

Summary
Determining the antigen specificities of the endogenous T-cell repertoire is important for screening naturally occurring or therapy-induced T-cell immunity and may help identify novel targets for T-cell-based therapies. Here, we describe a rapid, sensitive, and high-throughput protocol for expanding antigen-specific T cells from human peripheral blood mononuclear cells in vitro following peptide stimulation and detecting antigen-specific effector cytokine formation by flow cytometry. Our approach can be applied to examining specific T-cell subsets from various tissues.
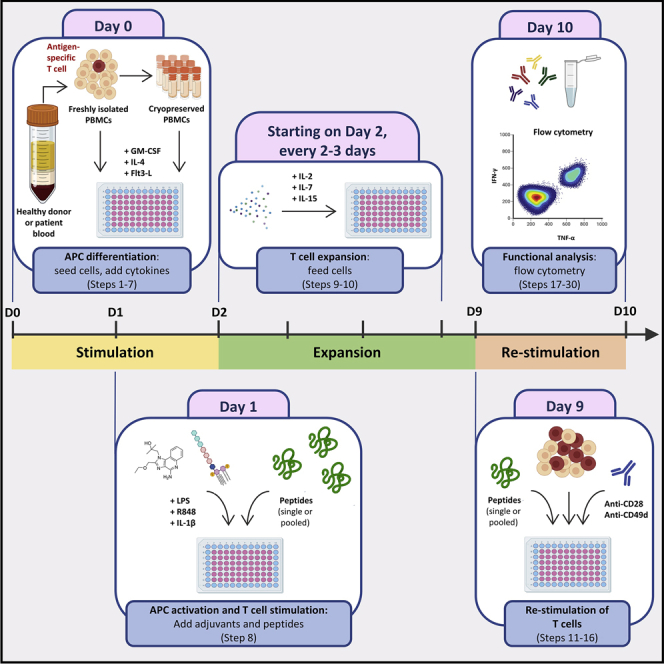
For complete details on the use and execution of this protocol, please refer to Roudko et al. (2020) and Cimen Bozkus et al. (2019).
Subjeact areas: Cell culture, Flow Cytometry/Mass Cytometry, Cell-based Assays, High Throughput Screening, Immunology
Graphical abstract
Highlights
-
•
Detailed protocol to expand human antigen-specific T cells from unfractionated PBMCs
-
•
Platform to prime naïve T cells and stimulate memory T cells
-
•
Platform to test cross-reactivity of T cells
-
•
Use of flow cytometry to easily detect reactive T cells
Determining the antigen specificities of the endogenous T-cell repertoire is important for screening naturally occurring or therapy-induced T-cell immunity and may help identify novel targets for T-cell-based therapies. Here, we describe a rapid, sensitive, and high-throughput protocol for expanding antigen-specific T cells from human peripheral blood mononuclear cells in vitro following peptide stimulation and detecting antigen-specific effector cytokine formation by flow cytometry. Our approach can be applied to examining specific T-cell subsets from various tissues.
Before you begin
The protocol below describes the specific steps for evaluating antigen-specific T cell responses following their expansion from human PBMCs. However, we have also successfully used this protocol with single cell suspensions isolated from human tumor tissues.
While the protocol we describe here is performed using bulk T cells in PBMCs, the same protocol can be used to evaluate antigen-reactivity within individual T cell subsets of interest by co-culturing sorted T cells with autologous or major histocompatibility complex (MHC)-matched antigen-presenting cells (APCs) at desired ratios. We have successfully performed such co-cultures with T cells and APCs seeded at 1:1 ratio. However, investigators should test varying ratios to determine the optimal T:APC ratio in their experiments.
All sterile procedures are performed in a Class II biological safety cabinet with standard aseptic technique. Cells are cultured in a humidified incubator at 37°C with 5% CO2.
Please ensure that the investigators have access to a flow cytometer with multi-parameter capabilities. Alternatively, other methods, such as the enzyme-linked immune absorbent spot (ELISpot) assay, can be utilized to detect T cell responses.
Peptide preparation
Timing: 1–3 h
This protocol uses synthetic peptides to stimulate T cells, thereby enabling antigen-specific T cell proliferation and detection of antigen-induced effector cytokine production by T cells. Therefore, peptides spanning the entire protein sequence being tested should be synthesized prior to experiment start.
-
1.Dissolve lyophilized peptides in an appropriate solvent and dilute to a desired concentration.
-
a.Assure that the peptides are completely dissolved, and no aggregates are formed.
-
a.
Note: Follow the manufacturer’s guidelines for how to dissolve peptides and the duration of storage.
Note: The final concentration of the peptide stock can be determined by the investigator. The solvent should not promote peptide degradation and be compatible with the experimental application. In general, we recommend DMSO as the initial solvent and a stock peptide concentration of 2–10 mM.
CRITICAL: It is important to limit the volume of DMSO used in peptide stocks. We recommend not exceeding a maximum final concentration of 0.1% DMSO in cell cultures.
-
2.
To avoid multiple freeze-thaw cycles, divide dissolved peptides into aliquots.
-
3.
Snap-freeze aliquoted peptides by immersion in liquid nitrogen and store at −80°C, protected from light.
Note: The assay can be performed using a single peptide or pooled peptides. To ensure successful T cell stimulation, please perform pilot experiments to identify the optimal peptide concentration and number of peptides to be pooled.
Media preparation
-
4.
Prepare complete RPMI media as outlined in the materials and equipment section.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC mouse anti-human CD3 | BioLegend | Clone: SK7, Cat# 344803 |
| APC mouse anti-human CD8 | BioLegend | Clone: RPA-T8, Cat# 301049 |
| Brilliant Violet 785 mouse anti-human CD4 | BioLegend | Clone: RPA-T4, Cat# 300554 |
| PE mouse anti-human IFN-γ | BioLegend | Clone: B27, Cat# 506507 |
| PE/Cyanine7 mouse anti-human TNF-α | BioLegend | Clone: Mab11, Cat# 502930 |
| Brilliant Violet 605 rat anti-human IL-2 | BioLegend | Clone: MQ1-17H12, Cat# 500332 |
| Purified mouse anti-human CD28 | BD Biosciences | Clone: CD28.2, Cat# 555726 |
| Purified mouse anti-human CD49d | BD Biosciences | Clone: 9F10, Cat# 555502 |
| Biological samples | ||
| Healthy human peripheral blood mononuclear cells | New York Blood Center | https://nybloodcenter.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| Custom peptides | GenScript | https://www.genscript.com/peptide.html |
| PepMix Human (MOG) | JPT | PM-MOG |
| CEFT pool | JPT | PM-CEFT-3 |
| Human granulocyte-macrophage colony-stimulating factor (GM-CSF) (LEUKINE® (sargramostim)) | Sanofi | NDC 0024-5843-05 |
| Human IL-4 | R&D Systems | Cat# 204-IL-010 |
| Human Flt-3 Ligand | R&D Systems | Cat# 308-FKN-025 |
| R848 | InvivoGen | Cat# tlrl-r848-5 |
| Ultrapure LPS, Salmonella minnesota | InvivoGen | Cat# tlrl-smlps |
| Human IL-1 beta | R&D Systems | Cat# 201-LB-010 |
| Human IL-2 | R&D Systems | Cat# 202-IL-050 |
| Human IL-7 | R&D Systems | Cat# 207-IL-025 |
| Human IL-15 | PeproTech | Cat# 200-15 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# 79346-1MG |
| Ionomycin calcium salt | Sigma-Aldrich | Cat# I0634-1MG |
| Software and algorithms | ||
| FACSDiva | BD Biosciences | N/A |
| FlowJo v10.7.1 | Tree Star | www.flowjo.com/ |
| Prism v9.1.0 | GraphPad | www.graphpad.com/ |
| Other | ||
| UltraComp eBead Plus Compensation Beads | Thermo Fischer Scientific | Cat# 01-3333-41 |
| ArC™ Amine Reactive Compensation Bead Kit | Thermo Fischer Scientific | Cat# A10346 |
| LIVE/DEAD Fixable Blue Dead Cell Stain Kit | Thermo Fischer Scientific | Cat# L34962 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| GolgiStop: Protein Transport Inhibitor (containing Monensin) | BD Biosciences | Cat# 554724 |
| GolgiPlug: Protein Transport Inhibitor (containing Brefeldin A) | BD Biosciences | Cat# 555029 |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | Cat# D4513-1VL |
| DPBS, calcium, magnesium | Gibco | Cat# 14040133 |
| X-VIVO 15 Serum-free Hematopoietic Cell Medium | Lonza | Cat# 04-418Q |
| RPMI 1640 Medium | Gibco | Cat# 11875-093 |
| Heat inactivated human serum | Gemini Bio-Products | Cat# 100-512 |
| Nalgene™ Rapid-Flow™ Sterile Single Use Vacuum Filter Units | Thermo Fischer Scientific | Cat# 566-0020 |
| 96-well Clear Round Bottom TC-treated Microplate | Corning | Cat# 3799 |
Materials and equipment
Cryopreservation solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Heat inactivated human serum | 90% | 45 mL |
| DMSO | 10% | 5 mL |
| Total | n/a | 50 mL |
Note: Heat inactivated human serum aliquots are stored at −20°C and thawed in 37°C water bath prior to use. Cryopreservation solution can be used freshly prepared or stored at 4°C up to a week.
DNase I stock solution
| Reagent | Final concentration | Amount |
|---|---|---|
| DPBS with Ca++ and Mg++ | n/a | 11 mL |
| DNase I | 1 mg/mL | 11 mg |
| Total | n/a | 11 mL |
Note: Aliquot and store DNase I solution at −80°C for up to 3 years. Thaw aliquots upon use.
2× cytokine solution (for 60 wells including 10% extra volume)
| Reagent | Final concentration | Amount |
|---|---|---|
| X-VIVO 15 | n/a | ~6.6 mL |
| Human GM-CSF | 2000 IU/mL | 4.4 μL |
| Human IL-4 | 1000 IU/mL | 3.3 μL |
| Human Flt3-L | 100 ng/mL | 6.6 μL |
| Total | n/a | 6.6 mL |
Note: X-VIVO 15 media is stored at 4°C. All other reagent stock solutions are stored at −80°C. 2× cytokine solution is prepared fresh upon use. The amounts noted in the table are calculated for the following stock concentrations to serve as an example: 3×106 IU/mL, 2×106 IU/mL and 100 μL/mL for GM-CSF, IL-4 and Flt3-L, respectively.
2× adjuvant solution (for 60 wells including 10% extra volume)
| Reagent | Final concentration | Amount |
|---|---|---|
| X-VIVO 15 | n/a | ~6.6 mL |
| R848 | 20 μM | 13.2 μL |
| LPS | 200 ng/mL | 1.32 μL |
| IL-1β | 20 ng/mL | 26.4 μL |
| Total | n/a | 6.6 mL |
Note: X-VIVO 15 media is stored at 4°C. All other reagent stock solutions are stored at −80°C. 2× adjuvant solution is prepared fresh upon use. The amounts noted in the table are calculated for the following stock concentrations to serve as an example: 10 mM, 1 mg/mL and 5 μg/mL for R848, LPS and IL-1β, respectively.
Complete RPMI media (R10)
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 Medium | n/a | 435 mL |
| HEPES (1M) | 10 mM | 5 mL |
| Gentamicin (10 mg/mL) | 0.1 mg/mL | 5 mL |
| GlutaMAX-I (100×) | 1× | 5 mL |
| Human serum | 10% | 50 mL |
| Total | n/a | 500 mL |
Note: After mixing all the components above, filter complete RPMI media (R10) through a 0.20 μm filter. Store R10 at 4°C up to 2 weeks.
2× feeding solution (for 60 wells including 10% extra volume)
| Reagent | Final concentration | Amount |
|---|---|---|
| R10 media | n/a | ~6.6 mL |
| IL-2 | 20 IU/mL | 6.6 μL |
| IL-7 | 20 ng/mL | 6.6 μL |
| IL-15 | 20 ng/mL | 2.64 μL |
| Total | n/a | 6.6 mL |
Note: R10 media is stored at 4°C. All other reagent stock solutions are stored at −80°C. 2× feeding solution is prepared fresh upon use. The amounts noted in the table are calculated for the following stock concentrations to serve as an example: 2×105 IU/mL, 20 μg/mL and 50 μg/mL for IL-2, IL-7 and IL-15, respectively.
2× re-stimulation solution (for 60 wells including 10% extra volume)
| Reagent | Final concentration | Amount |
|---|---|---|
| R10 media | n/a | ~6.6 mL |
| Anti-CD28 (0.5 mg/mL) | 2 μg/mL | 26.4 μL |
| Anti-CD49d (0.5 mg/mL) | 2 μg/mL | 26.4 μL |
| Total | n/a | 6.6 mL |
Note: All reagents are stored at 4°C. 2× re-stimulation solution is prepared fresh upon use.
11× protein transport inhibition solution (for 60 wells including 10% extra volume)
| Reagent | Final concentration | Amount |
|---|---|---|
| R10 media | n/a | 1296 μL |
| GolgiStop: Protein Transport Inhibitor (Containing Monensin) | 7.33 μL/mL | 9.68 μL |
| GolgiPlug: Protein Transport Inhibitor (Containing Brefeldin A) | 11 μL/mL | 14.52 μL |
| Total | n/a | 1.32 mL |
Note: All reagents are stored undiluted at 4°C. 11× protein transport inhibition solution is prepared fresh upon use. Final concentrations are selected based on manufacturer’s recommendation.
Viability staining stock solution
Prepared according to the manufacturer’s recommendation in which 50 μL of DMSO is added into 1 vial of lyophilized LIVE/DEAD Fixable Blue Dead Cell Stain. Once suspended, this stock solution is fresh or kept at 4°C for short term storage. For longer term storage, up to 2 weeks, suspended viability stock solution can be stored at −20°C as recommended by the manufacturer.
Step-by-step method details
Seeding cells and supplementing with cytokines
The following steps are performed on “Day 0” and describe how to start T cell cultures using unfractionated PBMCs. PBMCs are stimulated by a cytokine mixture containing GM-CSF, IL-4, and Flt3-L, which support the development of distinct dendritic cell (DC) subsets (Parajuli et al., 2001). Sterile reagents and aseptic technique should be used while performing the following steps.
-
1.Cryopreserve PBMCs.
-
a.Prepare cryopreservation solution containing 90% human serum and 10% DMSO.
-
b.Suspend isolated PBMCs at 5–25 million/mL concentration in cryopreservation solution.
-
c.Aliquot 1 mL of suspended PBMCs into cryovials.
-
d.Place cryovials into a cell freezing container, such as Mr. Frosty, and store at −80°C for 24–48 h.
-
e.Transfer cryovials into liquid nitrogen for long term storage.
-
a.
-
2.Thaw cryopreserved PBMCs.
-
a.Remove cryovials containing cryopreserved PBMCs from liquid nitrogen storage.
-
b.Immediately place cryovials into 37°C water bath.
-
c.Remove vials from the water bath as soon as cells are almost thawed (typically <1 min) with a small pellet remaining.
-
d.Spray the outside of the vials with 70% ethanol.
-
e.Transfer the vials into a biological safety cabinet.
-
a.
-
3.Treat thawed cells with DNase I.
-
a.Add 25 μL of DNase I stock solution into 24 mL X-VIVO 15 media to obtain a final 1 μg/mL concentration (approximately 2 U/mL).
-
b.Dilute 1 mL thawed cells by adding drop by drop into 24 mL X-VIVO 15 media containing DNase I.
-
a.
-
4.Resuspend PBMCs at 106 cells/mL in X-VIVO 15.
-
a.Centrifuge diluted cells at 400×g for 10 min.
-
b.Aspirate supernatant and resuspend cells in 5 mL X-VIVO 15 media.
-
c.Pass the resuspended cells through a 70 μm cell strainer.
-
d.Take an aliquot and count the total number of cells.
-
e.Adjust the volume of X-VIVO 15 media to obtain a cell concentration of 106 cells/mL.
-
a.
Note: If cells still appear clumpy, higher DNase I concentrations and/or longer incubation times may be adopted.
-
5.Seed cells.
-
a.Transfer 100 μL of the resuspended cells per well into 96-well U bottom plates to seed 105 cells/well.
-
a.
Optional: Freshly isolated PBMCs may also be used instead of cryopreserved PBMCs. We observed comparable assay performance using fresh or cryopreserved cells isolated from healthy donors and cancer patients, except, for certain cancer types freshly isolated cells yielded better results in which we detected greater IFN-γ or TNF-α expressing antigen-specific T cell frequencies after expansion. Therefore, while working with patient PBMCs, we recommend using freshly isolated cells, if available.
Note: The total number of wells to be seeded can be determined by the user. We recommend seeding at least 10 wells (1×106 cells) per group to enable detection of rare T cell clones.
Pause point: Seeded cells can be kept 12–24 h at a humidified incubator at 37°C with 5% CO2.
-
6.Add cytokines.
-
a.Prepare 2× cytokine solution.
-
b.Transfer 100 μL of the 2× cytokine solution into the wells containing 100 μL of cells, yielding the following final concentrations: 1000 IU/mL GM-CSF, 500 IU/mL IL-4, and 50 ng/mL Flt3-L.
-
c.Mix gently by pipetting.
-
a.
-
7.
Culture cells for 24 h at a humidified incubator at 37°C with 5% CO2.
Stimulating cells
Timing will change based on the number of the antigens being tested.
The following steps are performed on “Day 1”, using sterile reagents and aseptic technique. Here the goals are to induce the activation of antigen presenting cells (APCs) and to stimulate antigen-specific T cells. Adjuvants, namely Toll-like receptor agonists lipopolysaccharide (LPS) and R848, as well as the pro-inflammatory cytokine IL-1β, are used to induce APC activation, thereby enhancing their capacity to stimulate T cells (Lovgren et al., 2017, Wesa and Galy, 2001). Antigen-specific T cell expansion is achieved through stimulation of T cells by synthetic peptides. A single peptide or pooled peptides can be used depending on the experimental design. We have successfully used both short (8–12mers) and long (14-25mers) peptides. Routinely, we use pools of overlapping 15mers spanning the protein sequences of interest, which can elicit both CD4+ and CD8+ T cell responses, suggesting the feasibility of antigen cross-presentation in this assay system (Cimen Bozkus et al., 2019). In general, we pool less than 20 peptides in order to avoid potential peptide competition. However, we have successfully detected antigen-specific T cell responses with pools containing up to 125 peptides.
-
8.Stimulate cells.
-
a.Transfer the plates into a biological safety cabinet.
-
b.Remove 100 μL of solution from each well.
-
c.Add 100 μL of 2× adjuvant solution containing peptides into appropriate wells to obtain a final concentration of 10 μM R848, 100 ng/mL LPS, 10 ng/mL IL-1β and 1 μM of each peptide.
-
i.Calculate the total volume of 2× adjuvant solution needed based on your plate design and prepare 2× adjuvant solution.
-
ii.Divide 2× adjuvant solution into separate tubes for each condition you are testing.
-
iii.Add the peptides and the vehicle control into each tube.
-
iv.Mix well.
-
i.
-
d.Mix gently by pipetting.
-
e.Culture cells for 24 h at a humidified incubator at 37°C with 5% CO2.
-
a.
Note: We recommend using a multichannel pipettor while removing media or mixing the contents of the wells. Fresh tips should be used to avoid contamination between test conditions.
Note: We recommend using 1 μM final concentration of each peptide even when added as pooled, though lower peptide concentrations may also yield successful results. We recommend not exceeding 20 peptides per pool to avoid peptide competition. However, investigators should conduct pilot experiments to identify the optimal peptide concentrations and number of peptides to be pooled.
Note: To pool peptides, we combine equal volumes of individual peptides in a separate tube and mix well. It is important that each individual peptide included in the pool has the same concentration to assure equal representation within the pool.
Note: In addition to negative controls to assess background, such as media alone or vehicle control (e.g., DMSO), and other experimental controls determined by the investigator, we recommend using myelin-oligodendrocyte glycoprotein (MOG) and CEFT (previously defined MHC-I or II-restricted T cell epitopes from Cytomegalovirus, Epstein-Barr virus, Influenza virus or Clostridium tetani) pools as negative and positive peptide stimulation controls, respectively.
Note: If testing for the role of immune checkpoint blockade, blocking antibodies can be added at this stage supplemented in the 2× adjuvant solution together with the peptides.
Feeding cells
Timing will change based on the number of plates being fed.
The following steps describe how to feed T cells throughout the culture period using a cytokine cocktail containing IL-2, IL-7, and IL-15. This cytokine combination supports in vitro T cell expansion (Geginat et al., 2001). Notably, here we switch to using R10 media, RPMI supplemented with 10% human serum, which aids T cell proliferation. Feedings are performed using sterile reagents and aseptic technique on “Day 2, Day 4 and Day 7”. However, feedings after “Day 2” can be performed on alternative days. We recommend feeding the cells every 2–3 d.
-
9.Feed cells.
-
a.Transfer the plates into a biological safety cabinet.
-
b.Remove 100 μL of solution from each well.
-
c.Add 100 μL of 2× feeding solution to obtain a final concentration of 10 IU/mL IL-2, 10 ng/mL IL-7, 10 ng/mL IL-15.
-
i.Calculate the total volume of feeding solution needed based on your plate design and prepare 2× feeding solution.
-
i.
-
d.Mix gently by pipetting.
-
a.
-
10.
Culture cells for 48–72 h at a humidified incubator at 37°C with 5% CO2.
Note: For the last feeding, we recommend using R10 media without the cytokines in the feeding solution. This might help reduce the background.
Re-stimulating cells
Timing will change based on the number of antigens and donors being tested.
The steps below are performed on “Day 9” using sterile reagents and aseptic technique. In this section expanded T cells are re-stimulated with peptides they were initially stimulated with to induce effector cytokine production, which enables the identification and enumeration of antigen-specific T cells. If pooled peptides were used for the initial stimulation, here pools can be deconvoluted by re-stimulating the cells with the individual peptides constituting each pool, thereby demonstrating the immunogenic peptides within pools. For example, if on “Day 1” cells were stimulated with a peptide pool containing peptides a, b, and c, to deconvolute on “Day 9”, we re-stimulate these expanded cells with the individual a, b or c peptides in separate wells to evaluate the individual contribution of each peptide to elicited T cell responses. Furthermore, these steps can be leveraged to study cross-reactivity of T cells, for example T cells expanded following initial stimulation with mutant peptides can be re-stimulated with corresponding wild type peptides.
-
11.Harvest cells.
-
a.Label tubes with the names of each test condition.
-
b.Transfer the plates into a biological safety cabinet.
-
c.Mix the contents of each well vigorously by pipetting.
-
d.Collect and transfer the contents of wells into appropriate tubes.
-
i.Combine all replicate wells for each test condition.
-
i.
-
e.Centrifuge cells at 400×g for 8 min.
-
f.Aspirate supernatant and resuspend the cells in R10 media.
-
g.Take an aliquot and count the number of cells for each test condition.
-
h.Adjust the volume of R10 media to obtain a cell concentration of 2×106 cells/mL.
-
a.
Note: We routinely resuspend cells in each test condition in 0.5–1 mL R10 media. To count, we make 1:10–1:100 dilutions of resuspended cells in trypan blue, depending on cell density. However, these suggested volumes can be modified according to total harvested cell numbers, which will depend on the initially seeded number of replicate wells and the rate of T cell proliferation.
Note: Please refer to the troubleshooting section (problem 2) for concerns regarding low cell numbers after expansion.
-
12.Seed cells.
-
a.Transfer 100 μL of cells per well into a new 96-well U bottom plate in triplicates for each test group to obtain 2×105 cells/well.
-
a.
-
13.Re-stimulate cells.
-
a.Calculate the total volume of 2× re-stimulation solution needed based on your plate design with an extra amount to account for volume loss during transfer.
-
b.Prepare 2× re-stimulation solution.
-
c.Divide 2× re-stimulation solution into separate tubes for each test condition.
-
d.Add the peptides and control reagents into each tube appropriately and mix well.
-
e.Add 100 μL of 2× re-stimulation solution supplemented with peptides or control reagents onto appropriate wells containing cells.
-
f.Mix by pipetting.
-
a.
Note: Although the contents of 2× re-stimulation solution will be further diluted by the addition of peptides at Step 13.d., we consider this negligible since the added peptide volumes are exceedingly small (e.g., 1:5000 dilution or 0.02%).
Note: We recommend using 1 μM final concentration of each peptide, same as in initial stimulation described in step 8, both when used in a pool or individually. However, lower or greater peptide concentrations may also yield successful results.
Note: As an additional positive control, we recommend stimulating cells with phorbol myristate acetate (PMA) and ionomycin, which potently activates T cells independent of T cell receptor stimulation. We typically use PMA and ionomycin at a final concentration of 50 ng/mL and 1 μg/mL, respectively.
-
14.
Culture cells for 1 h at a humidified incubator at 37°C with 5% CO2.
-
15.Add protein transport inhibitors.
-
a.Prepare 11× protein transport inhibition solution and add 20 μL into each well.
-
b.Mix by pipetting.
-
a.
Optional: Although we utilize both monensin and brefeldin containing reagents in the protein transport inhibition solution, investigators can choose a single reagent to block protein transport that provides the optimal flow cytometric detection for their cytokines of interest. Furthermore, protein transport inhibitors may be supplemented in the 2× re-stimulation solution.
-
16.
Culture cells for 8 h at a humidified incubator at 37°C with 5% CO2.
Note: Investigators should perform pilot experiments to determine the optimal incubation time following peptide stimulation. We found that shorter incubation times yield better cell viability. However, detection of certain activation markers may require longer incubation times.
Optional: If the minimal epitope and the restricting MHC information are known, tetramer staining can be utilized to detect reactive T cells. If performing tetramer experiments, re-stimulation steps can be skipped.
Alternatives: If the goal is to determine the restricting MHC alleles for test peptides, alternative re-stimulation methods should be pursued. For example, cells can be re-stimulated by co-culturing them with peptide-pulsed antigen presenting cells that can be engineered to express desired MHC allele(s).
Staining for flow cytometry
The following details the staining method for flow cytometry to detect cytokine production by T cells. Here we investigate antigen-induced IFN-γ, TNF-α and IL-2 production by CD4+ and CD8+ T cells. However, expression of various activation markers by different T cell subsets can be evaluated.
-
17.
Mix pelleted cells by pipetting and transfer to V bottom 96-well plates.
-
18.
Centrifuge plates at 400×g for 5 min.
-
19.Remove the supernatant.
-
a.Rapidly decant the supernatant by inverting the plates over a waste container.
-
b.Gently tap the plates on a paper towel to remove excess fluid.
-
a.
-
20.Stain cells for viability and extracellular markers.
-
a.Prepare the extracellular staining solution.
-
i.Calculate the total volume of solution required including an extra amount to account for volume loss during transfer.
-
ii.Aliquot the determined volume of PBS.
-
iii.Add antibodies,anti-CD3, anti-CD4, and anti-CD8 at 1:200 dilution to obtain a final concentration of 1 μg/mL, 0.4 μg/mL, and 0.25 μg/mL, respectively.
-
iv.Add viability dye at 1:1000 dilution.
-
i.
-
b.Resuspend cells in 100 μL extracellular staining solution.
-
c.Mix well by pipetting.
-
d.Incubate at 4°C for 20 min protected from light.
-
a.
-
21.Wash cells.
-
a.Add 150 μL of PBS into each well.
-
b.Centrifuge plates at 400×g for 5 min.
-
c.Remove the supernatant.
-
i.Rapidly decant the supernatant by inverting the plates over a waste container.
-
ii.Gently tap the plates on a paper towel to remove excess fluid.
-
i.
-
a.
-
22.Fix and permeabilize the cells.
-
a.Add 100 μL of Fixation/Permeabilization solution (BD) into each well.
-
b.Mix well by pipetting.
-
c.Incubate at 4°C for 30 min protected from light.
-
a.
-
23.Wash cells.
-
a.Add 150 μL of 1× Perm/Wash Buffer (BD) into each well.
-
b.Centrifuge plates at 400×g for 5 min.
-
c.Remove the supernatant.
-
i.Rapidly decant the supernatant by inverting the plates over a waste container.
-
ii.Gently tap the plates on a paper towel to remove excess fluid.
-
i.
-
a.
-
24.Stain cells for intracellular proteins.
-
a.Prepare the intracellular staining solution.
-
i.Calculate the total volume of solution required including an extra amount to account for volume loss during transfer.
-
ii.Aliquot the determined volume of 1× Perm/Wash Buffer (BD).
-
iii.Add antibodies, anti-IFN-γ, anti-TNF-α, and anti-IL-2 at 1:200 dilution to obtain a final concentration of 0.25 μg/mL, 1 μg/mL, and 0.15 μg/mL, respectively.
-
i.
-
b.Resuspend cells in 100 μL intracellular staining solution.
-
c.Mix well by pipetting.
-
d.Incubate at 4°C for 30 min protected from light.
-
a.
-
25.Wash cells.
-
a.Add 150 μL of 1× Perm/Wash Buffer (BD) into each well.
-
b.Centrifuge plates at 400×g for 5 min.
-
c.Remove the supernatant.
-
i.Rapidly decant the supernatant by inverting the plates over a waste container.
-
ii.Gently tap the plates on a paper towel to remove excess fluid.
-
i.
-
a.
-
26.
Resuspend cells in 200 μL PBS.
-
27.
Store cells at 4°C protected from light until ready to acquire by flow cytometer.
-
28.Prepare single color compensation controls.
-
a.Label separate test tubes for each antibody used in staining for flow cytometry.
-
b.Vigorously vortex the beads.
-
c.Combine 1 drop of beads, 200 μL PBS and 1 μL antibody in a test tube.
-
d.Mix and incubate at 4°C protected from light for 15–30 min.
-
e.Add 1 mL of PBS and centrifuge tubes at 400×g for 5 min.
-
f.Decant supernatant and resuspend pellet in 250 μL PBS.
-
g.Store cells at 4°C protected from light until ready to acquire by flow cytometer.
-
a.
-
29.
Acquire and record data from experimental samples using a multi-color flow cytometer following fluorescence detector voltage and compensation setup.
-
30.
Use compatible software to analyze flow data, such as FlowJo.
Note: Please refer to the troubleshooting section (problem 3) for concerns if a high background signal for cytokine production is detected in negative control cells.
Alternatives: Here we use human antibodies from BioLegend and viability dye from Thermo Fischer Scientific for flow cytometry staining. However, antibodies and fixable viability dyes from other manufacturers may also be utilized.
Expected outcomes
Here we describe a rapid T cell assay protocol for the expansion of antigen-specific T cells from unfractionated human PBMCs. The use of unfractionated PBMCs in this robust and sensitive assay provides multiple advantages. It enables the testing of antigen-specific T cell responses even when the starting material is limited. We have successfully detected antigen-specific T cell responses by this assay using only 105 PBMCs. Furthermore, PBMCs include multiple antigen-presenting cell (APC) populations, such as monocytes and dendritic cells (DCs), including conventional (cDCs) and plasmacytoid (pDCs) subsets (Collin and Bigley, 2018). Although, their frequencies vary across individuals, monocytes and DCs make up 10%–20% and 1%–2% of PBMCs, respectively (Kleiveland, 2015). The presence of multiple APC populations within PBMCs is leveraged in this assay by treating PBMCs with a cytokine cocktail (GM-CSF, IL-4, and Flt3-L) that supports the development of distinct dendritic cell subsets (Parajuli et al., 2001), and maintains myeloid cell viability (Flt3-L), which together with adjuvants (LPS, R848, and IL-1β) helps elicit a greater magnitude of antigen-specific T cell responses compared to standard T cell assays lacking such components (Figure 1).
Figure 1.

Role of cytokine and adjuvant addition
PBMCs from HLA-A∗02+ healthy donors (n=4) were expanded following stimulation with MART-1 (ELAGIGILTV) or CEFT peptides with or without the addition of cytokines (GM-CSF, IL-4, Flt3-L) and adjuvants (LPS, R848, IL-1β) on Day 0 and Day 1, respectively. Percentage of IFN-γ+ CD8 T cells was measured by flow cytometry on Day 10.
Following peptide stimulation and feeding with IL-2, IL-7, and IL-15, T cells start proliferating. Typically, we observe a reduction in pellet size around day 2–5 of culture, likely due a reduction in non-T cells within PBMCs, followed by an increase in pellet size thereafter. Although the rate of expansion varies between donors, by day 10 the cultures predominantly consist of T cells (Figure 2), which are enriched for antigen-specific T cells of interest. Antigen-specific reactive T cells can be identified by flow cytometric detection of antigen-induced effector cytokine production (Figure 3A). As antigen-specific T cell populations reach to sufficient levels detectable by flow cytometry, we typically perform functional analysis on day 10 (Figure 3B). Another study that expanded CD8+ T cells from antigen-specific naïve precursors, which utilized a similar culture system involving dendritic cell mobilization prior to peptide stimulation, also found that antigen-specific T cell frequency peaked on day 10 of culture (Lissina et al., 2016). To detect antigen-specific T cells on day 10, we re-stimulate expanded cells with antigens to induce cytokine secretion. We expect that reactive T cells will produce effector cytokines, namely IFN-γ, TNF-α and IL-2. Reactive T cells typically produce a combination of these cytokines, but it is possible they will produce a single cytokine. Reactive T cells can also be identified using other activation markers, such as CD137, CD154, and CD107a. To ensure that cytokine formation is antigen-specific, we use multiple negative controls. DMSO stimulation serves as the background or non-specific activation control and may be subtracted from test groups to normalize responses across multiple donors. Re-stimulation with irrelevant peptides can serve as a negative control too. Since cells were not expanded with these peptides, detection of irrelevant peptide-specific T cells is not expected. Alternatively, self-peptides, which should not induce immune responses, such as myelin-oligodendrocyte glycoprotein (MOG), can be utilized. Peptides with known immunogenicity, such as viral epitope pool CEFT, can control for assay conditions and antigen-specific T cell expansion. Additionally, stimulation with PMA and ionomycin can be used to confirm the capacity of T cells to produce effector cytokines and serves as positive control for flow cytometry staining.
Figure 2.

Cell viability and total T cell frequencies following T cell expansion
(A) Example of cell numbers at the end of a T cell assay. Data is pooled from 2 independent experiments. PBMCs from healthy donors (106 cells/donor) were stimulated with either MART-1 (ELAGIGILTV) or CEFT peptides. Expanded cells were counted using trypan blue and a hemocytometer.
(B) Frequency of T cells among expanded cultures as determined by flow cytometry.
Figure 3.

Analysis of flow cytometry data
(A) Example gating strategy used to analyze flow cytometry data acquired at the end of the assay. Red arrows indicate gating hierarchy in which T cells are identified as CD3+, live, single cells. T cells are further divided into CD4+ and CD8+ subsets. Expression of effector cytokines, namely IL-2, IFN-γ, and TNF-α, is analyzed under CD4+ T cell gate. Numbers on each graph indicate the frequency (%) of gated events.
(B) Representative results demonstrating detection of reactive T cells following expansion using the described T cell assay protocol. PBMCs from a healthy donor were expanded following stimulation with the vehicle control DMSO, CEFT, which contains known viral epitopes, and test peptides, a pool of five 15mer overlapping peptides spanning mutated SLC35F5 (AKISFFFALCGFWQICHIKKHFQTHKLL) as follows: 1) AKISFFFALCGFWQI, 2) FFFALCGFWQICHIK, 3) LCGFWQICHIKKHFQ, 4) FWQICHIKKHFQTHK, 5) QICHIKKHFQTHKLL (as reported in Roudko et al., 2020). Expanded cells were re-stimulated as indicated with either DMSO, myelin-oligodendrocyte glycoprotein (MOG), which is a self-protein and used as a negative control, test peptides, CEFT, and PMA/Ionomycin, used as positive controls. Percentage of IFN-γ+ CD8 T cells was measured by flow cytometry. Data were represented as mean ± standard deviation (SD).
This is a highly versatile and scalable T cell assay protocol that can be easily modified to study various aspects of T cell biology. For example, the re-stimulation step enables the testing of T cell cross-reactivity. Moreover, numerous peptides can be tested in the same assay with limited material through the use of pooled peptides followed by deconvolution. The role of checkpoint inhibition can be studied by adding specific blocking antibodies during the initial peptide stimulation (Cimen Bozkus et al., 2019). Similarly, MHC class I and class II blocking antibodies can be utilized by pre-treating cells prior to peptide stimulation. Notably, in addition to expanding memory T cells, this protocol allows priming of naïve T cells (Cimen Bozkus et al., 2019). Therefore, disease-specific antigens, such as neoantigens, can be studied using healthy donor cells without the bias of disease-associated factors such as an immunosuppressive microenvironment. Furthermore, this assay can also be utilized to monitor Th2 and Th17 cell responses, as well as Th1 cells. Finally, in addition to being a beneficial screening tool to evaluate the immunogenicity of antigens and to characterize antigen-specific T cells, this protocol may be modified to yield large scale expansions of reactive T cells under GMP-grade culture conditions, which can be utilized in adoptive T cell therapies.
Quantification and statistical analysis
FlowJo v10.7.1 was used to analyze flow cytometry data. Reactive T cells were presented as the percentage of CD8+ or CD4+ T cells that express IFN-γ, TNF-α or IL-2. T cells are identified according to the gating strategy shown in Figure 3.
Statistical significance was evaluated using a t test to compare the frequency of T cells that expressed cytokines upon peptide stimulation vs background, no peptide controls. All statistical analyses were performed using GraphPad Prism v9.1.0 and p values <0.05 were considered significant.
Limitations
This assay is optimized for the use of peptides while determining antigen-specific T cell responses. If using other sources of antigens, such as full-length proteins, further optimizations will likely be necessary.
As an in vitro assay, this protocol may not reflect the complexity of antigen-presentation and T cell activation that occur in vivo. This assay utilizes external addition of synthetic peptides. Therefore, it relies on cross-presentation to elicit CD8+ T cell responses, whereas in vivo CD8+ T cell responses can also be generated by direct presentation through endogenous route. Moreover, processing of folded full-length proteins and shorter peptide fragments may differ, leading to differential antigen presentation. To overcome these problems, other sources of antigens may be utilized instead of peptides, such as minigenes encoding the antigens of interest.
Since naïve T cell precursors can be primed and activated in vitro, the detection of reactive T cells after expansion does not inform the presence of pre-existing memory responses due to in vivo T cell priming. This could be addressed by setting up the assay with sorted memory and naïve T cell populations instead of PBMCs. We anticipate that while working with memory T cells, shortened culture times may be sufficient to detect reactive T cells.
It is important to consider MHC-restricted antigen recognition while interpreting the immunogenicity of test antigens. Lack of reactive T cells may be due to low binding affinity of the test peptides to the donors’ MHC alleles. It may be necessary to perform MHC allele typing for appropriate selection of donors while testing antigens with known MHC-restrictions. Furthermore, detection of reactive cells may be limited by low precursor T cell frequencies, thereby requiring additional steps to enrich target T cell populations.
Heterogeneity between donors in response to test antigens is commonly observed, likely due to biology as donors have diverse MHC alleles and varying frequencies of precursor T cells and APCs, as well as differences in antigen processing. The biology is further complicated in patients due to the presence of immunosuppressive mechanisms, such as T cell exhaustion and the immune modulatory effects of the treatments they are receiving. Technical factors may also contribute to heterogeneity in response, such as the quality, e.g., viability, of cryopreserved PBMCs upon thawing. Additionally, the timing of each step, especially the expansion and re-stimulation may lead to fluctuations in the percentage of reactive T cells detected.
Troubleshooting
Problem 1
Insufficient number of PBMCs to test all desired conditions
Potential solution
Obtaining enough PBMCs to test various peptides may be challenging, especially with the patient samples. In such cases, we recommend stimulating PBMCs with pooled peptides, which can be prepared as an equimolar mixture of desired numbers of peptides. DMSO levels added to the cell culture should be monitored not to exceed 0.1%, especially when pooling many peptides. Pooled peptides can be deconvoluted in the re-stimulation step to evaluate the individual contribution of each peptide to elicited T cell responses, as detailed before step 11.
If the available PBMCs are limited even to test pooled peptides, it is possible to expand APCs and T cells prior to assay start using standard cultures, such as in vitro cultures of hematopoietic stem cells generating high yields of DCs (Balan et al., 2018) and anti-CD3/anti-CD28 stimulation and expansion of T cells.
Problem 2
Inadequate cell numbers after expansion
Potential solution
There may be several reasons underlying low cell numbers after expansion. 1) The rate of cell expansion varies considerably between different donors. Hence, multiple donors should be tested. 2) Cells may be lost during media exchanges. Noted to be critical after step 8, media removal should be performed carefully to avoid introducing any liquid turbulence. We recommend tilting the plates and slowly aspirating the media from the side of the wells using a multichannel pipette. 3) Excessive cell death may limit the number of live cells. Although we do not typically split the cells during the 10-day culture period, fast growth rates may result in a high cell density leading to increased cell death. In such cases, cells can be split to reduce density. If excessive cell death is observed in the absence of cell growth, cryopreservation methods may need to be optimized. Alternatively, fresh cells may be used. Furthermore, cell death may occur during re-stimulation (steps 13–16), yielding a high dead to live cell ratio. To this end, incubation times following re-stimulation should be optimized by the users. We found that longer incubation times are associated with a greater dead:live cell ratio. Elongated incubations with monensin and BFA can be toxic to cells. It is not recommended to culture cells with these reagents longer than 12 h. If longer incubation times are necessary, monensin and BFA concentrations may be reduced.
If cells did not expand well by day 10, yielding inadequate numbers for downstream functional analysis, the assay can be continued past day 10 with additional feeding steps. Furthermore, instead of 2×105 cells per well (step 12), lower numbers, such as 105 cells per well, can be seeded for downstream functional analysis. We recommend working with equal numbers of cells between groups for accurate comparisons. Alternatively, ELISpot assays may be performed as they require a smaller number of cells than flow cytometry to analyze effector cytokine formation.
Problem 3
Non-specific background activation
Potential solution
To eliminate the possibility that cytokine detection in negative control groups is caused by technical issues during flow cytometry analysis, fluorescent minus one (FMO) controls should be utilized, which will help correct compensation-related problems and improve the accuracy of gating to allow successful discrimination of negative and positive populations.
It is possible that the background cytokine detection is not due to technical issues. Although very rarely, we detected cytokine formation by up to 5% of T cells treated with vehicle control. This observation was donor-dependent and in such cases a positive peptide stimulation still yielded significantly greater cytokine formation in test groups compared to the control. As noted after step 9, we recommend avoiding cytokine addition in the last feeding prior to re-stimulation to reduce background activation. We also found that resting cells after harvesting and seeding (steps 11 and 12), prior to re-stimulation, may reduce background activation. Finally, to further minimize background cytokine formation, IL-2, IL-7, and IL-15 addition during feedings can be eliminated. Of note, eliminating IL-2, IL-7, and IL-15 will reduce growth rates and yield lower total cell numbers at harvest.
Problem 4
Cytokine formation is not detected
Potential solution
A lack of cytokine detection may be due to that the test peptides are not immunogenic, the donors lack the MHC alleles binding to the antigenic epitopes with high affinity, or the precursor T cells are of low frequency. If the test peptide has a known MHC-restriction, donors should be confirmed to carry the specific MHC allele. For detecting low-level T cell responses, ELISpot assays may perform better than flow cytometry (Karlsson et al., 2003).
Problem 5
MHC-restriction of immunogenic peptides cannot be determined
Potential solution
If the goal is to identify the restricting MHC alleles for the test peptides, we recommend working with PBMCs from MHC-typed donors. After expansion, tetramer experiments can be conducted to identify the restricting MHCs. Alternatively, at re-stimulation (step 13), instead of directly supplementing peptides, expanded cells can be co-cultured with engineered monoallelic cells that are pulsed with the antigen of interest. In silico MHC-peptide binding affinity predictions can be utilized to narrow down potential MHC-peptide pairs.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nina Bhardwaj (nina.bhardwaj@mssm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate or analyze any datasets or code.
Acknowledgments
This work was supported by the funds provided by the Myeloproliferative Neoplasms Research Foundation and the Parker Institute for Cancer Immunotherapy. Flow cytometry analyses were performed using instruments in the Tisch Cancer Institute Flow Cytometry Shared Resource Facility at the Icahn School of Medicine at Mount Sinai. Components of the graphical abstract were generated with BioRender.com.
Author contributions
C.C.B., A.B.B., and N.B. conceived and designed the project. C.C.B., A.B.B., and T.E. performed experiments, analyzed and interpreted data, and optimized the protocol. C.C.B. wrote the manuscript. A.B.B., T.E., and N.B. reviewed and edited the manuscript. N.B. supervised the study and provided funding.
Declaration of interests
N.B. is an extramural member of the Parker Institute for Cancer Immunotherapy, receives research funds from Regeneron, Harbor Biomedical, DC Prime, and Dragonfly Therapeutics, and is on the advisory boards of Neon Therapeutics, Novartis, Avidea, Boehringer Ingelheim, Rome Therapeutics, Roswell Park Comprehensive Cancer Center, BreakBio, Carisma Therapeutics, CureVac, Genotwin, BioNTech, Gilead Therapeutics, Tempest Therapeutics, and the Cancer Research Institute. C.C.B. is a Bridge Scholar of the Parker Institute for Cancer Immunotherapy. N.B. and C.C.B. have a patent application (application no. 17/290,128), which utilizes the T-cell immunogenicity assay protocol described here. A.B.B. is currently an employee of Bristol Myers Squibb. T.E. declares no interests.
Contributor Information
Cansu Cimen Bozkus, Email: cansu.cimenbozkus@mssm.edu.
Nina Bhardwaj, Email: nina.bhardwaj@mssm.edu.
References
- Balan S., Arnold-Schrauf C., Abbas A., Couespel N., Savoret J., Imperatore F., Villani A.C., Vu Manh T.P., Bhardwaj N., Dalod M. Large-scale human dendritic cell differentiation revealing notch-dependent lineage bifurcation and heterogeneity. Cell Rep. 2018;24:1902–1915.e6. doi: 10.1016/j.celrep.2018.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimen Bozkus C., Roudko V., Finnigan J.P., Mascarenhas J., Hoffman R., Iancu-Rubin C., Bhardwaj N. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. 2019;9:1192–1207. doi: 10.1158/2159-8290.CD-18-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M., Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154:3–20. doi: 10.1111/imm.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geginat J., Sallusto F., Lanzavecchia A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J. Exp. Med. 2001;194:1711–1719. doi: 10.1084/jem.194.12.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A.C., Martin J.N., Younger S.R., Bredt B.M., Epling L., Ronquillo R., Varma A., Deeks S.G., Mccune J.M., Nixon D.F. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T cells. J. Immunol. Methods. 2003;283:141–153. doi: 10.1016/j.jim.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Kleiveland C.R. Peripheral blood mononuclear cells. In: Verhoeckx K., Cotter P., Lopez-Exposito I., Kleiveland C., Lea T., Mackie A., Requena T., Swiatecka D., Wichers H., editors. The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models. Springer; 2015. pp. 161–167. [PubMed] [Google Scholar]
- Lissina A., Briceno O., Afonso G., Larsen M., Gostick E., Price D.A., Mallone R., Appay V. Priming of qualitatively superior human effector CD8+ T cells using TLR8 ligand combined with FLT3 ligand. J. Immunol. 2016;196:256–263. doi: 10.4049/jimmunol.1501140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovgren T., Sarhan D., Truxova I., Choudhary B., Maas R., Melief J., Nystrom M., Edback U., Vermeij R., Scurti G. Enhanced stimulation of human tumor-specific T cells by dendritic cells matured in the presence of interferon-gamma and multiple toll-like receptor agonists. Cancer Immunol. Immunother. 2017;66:1333–1344. doi: 10.1007/s00262-017-2029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli P., Mosley R.L., Pisarev V., Chavez J., Ulrich A., Varney M., Singh R.K., Talmadge J.E. Flt3 ligand and granulocyte-macrophage colony-stimulating factor preferentially expand and stimulate different dendritic and T-cell subsets. Exp. Hematol. 2001;29:1185–1193. doi: 10.1016/s0301-472x(01)00722-6. [DOI] [PubMed] [Google Scholar]
- Roudko V., Bozkus C.C., Orfanelli T., Mcclain C.B., Carr C., O'donnell T., Chakraborty L., Samstein R., Huang K.L., Blank S.V. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell. 2020;183:1634–1649.e17. doi: 10.1016/j.cell.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesa A.K., Galy A. IL-1 beta induces dendritic cells to produce IL-12. Int. Immunol. 2001;13:1053–1061. doi: 10.1093/intimm/13.8.1053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate or analyze any datasets or code.