

Abstract
This study evaluated the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of single and multiple oral doses of enpatoran (formerly named M5049), a new toll‐like receptor (TLR) 7 and 8 dual antagonist, and the effect of food on a single dose in healthy participants. In this single phase 1, randomized (3:1), double‐blind, placebo‐controlled study, 96 participants received single and multiple ascending oral doses of enpatoran. Participants in single‐dose cohorts received one dose of enpatoran (1, 3, 9, 25, 50, 100, or 200 mg) or placebo using a sentinel dosing strategy. Multiple‐dose cohorts received enpatoran (9, 25, or 200 mg once daily, or 25 or 50 mg twice daily) or placebo for 14 days. Safety, tolerability, PK, and PD (ex vivo‐stimulated cytokine secretion) were assessed in both parts. The effect of food was assessed in an open‐label, one‐way crossover study in the 25 mg single‐dose cohort. Single‐ and multiple‐oral doses of enpatoran up to 200 mg were well tolerated and no significant dose‐limiting adverse events or safety signals were observed under fasting or fed conditions. PK parameters were linear and dose‐proportional across the dose range evaluated, with a slightly delayed absorption and lower peak concentration observed at 25 mg with food. Exposure‐dependent inhibition of ex vivo‐stimulated interleukin‐6 secretion was observed, with maximum inhibition at 200 mg. Enpatoran was well tolerated at doses up to 200 mg. Further investigation of enpatoran is warranted as a potential treatment for diseases driven by TLR7/8 overactivation, such as systemic lupus erythematosus and COVID‐19 pneumonia.
Keywords: autoimmune diseases, pharmacodynamics, pharmacokinetics, safety, toll‐like receptors
The graphical abstract shows the study design and key outcomes from the study.

Abbreviations
- AEs
adverse events
- AUC0 –12
area under the curve during 12 h
- AUC0–∞
area under the curve extrapolated to infinity
- AUC0 – t
area under the plasma concentration–time curve from time zero (dosing time) to the last sampling time
- AUC0– τ
area under the curve during 24 h for QD and 12 h for BID
- BMI
body mass index
- CL/F
apparent clearance
- C max
maximum plasma concentration
- COVID‐19
coronavirus disease 2019
- FIH
first‐in‐human
- IFN
interferon
- IFN‐α
interferon‐alpha
- IL‐6
interleukin‐6
- IRF
interferon regulatory factor
- MAD
multiple ascending dose
- max
maximum
- NF‐κB
nuclear factor‐kappa B
- PD
pharmacodynamics
- PK
pharmacokinetics
- QD
once daily
- Racc AUCτ
accumulation factor to assess the increase in area under the curve until steady state is reached
- R acc C max
accumulation factor to assess the increase in maximum concentration until steady state is reached
- SAD
single ascending dose
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- SLE
systemic lupus erythematosus
- t 1/2
terminal half‐life
- TEAEs
treatment‐emergent adverse events
- TLR
toll‐like receptor
- t max
time to reach maximum plasma concentration
1. INTRODUCTION
Toll‐like receptors (TLRs) are a family of conserved transmembrane pattern recognition receptors that are widely expressed in a variety of immune cells and play a key role in both the innate immune response and development of antigen‐specific acquired immunity against a range of foreign molecules. TLR7 and TLR8 are intracellular receptors expressed in the cell endosome that bind single‐stranded ribonucleic acids (RNAs) from viruses, and are involved in the normal defense response against these pathogens.1
Studies suggest that TLR7 and TLR8 may each have specialized roles and induce an immune response via different mechanisms.2, 3, 4, 5 In addition to having slightly different ligand binding characteristics and cellular expression patterns, TLR7 and TLR8 also differentially activate the pro‐inflammatory mediators interferon (IFN) regulatory factor (IRF) and nuclear factor‐kappa B (NF‐κB), eliciting cell type‐specific responses.2 Whilst TLR7 shows dual activation of both the IRF and NF‐κB pathways and the production of interferon‐alpha (IFN‐α) and interleukin‐6 (IL‐6), TLR8 shows a bias toward greater activation of the NF‐κB pathway and the production of IL‐6.2
Aberrant activation of the TLR7 and TLR8 pathways by endogenous RNA molecules may be pathogenic, resulting in the production of Type I IFN, IL‐6 and other pro‐inflammatory mediators, and the production of autoantibodies, thus leading to the development of certain autoimmune diseases.6, 7, 8, 9, 10 For example, single nucleotide polymorphisms in both the TLR7 and TLR8 genes have been identified that are associated with systemic lupus erythematosus (SLE),11 and TLR7 overexpression and knockout studies suggest that TLR7 may promote SLE in mice.12, 13, 14, 15 Inhibition of TLR7 and TLR8 activation therefore represents a potential therapeutic target for patients with autoimmune diseases that are characterized by overactivation of these receptors, including SLE.6, 7
It is now understood that TLR signaling is an important mediator of coronavirus disease 2019 (COVID‐19) immunopathogenesis.16 Molecular docking studies suggest that spike protein, the major infective protein of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) virus, binds to various cell surface TLRs (TLR1, TLR4, and TLR6) that are subsequently involved in the induction of pro‐inflammatory responses.17 In addition, it is possible that activation of TLR7 and TLR8 by SARS‐CoV‐2 may contribute to the overt inflammatory response that can lead to a cytokine storm in COVID‐19 pneumonia, which is a major factor in COVID‐19 morbidity and mortality.18, 19, 20 TLR antagonists are therefore of interest for the treatment of COVID‐19 since they may reduce the cytokine storm in infected individuals through inhibition of TLR‐mediated pro‐inflammatory signaling.16, 19
Due to the specific and differing expression patterns and molecular functions of TLR7 and TLR8, it has been suggested that a dual TLR7/8 inhibitor may provide superior efficacy in reducing inflammation and autoimmune disease activity than agents targeting either TLR isotype alone.2 Enpatoran, previously known as M5049, is a potential first‐in‐class small molecule dual TLR antagonist having activity against both TLR7 and TLR8, which has been shown to inhibit the activation of TLR7/8 in vitro and ex vivo and to suppress disease activity in mouse models of SLE.21 Enpatoran, therefore, has the potential to inhibit the pathological activity of immune complexes and the progression of autoimmune diseases in the clinical setting.
The aim of this phase 1, first‐in‐human (FIH) study was to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of single and multiple doses of enpatoran, and to explore the effect of food on a single dose of enpatoran in healthy participants.
2. MATERIALS AND METHODS
2.1. Study design
This was a phase 1, double‐blind, randomized, placebo‐controlled study, and the first to use enpatoran in humans (ClinicalTrials.gov NCT03676322). Healthy participants were randomized (3:1) to receive single and multiple ascending doses (SAD and MAD, respectively) of orally administered enpatoran solution or placebo. The study comprised three distinct parts (Figure 1). See the Data S1 for information on the starting dose justification, dose escalation criteria, and blinding.
FIGURE 1.
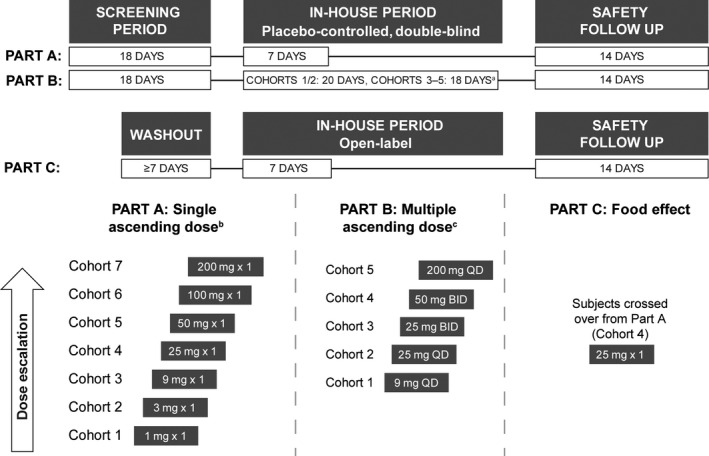
Study design and dose escalation scheme. Eight participants were randomized to each cohort; six to active treatment and two to placebo. Eight participants in Part A, Cohort 4, crossed over to Part C. aThe primary observation period was reduced to 17 days for Cohorts 3–5 of Part B, since available clinical data showed this timeframe provided sufficient safety and tolerability monitoring; bSingle‐dose cohort escalation (Part A): safety data from a single dose plus 1 week in‐house; cMultiple‐dose cohort escalation (Part B): safety data from 2 weeks of treatment until the end of the in‐house period. BID, twice daily; QD, once daily
In Part A, participants were randomized to single doses of enpatoran (1, 3, 9, 25, 50, 100, and 200 mg; named Part A Cohorts 1–7, respectively) or placebo. Two participants in each cohort served as sentinel participants; one received placebo and the other received enpatoran. The remaining six participants in each cohort were randomized following review of 24‐h safety data by blinded investigators. Part A consisted of an 18‐day screening period, a 7‐day in‐house period with a single dose on Day 1, and a 14‐day safety follow‐up period. Each dose was administered after an overnight fast of at least 10 h and was followed by a 4‐h post‐dose fast.
In Part B, participants were randomized to 9, 25, or 200 mg enpatoran once daily (QD) for 14 days (Cohorts 1, 2, and 5), 25 or 50 mg enpatoran twice daily (BID) for 13 days plus a single dose on Day 14 (Cohorts 3 and 4), or placebo for 14 days. Part B comprised an 18‐day screening period, a 20‐day (Cohorts 1 and 2) or 18‐day (Cohorts 3–5) in‐house period with dosing from Days 1 to 14, and a 14‐day safety follow‐up period. The morning dose was administered after an overnight fast of at least 8 h and the evening dose was administered after a fast of at least 1 h; participants continued to fast for 2 h post‐dose.
Part C was an open‐label, one‐way crossover study. Following a minimum 7‐day washout period, all participants from Part A Cohort 4 (25 mg dose) crossed over to Part C. This consisted of a 7‐day in‐house period with a single dose on Day 1, and a 14‐day safety follow‐up period. Following an overnight fast of at least 10 h, all participants received a single dose of enpatoran 30 min after the start of a high‐fat, high‐calorie breakfast. No food was allowed for 4 h post‐dose.
No participants were included in more than one cohort in any part of the study except for those enrolled in SAD Cohort 4, who crossed over to Part C.
The study was performed in accordance with the ethical principles of international guidelines, including the Declaration of Helsinki and Council for International Organization of Medical Sciences International Ethical Guidelines. The study was conducted at a single center in Germany (Clinical Pharmacology Unit, Nuvisan GmbH) and was approved by the Ethics Committee of the Bavarian Chamber of Physicians, Munich, Germany. All participants gave informed consent.
2.2. Study objectives
The primary study objectives were to evaluate the safety and tolerability of single and multiple oral doses of enpatoran and explore the effect of food on enpatoran PK parameters in healthy participants. Secondary objectives included the assessment of enpatoran PK parameters following single‐ and multiple‐dose administration, and evaluation of the safety and tolerability of enpatoran under fed conditions. Exploratory objectives included investigation of the PD effects (ex vivo‐stimulated cytokine secretion) of single and multiple doses of enpatoran compared with placebo.
2.3. Study participants
Inclusion and exclusion criteria were standard for a healthy volunteer study. Healthy male and female participants aged 18–45 years, with body mass index (BMI) 18.5–29.9 kg/m2 inclusive, and body weight 50–100 kg were eligible. All relevant medical and nonmedical conditions were taken into consideration when deciding whether an individual was eligible for the study. Participants were required to be nonsmokers for at least 90 days before screening. Female participants were only eligible if they were not of childbearing potential. Male participants had to agree to have their female partners use a highly effective contraception method 2 days before the first dose of study intervention until at least 14 weeks after the last dose. Key exclusion criteria included completion of oral anti‐infectives within 2 weeks prior to screening, any clinically significant infection or travel to regions with Zika virus or yellow fever within 4 weeks prior to screening, and vaccination within 3 months prior to the first treatment administration.
2.4. Safety and tolerability assessments
The nature, incidence, and severity of treatment‐emergent adverse events (TEAEs) were assessed and recorded continuously throughout the study and at the end of the 14‐day safety follow‐up period. Safety assessments included physical and neurological examination, vital signs, 12‐lead safety electrocardiograms (ECGs), QT and concentration‐QT analysis (Holter ECGs), and clinical laboratory parameters.
In Parts A and C, 12‐lead ECGs were recorded during screening, Day −1 to Day 4 of the in‐house period and during the safety follow‐up period. In Part B, 12‐lead ECGs were recorded during screening, on Days −1, 1, 3, 5, 8, 11, and 14 of the in‐house period and during the safety follow‐up period. In Parts A and B, digital Holter ECGs were performed from baseline (prior to the first dose of study intervention) through 24 h. In Part B, Cohorts 3, 4, and 5, Holter ECGs were also recorded on Day 14.
Concentration‐QT analysis was conducted using data collected by digital Holter ECGs and time‐matched enpatoran concentrations. A linear mixed effects model with fixed and random slope for concentration, as well as fixed and random intercept and the ΔQTcF (delta QT interval corrected using Fridericia's formula) as the response was fitted to explore the relationship between enpatoran exposure and the corrected QT interval (QTc).
2.5. Pharmacokinetic assessments
Concentrations of enpatoran in the plasma were analyzed using a validated liquid chromatography–tandem mass spectrometry method. Depending on the expected concentration of enpatoran, samples were analyzed using either a low range method (20–50,000 pg/ml) or high range method (1–10,000 ng/ml). The lower limits of quantification (LLOQs) were 20 pg/ml and 1 ng/ml for the respective methods. Accuracy ranged between 93.1% and 105.5%, and the acceptance criteria for precision was ≤15.0%.
A range of PK parameters were calculated using standard non‐compartmental methods under both fasting (Parts A and B) and fed (Part C) conditions, including: maximum plasma concentration (C max), area under the plasma concentration–time curve (AUC) from time zero (dosing time) to the last sampling time (AUC0– t), terminal half‐life (t 1/2), apparent clearance (CL/F), AUC during 24 h for QD and 12 h for BID (AUC0– τ) and time to reach maximum plasma concentration (t max). Accumulation ratios were calculated by comparing C max and AUCτ on Day 14 versus Day 1 of Part B. The non‐compartmental analyses of PK parameters were performed using Phoenix®/WinNonlin® Version 7.0 (Certara, L.P.).
In Parts A and C of the study, blood samples were obtained for analysis of enpatoran concentration and PK parameters pre‐dose and 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, and 120 h post‐dose. In Part B, blood samples were obtained pre‐dose and 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h post‐first dose on Day 1, and pre‐dose on Days 9, 10, 11, 12, and 13. On Day 14 in Part B, blood samples were taken pre‐dose, 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, and 120 h post‐last dose for Cohorts 1 and 2, and 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, and 72 h post‐last dose for Cohorts 3, 4, and 5. For BID dosing, the doses were given 12 h aside and the evening PK blood sample was carried out before the second dose.
2.6. Pharmacodynamic assessments
To assess TLR7/8 target modulation by enpatoran, blood samples were collected for cytokine immunoassays 24, 22, 20, and 16 h before first dosing (Day −1), at pre‐dose and 2, 4, 8, and 24 h post‐first dose (Day 1) for Parts A and B, as well as pre‐dose on Day 9, and pre‐dose and 2, 4, 8 and 24 h post‐dose on Day 14 for Part B. Time‐matched pre‐dose samples on Day −1 were collected and used to normalize the variability in the level of cytokine stimulation potentially associated with circadian rhythm.
As the TLR7/8 pathway is not active in normal tissues, TLR7/8 target modulation was assessed by ex vivo cytokine release immunoassays under stimulated (using the TLR7/8 agonist, R84822) and unstimulated conditions. Whole blood was collected in TruCulture tubes (Myriad RPM) and incubated overnight with or without R848 (Invivogen). IL‐6 was the primary PD biomarker assessed, but other cytokines, including IFN‐α, were also evaluated. Secretion of IL‐6 and IFN‐α into the culture medium was measured using the OptiMAP Luminex panel and Simoa assay, respectively (Myriad RPM).
Precision was determined by measuring all levels of controls in replicate over a minimum of 3 days. The acceptance criteria for precision were ≤20% coefficient of variation of quality control samples (n = 3). The LLOQ was determined by performing twofold serial dilutions of standard solutions for a total of eight dilutions; the standards were tested in triplicate over three runs and the LLOQ threshold was determined as the concentration at which the coefficient of variation for the dilution was 30%.
2.7. Statistical analyses
For demographic, baseline and safety assessments, continuous measurements were summarized using descriptive statistics and, for categorical data, by means of frequency tables. The safety analysis set comprised all study participants who received at least one dose of enpatoran or placebo.
Enpatoran concentrations in plasma and PK parameters were summarized using descriptive statistics. Dose proportionality was assessed graphically and using a power model for both Part A and Part B. The power model related the logarithm of dose as the independent variable to the logarithm of the respective PK parameter (ln[PK parameter] = α + β × ln[dose]). An estimate for the slope of the regression line with a 95% confidence interval (CI) is presented. For Part B, the power model was applied separately for the QD and BID groups. The PK analysis set for Parts A and B comprised all participants who received at least one dose of enpatoran, who had no clinically important protocol deviations or events likely to affect PK, and at least one measurable post‐dose enpatoran plasma concentration. The PK analysis set for Part C comprised all participants who received one dose of enpatoran in both Parts A and C and had at least one evaluable primary PK parameter in both periods.
Absolute values from cytokine immunoassays were normalized by subtracting the unstimulated value from the value achieved following R848 stimulation, and the normalized values were used to calculate the percent inhibition of each cytokine under enpatoran exposure (with normalized baseline values as reference). The PD analysis set comprised all participants who received at least one dose of enpatoran or placebo, had at least one measurable PD endpoint at a scheduled PD time point pre‐ and post‐dose, and no clinically important protocol deviations likely to affect PD.
Food effect was analyzed using a linear mixed model, with treatment group as a fixed effect and subject as a random effect, for log‐transformed C max and AUC0– t.
3. RESULTS
3.1. Participant disposition
This study was conducted between October 15, 2018 and August 1, 2019. A total of 332 individuals were screened and 96 were considered eligible to enter the study: 56 participated in Part A, 40 in Part B, and 8 of those in Part A crossed over to Part C. Eight participants were randomized to each dose cohort, with six receiving active treatment and two receiving placebo. All participants received the assigned treatment and completed the study.
Overall, the mean (range) age was 31 (18–45) years and the mean BMI was 24.6 (18.9–29.8) kg/m2; distributions of age and BMI were comparable across the different parts of the study and the placebo groups (Table 1). Two participants were female and 94 were male, and all were white. No relevant medical history was recorded.
TABLE 1.
Participant demographic characteristics per study part (safety analysis set)
| Part A | Part B | Part C | |||||
|---|---|---|---|---|---|---|---|
|
Overall n = 56 |
Placebo n = 14 |
Enpatorana n = 42 |
Overall n = 40 |
Placebo n = 10 |
Enpatorana n = 30 |
Enpatoranb n = 8 |
|
| Sex, n (%) | |||||||
| Male | 55 (98.2) | 14 (100.0) | 41 (97.6) | 39 (97.5) | 10 (100.0) | 29 (96.7) | 7 (87.5) |
| Female | 1 (1.8) | 0 (0.0) | 1 (2.4) | 1 (2.5) | 0 (0.0) | 1 (3.3) | 1 (12.5) |
| Age, y | |||||||
| Meanc ± SD | 31 ± 6.8 | 30 ± 6.5 | 32 ± 6.9 | 32 ± 8.0 | 30 ± 8.4 | 33 ± 7.9 | 34 ± 9.3 |
| Minc, max | 18, 45 | 18, 43 | 20, 45 | 19, 45 | 19, 42 | 21, 45 | 18, 45 |
| BMId, kg/m2 | |||||||
| Meanc ± SD | 24.5 ± 2.55 | 24.4 ± 3.16 | 24.5 ± 2.35 | 24.7 ± 2.93 | 23.5 ± 3.48 | 25.1 ± 2.68 | 24.0 ± 2.74 |
| Min, max | 18.9, 29.8 | 18.9, 29.8 | 19.9, 29.1 | 19.0, 29.3 | 19.0, 28.2 | 20.1, 29.3 | 18.9, 26.4 |
Abbreviations: BMI, body mass index; max, maximum; min, minimum; SD, standard deviation.
All doses.
Participants crossed over from Part A (Cohort 4, 25 mg) to Part C and all received a single 25 mg dose of enpatoran.
Arithmetic mean.
At screening.
3.2. Safety and tolerability
During Parts A and B, 13 of the 96 participants reported a total of 21 TEAEs; no TEAEs were reported during Part C of the study. The incidence of TEAEs in single‐ and multiple‐dose cohorts were similar (Tables 2 and 3). All TEAEs were mild to moderate and resolved without sequelae. No dose‐dependent TEAEs were observed.
TABLE 2.
Treatment‐emergent adverse events following enpatoran single‐dose administration (safety analysis set)
| Number (%) of participants |
Placeboa n = 14 |
Enpatoran dose |
Enpatoranb n = 42 |
Overall n = 56 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
1 mg n = 6 |
3 mg n = 6 |
9 mg n = 6 |
25 mg n = 6 |
50 mg n = 6 |
100 mg n = 6 |
200 mg n = 6 |
||||
| Any TEAE | 2 (14.3) | — | 4 (66.7) | — | — | — | — | 1 (16.7) | 5 (11.9) | 7 (12.5) |
| Any study‐treatment‐related TEAE | 1 (7.1) | — | 3 (50.0) | — | — | — | — | 1 (16.7) | 4 (9.5) | 5 (8.9) |
| At least one event | 2 (14.3) | — | 4 (66.7) | — | — | — | — | 1 (16.7) | 5 (11.9) | 7 (12.5) |
| Gastrointestinal disorders | ||||||||||
| Flatulence | — | — | 4 (66.7) | — | — | — | — | — | 4 (9.6) | 4 (7.1) |
| Diarrhea | — | — | 1 (16.7) | — | — | — | — | — | 1 (2.4) | 1 (1.8) |
| Nausea | 1 (7.1) | — | — | — | — | — | — | — | — | 1 (1.8) |
| Vomiting | 1 (7.1) | — | — | — | — | — | — | — | — | 1 (1.8) |
| General disorders and administration site conditions | ||||||||||
| Fatigue | — | — | 1 (16.7) | — | — | — | — | — | 1 (2.4) | 1 (1.8) |
| Vessel puncture site pain | 1 (7.1) | — | — | — | — | — | — | — | — | 1 (1.8) |
| Infections and infestations | ||||||||||
| Nasopharyngitis | — | — | 1 (16.7) | — | — | — | — | — | 1 (2.4) | 1 (1.8) |
| Nervous system disorders | ||||||||||
| Dysgeusia | — | — | — | — | — | — | — | 1 (16.7) | 1 (2.4) | 1 (1.8) |
Abbreviation: TEAE, treatment‐emergent adverse event.
Pooled placebo.
Enpatoran all doses.
TABLE 3.
Treatment‐emergent adverse events following enpatoran multiple‐dose administration (safety analysis set)
| Enpatoran dose | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number (%) of participants |
Placeboa n = 10 |
9 mg QD n = 6 |
25 mg QD n = 6 |
200 mg QD n = 6 |
25 mg BID n = 6 |
50 mg BID n = 6 |
Enpatoranb n = 30 |
Overall n = 40 |
||
| Any TEAE | 1 (10.0) | 2 (33.3) | 2 (33.3) | — | 1 (16.7) | — | 5 (16.7) | 6 (15.0) | ||
| Any study‐treatment‐related TEAE | — | 1 (16.7) | 1 (16.7) | — | — | — | 2 (6.7) | 2 (5.0) | ||
| At least one event | 1 (10.0) | 2 (33.3) | 2 (33.3) | — | 1 (16.7) | — | 5 (16.7) | 6 (15.0) | ||
| Infections and infestations | ||||||||||
| Nasopharyngitis | — | 1 (16.7) | — | — | — | — | 1 (3.3) | 1 (2.5) | ||
| Rhinitis | — | — | 1 (16.7) | — | — | — | 1 (3.3) | 1 (2.5) | ||
| Urinary tract infection | — | — | — | — | 1 (16.7) | — | 1 (3.3) | 1 (2.5) | ||
| Injury, poisoning and procedural complications | ||||||||||
| Contusion | — | — | 1 (16.7) | — | — | — | 1 (3.3) | 1 (2.5) | ||
| Musculoskeletal and connective tissue disorders | ||||||||||
| Myalgia | 1 (10.0) | — | — | — | — | — | — | 1 (2.5) | ||
| Nervous system disorders | ||||||||||
| Headache | — | 1 (16.7) | — | — | — | — | 1 (3.3) | 1 (2.5) | ||
Abbreviations: BID, twice daily; QD, once daily; TEAE, treatment‐emergent adverse event.
Pooled placebo.
Enpatoran all doses.
Flatulence occurred in 7.1% of participants in Part A and was the most frequently reported TEAE (Table 2). All other TEAEs occurred in only one participant each (Tables 2 and 3).
Fourteen TEAEs were considered by the Investigator to be related to the study intervention. The nature and incidence of study intervention‐related TEAEs was similar in the SAD (n = 5, 9%) and MAD (n = 2, 5%) cohorts, and included nausea and vomiting (in the same participant) in the placebo group, flatulence (three participants) and diarrhea (one participant) in the SAD cohorts, and headache (five events in one participant) and rhinitis (one participant) in the MAD cohorts.
No serious AEs occurred during the study, there were no discontinuations due to TEAEs and no deaths. No clinically relevant changes in laboratory values, vital signs or ECGs were observed in any participants over the study period. Enpatoran administered as SAD or MAD up to 200 mg did not result in any prolongation of QT interval and concentration‐QT analyses did not show any effect of enpatoran concentration on QTc (Figure S1).
3.3. Pharmacokinetics of enpatoran
The plasma concentration of enpatoran was found to increase rapidly in a dose‐dependent manner after both single‐ and multiple‐dose administration over the evaluated range of 1–200 mg (Figure 2).
FIGURE 2.
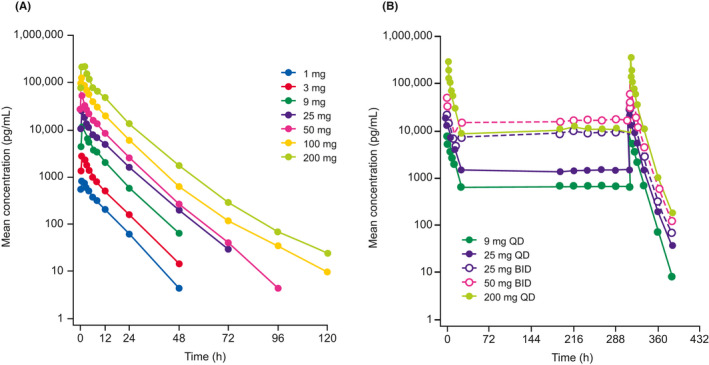
Mean plasma concentration‐time profiles for enpatoran following (A) single and (B) multiple (Days 1–14) dosing on a log‐linear scale (PK analysis set). BID, twice daily; PK, pharmacokinetic; QD, once daily
Tmax of enpatoran was reached between 1 and 1.5 h across all single‐dose levels, and C max and AUC0–∞ were dose‐proportional from 1 to 200 mg. The t 1/2 of enpatoran after a single dose ranged from 6.81 to 10.6 h (Table 4). The CL/F ranged from 120 to 162 L/h and was dose independent from 1 to 200 mg.
TABLE 4.
Enpatoran PK parameters following single administration (PK analysis set)
| Parameter | Part A (fasting) | Part C | ||||||
|---|---|---|---|---|---|---|---|---|
|
1 mg n = 6 |
3 mg n = 6 |
9 mg n = 6 |
25 mg n = 6 |
50 mg n = 6 |
100 mg n = 6 |
200 mg n = 6 |
25 mg fed n = 6 |
|
| Cmax, ng/ml | 0.834 (30.4) | 2.64 (20.2) | 11.4 (31.5) | 22.5 (42.0) | 50.6 (20.5) | 121 (50.9) | 230 (28.8) | 20.0 (21.2) |
| AUC0–∞, h·ng/ml | 7.02 (29.4) | 18.6 (31.6) | 75.0 (28.0) | 163 (43.3) | 317 (17.1) | 744 (28.9) | 1580 (39.2) | 216 (26.3) |
| AUC0–12, h·ng/ml | 4.99 (24.7) | 13.5 (25.6) | 55.0 (27.0) | 113 (38.0) | 231 (13.6) | 537 (35.3) | 1143 (31.0) | 149 (23.3) |
| tmax, h, median (min, max) |
1.50 (1.00, 3.00) |
1.00 (1.00, 2.00) |
1.00 (1.00, 1.00) |
1.00 (1.00, 2.00) |
1.00 (1.00, 1.00) |
1.00 (0.583, 2.00) |
1.50 (1.00, 2.00) |
3.00 (1.00, 4.00) |
| t1/2, h | 6.81 (19.2) | 6.83 (21.4) | 7.11 (11.8) | 7.87 (14.0) | 7.45 (13.0) | 10.1 (41.6) | 10.6 (42.2) | 8.42 (18.4) |
All values geometric mean (geometric coefficient of variation in percent) unless otherwise stated.
Abbreviations: AUC0–12, area under the curve during 12 h; AUC0–∞, area under the curve from time of dosing extrapolated to infinity; C max, maximum plasma concentration; max, maximum; min, minimum; PK, pharmacokinetic; t 1/2, terminal half‐life; t max, time to reach maximum plasma concentration.
Enpatoran PK parameters following the first dose in Part B (Day 1) were comparable with those found for single dosing (Tables 4 and 5). The geometric mean accumulation ratios for C max and AUCτ were 1.12–1.20 for QD dosing and 1.37–1.67 for BID dosing. There were no notable differences between PK parameters for multiple doses of 25 mg QD and 25 mg BID.
TABLE 5.
Enpatoran PK parameters following multiple administration (PK analysis set)
| Parameter |
9 mg QD n = 6 |
25 mg QD n = 6 |
200 mg QD n = 6 |
25 mg BID n = 6 |
50 mg BID n = 6 |
|---|---|---|---|---|---|
| DAY 1 | |||||
| Cmax, ng/ml | 11.0 (23.4) | 24.8 (20.1) | 274 (24.6) | 26.4 (47.7) | 50.9 (39.5) |
| AUC0– τ, h·ng/ml | 65.0 (22.2) | 148 (11.7) | 1247 (20.4) | 120 (37.2) | 221 (25.6) |
| AUC0–∞, h·ng/ml | 71.5 (22.9) | 163 (11.9) | 1315 (20.5) | 153 (40.8) | 298 (26.9) |
| tmax, h, median (min, max) | 1.00 (1.00, 2.00) | 1.00 (1.00, 1.00) | 1.00 (1.00, 1.00) | 1.00 (1.00, 2.00) | 1.00 (0.50, 1.00) |
| DAY 14 | |||||
| Cmax, ng/ml | 13.2 (30.8) | 29.8 (33.7) | 325 (19.6) | 37.0 (29.9) | 70.0 (38.4) |
| AUC0– τ, h·ng/ml | 78.2 (17.4) | 178 (26.9) | 1392 (20.7) | 190 (38.3) | 368 (22.2) |
| tmax, h, median (min, max) | 1.00 (1.00, 2.02) | 1.00 (1.00, 1.00) | 1.00 (1.00, 1.00) | 1.00 (1.00, 2.00) | 1.00 (1.00, 2.05) |
| t1/2, h | 7.46 (7.9) | 8.14 (16.5) | 7.69 (5.8) | 8.27 (4.7) | 8.74 (8.6) |
| R acc C max | 1.19 (27.2) | 1.20 (25.3) | 1.19 (10.9) | 1.40 (20.6) | 1.37 (25.4) |
| Racc AUCτ | 1.20 (15.6) | 1.20 (16.9) | 1.12 (10.2) | 1.58 (7.2) | 1.67 (10.4) |
All values geometric mean (geometric coefficient of variation in percent) unless otherwise stated.
Abbreviations: AUC0–12, area under the curve during 12 h; AUC0–∞, area under the curve from time of dosing extrapolated to infinity; AUC0– τ , area under the curve during 24 h for QD and 12 h for BID; BID, twice daily; C max, maximum plasma concentration; max, maximum; min, minimum; PK, pharmacokinetic; QD, once daily; R acc AUCτ, accumulation factor to assess the increase in exposure until steady state is reached; R acc C max, accumulation factor to assess the increase in maximum concentration until steady state is reached; t 1/2, terminal half‐life; t max, time to reach maximum plasma concentration.
Enpatoran plasma exposure (C max and AUC0–∞/AUC0– τ) appeared to be dose‐proportional over the SAD range of 1–200 mg and the MAD range of 9–200 mg QD and 25–50 mg BID (Tables 4 and 5; Figure 3). Dose proportionality was further confirmed using a power model; regression analysis revealed estimations of the slope of 1.06 (95% CI: 1.00, 1.11) and 1.02 (95% CI: 0.96, 1.07) for C max and AUC0–∞, respectively, after single dosing. On Day 14 of repeated dosing, estimates for C max and AUC0– τ of 1.05 (95% CI: 0.94, 1.17) and 0.94 (95% CI: 0.85, 1.02) were obtained for QD dosing. The respective estimates for the two BID groups on Day 14 were 0.92 (95% CI: 0.30, 1.54) and 0.96 (95% CI: 0.39, 1.52).
FIGURE 3.
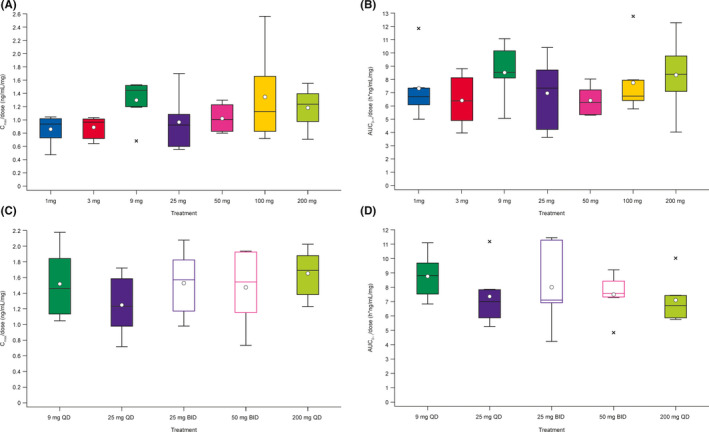
Dose‐normalized enpatoran PK parameters (A) C max and (B) AUC0–∞ following a single dose, and (C) C max and (D) AUC0– τ on Day 14 of multiple‐dose administration (PK analysis set). Box plots show medians, with minimum and maximum values. Circles represent the arithmetic means and crosses represent outliers. AUC0– τ, area under the curve during 24 h for QD and 12 h for BID; AUC0–∞, area under the curve extrapolated to infinity; BID, twice daily; C max, maximum plasma concentration; PK, pharmacokinetic; QD, once daily
Enpatoran oral solution was rapidly absorbed when given as single or multiple doses, with a median time to reach the t max of 1–1.5 h under both fed and fasting conditions (Tables 4 and 5).
3.4. The effect of food on enpatoran pharmacokinetics
When a single dose of 25 mg enpatoran was given 30 min after participants ingested a high‐fat, high‐calorie meal, mean plasma concentration–time curves showed delayed absorption and a slightly lower peak concentration compared with those observed under fasting conditions (Figure 4). Following ingestion of food, AUC0– t increased by 33% (fed versus fasting, 90% CI: 1.12, 1.59) and C max decreased by 11% (fed versus fasting, 90% CI: 0.73, 1.09). In addition, CL/F decreased by approximately 25% and t max was delayed (3 h under fed compared with 1 h under fasting conditions).
FIGURE 4.
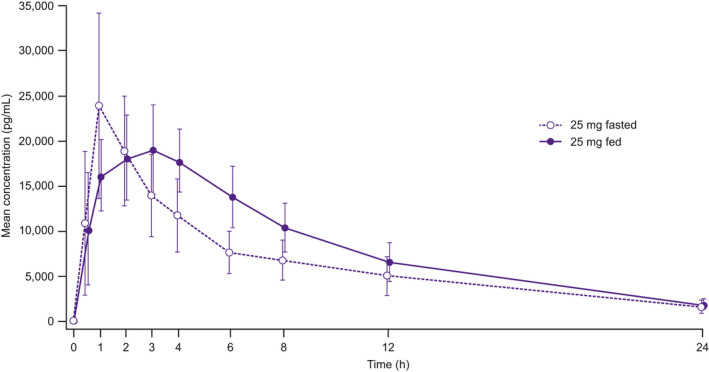
Mean (SD) enpatoran plasma concentration‐time profiles following single administration in fasting (Study Part A) and fed (Study Part C) states (PK analysis set). PK, pharmacokinetic; SD, standard deviation
3.5. Pharmacodynamics of enpatoran
Results of cytokine immunoassays showed exposure‐dependent inhibition of ex vivo‐stimulated cytokine secretion. Peak TLR7/8 target modulation by enpatoran was observed at 2 h post‐dose and diminished over 24 h, with maximum inhibition at a dose of 200 mg (Figure 5A). At least 60% inhibition of IL‐6 secretion was maintained throughout the dosing interval with enpatoran doses of 200 mg QD and 25 and 50 mg BID.
FIGURE 5.
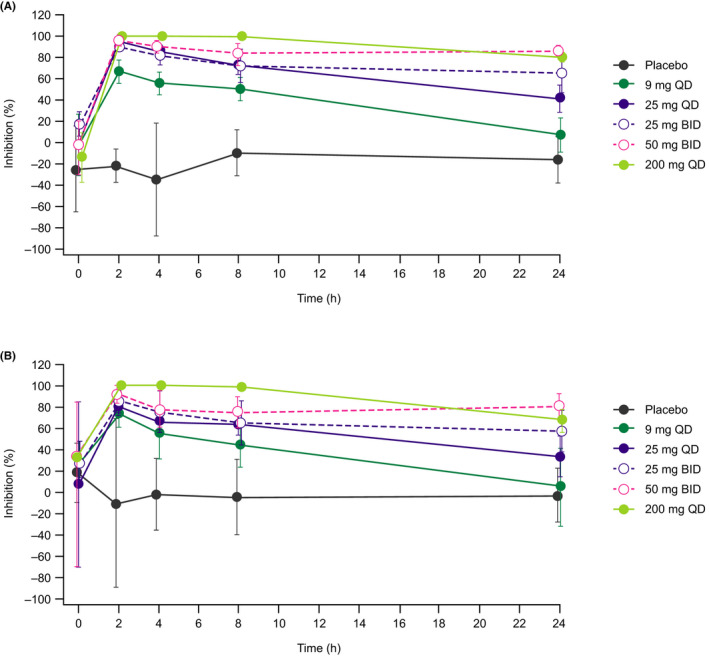
Mean (SD) percentage inhibition–time profile of (A) interleukin‐6 and (B) interferon α assessed using an ex vivo‐stimulated cytokine immunoassay on Day 1 of enpatoran multiple‐ascending‐dose administration (PD analysis set). BID, twice daily; PD, pharmacodynamic; QD, once daily; SD, standard deviation
A similar trend was observed for enpatoran inhibition of IFN‐α (Figure 5B), although the inhibitory effect of enpatoran on IFN‐α was less potent than on IL‐6.
4. DISCUSSION
The TLR family of pattern recognition receptors are now recognized as having an important role in both innate and adaptive immunity and the body's normal defense against foreign molecules. However, evidence suggests that overactivation of TLR7 and TLR8 in response to endogenous RNA may drive the development and progression of certain autoimmune diseases, such as SLE, and therefore inhibition of such activation represents a possible therapeutic target.6, 7 Also, inhibition of TLR7/8 at a critical point in the progression of COVID‐19 may suppress the cytokine storm and improve survival outcomes.18
Enpatoran is a small‐molecule, dual TLR7 and TLR8 antagonist which has been shown in a range of in vitro assays and preclinical studies, including mouse models of SLE, to specifically inhibit TLR7/8 signaling.21 In light of these positive preclinical findings, enpatoran warrants further investigation in human subjects to evaluate whether it similarly inhibits TLR7 and TLR8 activation, reduces cytokine secretion and modulates disease activity in patients with autoimmune diseases, such as SLE, and those with viral infections that stimulate this pathway.2
As an initial step in clinical investigation, this FIH, phase 1, randomized, placebo‐controlled study was undertaken to assess the safety, tolerability, PK, and PD of single and multiple daily doses of enpatoran up to 200 mg administered as a solution for 14 days and to explore the effect of food on the PK of enpatoran.
Our results showed that enpatoran was well tolerated across the dose ranges tested, with no safety signals observed. There were no serious or dose‐limiting TEAEs. TEAEs occurred in 14% of participants, all of which were classified as mild or moderate. The most common TEAEs reported were flatulence and headache. All TEAEs resolved and none required participants to withdraw from the study.
PK parameters were found to be linear and dose‐proportional over the evaluated range of 1–200 mg after both single and multiple dosing, and enpatoran was rapidly absorbed. The terminal half‐life of enpatoran (6.81–10.6 h after a single dose) was found to be shorter than predicted from a preclinical model (based on in vitro to in vivo extrapolation and allometric scaling), with a higher apparent clearance (120–162 L/h after a single dose); this is well‐manageable by increasing the dosing frequency to twice daily. The seeming trend of increased enpatoran half‐life with increasing single doses is due to the different number of sampling times used to derive PK parameters. At higher doses, more samples were above the LLOQ, therefore contributing to a higher mean half‐life. This is supported by the fact that the same trend was not observed with multiple dosing. Terminal half‐life after 14 days dosing ranged from 7.46–8.74 h.
It is recognized that the PK of a compound administered orally may be affected by food. The exploratory assessment of high‐fat, high‐calorie food consumption on enpatoran exposure (in the 25 mg dose cohort) revealed a modest increase in AUC0– t, a delay in t max and a slight decrease in C max.
Single and multiple ascending doses of enpatoran up to 200 mg showed effective TLR7/8 target modulation in an exposure‐dependent manner, as assessed by ex vivo cytokine‐release immunoassays. The lowest efficacious dose of enpatoran in mouse models of SLE achieved 60% sustained inhibition of IL‐6. The highest three doses of enpatoran tested (200 mg QD, 25 mg BID, and 50 mg BID) maintained at least 60% inhibition of IL‐6 secretion throughout the dosing interval. Enpatoran was less potent at inhibiting IFN‐α than IL‐6, which was consistent with findings in preclinical studies and the proposed mode of action of enpatoran.21 Population PK/PD modeling based on PK and PD data from this study was used to inform dose selection for further clinical trials of enpatoran, supporting the use of enpatoran BID dosing regimens.23
Investigation of TLR antagonists and their signaling pathways as potential therapeutic targets for autoimmune diseases continues to progress and a number of single and dual TLR7 and/or TLR8 inhibitors are currently in early development. Synthetic oligonucleotide‐based antagonists, including TLR7/9 and TLR7/8/9 inhibitors, have been developed, but they require frequent subcutaneous injections that are not practical clinically.24, 25 Phase 1 results of a dual TLR7/8 antagonist have been reported, but there is currently a lack of information on its potential.26 Small molecules specifically targeting TLR7 or TLR8 have also been reported, although they have not yet been tested in vivo.27, 28
The aim is to develop more effective treatments for notably difficult‐to‐manage autoimmune diseases, where there are often contraindications to currently available therapies. In the case of SLE, antimalarials have long been the cornerstone of treatment. Therapeutic options are sparse and off‐label treatments are commonly used, including corticosteroids, mycophenolate mofetil, calcineurin inhibitors, and cyclophosphamide for extremely severe renal disease.29 The monoclonal antibody belimumab is the only biologic agent approved for SLE to date. The selection of treatment depends on the patient's disease activity and comorbidities, and is most often limited by drug toxicities.29 Through dual inhibition of TLR7 and TLR8, enpatoran has a broad inhibitory profile that differentiates it from existing SLE treatments, including belimumab which only targets B cells. There is also great interest in the use of TLR antagonists at a critical point in COVID‐19 to reduce the hyper‐inflammatory response and prevent progression to a cytokine storm.
It is worth noting that, because the study population was limited to healthy volunteers, the results may not necessarily be generalizable to patients with SLE who have elevated cytokines correlating with active disease. In addition, the study was not designed to assess safety parameters over the long term and the preliminary assessment of dose proportionality is limited for the BID groups by the inclusion of only two dose levels.
Evaluation of the safety and tolerability of enpatoran, which has dual TLR7/8 antagonist activity, and assessment of its PK and PD parameters in healthy individuals gives a clear indication that this molecule warrants further investigation as a potential treatment for diseases characterized by overactivation of TLR7 and TLR8. In light of these results, a phase 2 study of enpatoran in COVID‐19 pneumonia (NCT04448756) and a phase 1b study of enpatoran in SLE (NCT04647708) are ongoing, both of which are exploring BID dosing regimens.
DISCLOSURE
AP, LKS, ABy, Aba, and CV are employees of the healthcare business of Merck KGaA, Darmstadt, Germany. JS and KG are employees of EMD Serono, Billerica, MA, USA. At the time of the study, VO was an employee of EMD Serono, Billerica, MA, USA. NM is an employee of Cytel, Paris, France. CR and DS are employees of Nuvisan GmbH, Neu‐Ulm, Germany. EH is a consultant for Nuventra, Inc.
AUTHOR CONTRIBUTION
AP, JS, LKS, EH, NM, and KG contributed to study design and/or conception. AP, JS, LKS, CV, EH, NM, and KG contributed to the acquisition, analysis or interpretation of data. All authors reviewed the manuscript, approved the final version and agree to be accountable for all aspects of the work.
Supporting information
Data S1
Figure S1
ACKNOWLEDGEMENTS
The authors thank those who took part in the study. The study was funded by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945). Medical writing support was funded by the healthcare business of Merck, KGaA, Darmstadt, Germany and provided by Bioscript Science, Macclesfield, UK, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Port A, Shaw JV, Klopp‐Schulze L, et al. Phase 1 study in healthy participants of the safety, pharmacokinetics, and pharmacodynamics of enpatoran (M5049), a dual antagonist of toll‐like receptors 7 and 8. Pharmacol Res Perspect. 2021;9:e00842. 10.1002/prp2.842
Funding information
The study and medical writing support were funded by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945).
DATA AVAILABILITY STATEMENT
Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to the healthcare business of Merck KGaA, Darmstadt, Germany's Data Sharing Policy. All requests should be submitted in writing to the healthcare business of Merck KGaA, Darmstadt, Germany's data sharing portal. When the healthcare business of Merck KGaA, Darmstadt, Germany, has a co‐research, co‐development, or co‐marketing or co‐promotion agreement, or when the product has been out licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, the healthcare business of Merck KGaA, Darmstadt, Germany, will endeavor to gain agreement to share data in response to requests.
REFERENCES
- 1.Zhang Z, Ohto U, Shibata T, et al. Structural analyses of toll‐like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. 2018;25:3371‐3381.e5. [DOI] [PubMed] [Google Scholar]
- 2.Bender AT, Tzvetkov E, Pereira A, et al. TLR7 and TLR8 differentially activate the IRF and NF‐kappaB pathways in specific cell types to promote inflammation. Immunohorizons. 2020;4:93‐107. [DOI] [PubMed] [Google Scholar]
- 3.Gorden KB, Gorski KS, Gibson SJ, et al. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259. [DOI] [PubMed] [Google Scholar]
- 4.Larangé A, Antonios D, Pallardy M, Kerdine‐Römer S. TLR7 and TLR8 agonists trigger different signaling pathways for human dendritic cell maturation. J Leukoc Biol. 2009;85:673‐683. [DOI] [PubMed] [Google Scholar]
- 5.de Marcken M, Dhaliwal K, Danielsen AC, Gautron AS, Dominguez‐Villar M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci Signal. 2019;12:eaaw1347. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y‐W, Tang W, Zuo J‐P. Toll‐like receptors: potential targets for lupus treatment. Acta Pharmacol Sin. 2015;36:1395‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Celhar T, Fairhurst A‐M. Toll‐like receptors in systemic lupus erythematosus: potential for personalized treatment. Front Pharmacol. 2014;5:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrivastav M, Niewold TB. Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front Immunol. 2013;4:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng Y, Zou L, Yan D, et al. Extracellular microRNAs induce potent innate immune responses via TLR7/MyD88‐dependent mechanisms. J Immunol. 2017;199:2106‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vollmer J, Tluk S, Schmitz C, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves toll‐like receptors 7 and 8. J Exp Med. 2005;202:1575‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee YH, Choi SJ, Ji JD, Song GG. Association between toll‐like receptor polymorphisms and systemic lupus erythematosus: a meta‐analysis update. Lupus. 2016;25:593‐601. [DOI] [PubMed] [Google Scholar]
- 12.Lee PY, Kumagai Y, Li Y, et al. TLR7‐dependent and FcgammaR‐independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995‐3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell Richard A, Shlomchik MJ. Toll‐like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417‐428. [DOI] [PubMed] [Google Scholar]
- 14.Fairhurst AM, Hwang SH, Wang A, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. 2008;38:1971‐1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savarese E, Steinberg C, Pawar RD, et al. Requirement of toll‐like receptor 7 for pristane‐induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107‐1115. [DOI] [PubMed] [Google Scholar]
- 16.Patra R, Chandra Das N, Mukherjee S. Targeting human TLRs to combat COVID‐19: a solution? J Med Virol. 2021;93:615‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS‐CoV‐2 spike glycoprotein with ACE‐2 receptor homologs and human TLRs. J Med Virol. 2020;92:2105‐2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun X, Wang T, Cai D, et al. Cytokine storm intervention in the early stages of COVID‐19 pneumonia. Cytokine Growth Factor Rev. 2020;53:38‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khanmohammadi S, Rezaei N. Role of toll‐like receptors in the pathogenesis of COVID‐19. J Med Virol. 2021;93(5):2735‐2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vlach J, Bender AT, Przetak M, et al. Discovery of M5049: a novel selective TLR7/8 inhibitor for treatment of autoimmunity. J Pharmacol Exp Ther. 2021;376:397‐409. [DOI] [PubMed] [Google Scholar]
- 22.Jurk M, Heil F, Vollmer J, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R‐848. Nat Immunol. 2002;3:499‐599. [DOI] [PubMed] [Google Scholar]
- 23.Klopp‐Schulze L, Shaw J, Dong J, Khandelwal A, Adams E, Goteti K. P1–050 Model‐informed dose selection of dual toll‐like receptor 7/8 inhibitor M5049 for the treatment of COVID‐19 pneumonia. Clin Pharmacol Ther. 2021;109(S1):S20. [Google Scholar]
- 24.Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of toll‐like receptor‐7 (TLR‐7) or TLR‐7 plus TLR‐9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721‐1731. [DOI] [PubMed] [Google Scholar]
- 25.Balak DM, van Doorn MB, Arbeit RD, et al. IMO‐8400, a toll‐like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo‐controlled trial in patients with moderate‐to‐severe plaque psoriasis. Clin Immunol. 2017;174:63‐72. [DOI] [PubMed] [Google Scholar]
- 26.Nakai K, Yasadua S, Chang M, et al. Safety, pharmacokinetics, and pharmacodynamics in first‐in‐human study of a novel compound E6742, a toll‐like receptor 7 and 8 antagonist. In: 18th World Congress of Basic and Clinical Pharmacology, Kyoto, Japan. 2018. https://www.jstage.jst.go.jp/article/jpssuppl/WCP2018/0/WCP2018_PO1‐11‐13/_pdf/‐char/ja. [Google Scholar]
- 27.Zhang S, Hu Z, Tanji H, et al. Small‐molecule inhibition of TLR8 through stabilization of its resting state. Nat Chem Biol. 2018;14:58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bou Karroum N, Moarbess G, Guichou JF, et al. Novel and selective TLR7 antagonists among the imidazo[1,2‐a]pyrazines, imidazo[1,5‐a]quinoxalines, and pyrazolo[1,5‐a]quinoxalines series. J Med Chem. 2019;62:7015‐7031. [DOI] [PubMed] [Google Scholar]
- 29.Basta F, Fasola F, Triantafyllias K, Schwarting A. Systemic lupus erythematosus (SLE) therapy: the old and the new. Rheumatol Ther. 2020;7:433‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figure S1
Data Availability Statement
Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to the healthcare business of Merck KGaA, Darmstadt, Germany's Data Sharing Policy. All requests should be submitted in writing to the healthcare business of Merck KGaA, Darmstadt, Germany's data sharing portal. When the healthcare business of Merck KGaA, Darmstadt, Germany, has a co‐research, co‐development, or co‐marketing or co‐promotion agreement, or when the product has been out licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, the healthcare business of Merck KGaA, Darmstadt, Germany, will endeavor to gain agreement to share data in response to requests.