-hoc test (I). For statistical comparisons, ***p < 0.001; NS, not significant.
Data are presented as mean ± SEM.
Given that AD and AV have distinct molecular markers and connectivity patterns, we used ex vivo electrophysiology in ATN slices to compare these subdivisions. AD neurons projecting to PreSub were labeled by a retrograde RV expressing green fluorescent protein (GFP) (Figures 3E–3F). Within AD, GFP+ and GFP− neurons had similar properties (Figures 3G–3I). However, we observed striking differences between AD and AV (Figures 3G–3I, and see Figures S3A–S3H). We next characterized the two major AD output circuits (Figure S3I). Ex vivo electrophysiological recordings showed that optogenetic stimulation of AD neurons resulted in larger excitatory post-synaptic currents in PreSub as compared to RSC neurons (Figures S3J–S3K). In addition, these two circuits were different in their short-term plasticity (Figure S3L). Interestingly, when we injected a ChR2-eYFP virus in the PreSub region and a ChR2-mCherry virus in RSC, we observed that their axonal terminals showed distinct patterns of projections back to ATN: AV received stronger input from the PreSub region as compared to AD, whereas both AD and AV received strong input from RSC (Figures 3J–3K, and see Figure S3M). These experiments revealed distinct properties between AD and AV thalamus.
The AD→RSC circuit is necessary for contextual memory encoding
Although our risk gene KD experiments clearly link AD thalamus to CFC memory, the precise role of this ATN subdivision in wild type mouse behavior remains unclear. We first injected a retrograde RV expressing Cre in PreSub and a Cre-dependent inhibitory DREADDs hM4Di-mCherry virus in AD (Figure 4A), and subsequently validated that the chemogenetic ligand compound 21 (C21) reversibly decreased AD neuronal firing (Figure S3N). In the CFC paradigm, inhibiting AD during training did not alter foot shock-induced freezing, however LTM recall was impaired (Figure 4B). Neither control nor AD inhibited mice displayed increased freezing behavior in a neutral context (Figure 4B), and motor behaviors were normal in these mice (Figures S3O–S3P). To determine whether our observation that AD plays an important role in contextual memory encoding extended to another memory paradigm, we performed the inhibitory avoidance (IA) task. AD inhibition during encoding also impaired performance in the IA memory task (Figure S3Q). In contrast, inhibition of AD immediately after CFC encoding (referred to as the cellular consolidation phase) or during CFC LTM recall did not affect performance (Figures S3R–S3S), and AD was not necessary for innate avoidance or tone fear encoding (Figures S3T–S3U). We also noted that AD plays an important role in a demanding version of the spatial working memory paradigm (Figure S3V).
Figure 4. The AD→RSC→EC Circuit is Necessary for Contextual Memory Encoding.

(A) hM4Di expression in AD.
(B) CFC behavior (n = 9 mice per group). mCherry control (mCh) mice received a Cre-dependent mCherry virus in place of the hM4Di virus.
(C) mEPSCs of AD neurons from home cage (16 neurons) or CFC training (18 neurons) groups (n = 3 mice per group).
(D) Activity of AD neurons using Fos-TRAP mice (n = 6 mice per group). Immediate shock (Imm. Shk.). AD neurons revealed by C1QL2 staining.
(E-F) LFP traces before (Pre) vs. after (Post) CFC training, change in LFP power after training
(E), change in power for individual frequency bands (F) (n = 15 mice).
(G-H) AMPA/NMDA ratio recordings of AD circuits, representative traces (G), quantification
(H) (AD→PreSub: 29 neurons per group, AD→RSC: 27 home cage and 26 training neurons, n = 3 mice per group).
(I) Optogenetic terminal inhibition (eArch-eYFP, n = 12 mice) or activation (ChR2-eYFP, n = 7 mice) during CFC training. Control (eYFP, n = 14 mice). LTM test is plotted.
(J-K) cFos staining in RSC using home cage (n = 7 mice), training control (mCherry or mCh, n = 7 mice), training AD hM4Di-mCh (n = 8 mice) groups (J), cFos staining in hippocampal CA1
(K) (n = 6 mice per group). Both mCh and hM4Di-mCh groups received C21 injections prior to training. Dentate gyrus (DG).
(L) Two-step RV tracing showing AD, AV inputs to entorhinal cortex (EC)-projecting RSC neurons. Starters (yellow) in RSC (left image), upstream ATN labeling (right image).
(M) Optogenetic terminal inhibition of EC-projecting RSC neurons, which receive ATN input, during training (eYFP n = 9 mice, eArch-eYFP n = 11 mice).
Two-tailed unpaired t test (B-C, H, M), paired t test (F), and one-way ANOVA followed by Bonferroni post-hoc test (D, I-K). For statistical comparisons, *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
Data are presented as mean ± SEM.
In search of cellular correlates of memory encoding in AD, we found that the frequency of miniature excitatory post-synaptic currents (mEPSCs) was increased post-CFC training (Figure 4C, and see Figure S4A). This increase correlated with an increase in the active cFos+ ensemble size in AD (Figure 4D, and see Figure S4B). In vivo local field potential (LFP) recordings from AD showed significant increases in the power of theta and gamma oscillations following encoding (Figures 4E–4F, and see Figure S4C), which was not observed when AD was chemogenetically inhibited during encoding (Figure S4D). To determine if one or both of the major AD outputs play a role in memory encoding, we measured synaptic strengthening in these circuits post-encoding. Encoding increased the AMPA/NMDA ratio of the AD→RSC circuit, but not the AD→PreSub circuit (Figures 4G–4H). Consistently, in vivo LFP coherence between AD and RSC, but not AD and PreSub, exhibited enhancements post-encoding (Figures S4E–S4G), and enhanced theta-gamma cross-frequency coupling (Figure S4H). Since these in vivo electrophysiological correlates have been consistently linked to cognitive processes (Colgin, 2015), these data support the idea that AD neurons and their projections to RSC in particular play an important role in memory encoding. To directly test this idea, we performed chemogenetic inhibition of RSC or PreSub excitatory neurons during CFC encoding. Inhibition of RSC neurons, but not PreSub neurons, led to a recall deficit, which mimicked the effect of AD inhibition (Figures S4I–S4K). Further, optogenetic terminal inhibition in RSC directly demonstrated that the AD→RSC circuit is necessary for encoding, and optogenetic activation of this circuit during encoding is sufficient to enhance LTM recall (Figure 4I, and see Figures S4L–S4M). These observations are further supported by optogenetic terminal inhibition experiments using C1ql2-Cre mice, which revealed that the AD→RSC circuit, but not the AD→PreSub circuit, is necessary for encoding (Figure S5A, and see Figure S4M).
We next examined the effect of chemogenetic AD inhibition during encoding on neural activity in downstream structures such as RSC and hippocampal CA1. We found that AD inhibition impaired the learning-induced enhancement of CFOS+ ensembles in both RSC and CA1 (Figures 4J–4K, and see Figures S5B–S5C). Because this suggested that manipulating AD contributed to changes in hippocampal activity, we wanted to identify the circuit basis for this observation. We started by confirming that RSC projects to entorhinal cortex (EC) (Witter et al., 2017) (Figures S5D–S5E), which serves as the major input to the hippocampus. We hypothesized that AD→RSC→EC may underlie the important contribution of AD→RSC in encoding. To directly visualize connectivity between AD, RSC, and EC, we injected a retrograde Cre virus in EC and Cre-dependent RV in RSC. In support of our idea, we showed that more AD neurons, in comparison to AV, project to the EC-projecting RSC neurons (Figure 4L, and see Figure S5F). The finding that AD inhibition during encoding decreased CFOS activation of EC-projecting RSC neurons (Figure S5G) further strengthened this idea. To link these tracing and neural activity data to behavior, we injected an anterograde virus expressing Cre (Zingg et al., 2017) in ATN, a Cre-dependent eArch-eYFP virus in RSC, and implanted optic fibers in EC (Figure S5H). Optogenetic terminal inhibition in EC of the ATN→RSC→EC circuit during encoding impaired CFC LTM recall (Figure 4M, and see Figures S4M and S5I). These experiments uncovered the neural circuit mechanism by which AD thalamus contributes to the cortico-hippocampal memory network.
The AV→RSC circuit regulates memory specificity
Since AV thalamus also projects to RSC, we wanted to investigate their role in CFC memory. By expressing Cre in AD through injection of a retrograde RV expressing Cre in PreSub and a Cre-Off halorhodopsin (NpHR-eYFP) virus (Saunders et al., 2012) in ATN, we confirmed specific AV thalamus labeling and light-induced neuronal inhibition (Figure 5A, and see Figure S5J). In contrast to AD thalamus, inhibition of AV cell bodies or AV→RSC terminals during CFC training had no effect on LTM recall, however these mice displayed robust generalization in the neutral context test (Figure 5B, and see Figures S4M and S5K). This phenotype correlated with a post-training decrease in the frequency of mEPSCs on AV neurons (Figures S5L–S5M).
Figure 5. The AV→RSC Circuit Regulates Memory Specificity.

(A) Halorhodopsin (NpHR) expression in AV, C1QL2 staining (red).
(B) AV cell bodies or AV→RSC terminal inhibition during CFC training (day 1) followed by LTM recall and neutral context tests (control eGFP n = 8 mice, AV NpHR n =10 mice, AV→RSC NpHR n = 8 mice).
(C) cFos staining in RSC using home cage (n = 4 mice), training control (eGFP, n = 5 mice), training AV→RSC NpHR-eYFP (n = 5 mice) groups.
(D) Retrograde RV tracing in PV-Cre, somatostatin (SST)-Cre, or VIP-Cre mice. Images show RV labeling in AV thalamus (left), quantification of RV+ cells in AV (n = 4 mice per group) (right). Normalized starters across groups.
(E-G) cFos activation of PV, VIP cell types in RSC during CFC training, representative images (E-F), overlap quantification (G) (PV-Cre: home cage n = 7 and training n = 8 mice, VIP-Cre: home cage n = 5 and training n = 7 mice). Cre mice were prepared by injecting a Cre-dependent eYFP virus in RSC.
(H) Fold change plotted relative to average home cage counts (n = 8 PV-Cre training mice, n = 7 VIP-Cre training mice).
(I-J) AV→RSC inhibition with PV or VIP activation in RSC during training, viral injection schematic (I), neutral context test (J) (PV-Cre: C21 n = 8 and C21+light n = 6 mice, VIP-Cre: C21 n = 7 and C21+light n = 6 mice).
(K-L) AD→RSC or AV→RSC terminal inhibition during training in the cocaine-induced conditioned place preference behavior. Preference for the cocaine (Coc) vs. the saline (Sal) side is plotted within animal for the recall test (K), and the modified chamber test (L) (n = 12 mice per group).
One-way ANOVA followed by Bonferroni post-hoc test (B-D), two-tailed unpaired t test (G-H, J), and paired t test (K-L). For statistical comparisons, *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
Data are presented as mean ± SEM.
To further examine this generalization phenotype, we optogenetically inhibited AV→RSC terminals during encoding and quantified activated ensembles in RSC (Figure 5C, and see Figures S5N–S5O). Strikingly, AV→RSC inhibited mice showed increased levels of learning-induced CFOS+ ensembles in RSC, which hinted at the possibility that the role of AV during encoding requires inhibitory neurons in RSC. Pursuing this possibility, using Cre-dependent RV injected in RSC of different inhibitory neuron-specific Cre mouse lines we found that AV neurons primarily project to parvalbumin (PV) and vasoactive intestinal polypeptide (VIP) inhibitory neurons (Figure 5D, and see Figure S6A). Though both PV and VIP populations, which were labeled using a Cre-dependent eYFP virus in PV-Cre and VIP-Cre mice, exhibited an increase in CFOS activation post-training (Figures 5E–5G), VIP neurons had a greater fold change (Figure 5H). We next prepared mice in which AV→RSC terminals could be inhibited optogenetically with simultaneous activation of either PV or VIP neurons in RSC chemogenetically (Figure 5I). AV→RSC inhibition with VIP, but not PV activation, during encoding prevented the generalization phenotype in AV inhibited mice (Figure 5J, and see Figures S6B–S6C). In addition, using the cocaine-induced conditioned place preference (CPP) paradigm, we showed that the AD→RSC circuit is necessary for effective memory encoding (Figure 5K, and see Figure S6D). However, in a modified CPP chamber, although the control eYFP group no longer exhibited any behavioral preference, the AV→RSC inhibited group showed significant preference (i.e., generalization behavior) (Figure 5L), which demonstrated that the differential roles of AD (encoding) and AV (specificity) inputs to RSC in a negative-valence CFC memory task extends to a positive-valence CPP memory task.
Normalizing hyperexcitability of AD neurons rescues memory deficits in ASD and schizophrenia models
With a better understanding of AD circuits underlying memory in wild type mice, we wanted to examine how PTCHD1 KD alters AD neuronal properties. Using ex vivo electrophysiology, PTCHD1 KD revealed a decrease in action potential (AP) half width, which correlated with an increase in the excitability of AD neurons (Figure 6A, and see Figure S6E), consistent with our previous findings in the TRN (Nakajima et al., 2019). To determine whether PTCHD1 KD has any impact on CFC training-induced AMPA/NMDA ratio increases in the AD→RSC circuit, we prepared KD mice that included a Cre-dependent ChR2-eYFP virus in AD for recordings (Figure 6B). We observed a lack of CFC training-induced synaptic strengthening (AMPA/NMDA ratio) in the AD→RSC circuit of KD mice (Figure 6C, and see Figure S6F).
Figure 6. Normalizing Neuronal Hyperexcitability Rescues Memory Deficits in PTCHD1 KD Mice.

(A) Ex vivo recordings from control (mCherry or mCh) vs. KD mice showing action potential (AP) threshold, AP half width, and neuronal excitability (24 mCh neurons, 23 KD neurons, n = 3 mice per group).
(B-C) Viral injection schematic for electrophysiological recordings (B), AMPA/NMDA ratio recordings of the AD→RSC circuit (C) in wild type (data from Figure 4H) or KD (17 neurons per group, n = 3 mice each) animals.
(D-F) Viral approach to chemogenetically normalize excitability in KD mice (D), AD neuronal excitability rescue ex vivo (E) (mCh control data from panel A, 14 neurons each for KD C21 low dose and KD C21 regular dose from n = 3 mice per group), AMPA/NMDA (A/N) ratio rescue in the AD→RSC circuit of KD mice (F) (PTCHD1 KD home cage and training data from panel C, 18 neurons for training low dose and 19 neurons for training regular dose from n = 3 mice per group).
(G-H) cFos activation in RSC during CFC training for KD and rescue groups (G) (mCh controls n = 4 mice per group, KD home cage and training n = 4 mice per group, KD hM4Di groups n = 8 mice per group). CFC LTM test in KD and rescue groups (H) (mCh control and PTCHD1 KD data from Figure 1I, KD low n = 9 mice, KD regular n = 8 mice).
Two-way ANOVA with repeated measures followed by Bonferroni post-hoc test (excitability data in A, E), two-tailed unpaired t test (AP threshold/half width in A, C, mCh control in G), and one-way ANOVA followed by Bonferroni post-hoc test (F, PTCHD1 KD in G, H). For statistical comparisons, *p < 0.05, **p < 0.01; NS, not significant.
Data are presented as mean ± SEM.
We hypothesized that the increased excitability of AD neurons in KD mice may prevent synaptic strengthening during CFC training, which is necessary for efficient encoding. Specifically, in control mice the excitability of AD neurons would increase during training, which leads to strengthening of the AD→RSC circuit, but in KD mice due to the increased excitability of AD neurons before training there will not be the important training-induced increase in excitability and corresponding synaptic strengthening. By recording from AD neurons before and after CFC training in control and KD groups, we obtained experimental evidence to support this idea (Figure S6G). These findings are consistent with previous reports showing a correlation between neuronal hyperexcitability and impairments in long-term potentiation (Speca et al., 2014; Gruter et al., 2015). We next developed a dose-dependent chemogenetic approach to normalize the excitability of AD neurons in KD mice (Figure 6D). When the excitability was returned to physiological levels (i.e., using a low dose of C21) (Figure 6E), training-induced strengthening of the AD→RSC circuit (Figure 6F), training-induced CFOS+ ensemble size in RSC (Figure 6G, and see Figure S6H), and LTM recall were all rescued (Figure 6H, and see Figure S6I).
We wanted to know whether the KD of risk genes other than PTCHD1 might also lead to neuronal excitability alterations in AD (Figure S6J). In contrast to PTCHD1 KD, YWHAG KD in AD neurons did not have an effect on AP half width, but resulted in a decreased AP threshold (Figure 7A). Similar to PTCHD1 KD, YWHAG KD neurons also showed hyperexcitability (Figure 7B), which prevented training-induced strengthening of the AD→RSC circuit (Figure 7C). Therefore, we applied our excitability normalization strategy (Figure 7D) and found that the hyperexcitability of YWHAG KD neurons could be returned to physiological levels (Figure 7E). YWHAG KD mice with normalized AD excitability showed control levels of behavioral performance in the CFC paradigm (Figure 7F). HERC1 KD mice also exhibited AD neuronal hyperexcitability (Figures 7G–7H), and lacked training-induced strengthening of the AD→RSC circuit (Figure 7I). Normalizing the excitability of AD neurons in HERC1 KD mice rescued their CFC memory (Figure 7J). We further demonstrated that normalizing the excitability of AD rescues performance of PTCHD1, YWHAG, and HERC1 KD mice in the spatial working memory task (Figures S6K–S6M). These experiments show that the KD of different disease risk genes from AD thalamus leads to a common alteration in neuronal excitability, which if treated is sufficient to rescue memory deficits.
Figure 7. Normalizing Neuronal Hyperexcitability Rescues Memory Deficits in YWHAG and HERC1 KD Mice.

(A-B) Ex vivo recordings from control (mCherry or mCh) vs. YWHAG KD mice showing AP threshold and AP half width (A), neuronal excitability (B) (15 mCh neurons, 16 KD neurons, n = 3 mice per group).
(C) AMPA/NMDA ratio recordings of the AD→RSC circuit in YWHAG KD mice (14 neurons per group, n = 3 mice each).
(D) Viral approach to chemogenetically normalize excitability in YWHAG KD mice.
(E) AD neuronal excitability rescue ex vivo (mCh control and YWHAG KD data from panel B, 15 neurons for KD C21 low dose group from n = 3 mice).
(F) CFC training and LTM recall test in KD and rescue groups (mCh control and YWHAG KD data from Figure 2B, KD low n = 9 mice).
(G-H) Ex vivo recordings from mCh control vs. HERC1 KD mice showing AP threshold and AP half width (G), neuronal excitability (H) (15 mCh neurons, 23 KD neurons, n = 3 mCh mice, n = 4 KD mice).
(I) AMPA/NMDA ratio recordings of the AD→RSC circuit in HERC1 KD mice (12 home cage, 13 training neurons, n = 3 mice each).
(J) CFC training and LTM recall test in KD and rescue groups (mCh control and HERC1 KD data from Figure 2K, KD low n = 9 mice).
(K-M) KIR2.2 (K) (11 mCh neurons from 5 mice, 10 PTCHD1 KD neurons from 5 mice, 11 YWHAG KD neurons from 5 mice, 12 HERC1 KD neurons from 6 mice), CAV2.1 (L) (9 mCh neurons from 6 mice, 8 PTCHD1 KD neurons from 5 mice, 8 YWHAG KD neurons from 5 mice, 8 HERC1 KD neurons from 6 mice), and CAV2.2 (M) (9 mCh neurons from 6 mice, 8 PTCHD1 KD neurons from 5 mice, 8 YWHAG KD neurons from 5 mice, 8 HERC1 KD neurons from 6 mice) ex vivo current recordings. Current-voltage plotted for KIR2.2, current density-voltage plotted for CAV2.1 and CAV2.2.
Two-tailed unpaired t test (A, C, G, I), two-way ANOVA with repeated measures followed by Bonferroni post-hoc test (B, E, H, K-M), and one-way ANOVA followed by Bonferroni post-hoc test (F, J). For statistical comparisons, *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
Data are presented as mean ± SEM.
We wanted to identify molecular alterations underlying hyperexcitability in AD neurons of PTCHD1, YWHAG, and HERC1 KD mice. We focused on channels that are necessary for maintaining AP threshold and AP half width in thalamic neurons (Kasten et al., 2007), and among these, ones that are robustly expressed in AD (Lein et al., 2007). We narrowed down to two channels that may underlie AP threshold changes (potassium voltage-gated channel subfamily A member 1 or KV1.1, potassium inwardly rectifying channel subfamily J member 12 or KIR2.2) and three channels that may underlie AP half width changes (potassium voltage-gated channel subfamily Q member 2 or KV7.2, calcium voltage-gated channel subunit alpha-1A or CAV2.1, calcium voltage-gated channel subunit alpha-1B or CAV2.2). FISH staining revealed that three out of the five candidate channels, specifically KIR2.2, CAV2.1, and CAV2.2, are decreased in at least one KD mouse model (Figures S7A–S7B). To directly measure these individual currents in KD mice, we performed ex vivo recordings. We found that the KIR2.2 current amplitude is decreased in YWHAG and HERC1 KD mice, whereas the CAV2.1 and CAV2.2 current amplitudes are decreased in PTCHD1 and HERC1 KD mice (Figures 7K–7M). These studies identified individual channel subtypes that may underlie AD neuronal hyperexcitability in different KD models.
DISCUSSION
We have shown that anterior thalamic dysfunction, in particular impairments in the AD subdivision, is a shared feature across a subset of ASD and schizophrenia models that exhibit ID-like memory defects. At the cellular level, three different ASD and schizophrenia KD models exhibited hyperexcitability of AD neurons, through different mechanisms. Furthermore, neuronal hyperexcitability was causally related to cognitive deficits in these KD mice because normalization of this physiological property rescued memory deficits in all three models. These observations suggest that a subset of different human disorders with ID may involve anterior thalamic dysfunction.
Our interest in understanding the role of PTCHD1 in the context of cognitive impairments led to the discovery that AD thalamus underlies memory phenotypes in a subset of different neuropsychiatric models. For two reasons, we examined the role of AD and neighboring AV in wild type mice. First, in the literature, the precise role of these two ATN subdivisions has not been reported, primarily due to the lack of precise manipulation strategies. This is important to help explain how dysfunction in these nuclei contribute to disease phenotypes. Second, in addition to AD, several ASD and schizophrenia risk genes are expressed in AV thalamus. Therefore, we wanted to know whether these two nuclei support the same or different cognitive processes. We found that the AD→RSC circuit is necessary for memory encoding, whereas the AV→RSC circuit regulates memory specificity. These findings indicate that neighboring ATN subdivisions differentially contribute to a cognitive task.
AD thalamus is specifically important for contextual encoding processes, as evidenced by loss of function phenotypes observed in contextual fear conditioning and inhibitory avoidance paradigms, but not in tone fear encoding. Further support for this role of AD comes from the fact that it is the only ATN subdivision that directly receives visual input (Jankowski et al., 2013). Regarding the AD→PreSub circuit, since we did not observe a significant contribution to our memory behavioral paradigm, it is likely that this circuit plays a bigger role in head direction coding (Winter et al., 2015). The function of AV thalamus in memory specificity is strengthened by the findings that AV but not AD receives prefrontal cortex (PFC) inputs (Figure 3D), and that PFC is important for generalization behavior (Xu and Sudhof, 2013). Interestingly, their study showed that the PFC→nucleus reuniens (RE) circuit is important for memory specificity, based on which we found that PFC neurons projecting to RE also send collaterals to AV but not AD thalamus (Figure S7C).
Given that AD and AV converge on the same cortical region, it is important to understand how these two excitatory inputs give rise to distinct behavioral phenotypes at the level of RSC neurons. One possibility is that distinct RSC ensembles receive input from AD or AV neurons, for which we have obtained some cellular-level evidence (Figures S7D–S7E). Another possibility is that AD and AV together control the level of activation of EC-projecting RSC (i.e., RSC→EC) neurons during encoding within a physiological range. Specifically, if the neural activity of RSC→EC neurons were below a minimal threshold, memory encoding would be impaired, whereas if their activity level exceeded an upper limit, memory encoding would be unaffected but there would be a decrease in specificity. Our data supports this second possibility because we found that AD but not AV provides the major excitatory drive to RSC→EC neurons, and AV provides important excitatory drive to VIP+ inhibitory neurons in RSC that are capable of regulating the overall activity of RSC→EC neurons.
Our work provides a better understanding of how anterior thalamus regulates cortico-entorhinal-hippocampal circuits during memory formation. Our studies also reveal an important link between anterior thalamic dysfunction and cognitive impairments in a subset of ASD and schizophrenia models, which may provide the foundation for developing therapeutic strategies capable of treating cognitive impairments in multiple disorders.
STAR METHODS
Lead contact
Further information and requests for reagents will be fulfilled by the lead contact Guoping Feng (fengg@mit.edu).
Materials availability
Plasmids and C1QL2-IRES-Cre mice are available from the corresponding authors on request.
Data and code availability
Data are available from the corresponding authors on reasonable request. This study did not generate any new code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
C57BL/6J wild type male mice were obtained from Jackson Laboratory. Experiments using CaMKII-Cre mice employed the T29–1 transgenic line (Stock No. 005359, Jackson Laboratory). Experiments using GAD2-Cre mice employed the GAD2-IRES-Cre knock-in line (Stock No. 028867, Jackson Laboratory). Experiments using PV-Cre mice employed the B6 PVCre knock-in line (Stock No. 017320, Jackson Laboratory). Experiments using SST-Cre mice employed the SST-IRES-Cre knock-in line (Stock No. 028864, Jackson Laboratory). Experiments using VIP-Cre mice employed the VIP-IRES-Cre knock-in line (Stock No. 031628, Jackson Laboratory). For AD neural activity labeling based on the c-fos promoter, we used the previously described c-fos-CreERT2 mouse line (Guenthner et al., 2013). These mice are also known as FosCreER or Fos-TRAP mice in which cFos-positive neurons can be labeled by the intraperitoneal injection of 4-hydroxytamoxifen (4-OHT) within a user-defined time-window. For our experiments, Fos-TRAP mice were crossed with the Cre-dependent tdTomato reporter mouse line Ai14, which were obtained from Jackson Laboratory (Stock No. 007908). All transgenic and knock-in mouse lines were maintained as hemizygotes. Mice had access to food and water ad libitum and were socially housed in numbers of two to five littermates until surgery. Following surgery, mice were single housed. For behavioral experiments, all mice were male and 3–5 months old. All experiments were conducted in accordance with U.S. National Institutes of Health (NIH) guidelines and the Massachusetts Institute of Technology Department of Comparative Medicine and Committee on Animal Care.
Generation of C1ql2-Cre mice.
C1ql2-IRES-Cre knock-in mice were generated using cloning- free CRISPR as previously described (Aida et al., 2015). Briefly, a C1ql2-IRES-Cre targeting vector was constructed by Gibson assembly (NEB E2621X) using IRES-Cre-pA cassette (from PL450-IRES-Cre-pA plasmid, a kind gift from Z. Josh Huang at Cold Spring Harbor Laboratory), PCR amplified 2 kb C1ql2 homology arms, and a pBluescript plasmid backbone. Synthetic crRNA and tracrRNA were purchased from IDT, Synthego, and Fasmac. Injection mixtures were prepared by mixing crRNA (CGCCCUCUAGGCCCCUAAUC for protospacer sequence, final concentration 1.22 μM) and tracrRNA (final concentration 1.22 μM) in nuclease-free water and Tris-HCl pH 7.39 (final concentration 10 mM). The mixture was heat denatured at 94°C for 5 min, followed by re-annealing at room temperature for 10 min. EnGen Cas9 NLS, S. pyogenes (New England Biolabs, final concentration 60 ng μl−1) was added and the mixture was incubated at 37°C for 15 min, then mixed with the C1ql2-IRES-Cre targeting vector (final concentration 5 ng μl−1) and RAD51 protein (Abcam ab63808, final concentration 10 ng μl−1). The injection mixture was kept on ice and briefly heated to 37°C prior to injections. Female mice (4–5 weeks old, C57BL/6NTac) were super-ovulated by intraperitoneal injection of PMS (5 IU per mouse, three days prior to microinjections) and hCG (5 IU per mouse, 47 hr after PMS injections) and then paired with males. Pregnant females were sacrificed by cervical dislocation at day 0.5 pcd, and zygotes were collected into 0.1% hyaluronidase/FHM (Sigma). Zygotes were washed in drops of FHM, and cumulus cells were removed. Zygotes were cultured in KSOM-AA for one hour and then used for microinjections. Pronuclear microinjections were performed using a Narishige micromanipulator, Nikon Eclipse TE2000-S microscope, and Eppendorf 5242 microinjector. Individual zygotes were injected with 1–2 pl of the injection mixture using an automatic injection mode set according to needle size and adjusted for a visible increase in pronuclear volume. Following injections, cells were cultured in KSOM-AA overnight, then embryos were surgically implanted into pseudopregnant CD-1 females (Charles River Laboratories, strain code 022) 24 hr post-injection, and allowed to develop normally until natural birth. Genomic DNA was purified from tail samples and PCR genotyped. Cre activity and specificity were tested by injection of AAV9-EF1α-DIO-eYFP into ATN and eYFP fluorescence localized to C1QL2+ AD neurons.
Marmosets.
Common marmoset (Callithrix jacchus) monkeys were used for fluorescent in situ hybridization (FISH) experiments. Marmosets had access to food and water ad libitum and were socially housed in numbers of two to three cage mates. Male marmosets ranging from 4–6 years old were used for all experiments. All experiments were conducted in accordance with U.S. National Institutes of Health (NIH) guidelines and the Massachusetts Institute of Technology Department of Comparative Medicine and Committee on Animal Care.
METHOD DETAILS
DropViz RNA-sequencing dataset
Single-cell suspensions were generated from adult male C57BL/6J mice (60–70 days old). Mouse thalamic excitatory (VGLUT2+) neuron single-cell RNA-sequencing data is based on 89,027 cells (n = 6 mice). Detailed information regarding cell suspensions, cell recovery rates, cell type and subtype acquisition, Drop-seq library preparation and sequencing, and quantitative analyses has been previously described (Saunders et al., 2018).
Fluorescent in situ hybridization.
Experiments used C57BL/6J mouse brain samples, virus- injected mouse brain samples, or common marmoset brain samples. These mouse and marmoset brain samples were extracted, embedded in OCT compound (Tissue-Tek), and flash frozen in liquid nitrogen. A normal human donor thalamus brain sample containing ATN was obtained from Cureline Inc. This human sample was also embedded in OCT compound and flash frozen in liquid nitrogen. Coronal sections (16 μm thickness) were prepared on a cryostat (Leica) and stored at −80°C. FISH mRNA staining was performed using the ACD RNAScope multiplex fluorescent protocol for fresh frozen tissue. Briefly, charged slides with mouse, marmoset, or human tissue sections were fixed in pre-chilled paraformaldehyde (PFA) for 30 min, followed by a series of dehydration steps using 50%, 70%, and 100% ethanol. Sections were then permeabilized with ACD protease IV for 30 min, followed by probe hybridization for 2 hr at 40°C. Fluorescent labeling of up to 3 probes per section was performed using four steps of Amp 1-FL to Amp 4-FL. Sections were stained with DAPI and stored at 4°C. Mouse ACD probes for Cntnap2 (Cat. No. 449381), Atp1a3 (Cat. No. 432511), Gria3 (Cat. No. 426251), Mtor (Cat. No. 451651), Ywhag (Cat. No. 812981), Herc1 (Cat. No. 871341), Cacna1g (Cat. No. 459761), C1ql2 (Cat. No. 480871), PV (Cat. No. 421931), Col25a1 (Cat. No. 538511), rabies virus (Cat. No. 456781), Ptchd1 (Cat. No. 489651), Slc17a6 (Cat. No. 319171), Kcnj12 (Cat. No. 525171), Kcnq2 (Cat. No. 444251), Kcna1 (Cat. No. 481921), Cacna1a (Cat. No. 493141), and Cacna1b (Cat. No. 468811) were used. Marmoset ACD probes for C1ql2 (Cat. No. 525821) and Col25a1 (Cat. No. 557651) were used. Human ACD probe for C1ql2 (Cat. No. 478011) was used. Stained sections were imaged with a 20X magnification objective on a Leica confocal microscope. Images were processed using ImageJ, and quantifications were performed manually from 3–5 sections per animal. All counting experiments were conducted blind to experimental group.
Viral constructs.
The following viruses were acquired from Addgene: AAVretro-Cre (specifically AAVretro-hSyn-Cre, catalog #105553-AAVrg, 7 × 1012 GC ml−1 titer), AAV9-EF1 α-DIO-ChR2-eYFP (catalog #20298-AAV9, 7 × 1012 GC ml−1 titer), AAV9-CaMKIIα-ChR2-eYFP (catalog #26969-AAV9, 1 × 1013 GC ml−1 titer), AAV9-CaMKIIα-ChR2-mCherry (catalog #26975-AAV9, 7 × 1012 GC ml−1 titer), AAV8-hSyn-DIO-hM4Di-mCherry (catalog #44362-AAV8, 1 × 1013 GC ml−1 titer), AAV8-hSyn-DIO-mCherry (catalog #50459-AAV8, 7 × 1012 GC ml−1 titer), AAV1-hSyn-Cre (anterograde virus, catalog #105553-AAV1, 1 × 1013 GC ml−1 titer), AAV9- hSyn-DIO-hM3Dq-mCherry (catalog #44361-AAV9, 1 × 1013 GC ml−1 titer), AAV8-hSyn- mCherry (catalog #114472-AAV8, 1 × 1013 GC ml−1 titer), AAV8-hSyn-DIO-hM4Di-mCitrine (catalog #50455-AAV8, 1 × 1013 GC ml−1 titer), and AAV8-CaMKIIα-hM4Di-mCherry (catalog #50477-AAV8, 2 × 1012 GC ml−1 titer). The following Cre-Off (DO) AAV constructs were acquired from Addgene: AAV-EF1α-DO-NpHR3.0-eYFP (plasmid #37087), AAV-EF1α-DO-eGFP (plasmid #37085), and AAV-EF1α-DO-ChETA-tdTomato (plasmid #37756). The AAV-EF1α-DIO-C1V1-eYFP construct (plasmid #35497) was also acquired from Addgene. All these plasmids were serotyped with AAV5 coat proteins and packaged by the Viral Core at Boston Children’s Hospital (2 × 1013 GC ml−1 viral titers). The AAV-CaMKIIα-mCherry construct (plasmid #114469) was obtained from Addgene, serotyped with AAV8 coat proteins, and packaged by the Viral Core at Boston Children’s Hospital (4 × 10 GC ml−1 viral titer). The AAV-cFos-CreERT2 construct was a gift from Karl Deisseroth, which was serotyped with AAV9 coat proteins and packaged by the Viral Core at Boston Children’s Hospital (5 0 × 1012 GC ml−1 viral titer). The AAV9-EF1α-DIO-eYFP (1.2 × 1013 GC ml−1 viral titer) and AAV9-EF1α-DIO-eArch3.0-eYFP (1.6 × 1013 GC ml−1 viral titer) viruses were acquired from the University of North Carolina (UNC) at Chapel Hill Vector Core.
Cholera toxin subunit B.
To characterize neuronal populations in AD, AV, and RSC based on their projection targets, we used cholera toxin subunit B (CTB) conjugated to Alexa-488, Alexa-555, or Alexa-647 diluted in phosphate buffered saline (PBS) solution at a final concentration of 1% wt vol−1. Diluted CTB was aliquoted and stored at −20°C. For mouse circuit tracing experiments, 80–300 nl CTB was unilaterally injected into target sites. Six days after injections, mice were perfused for histology followed by coronal/sagittal sectioning (50 μm thickness) using a vibratome (Leica). For circuit-specific neuronal activity (i.e., cFos) experiments using mice, CTB only-, CTB and AD hM4Di-mCh virus-, or CTB and AV NpHR-eYFP virus-injected animals went through the contextual fear conditioning (CFC) behavior protocol 30 days after injections followed by timed perfusions 60 min after behavior. For AD and AV manipulation mice, details are provided in the rabies virus sub-heading. CTB sections were imaged with a 20X magnification objective on a Leica confocal microscope. Images were processed using ImageJ, and quantifications were performed manually from 3–5 sections per animal. All counting experiments were conducted blind to experimental group.
Rabies virus.
To label ATN inputs to PreSub and RSC, 150 nl first generation rabies virus (RV) expressing GFP was injected into each of these downstream targets. Five days after injections, these mice were used for FISH staining as described above. For mouse ex vivo electrophysiological recordings from AD vs. AV thalamic neurons, RV-GFP was injected into PreSub followed by recordings five days later. For recordings, details are provided in the “Ex vivo electrophysiology” sub-heading. To identify inputs to Cre+ neurons, we used a monosynaptic retrograde tracing approach via a Cre-dependent helper virus combined with RV technology. The first component was an AAV vector that allowed simultaneous expression of three genes: TVA, eGFP, and RV glycoprotein (G). Briefly, this vector was constructed by deleting the sequence between the inverse terminal repeats of pAAV-MCS (Stratagene), and replacing it with a cassette containing the following: human synapsin-1 promoter (Syn, Genbank NG_008437); the Kozak sequence; a FLEX cassette containing the transmembrane isoform of TVA (lacking a start codon), eGFP, and G separated by the highly efficient porcine teschovirus self-cleaving 2A element; the woodchuck post-transcriptional regulatory element (WPRE) and a bovine growth hormone polyadenylation site. This vector was termed pAAV-synP-FLEX-sTpEpB (i.e., the helper virus) and serotyped with AAVrh8 coat proteins. The second component was a deletion-mutant RV produced by replacing the eGFP gene in cSPBN-4GFP with the mCherry gene (i.e., the RVΔG-mCherry virus, also known as the Rabies-mCh virus), which was packaged with the ASLV-A envelope protein. For tracing experiments using different Cre mouse lines, 100 nl of the Cre-dependent helper virus was unilaterally injected into PreSub or RSC. One week later, 100 nl of RVΔG-mCherry virus was unilaterally injected into the same PreSub or RSC. Six days after the second viral injection, mice were perfused for histology and imaging. To map brain-wide inputs to AD vs. AV, 150 nl AAVretro-Cre virus was unilaterally injected into PreSub (for AD) or RSC (for AV) combined with 100 nl Cre-dependent helper virus injections into ATN. Three weeks later, 100 nl of RVΔG-mCherry virus was unilaterally injected targeting AD (PreSub injected mice) or AV (RSC injected mice). One week after the second viral injection, mice were perfused for histology and imaging. To identify ATN neurons that project to EC-projecting RSC neurons, 250 nl AAVretro-Cre virus was unilaterally injected into EC combined with 100 nl Cre-dependent helper virus injections into RSC. Three weeks later, 100 nl of RVΔG-mCherry virus was unilaterally injected into the same RSC. One week after the second viral injection, mice were perfused for histology and imaging. RV+ coronal sections (50 μm) were imaged with a 10X or 20X magnification objective on an Olympus epifluorescent microscope. Images were processed using ImageJ, and quantifications were performed manually from 3–5 sections per animal.
For brain-wide inputs to AD vs. AV, tiled images were taken for entire coronal sections (every 4th section from each brain sample), which were needed for manual atlas alignment using an electronic version of the Franklin and Paxinos ‘Mouse Brain in Stereotaxic Coordinates’ (3rd edition). Quantifications for these brain-wide input mapping experiments were performed manually. For each RV experiment, starter cell counts across mice were normalized, which has also been indicated in the respective figure legends. All counting experiments were conducted blind to experimental group. A third type of RV, referred to as the second generation RV, has been used for ex vivo electrophysiology and behavioral experiments. Specifically, this RV expresses Cre recombinase (i.e., RVdGL-Cre) in upstream neurons. For cell body electrophysiology, RVdGL-Cre was injected into PreSub combined with a Cre-dependent ChR2-eYFP virus in ATN, which allowed labeling of only AD neurons within ATN with high specificity. This strategy to label AD neurons was employed for CFC behavioral manipulations with a Cre-dependent hM4Di-mCherry virus, AD circuit electrophysiology with a Cre-dependent ChR2-eYFP virus, AD→RSC circuit manipulations during behavior with either a Cre-dependent ChR2-eYFP virus or a Cre-dependent eArch-eYFP virus, AD manipulations during behavior with a Cre-dependent hM4Di-mCherry virus for cFos analyses, AD manipulations during behavior with a Cre-dependent hM4Di virus for cFos analyses in EC-projecting RSC neurons that have been labeled with CTB, AD-specific gene knockdown (KD) experiments, AD circuit electrophysiology with a Cre-dependent ChR2-eYFP virus in KD mice, rescue experiments in KD mice, AD behavioral manipulations with a Cre-dependent hM4Di virus for in vivo local field potential (LFP) recordings, and simultaneous AD and AV labeling experiments. The RVdGL-Cre virus injected into PreSub combined with a Cre-Off (DO) NpHR-eYFP virus injected in ATN allowed labeling of only AV neurons within ATN with high specificity (i.e., because AD but not AV projects to PreSub, RVdGL-Cre in AD neurons turns off viral expression). This strategy to label AV neurons was employed for behavioral manipulations, AV manipulations during behavior for cFos analyses in RSC neurons, AV→RSC inhibition with PV or VIP activation in RSC during behavior, AV manipulations during behavior for cFos analyses in EC-projecting RSC neurons that have been labeled with CTB, and simultaneous AD and AV labeling experiments.
In vivo genome editing.
In vivo knockdown experiments targeting AD thalamus or hippocampal CA1 employed an AAV CRISPR/Cas9 approach. Single guide RNA (sgRNA) candidates targeting Ptchd1, Ywhag, Gria3, Herc1, Atp1a3, Mtor, and Cntnap2 with high specificity and high efficiency were computationally identified from sgRNA libraries for genome-wide CRISPR knockout screening (Doench et al., 2016). Three U6-sgRNA(FE) gene fragments with the F+E tracrRNA backbone were synthesized by Integrated DNA Technologies (sequences are provided below, spacer sequences are capitalized). These fragments were cloned into the pX552-mCherry plasmid (EGFP in pX552 plasmid was replaced with mCherry, pX552 was obtained from Addgene, plasmid #60958) by Gibson assembly (NEB E2621X) to construct pX552–3xsgRNA(FE)-mCherry. We used a previously reported sgRNA plasmid targeting Cacna1g (Li et al., 2020). These constructs were functionally validated in Neuro2A cells. The AAV vectors were serotyped with AAV9 coat proteins and packaged in-house or by the Viral Core at Boston Children’s Hospital (8 × 1012 genome copy (GC) ml−1 viral titers for Ptchd1, Cacna1g).
In-house AAV production followed a previously described method (Challis et al., 2019). Briefly, sgRNA plasmids, pAdDeltaF6 (Addgene, plasmid #112867), and pAAV2/9 (Addgene, plasmid #112865) were co-transfected into HEK293T cells using polyethylenimine (Cat. No. 23966–1, Polysciences). Cells were cultured in Dulbecco’s modified essential medium (DMEM, Invitrogen) containing 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco) at 37°C with 5% CO2. Cells were harvested 72 hr post transfection by 4,000×g centrifugation at 4°C for 10 min. Virus in media was precipitated by 8% PEG8000 (Sigma). Cell pellets and virus precipitated from media were re-suspended in digestion buffer containing 500 mM NaCl, 40 mM Tris base, and 10mM MgCl2. Benzonas nuclease (100U, Sigma) was added in the digestion buffer and incubated at 37°C water bath for 1 hr. Next, we performed centrifugation at 2000×g for 15 min, and the supernatant was used on a discontinuous gradient of 15%, 25%, 40%, and 60% iodixanol in a 36.2 ml ultracentrifuge tube (Optiseal Seal, Cat. No. 362183, Beckman). Ultracentrifugation was performed at 350,000×g, 18°C for 2.5 hr. 5 ml fractions in 40% layer and 40%−60% interface was collected. These fractions were desalted using a 100 kDa cutoff ultrafiltration tube (15 ml, Millipore). Buffer was exchanged 4 times with 1x PBS with 0.001% Pluronic F-68. AAV titers were determined by real-time quantitative PCR (qPCR) using the primers of mCherry. Forward primer: 5’ GAGTTCATCTACAAGGTGAAGCTGCGC 3’, reverse primer: 5’ CTTGTAGGTGGTCTTGACCTCAGCGTC 3’ (1–2.5 × 1012 GC ml−1 for Ywhag, Gria3, Herc1, Atp1a3, Mtor, Cntnap2).
For AD targeting, these sgRNA AAVs were combined with a Cre-dependent SpCas9 AAV, which was developed by Jie Xu and Dong Kong. The AAV-DIO-SpCas9 plasmid was serotyped with AAV9 coat proteins and packaged by the Viral Core at Boston Children’s Hospital (2 × 1013 GC ml−1 viral titer). For these in vivo experiments, RVdGL-Cre was injected into PreSub and a 1:1 mix of AAV9-sgRNA-mCherry:AAV9-DIO-SpCas9 was injected into ATN, which allowed for AD-specific knockdown of target genes. For CA1 targeting, sgRNA AAVs were combined 1:1 with a constitutive AAV9-CMV-SpCas9 virus (4 × 1012 GC ml−1 viral titer, Vector Biolabs). FISH was used for in vivo knockdown validation.
U6-sgPtchd1–1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgcTACCAGGTCGAAGAGAGCGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgPtchd1–2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccggGTGCTTGGAGCGGTTGACCgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgPtchd1–3(FE) sequence
agcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgTATAATGGGCACCAACTCGGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
U6-sgYwhag-1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgGATGGTGGACCGCGAGCAACgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgYwhag-2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCAGGAGGTTCCGTTCCTCATgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgYwhag-3(FE) sequence
agcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatga ttccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgGAGCAGAAGACGTCTGCGGAgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
U6-sgGria3–1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgACGTGGTAGTTCAAATGGAAgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgGria3–2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCCTTTAAGGCTGGGCGCATCgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgGria3–3(FE) sequence
agcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCAAAGCCATTTATGAGCCTGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
U6-sgHerc1–1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCATCTGCTGATCGGAGTCAGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgHerc1–2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggAaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgACTATGGGAAACTAGGACATgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgHerc1–3(FE) sequence
AgcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCCGAGATAGAATGAACAGTGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
U6-sgAtp1a3–1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgAGGAGATCCTAGCCCGGGATgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgAtp1a3–2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggAaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgGCCGGTGATGATCACGACTGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgAtp1a3–3(FE) sequence
agcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCGGGGTGTGGTGGTAGCCACgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
U6-sgMtor-1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgAAGTGTCCCCTGCCATCGCCgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgMtor-2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggAaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgCTGCGTGGTGAGAATCAGACgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgMtor-3(FE) sequence
agcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgGACGTTGATGCAGAAGGTAGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
U6-sgCntnap2–1(FE) sequence
agtggccaactccatcactaggggttcctgcggccgcacgcgtaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgGAGCTGCTGAAGGACACATGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactatagg
U6-sgCntnap2–2(FE) sequence
cccagtcacgacgttgtaaaacgacggccagtgagcgcgcgtaatacgactcactataggAaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgATCAGTGCCATTGCAACCCAgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttagcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgc
U6-sgCntnap2–3(FE) sequence
agcggataacaatttcacacaggaaacagctatgaccatgattacgccaagcgcgcaaggtcgggcaggaagagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgAGGTCACATCGGGCTGCGTGgtttaagagctatgctggaaacagcatagcaagtttaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttctagactgcagagggccctgcgtatgagtgcaagtgggttttaggaccaggatgaggcggggtg
Mouse surgery and optic fiber implants.
Animals were anesthetized with isoflurane for stereotaxic injections, and were given 1 mg kg−1 meloxicam as analgesic prior to incisions. Injections were targeted to PreSub (−3.8 mm AP, +/− 1.75 mm ML, −1.7 mm DV), RSC (−2.46 mm AP, +/− 0.25 mm ML, −0.9 mm DV), ATN (−0.55 mm AP, +/− 0.9 mm ML, −3.15 mm DV), AD (−0.7 mm AP, +/− 0.75 mm ML, −2.75 mm DV), AV (−0.58 mm AP, +/− 1.1 mm ML, −3.25 mm DV), EC (−4.75 mm AP, +/− 3.35 mm ML, −3 mm DV), hippocampal CA1 (−2.1 mm AP, +/− 1.5 mm ML, −1.4 mm DV), RE (−0.58 mm AP, +/− 0.25 mm ML, −4.25 mm DV), and PFC (+1.94 mm AP, +/− 0.4 mm ML, −2.9 mm DV). Standard injection volumes were 200 nl for PreSub and RSC, 300 nl for ATN, 125 nl for AD and AV, 300 nl for EC, 400 nl for CA1, 250 nl for RE, and 300 nl for PFC. Except for certain retrograde tracing experiments (listed in the rabies virus sub-heading), all other experiments employed these standard injection volumes. CTB/viruses were injected at 70 nl min−1 using a glass micropipette attached to a 10 ml Hamilton microsyringe. The needle was lowered to the target site and remained for 5 min before beginning the injection. After the injection, the needle stayed for 10 min before it was withdrawn. For behavioral manipulation experiments using optogenetics, single mono-fiber implants (200 μm core diameter, Newdoon) were lowered either above injection sites or terminals bilaterally (AV, −0.58 mm AP, +/− 1.1 mm ML, −3.1 mm DV; RSC, −2.46 mm AP, +/− 0.25 mm ML, −0.7 mm DV; PreSub (−3.8 mm AP, +/− 1.75 mm ML, −1.85 mm DV); EC, −4.65 mm AP, +/− 3.35 mm ML, −2.25 mm DV). The implant was secured to the skull with two jewelry screws, adhesive cement (C&B Metabond), and dental cement. Mice were given 1–2 mg kg−1 sustained-release buprenorphine as analgesic after surgeries and allowed to recover for at least 2 weeks before behavioral experiments. All injection sites were verified histologically. As criteria, we only included mice with virus expression limited to the targeted regions.
Immunohistochemistry.
Mice were dispatched using an overdose of isoflurane and transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA). Brains were extracted and incubated in 4% PFA at room temperature overnight. Brains were transferred to PBS and 50 μm coronal slices were prepared using a vibratome. For immunostaining, each slice was placed in PBS + 0.2% Triton X-100 (PBS-T), with 5% normal goat serum for 1 hr and then incubated with primary antibody at 4°C for 24 hr. Slices then underwent three wash steps for 10 min each in PBS-T, followed by a 2 hr incubation with secondary antibody. After three more wash steps of 10 min each in PBS-T, slices were mounted on microscope slides. Antibodies used for staining were as follows: rabbit anti-C1QL2 (1:500, Thermo Fisher) and anti-rabbit Alexa-488 (1:500), chicken anti-GFP (1:1000, Life Technologies) and anti-chicken Alexa-488 (1:1000), rabbit anti-RFP (1:1000, Rockland) and anti-rabbit Alexa-555 (1:500), rabbit anti-cFos (1:500, Cell Signaling Technology) and anti-rabbit Alexa-488 or Alexa-555 (1:300), and nuclei were stained with DAPI (1:3000, Sigma). To visualize rabies virus starter cells, GFP antibody staining was performed. To visualize ChR2-expressing terminals in ATN, both GFP and RFP antibody staining was performed. To visualize AD hM4Di-mCherry terminals in RSC, RFP antibody staining was performed. To visualize ChR2-eYFP terminals in ATN, GFP antibody staining was performed. To visualize AD and AV cell body labeling in ATN, both GFP and RFP antibody staining was performed. All analyses were performed blind to the experimental conditions.
Fos-TRAP activity-dependent labeling.
For activity-dependent labeling experiments, as mentioned above FosTRAP mice crossed to Ai14 reporter mice were employed. 4-hydroxytamoxifen (4-OHT, Sigma-Aldrich) was dissolved in 100% ethanol solution by shaking at 37°C for 20–30 min. One-part castor oil to four parts sunflower oil was combined to prepare the oil mixture that would eventually be injected intraperitoneally (IP) into the mouse. Dissolved 4-OHT was combined with the oil mixture, followed by ethanol evaporation using a centrifuge. The final concentration of 4-OHT dissolved in the oil mixture was 10 mg ml−1. For each mouse, optimal activity-dependent labeling was achieved using a target concentration of 30–40 mg kg−1. One hour prior to the behavioral epoch of interest, mice were injected with 4-OHT. Following behavior experiments, mice were returned to their home cages and remained undisturbed for at least 72 hours, after which they were perfused for histological analyses.
Chemogenetic and optogenetic experiments.
For chemogenetic (i.e., hM4Di or hM3Dq) neuronal activity manipulation experiments, we used the second-generation agonist known as compound 21 (C21). This agonist was purchased in a water-soluble dihydrochloride form (Hello Bio). For each mouse, optimal chemogenetic activity was achieved using a target concentration of 2 mg kg−1 (injected IP), 45 min before the behavioral epoch of interest. The exception to this target concentration was for low (0.6 mg kg−1) vs. regular (2 mg kg−1) dose experiments in PTCHD1 KD mice, and low dose experiments in YWHAG and HERC1 KD mice. For optogenetic neuronal activity manipulation experiments, ChR2 was activated at 20 Hz (15 ms pulse width) with a 473 nm laser (10–15 mW, blue light), eArch and NpHR was activated with a 570 nm laser (10 mW, constant green light), C1V1 was activated at 20 Hz (15 ms pulse width) with a 570 nm laser (10 mW, green light), and ChETA was activated at 20 Hz (15 ms pulse width) with a 410 nm laser (10 mW, blue light).
Cell counting.
For details regarding quantification of RV tracing experiments, please refer to the rabies virus sub-heading. Unless specified, brain sections were imaged with a 20X magnification objective on a Leica confocal microscope. Images were processed using ImageJ, and quantifications were performed manually from 3–5 sections per animal. All counting experiments were conducted blind to experimental group. Researcher 1 trained the animals, prepared slices, and randomized images, while Researcher 2 performed cell counting. Percentage of PreSub- projecting (CTB555) AD neurons that send collaterals to RSC (CTB488) was calculated as ((CTB488+ CTB555+) / (Total CTB555+)) × 100. The percentage of retrogradely-labeled (by RV) AD neurons that express the marker C1ql2 was calculated as ((RV+ C1ql2+) / (RV+)) × 100. To quantify the number of neurons in each brain region projecting to AD or AV neurons, RV-mCherry+ neurons in each upstream target structure were counted from all coronal slices containing the structure per mouse. To quantify the number of activated (cFos+) neurons in Fos-TRAP/Ai14 mice, tdTomato+ neurons in AD thalamus were manually counted from home cage, CFC training, and immediate shock groups. To quantify neuronal activity in RSC/CA1, cFos+ neurons were manually counted from specified behavioral groups. These active neuron cell counts were normalized to the number of DAPI+ cells in the field of view. Percentage of retrogradely-labeled (CTB+) neurons that are activated (cFos+) was calculated as ((CTB+ cFos+) / (Total CTB555+)) × 100. Percentage of PV-Cre or VIP-Cre (eYFP+) neurons that are activated (cFos+) was calculated as ((eYFP+ cFos+) / (Total eYFP+)) × 100. To quantify the number of neurons in AV thalamus that project to PV+, SST+, or VIP+ inhibitory neurons in RSC, RV-mCherry+ neurons in AV were manually counted. Percentage of Slc17a6+ neurons in AD that express the marker C1ql2 was calculated as ((Slc17a6+ C1ql2+) / (Total Slc17a6+)) × 100. Percentage of RSC neurons that receive both AD input (eYFP+) and AV input (cFos+) was calculated as ((eYFP+ cFos+) / (eYFP+)) × 100. Similarly, for risk gene KD experiments, fluorescence intensity was measured in AD or CA1 using ImageJ. These values were averaged and compared between mCh control and KD mice. Data were analyzed using Microsoft Excel with the Statplus plug-in or Prism 6 software.
Ex vivo electrophysiology
Slice preparation.
All ex vivo experiments were conducted blind to experimental group. Researcher 1 trained the animals and administered drug, while Researcher 2 dispatched the animals and conducted physiological recordings. Mice (8–12 weeks old) were anesthetized with isoflurane, decapitated, and brains were quickly removed. For AMPA/NMDA ratio recordings, coronal slices (300 μm thick) were prepared in an oxygenated cutting solution at 4°C by using a vibratome (Leica). The cutting solution contained (in mM): 30 NaCl, 4.5 KCl, 1.2 NaH2PO4, 194 sucrose, 26 NaHCO3, 10 D-glucose, 0.2 CaCl2, 8 MgSO4, and saturated with 95% O2 − 5% CO2 (pH 7.3, osmolarity of 350 mOsm). Slices were recovered in ACSF at 33°C (+/− 0.5°C) for 15 min and then kept at room temperature for 1 hr before recordings. The ACSF contained (in mM): 119 NaCl, 2.3 KCl, 2.5 CaCl2, 1.3 MgSO4, 26.2 NaHCO3, 1 NaH2PO4, 11 D-glucose, and saturated with 95% O2 - 5% CO2 (pH 7.3, osmolarity of 300 mOsm). For all other recordings, the brain was quickly removed and placed in ice-cold ACSF consisting of (in mM): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 25 NaHCO3, and 10 D-glucose. Slices were stored for 30 min at 33°C (+/− 0.5°C) and then kept at room temperature until recording.
Electrophysiological recordings.
Whole cell recordings in current clamp- or voltage clamp-mode were performed using an IR-DIC microscope (Olympus) with a water immersion 40X objective (NA 0.8), equipped with four automatic manipulators (Luigs and Neumann) and a CCD camera (Hamamatsu Co). For all recordings, borosilicate glass pipettes were fabricated (Sutter Instrument) with resistances of 3.5 to 5 MΩ. The AMPA/NMDA ratio measurements were performed by adding 100 μM picrotoxin (Tocris) in the extracellular solution, and voltage clamp recordings were performed using the following intracellular solution (in mM): 120 cesium methansulfonate, 10 HEPES, 1.1 EGTA, 5 NaCl, 1.1 TEA-Cl, 4 Mg-ATP, 0.3 Na-GTP, 4 QX314, and 0.5% biocytin. The osmolarity of this intracellular solution was 298 mOsm and the pH was 7.2. AMPA/NMDA ratio is defined as the ratio of the EPSC peak at −70 mV to the EPSC magnitude at +40 mV (50 ms following stimulation). To measure calcium currents (CaV2.1, CaV2.2), patch pipettes were filled with a solution containing the following (in mM): 103 CsCl, 12 CsOH, 12 methanesulfonic acid, 4 NaCl, 5 TEA-Cl, 10 HEPES, 0.5 EGTA, 10 phosphocreatine, 5 lidocaine N-ethyl chloride, 4 ATP magnesium salt, and 0.3 GTP sodium salt. pH was adjusted to 7.2–7.4 with KOH, and osmolarity was adjusted to 298–300 mOsm with K2SO4. Neurons were held at −80 mV and stepped from −60 mV to +20 mV in 10 mV increments, in the presence of TTX (1 μM), picrotoxin (100 μM), 4AP (1 mM), tetraethylammonium chloride (10 mM), and cesium chloride (2 mM). Calcium currents were recorded before and after application of ω-Conotoxin GVIA (200 nM), and further addition of ω- Agatoxin IVA (100 nM). CaV2.1 currents were the component blocked by ω-Agatoxin IVA, while CaV2.2 currents were the component blocked by ω-Conotoxin GVIA. For other recordings, pipettes were filled with the following intracellular solution (in mM): 110 K-gluconate, 40 KCl, 10 HEPES, 3 ATP, 0.5 GTP, 0.2 EGTA, and 0.5% biocytin. The osmolarity of this intracellular solution was 290 mOsm and the pH was 7.25. To measure potassium current (Kir2.2), neurons were held at −60 mV and stepped to −140 mV in 10 mV increments, in the presence of TTX (1 μM), before and after Ba2+ (10 μM) application. Kir2.2 currents were the component blocked by Ba2+. For mEPSC recordings, neurons were clamped at −70 mV in the presence of TTX (1 μM) and picrotoxin (100 μM). Synaptic currents were analyzed with the Mini Analysis Program (Synaptosoft). A series of 500 ms suprathreshold currents of 50–300 pA were used to quantify the excitability with holding at −55 mV. Membrane time constant (tau) was measured with a single exponential fit of the voltage deflection produced by a small hyperpolarizing current injection from the holding potential (−70 mV). Input resistance (Rin) was calculated as the slope of linear fits of current-voltage plots generated from a series of increasing current injection steps. Shape parameters were measured from the first action potential with 200 ms current injection (from the holding potential of −70 mV). Ih-induced sag currents were evoked by brief injections of hyperpolarizing currents in current clamp mode. To compare sag amplitudes between different groups, amplitudes of the current injections were adjusted in each cell to result in the same peak hyperpolarization, and the sag amplitude was determined as the repolarization from the peak to a steady state, during the entire length of current injection. Recordings were amplified using up to two dual channel amplifiers (Molecular Devices), filtered at 2 kHz, digitized (20 kHz), and acquired through an ADC/DAC data acquisition unit (Instrutech) using custom software running on Igor Pro (Wavemetrics). Access resistance (RA) was monitored throughout the duration of the experiment and data acquisition was suspended whenever RA was beyond 20 MΩ. For recordings after CFC training, anesthesia and slice preparation was initiated 45 min after the behavioral epoch. For recordings related to the excitability rescue in KD mice, C21 was injected IP 1 hr prior to anesthesia and slice preparation.
Optogenetic stimulation during recordings.
Optogenetic stimulation was achieved through Polygon400 (Mightex) with built-in LED sources (470 nm or 590 nm). Light power on the sample was 20 mW/mm2. To test ChR2 expression, slices were stimulated with 5 Hz blue light pulses. To test NpHR function, continuous green light was delivered to the slices. To test synaptic connections, slices were stimulated with a single light pulse of 1 s, repeated 10 times every 5 s, and the average response was computed. The monosynaptic glutamatergic nature of a connection was confirmed by sequential bath application of 1 μM TTX (Tocris), 100 μM 4AP (Tocris), and 10 μM CNQX (Tocris). Paired-pulse ratio refers to the ratio of the peak of the second EPSC to the peak of the first EPSC using a 50 ms interstimulus interval.
Post-hoc immunohistochemistry.
Recorded cells were filled with biocytin and subsequently recovered for brain region and/or cell type verification. Slices were first incubated with 4% PFA for 16 hr at 4°C. After washing with 0.5% Triton X-100 in PBS, slices were incubated in 5% normal goat serum for 2 hr. Following serum, slices were incubated in streptavidin CF555 (1:200, Biotium) for 2 hr at room temperature. Before mounting, slices were incubated with DAPI (1:3000) for 30 min.
In vivo LFP recordings
Surgical procedure.
C57BL/6J male mice (25–35 g, 10–16 weeks of age, Jackson Laboratory) were used for LFP recordings using chronically implanted electrodes. Mice were group housed before implantation surgeries but housed individually after in order to minimize damage to the implants. Animals were initially anesthetized with 5% isoflurane and maintained under anesthesia with 1–2% isoflurane during surgery. Implantable LFP electrodes made by teflon-coated tungsten microwires (50 μm, A-M Systems) were targeted to AD (−0.7 mm AP, +/− 0.75 mm ML, −2.75 mm DV), RSC (−2 mm AP, +/− 0.25 mm ML, −1.1 mm DV), and PreSub (−3.8 mm AP, +/− 1.75 mm ML, −1.7 mm DV). LFP electrodes were coated with DiI555 (Thermo Fisher Scientific) prior to implantation, which provided a fluorescent track for post-hoc electrode tip verification in brain sections. The reference and ground screws with wire lead (Pinnacle Technology) were targeted to the occipital skull. All electrodes were secured with dental cement and connected to a headmount (Pinnacle Technology) in combination with EMG leads for detecting sleep-wake status during LFP recordings. Animals recovered for at least 10 days post-surgery before LFP recordings during behavior. For pre vs. post CFC training LFP recordings, mice that were previously being recorded in their home cages were carefully unplugged, followed by behavioral procedures. Immediately after behavior (~5 min later), mice were plugged back into the LFP recording system for data collection. For circuit recordings, electrodes were targeted to AD and RSC or AD and PreSub on one hemisphere. As criteria, we only included mice with DiI555 electrode tip staining limited to the targeted regions.
Data acquisition and processing.
LFP signals were amplified, digitized continuously at 1 kHz using a tethered recording system with a differential amplifier (Pinnacle Technology) in awake, freely moving mice, and acquired (Pinnacle Sirenia acquisition software) for offline analysis using MATLAB (MathWorks). Spectral power was calculated in 0.5 Hz bins (fast Fourier transform with Hamming windows) with artifact-free LFP signals based on the following frequency bands: delta (1–4 Hz), theta (6–10 Hz), beta (12–30 Hz), and gamma (30–100 Hz). The coherence between two signals x(t) and y(t) were calculated as a function of the power spectral density of x and y (Pxx and Pyy), and the cross power spectral density of x and y (Pxy) with values between 0 and 1 for verifying x and y correspondence at each frequency. Inter-regional (AD→RSC) cross-frequency phase-amplitude coupling was calculated as previously described (Tort et al., 2010). The modulation index (MI) is a measure of the magnitude with which the phase of low-frequency rhythms (1–12 Hz) modulates the amplitude of high-frequency rhythms (20–100 Hz). MI was evaluated in 1 Hz frequency bins. Instantaneous phase and amplitude time series data were calculated by Hilbert transformation of band-pass-filtered LFP signals (zero phase filtering with a finite impulse response (FIR) filter of order 60, 3- and 2-Hz bandwidths for phase and amplitude frequencies, respectively).
Behavior assays
Experiments were conducted during the light cycle (7 am to 7 pm). Mice were randomly assigned to experimental groups for specific behavioral assays immediately after surgery. Mice were habituated to investigator handling for 1–2 minutes on three consecutive days. Handling took place in the holding room where the mice were housed. Prior to each handling session, mice were transported by wheeled cart to and from the vicinity of the behavior rooms to habituate them to the journey. All behavior experiments were analyzed blind to experimental group. Unpaired student’s t-tests were used for independent group comparisons, with Welch’s correction when group variances were significantly different, or ANOVA followed by Bonferroni post-hoc tests were used. Given behavioral variability, assays were performed using a minimum of 6–10 mice per group to ensure adequate power for any observed differences. Following behavioral protocols, brain sections were prepared to confirm efficient viral labeling in target areas. Animals lacking adequate labeling were excluded prior to behavior quantification.
Open field exploration.
Spontaneous motor activity was measured in an open field arena (40 × 40 × 30 cm) for 20 min. Mice were transferred to the testing room and acclimated for 30 min before the test session. During the testing period, lighting in the room was turned off. The apparatus was cleaned with quatricide before and between runs. Total movement (distance traveled) in the arena was quantified using an automated infrared (IR) detection system (Omnitech Digiscan, AccuScan Instruments). To test the effect of different optogenetic manipulations on locomotion, mice were plugged into the laser source and light was turned on once the animals were placed into the arena. Recordings were performed for 10 min. Raw data were extracted and analyzed using Microsoft Excel.
Rotarod motor coordination.
Controlled motor coordination was measured in a rotarod apparatus (Med Associates). Mice were transferred to the testing room and acclimated for 15 min before the test session. Mice were placed on the rod, which accelerated from 4–40 r.p.m., until they fell (this time was provided by the apparatus and recorded as latency to fall for each trial). Each mouse was tested for three trials in a single day, with about 15 min between trials. Raw data were recorded and analyzed using Microsoft Excel.
Contextual fear conditioning.
Two distinct contexts were employed. The conditioning context was a 29 × 25 × 22 cm chamber with grid floors, dim white lighting, and scented with 0.25% benzaldehyde. The neutral context consisted of a 29 × 25 × 22 cm chamber with white perspex floors, red lighting, and scented with 1% acetic acid. All mice were conditioned (120 s exploration, one 0.65 mA shock of 2 s duration at 120 s, 60 s post-shock period, second 0.65 mA shock of 2 s duration at 180 s, 60 s post-shock period), and tested (3 min) one day later. Twenty-four hours after the recall test on day 2, the neutral context test (3 min) was performed (i.e., neutral context tests were always on day 3). Experiments showed no generalization in the neutral context for wild type/control mice. Floors of chambers were cleaned with quatricide before and between runs. Mice were transported to and from the experimental room in their home cages using a wheeled cart. For immediate shock controls, animals were placed in the conditioning chamber, received a 2 s foot shock after the first 5 s and then were immediately removed from the chamber. For experiments that included optogenetic manipulations, the behavior chamber ceilings were customized to hold a rotary joint (Doric Lenses) connected to two 0.3 m optic fibers. All mice had optic fibers attached to their optic fiber implants prior to training and recall tests. Since optogenetic manipulations (i.e., optic fibers) interfered with automated motion detection, freezing behavior was manually quantified for all experiments.
Inhibitory avoidance.
A 29 × 25 × 22 cm unscented chamber with square ceilings and intermediate lighting was used. The chamber consisted of two sections, one with grid flooring and the other with a white platform. During the training session (1 min), mice were placed on the white platform, which is the less preferred section of the chamber (relative to the grid section). Once mice entered the grid section of the chamber (all four feet), 0.65 mA shocks of 2 s duration were delivered. On average, each mouse received 2–3 shocks per training session. After 1 min, mice were returned to their home cage. The next day, total time on the white platform was manually quantified (3 min test).
Innate avoidance.
Innate avoidance behavior in response to 2,3,5-trimethyl-3-thiazoline (TMT), a component of fox feces, was measured. Mice were placed in the center of a 40 × 30 cm Plexiglass arena, which contained four small dishes (3 cm diameter) in each of the corners. Mice were first habituated to the arena for 10 min. During trial 1, mice were allowed to explore the arena in which all four dishes contained 1x PBS (0.5 ml each) for 15 min. The preferred corner was recorded for the subsequent trial for each mouse. Approximately 30 min after trial 1, mice were returned to the arena in which their preferred corner now had 5% TMT (colorless) instead of 1x PBS (trial 2). Mice were once again allowed to explore the area for 15 min, after which they were returned to their home cages. Relative to the time spent in their preferred corner during trial 1, time spent in this same corner during trial 2 was manually quantified (i.e., avoidance behavior). The arena was rotated between mice, and to make sure that the TMT odor did not persist between mice these tests were performed in the fume hood.
Tone fear conditioning.
The conditioning context was a 29 × 25 × 22 cm chamber with grid floors, bright white lighting, and scented with 1% acetic acid. The recall test context consisted of a 30 × 25 × 33 cm chamber with white perspex floors, red lighting, and scented with 0.25% benzaldehyde. Mice were conditioned (120 s exploration, 10 s tone co-terminating with a 0.65 mA shock of 2 s duration, 60 s post-shock period, repeated 2 more times). Memory recall was tested (1 min exploration, 60 s tone, 60 s post-tone period, repeated 2 more times) one day later. The tone was calibrated to 75 dB SPL, with a frequency of 5 kHz. Experiments showed no generalization in the recall test context during the initial exploration period. Freezing behavior was manually quantified.
Spatial working memory: T-maze.
For spatial working memory behavior, we used the delayed non-match-to-place (DNMP) T-maze protocol. Mice selected for this paradigm were food deprived until they reached 85% of their initial body weight. During food deprivation, mice were habituated to the sugar pellets (20 mg), which would subsequently be used as a reward in the T-maze. Mice were habituated to the T-maze for 10 min. During habituation to the maze, sugar pellet rewards were placed in the reward cups (2.5 cm diameter) at the end of each arm, and were replaced as they were consumed. The behavioral training consisted of ten trials per day with each trial having two separate runs (Sample and Choice runs). The first run in each trial was the Sample run, in which mice were placed in the stem of the T-maze and allowed to run to the end of one arm of the maze (the other arm was closed off). This open arm was rewarded. After reward consumption, mice were returned to their home cage for ~30 s when the T-maze was quickly cleaned and both arms were opened. Mice were once again placed in the stem of the T- maze and during this Choice run mice were allowed to choose which of the two arms to visit. The opposite arm from the one visited during the previous Sample run was rewarded. If the mouse chose the incorrect arm (i.e., the previous arm), it was blocked in that arm for a 30 s punishment. Following this Sample and Choice run procedure for a single trial, each mouse performed nine more trials per day with an inter-trial interval of 20 min. Mice were manually scored on the percentage of time that they made a successful alternation and how many days until they reached a daily success rate of over 70% for two consecutive days (referred to as days to criterion). Once they reached criterion, the next two days were used for testing animals’ success rate when the delay between Sample and Choice runs was 10 s (ten trials per day). Their performance in the 10 s delay condition was an average of these two test days. Similarly, the following two days were used for testing animals’ success rate when the delay between Sample and Choice runs was 60 s (ten trials per day), which was a more demanding version of this task.
Cocaine-induced conditioned place preference.
The conditioned place preference (CPP) behavior chamber was a rectangular arena (42 × 15 cm), divided into three quadrants (left, middle, right). The left and right quadrants were 15 cm long, while the middle quadrant was 12 cm long. The left quadrant had wide grid floors and a pattern (series of parallel lines) on the wall. The right quadrant had white smooth polypropylene floors and a pattern (series of circles) on the wall. On day 1 (pre-exposure), mice were allowed to explore the entire arena for 30 min. Experiments showed no preference to any one quadrant. On day 2 (training), mice were confined to the left or right quadrants for 10 min following cocaine (20 mg kg−1) or saline intraperitoneal administration in addition to receiving optogenetic light activation for the entire session. This 10 min session was repeated twice with an inter-trial interval of 3 hr. On days 3–7 (training continued), mice were conditioned in opposite quadrants in an alternating manner (i.e., cocaine left-saline right-cocaine left, etc) until every mouse received 3 cocaine- and 3 saline-pairing days. For every behavioral cohort, half the mice were conditioned with cocaine in the left quadrant, while the remaining mice received cocaine in the right quadrant. On day 8, memory recall was measured by preference to the left or right quadrant (10 min), without optogenetic light activation. All sessions were performed with dim white lighting. On day 9, generalization behavior was measured using a modified CPP chamber (10 min), without optogenetic light activation. Specifically, in this modified chamber, the left quadrant had thin grid floors and a pattern (series of squares) on the wall, while the right quadrant had smooth black floors and a pattern (series of triangles) on the wall, and the room had bright white lighting. Mouse behavior, specifically position tracking and duration, was recorded using an automated infrared (IR) detection system (EthoVision XT, Noldus). Raw data were extracted and analyzed using Microsoft Excel.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean values accompanied by SEM. No statistical methods were used to predetermine sample sizes. Data analysis was performed blind to the conditions of the experiments. Data were analyzed using Microsoft Excel with the Statplus plug-in and Prism 6 software. One-way ANOVA followed by Bonferroni post-hoc tests, two-way ANOVA with repeated measures followed by Bonferroni post-hoc tests, two-tailed unpaired t tests, and paired t tests were used to test for statistical significance when appropriate. Statistical parameters including the exact value of n, precision measures (mean ± SEM), and statistical significance are reported in each main and supplementary figure legend. The significance threshold was placed at α = 0.05 (NS, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-C1QL2 | Thermo Fisher | PA5-101536 |
| Chicken anti-GFP | Thermo Fisher | A-10262 |
| Rabbit anti-RFP | Rockland | 600-401-379 |
| Rabbit anti-cFos | Cell Signaling | 2250 |
| Anti-rabbit Alexa-488 | Thermo Fisher | A-11008 |
| Anti-chicken Alexa-488 | Thermo Fisher | A-11039 |
| Anti-rabbit Alexa-555 | Thermo Fisher | A-21428 |
| Bacterial and virus strains | ||
| AAVretro-hSyn-Cre | Addgene | 105553-AAVrg |
| AAV9-EF1α-DIO-ChR2-eYFP | Addgene | 20298-AAV9 |
| AAV9-CaMKIIα-ChR2-eYFP | Addgene | 26969-AAV9 |
| AAV9-CaMKIIα-ChR2-mCherry | Addgene | 26975-AAV9 |
| AAV8-hSyn-DIO-hM4Di-mCherry | Addgene | 44362-AAV8 |
| AAV8-hSyn-DIO-mCherry | Addgene | 50459-AAV8 |
| AAV1-hSyn-Cre | Addgene | 105553-AAV1 |
| AAV9-hSyn-DIO-hM3Dq-mCherry | Addgene | 44361-AAV9 |
| AAV8-hSyn-mCherry | Addgene | 114472-AAV8 |
| AAV8-hSyn-DIO-hM4Di-mCitrine | Addgene | 50455-AAV8 |
| AAV8-CaMKIIα-hM4Di-mCherry | Addgene | 50477-AAV8 |
| AAV9-EF1α-DIO-eYFP | UNC Vector Core | N/A |
| AAV9-EF1α-DIO-eArch3.0-eYFP | UNC Vector Core | N/A |
| RV-GFP | Wickersham I., MIT | N/A |
| AAV5-synP-FLEX-sTpEpB | Wickersham I., MIT | N/A |
| RVΔG-mCherry | Wickersham I., MIT | N/A |
| RVdGL-Cre | Wickersham I., MIT | N/A |
| AAV9-sgRNAPTCHD1-mCh | This paper | N/A |
| AAV9-sgRNACACNA1G-mCh | Li et al., 2020 | N/A |
| AAV9-sgRNAYWHAG-mCh | This paper | N/A |
| AAV9-sgRNAGRIA3-mCh | This paper | N/A |
| AAV9-sgRNAHERC1-mCh | This paper | N/A |
| AAV9-sgRNAATP1A3-mCh | This paper | N/A |
| AAV9-sgRNAMTOR-mCh | This paper | N/A |
| AAV9-sgRNACNTNAP2-mCh | This paper | N/A |
| AAV9-CMV-SpCas9 | Vector Biolabs | N/A |
| Biological samples | ||
| Normal human donor anterior thalamus sample | Cureline, Inc. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | Sigma | D9542 |
| CTB 488 | Thermo Fisher | C34775 |
| CTB 555 | Thermo Fisher | C34776 |
| CTB 647 | Thermo Fisher | C34778 |
| Critical commercial assays | ||
| Deposited data | ||
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | Jackson Labs | N/A |
| C1QL2-IRES-Cre mice | This paper | N/A |
| CaMKII-Cre mice | Jackson Labs | 5359 |
| GAD2-IRES-Cre mice | Jackson Labs | 28867 |
| PV-Cre mice | Jackson Labs | 17320 |
| SST-IRES-Cre mice | Jackson Labs | 28864 |
| VIP-IRES-Cre mice | Jackson Labs | 31628 |
| FosTRAP mice | Guenthner et al., 2013 | N/A |
| Common marmosets | Feng G., MIT | N/A |
| Oligonucleotides | ||
| Cntnap2 mouse probe | ACD | 449381 |
| Atp1a3 mouse probe | ACD | 432511 |
| Gria3 mouse probe | ACD | 426251 |
| Mtor mouse probe | ACD | 451651 |
| Ywhag mouse probe | ACD | 812981 |
| Herc1 mouse probe | ACD | 871341 |
| Cacna1g mouse probe | ACD | 459761 |
| C1ql2 mouse probe | ACD | 480871 |
| PV mouse probe | ACD | 421931 |
| Col25a1 mouse probe | ACD | 538511 |
| Rabies virus probe | ACD | 456781 |
| Ptchd1 mouse probe | ACD | 489651 |
| Slc17a6 mouse probe | ACD | 319171 |
| Kcnj12 mouse probe | ACD | 525171 |
| Kcnq2 mouse probe | ACD | 444251 |
| Kcna1 mouse probe | ACD | 481921 |
| Cacna1a mouse probe | ACD | 493141 |
| Cacna1b mouse probe | ACD | 468811 |
| C1ql2 marmoset probe | ACD | 525821 |
| Col25a1 marmoset probe | ACD | 557651 |
| C1ql2 human probe | ACD | 478011 |
| Recombinant DNA | ||
| AAV-EF1α-DO-NpHR3.0-eYFP | Addgene | 37087 |
| AAV-EF1α-DO-eGFP | Addgene | 37085 |
| AAV-EF1α-DO-ChETA-tdTomato | Addgene | 37756 |
| AAV-EF1α-DIO-C1V1-eYFP | Addgene | 35497 |
| AAV-CaMKIIα-mCherry | Addgene | 114469 |
| AAV-cFos-CreERT2 | Ye et al., 2016 | N/A |
| AAV-DIO-SpCas9 | Xu et al., 2018 | N/A |
| Software and algorithms | ||
| ImageJ | NIH | imagej.nih.gov/ij |
| Prism 6 | GraphPad Software | graphpad.com/scientific-software/prism |
| EthoVision | Noldus | noldus.com/animal-behavior-research/products/ethovision-xt |
| MATLAB | MathWorks | mathworks.com/products/matlab.html |
| Other | ||
HIGHLIGHTS.
AD thalamus is necessary for memory encoding
AV thalamus regulates memory specificity
Knockdown of autism and schizophrenia risk genes from AD lead to cognitive deficits
Disease models exhibit converging cellular and circuit mechanisms in AD
ACKNOWLEDGMENTS
We thank Z. J. Huang for the IRES-Cre construct; P. Qi and D. Hee for help with mouse generation; E. Hwang for help with LFP analyses; C. Wang for viral packaging; M. Fleishman, B. Clear, J. Kim, H. Zaniewski, and K. Levandowski for technical assistance; and all members of the Feng laboratory for their support. This work was supported by the Stanley Center at the Broad Institute, Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT, James and Patricia Poitras Center for Psychiatric Disorders Research at MIT, and NIH BRAIN Initiative (U01MH114819) (to G.F.). MEXT, CNSI/NINS, Nakatani Foundation, SENSHIN Foundation, Mochida Foundation, Takeda Foundation, and Medical Research Institute of TMDU supported T.A. NSF EAGER 2035018 supported Q.C.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, and Tanaka K (2015). Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggleton JP, Keith AB, and Sahgal A (1991). Both fornix and anterior thalamic, but not mammillary, lesions disrupt delayed non-matching-to-position memory in rats. Behav. Brain Res. 44, 151–161. [DOI] [PubMed] [Google Scholar]
- Challis RC, Kumar SR, Chan KY, Challis C, Beadle K, Jang MJ, Kim HM, Rajendran PS, Tompkins JD, Shivkumar K, et al. (2019). Systemic AAV vectors for widespread and targeted delivery in rodents. Nat. Protoc. 14, 379–414. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Sullivan HA, MacLennan BJ, Xu R, Hou Y, Lavin TK, Lea NE, Michalski JE, Babcock KR, Dietrich S, et al. (2018). Nontoxic, double-deletion-mutant rabies viral vectors for retrograde targeting of projection neurons. Nat. Neurosci. 21, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Noor A, Begagne B, Baker K, Bok LA, Brady AF, Chitayat D, Chung BH, Cytrynbaum C, Dyment D, et al. (2015). Phenotypic spectrum associated with PTCHD1 deletions and truncating mutations includes intellectual disability and autism spectrum disorder. Clin. Genet. 88, 224–233. [DOI] [PubMed] [Google Scholar]
- Colgin LL (2015). Theta-gamma coupling in the entorhinal-hippocampal system. Curr. Opin. Neurobiol. 31, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden CEM, Buxbaum JD, and De Rubeis S (2018). Disrupted circuits in mouse models of autism spectrum disorder and intellectual disability. Curr. Opin. Neurobiol. 48, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruter T, Wiescholleck V, Dubovyk V, Aliane V, and Manahan-Vaughan D (2015). Altered neuronal excitability underlies impaired hippocampal function in an animal model of psychosis. Front. Behav. Neurosci. 9, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenthner CJ, Miyamichi K, Yang HH, Heller HC, and Luo L (2013). Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski MM, Ronnqvist KC, Tsanov M, Vann SD, Wright NF, Erichsen JT, Aggleton JP, and O’Mara SM (2013). The anterior thalamus provides a subcortical circuit supporting memory and spatial navigation. Front. Syst. Neurosci. 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten MR, Rudy B, and Anderson MP (2007). Differential regulation of action potential firing in adult murine thalamocortical neurons by Kv3.2, Kv1, and SK potassium and N-type calcium channels. J. Physiol. 584, 565–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB, Karayiorgou M, and Gogos JA (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. 105, 7076–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li Y, Lopez-Huerta VG, Adiconis X, Levandowski K, Choi S, Simmons SK, Arias- Garcia MA, Guo B, Yao AY, Blosser TB, et al. (2020). Distinct subnetworks of the thalamic reticular nucleus. Nature 583, 819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JL, and Shoemaker M (2009). Intellectual disability and its relationship to autism spectrum disorders. Res. Dev. Disabil. 30, 1107–1114. [DOI] [PubMed] [Google Scholar]
- Mitchell AS, and Dalrymple-Alford JC (2006). Lateral and anterior thalamic lesions impair independent memory systems. Learn. Mem. 13, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan VA, Leonard H, Bourke J, and Jablensky A (2008). Intellectual disability co-occurring with schizophrenia and other psychiatric illness: population-based study. Br. J. Psychiatry 193, 364–372. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Schmitt LI, Feng G, and Halassa M (2019). Combinatorial targeting of distributed forebrain networks reverses noise hypersensitivity in a model of autism spectrum disorder. Neuron 104, 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Tuathaigh CM, Babovic D, O’Sullivan GJ, Clifford JJ, Tighe O, Croke DT, Harvey R, and Waddington JL (2007). Phenotypic characterization of spatial cognition and social behavior in mice with ‘knockout’ of the schizophrenia risk gene neuregulin 1. Neuroscience 147, 18–27. [DOI] [PubMed] [Google Scholar]
- Saunders A, Johnson CA, and Sabatini BL (2012). Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Front. Neural Circuits 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Hall JM, and Vetreno RP (2011). Anterior thalamic lesions alter both hippocampal-dependent behavior and hippocampal acetylcholine release in the rat. Learn. Mem. 18, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Neale BM, Daly M, and the Schizophrenia Exome Meta-Analysis (SCHEMA) Consortium. (2020). Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. medRxiv 2019 2815 [Preprint]. 18 September 2020. [Google Scholar]
- Speca DJ, Ogata G, Mandikian D, Bishop HI, Wiler SW, Eum K, Wenzel HJ, Doisy ET, Matt L, Campi KL, et al. (2014). Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav. 13, 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DGR, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort AB, Komorowski R, Eichenbaum H, and Kopell N (2010). Measuring phase-amplitude coupling between neuronal oscillations of different frequencies. J. Neurophysiol. 104, 1195–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP, Linley SB, Groenewegen HJ, and Witter MP (2015). “Thalamus” in The Rat Nervous System, Paxinos G, Ed. (Academic Press; ). [Google Scholar]
- Vetere G, Xia F, Ramsaran AI, Tran LM, Josselyn SA, and Frankland PW (2021). An inhibitory hippocampal-thalamic pathway modulates remote memory retrieval. Nat. Neurosci. 24, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton EC, and Aggleton JP (1999). Differential deficits in the Morris water maze following cytotoxic lesions of the anterior thalamus and fornix transection. Behav. Brain Sci. 98, 27–38. [DOI] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, and Halassa M (2016). Thalamic reticular impairment underlies attention deficit in Ptchd1Y/− mice. Nature 532, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Lyon DC, Barnard RJO, Mori T, Finke S, Conzelmann KK, Young JAT, and Callaway EM (2007). Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SS, Clark BJ, and Taube JS (2015). Disruption of the head direction cell network impairs the parahippocampal grid cell signal. Science 347, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Doan TP, Jacobsen B, Nilssen ES, and O’Mara S (2017). Architecture of the entorhinal cortex: a review of entorhinal anatomy in rodents with some comparative notes. Front. Syst. Neurosci. 11, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, and Sudhof TC (2013). A neural circuit for memory specificity and generalization. Science 339, 1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bartolome CL, Low CS, Yi X, Chien CH, Wang P, and Kong D (2018) Genetic identification of leptin neural circuits in energy and glucose homeostasis. Nature 556, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki N, Li X, Lambot L, Ren LY, Radulovic J, and Shepherd G (2019). Long-range inhibitory intersection of a retrosplenial thalamocortical circuit by apical tuft-targeting CA1 neurons. Nat. Neurosci. 22, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Allen WE, Thompson KR, Tian Q, Hsueh B, Ramakrishnan C, Wang AC, Jennings JH, Adhikari A, Halpern C, et al. (2016). Wiring and molecular features of prefrontal ensembles representing distinct experiences. Cell 165, 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KA, Manaye KF, Liang C, Hicks PB, and German DC (2000). Reduced number of mediodorsal and anterior thalamic neurons in schizophrenia. Biol. Psychiatry 47, 944–953. [DOI] [PubMed] [Google Scholar]
- Zingg B, Chou XL, Zhang ZG, Mesik L, Liang F, Tao HW, and Zhang LI (2017). AAV-mediated anterograde transsynaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding authors on reasonable request. This study did not generate any new code.