

Abstract
Herein, we report a hydrodefluorination reaction of polyfluoroarenes catalyzed by bismuthinidenes, Phebox-Bi(I) and OMe-Phebox-Bi(I). Mechanistic studies on the elementary steps support a Bi(I)/Bi(III) redox cycle that comprises C(sp2)–F oxidative addition, F/H ligand metathesis, and C(sp2)–H reductive elimination. Isolation and characterization of a cationic Phebox-Bi(III)(4-tetrafluoropyridyl) triflate manifests the feasible oxidative addition of Phebox-Bi(I) into the C(sp2)–F bond. Spectroscopic evidence was provided for the formation of a transient Phebox-Bi(III)(4-tetrafluoropyridyl) hydride during catalysis, which decomposes at low temperature to afford the corresponding C(sp2)–H bond while regenerating the propagating Phebox-Bi(I). This protocol represents a distinct catalytic example where a main-group center performs three elementary organometallic steps in a low-valent redox manifold.
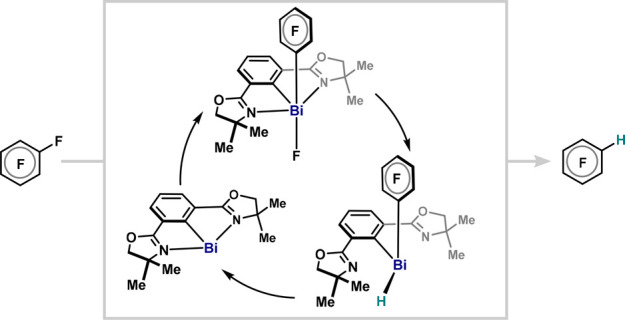
The elementary organometallic steps, oxidative addition (OA), ligand metathesis (LM), and reductive elimination (RE), define the innate capacity of transition-metal centers to revolve between different oxidation states in numerous catalytic processes (Figure 1A).1 With the aim of mimicking such reactivity by elements beyond the d-block, the past decades have witnessed prominent progress in low-valent main-group compounds exhibiting transition-metal-like reactivity, in particular, the cleavage of strong chemical bonds (e.g., N–H, O–H, H–H, C–H, C–F) through OA.2 However, the intrinsic difficulties posed by the regeneration of low-valent species via RE limited the development of efficient catalytic redox processes based on main-group catalysts.2b,2c Located in the middle of the p-block, group 15 elements have recently been identified as privileged candidates to unfold redox catalysis,3 as exemplified by the success of redox cycling using P and Bi redox couples in various catalytic reactions.4−6 In this endeavor, our group reported catalytic C(sp2)–F and C(sp2)–OTf/ONf bond formation proceeding through canonical cross-coupling steps in a Bi(III)/Bi(V) manifold (Figure 1B).5 However, in contrast to other pnictogens, Bi possesses additional low-valent redox manifolds to be exploited. Indeed, the Bi(I)/(III) redox couple has recently emerged and found applications in catalytic transfer hydrogenation of azo- and nitro-arenes, as well as in the catalytic activation of N2O.6 The low-valent Bi(I)/(III) redox manifold distinguishes itself from the high-valent and radical processes7 by its superior catalytic efficiency, and achieving catalytic redox transformations via the full triad of three elementary organometallic steps would be highly desirable.
Figure 1.

(A) Well-established transition-metal catalytic cycle. (B) Bi(III)/Bi(V) redox catalysis including elementary organometallic steps of OA/LM/RE. (C) HDF via Bi(I)/Bi(III) catalysis: elementary organometallic steps at low-valent main-group centers.
Hydrodefluorination (HDF) of polyfluoroarenes is a fundamental reaction that enables access to partially fluorinated building blocks from perfluorinated bulk chemicals.8 HDFs have largely been dominated by transition-metal catalysis,9,10 and a considerable number of these systems proceed through the catalytic steps depicted in Figure 1A.10 Recent progress in HDFs extended the available strategies to photoredox catalysis11 and main-group catalysis,12,13 which proceed through mechanistically distinct catalytic steps. In addition to its synthetic potential, HDF serves as a model reaction for studying the performance of main-group compounds in the elementary organometallic steps of a catalytic cycle. In this regard, C–F OA has been established for low-valent group 13/14 elements,14 and recently Radosevich has further shown an elegant synthetic cycle for HDF at a phosphorus triamide.15 Herein, we report that bismuthinidenes with a rationally designed N,C,N-pincer ligand scaffold unlock the catalytic HDF of a variety of polyfluoroarenes (Figure 1C). Mechanistic studies suggest a Bi(I)/Bi(III) cycle comprising C–F OA, F/H LM, and C–H RE steps, in a manner akin to a canonical catalytic cycle of transition-metal congeners.
Initially, we attempted the HDF of hexafluorobenzene (1a) using 5 mol% of Dostál’s bismuthinidene 3(16) as catalyst and 2.4 equiv. of Et2SiH2 as hydrogen source in THF at 60 °C (Figure 2A). Unfortunately, only a trace amount of HDF product (2a, <1%) was detected after 20 h. With the aim of tuning the electronics of the Bi(I) center, an alternative N,C,N-pincer scaffold was envisaged, where the imine arms are replaced with oxazoline groups. In this manner, two new bismuthinidenes supported by a 2,6-bis(oxazolinyl)phenyl (Phebox) ligand scaffold,17 Phebox-Bi(I) (4) and OMe-Phebox-Bi(I) (5), were synthesized via cobaltocene reduction of the parent bismuth chlorides 6 and 7.6b,18 When 4 and 5 were tested as catalysts for the HDF of 1a, 40% and 74% of 2a were obtained, respectively. In the case of 5, two-fold HDF (2a′) could also be detected in 16% yield. To gain more insights on the boosted reactivity, X-ray crystal structures of 4 and 5 were compared with that of 3, showing considerably more elongated Bi1–C1 distances [2.193(6) Å for 4,19 2.201(2) Å for 5, cf. 2.146(18) Å for 3,19Figure 2B]. These data suggest that electron delocalization of the 6pz2 lone pair of Bi to the ipso C(sp2) is diminished in the new bismuthinidenes, leading to the enhanced reactivity of the Phebox-based Bi(I) in HDFs.16,20
Figure 2.
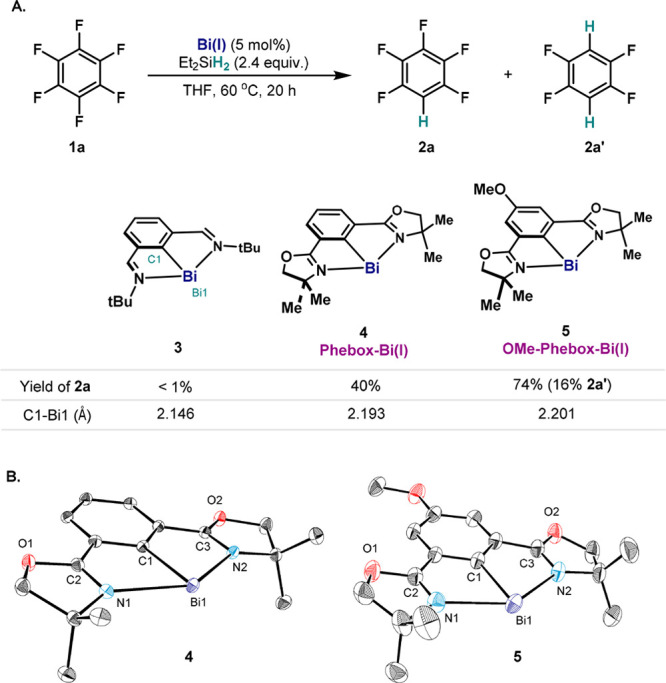
(A) HDF of 1a; 19F NMR yields are given. (B) ORTEP drawings of 4 and 5, with ellipsoids drawn at the 50% probability level. H atoms of 4 and 5, the second molecule in the asymmetric unit of 4, and disordered parts of 5 are omitted for clarity. Selected bond lengths (Å): for 4 (the bond lengths for the second molecule of 4 are given in brackets), Bi1–C1 2.189(3) [2.196(3)], Bi1–N1 2.525(3) [2.523(3)], Bi1–N2 2.503(3) [2.502(3)], N1–C2 1.288(4) [1.287(5)], N2–C3 1.288(4) [1.291(4)]; for 5, Bi1–C1 2.201(2), Bi1–N1 2.5359(19), Bi1–N2 2.5142(18), N1–C2 1.282(3), N2–C3 1.284(3).
With these Bi(I) catalysts in hand, HDFs of other polyfluoroarenes were evaluated (1b–1n, Table 1). In general, HDF proceeds in high yields; however, the reaction parameters varied significantly depending on the substituents of the substrates.18 Pentafluoropyridine (1b) and pentafluorobenzenes with strong electron-withdrawing groups (CF3, CO2Me, and CN, 1c–1f) underwent HDF readily at ambient temperature. Whereas 1b reached full conversion in 1 h using 3, the reaction finished within 2 min using 4 as catalyst. The high reactivity of 4 permitted lowering the catalyst loading to a remarkable 0.05 mol% while maintaining a high yield of 2b (1640 TON). Di-, tri-, and tetra-HDFs occurred for 1f–1h, 1j, and 1k when higher amount of Et2SiH2 (1.2–2.4 equiv.) were used. Several highly fluorinated phosphine compounds (1i–1k) utilized in various catalytic processes could be electronically fine-tuned through this HDF process.21 Partially fluorinated substrates (2a and 1n) and substrate with electron-neutral functionality (1l) were also amenable to HDF using 5 as catalyst. No directing effect was observed in HDF of 1m, thus providing orthogonal selectivity to transition-metal-catalyzed systems.10f It should be mentioned that, similar to the reported systems based on transition metals, the HDF becomes sluggish when applied to polyfluoroarenes bearing electron-donating groups.9e,10d,10i For instance, reaction of 2,3,4,5,6-pentafluorotoluene (1o) only delivered 2,3,5,6-tetrafluorotoluene (2o) in 3.5% yield after 3 days.
Table 1. Scope of the Bi(I)-Catalyzed HDFa.


Reactions performed on 0.25 mmol scale of 1b–1n.
Yields calculated by quantitative 19F NMR using 4-fluorotoluene as internal standard.
0.20 mmol scale of 1k.
In light of its demonstrated high reactivity, 1b was chosen as the model compound to study the mechanism of the Bi(I)-catalyzed HDF reaction. First, Phebox-Bi(I) (4) was subjected to 1.0 equiv. of 1b in THF-d8 (Figure 3A, path a). After 5 min, 19F NMR at 25 °C showed a distinct multiplet at −125.6 ppm, which is shifted dramatically compared to the meta-fluorines of 1b and 2b (1b, −163.0 ppm; 2b, −141.7 ppm). However, such chemical shift is consistent with the ortho-fluorines of 4-tetrafluoropyridyl attached to Bi in the reported Bi(4-C5F4N)3 (−120.7 ppm)22 and to other electropositive centers (e.g., Mg,14f Ni23). 1H–19F HOESY data at −40 °C further revealed the spatial proximity between these fluorines and two of the methyl groups of the Phebox backbone, suggesting the formation of Phebox-Bi(III)(4-tetrafluoropyridyl) fluoride (8a) via OA. However, the complex interconversions observed between 8a and other Bi species precluded its complete characterization.18 Nevertheless, when this mixture was treated with 2.0 equiv. of Et2SiH2, regeneration of 4 (>99%) and formation of the HDF product 2b (77%) were observed, manifesting the capacity of forging a C(sp2)–H bond through a Bi(I)/Bi(III) redox event.
Figure 3.
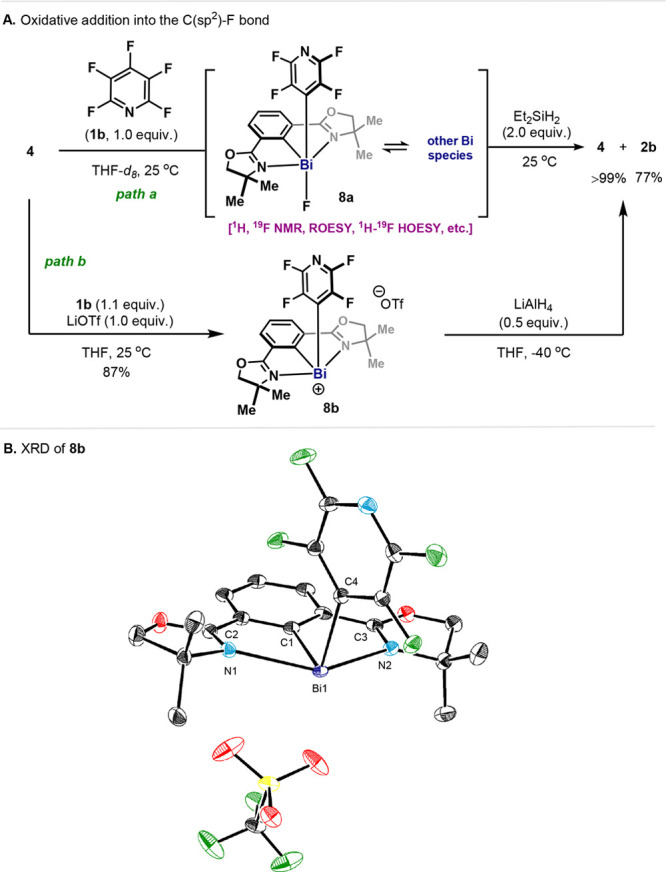
(A) OA of 4 with 1b; path a: 4 (20.8 μmol) and 1b (1.0 equiv.) in 0.5 mL of THF-d8 at 25 °C; path b: 4 (2.08 mmol), 1b (1.1 equiv.) and LiOTf (1.0 equiv.) in 7.0 mL of THF at 25 °C. (B) ORTEP drawing of 8b, with ellipsoids drawn at the 50% probability level. H atoms of 8b are omitted for clarity. Selected bond lengths (Å) and angles (°): Bi1–C1 2.225(2), Bi1–C4 2.294(2), Bi1–N1 2.450(2), Bi1–N2 2.4779(19), N1–C2 1.280(3), N2–C3 1.286(3); C1–Bi1–C4 93.60(8).
It was reasoned that the reactivity of the fluoride after C–F cleavage played an important role in the observed equilibriums. Hence, it was envisaged that fluoride abstraction after OA would lead to a well-defined cationic bismuth species with higher stability. Indeed, when the same reaction was performed in the presence of 1.0 equiv. of LiOTf, the triflate salt 8b was isolated in 87% yield (Figure 3A, path b). The attachment of the 4-tetrafluoropyridyl group to the Bi center results in the 19F signals of the ortho-fluorines appearing in a region (−121.4 ppm) similar to the observed shift of 8a. Moreover, the observation of diastereotopic methyl groups and methylene protons in the oxazolines of 8b by 1H NMR (CH3, 1.60 and 1.27 ppm; CH2, 4.59 and 4.56 ppm) confirms that the symmetry through the plane of Phebox ligand has been broken in 8b. The X-ray crystal structure of 8b confirms the weak interaction between the cationic Bi center and the triflate anion, as shown by the large distance between the closest oxygens of triflate and the Bi center (2.974 Å, ∑cov(Bi–O) = 2.14 Å,24Figure 3B). In spite of the cationic nature of 8b, the Bi1–C4 bond is still polarized [2.294(2) Å].25 As a result, 8b is highly moisture-sensitive, yielding [Phebox-Bi(OTf)]2O, 2b, and other oxo-bismuth species upon hydrolysis.18 Similar reactivity has been observed for Bi(4-C5F4N)322 and other perfluoro-aryl26 or -alkyl27 Bi(III) compounds. Although 8b showed no reactivity toward hydrosilanes due to the absence of fluoride anion, reduction of 8b with stronger metal hydrides (e.g., LiAlH4) readily yielded 4 and 2b (Figure 3A, path b).
At this point, it was hypothesized that a Ar2Bi(III)-H was generated via LM of 8a or 8b with hydrosilanes or metal hydrides. Organobismuth(III) hydrides are usually unstable species,28 prone to H2 release and formation of metallic Bi,29 Bi(I),6a,16 or dimetallic Bi(II)–Bi(II) compounds.7,30 Reported by Power in 2000, (2,6-Mes2H3C6)2BiH represents the only stable and well-defined organobismuth hydride until now.31 This compound indicated an alternative reaction pathway, namely C–H/D bond formation, yielding stable dibismuthene [Ar–Bi(I)=Bi(I)–Ar] and Ar-H/D (Ar = 2,6-Mes2H3C6). Later, the hydride signal of this bismuth hydride was located at a remarkably deshielded position (19.39 ppm),32 which resulted from the spin-orbital heavy-atom effect on the light atom (SO-HALA effect).30,33−35 Treatment of 8b with 0.5 equiv. of LiAlH4 at −78 °C resulted in instant formation of a new organobismuth species (Figure 4A, top). A broad singlet at 24.52 ppm in 1H NMR was detected (Figure 4B, top), suggesting that this species corresponds to Phebox-Bi(III)(4-tetrafluoropyridyl) hydride (9) with an electronic environment around Bi–H similar to that of the reported (2,6-Mes2H3C6)2BiH. In addition, 9 has an asymmetric and dynamic structure, as revealed by the considerably broadened NMR signals of the oxazolines (e.g., H-4, Figure 4B) and ortho-fluorines of the 4-tetrafluoropyridyl (−117.9 ppm).18 At –40 °C, 9 rapidly decayed into Phebox-Bi(I) (4) and HDF product (2b) in ca. 90% and 80% yields, indicating C(sp2)–H RE at the Bi center. Under catalytic conditions, 9 was the major species and remained relatively stable in concentration (Figure 4A and 4B, middle). Structural information on 9 was gathered from 2D NMR data of the reaction mixture. Particularly, C-5 (157.8 ppm) of 9 is noticeably more shielded than those of 4, 6, and 8b (4, 172.7 ppm; 6, 181.9 ppm; 8b, 182.3 ppm), but similar to that of the precursor Phebox-Br (10, 161.9 ppm). These electronic differences suggest that the oxazolines remain uncoordinated to the Bi center in 9, permitting the Bi center to adopt a trigonal pyramidal geometry. To further interrogate the nature of the unusual downfield proton signal, the catalytic reaction was performed using Et2SiD2. While all the signals assigned to 9 could be observed, the signal at 24.52 ppm did not appear in 1H NMR, suggesting the formation of corresponding bismuth deuteride 9-D (Figure 4A and 4B, bottom). As expected, decomposition of 9-D results in formation of 2b-D. It is important to point out that this is a distinct example where NMR spectroscopic data supports the involvement of an organobismuth hydride in a catalytic process, resulting in the formation of a C–H bond.
Figure 4.

(A) Proposed Bi-H/D intermediates (9/9-D) and C-H/D reductive elimination. (B) 1H NMR spectra of 9/9-D at −40 °C; top: LiAlH4 reduction of 8b; middle: catalytic HDF of 1b; bottom: catalytic HDF of 1b using Et2SiD2.
Taking 1b as an example, a Bi(I)/Bi(III) catalytic cycle can be proposed (Figure 5). Bismuthinidene 4 undergoes OA to 1b, delivering the Bi(III) intermediate 8a. Subsequent F/H LM between 8a and Et2SiH2 leads to the formation of diorganobismuth hydride (9) and fluorosilane. The catalytic redox loop is closed with RE from 9, releasing HDF product (2b) and regenerating Bi(I) (4).
Figure 5.

Proposed catalytic cycle for Bi(I)-catalyzed HDF.
In conclusion, we present that bismuthinidenes supported by a Phebox ligand scaffold facilitate catalytic HDF reaction of a variety of polyfluoroarenes under mild conditions. Mechanistic investigations enabled the identification of the intermediates involved, both after C–F cleavage (8b) and prior to C–H bond formation (9). These findings support a distinct Bi(I)/Bi(III) redox cycle where Bi centers manifest oxidative addition, ligand metathesis, and reductive elimination steps, conventionally exploited in transition-metal catalysis. The facile cycling through three elementary organometallic steps in the Bi(I)/Bi(III) redox manifold serves as a response to the long-standing challenge in the field of redox catalysis using low-valent main-group compounds, potentially enabling a myriad of catalytic redox processes beyond HDF.
Acknowledgments
Financial support for this work was provided by the Max-Planck-Gesellschaft, Max-Planck-Institut für Kohlenforschung and Fonds der Chemischen Industrie (FCI-VCI). Y.P. thanks CSC for a Ph.D. scholarship. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Agreement No. 850496 (ERC Starting Grant, J.C.). We thank Prof. Dr. A. Fürstner for insightful discussions and generous support. We thank the MS, GC, and X-ray departments of Max-Planck-Institut für Kohlenforschung for analytical support. We thank Dr. R. Goddard for X-ray crystallographic analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c06735.
Experimental procedures and analytical data (1H, 13C, 19F, and 31P NMR, HRMS and X-ray crystallographic details) for new compounds (PDF)
Accession Codes
CCDC 2091964–2091968 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Open access funded by Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- a de Meijere A.; Diederich F.. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH Verlag GmbH & Co. KGaA: Mörlenbach, Germany, 2004; pp 1–64. [Google Scholar]; b Crabtree R. H.The Organometallic Chemistry of the Transition Metals; John Wiley & Sons: Hoboken, NJ, 2005; pp 163–184. [Google Scholar]; c Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Mill Valley, CA, 2010; pp 877–965. [Google Scholar]
- a Power P. P. Main-Group Eements as Transition Metals. Nature 2010, 463, 171–177. 10.1038/nature08634. [DOI] [PubMed] [Google Scholar]; b Chu T.; Nikonov G. I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. 10.1021/acs.chemrev.7b00572. [DOI] [PubMed] [Google Scholar]; c Weetman C.; Inoue S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. 10.1002/cctc.201800963. [DOI] [Google Scholar]; d Melen R. L. Frontiers in Molecular p-Block Chemistry: From Structure to Reactivity. Science 2019, 363, 479–484. 10.1126/science.aau5105. [DOI] [PubMed] [Google Scholar]; e Spikes G. H.; Fettinger J. C.; Power P. P. Facile Activation of Dihydrogen by an Unsaturated Heavier Main Group Compound. J. Am. Chem. Soc. 2005, 127, 12232–12233. 10.1021/ja053247a. [DOI] [PubMed] [Google Scholar]; f Welch G. C.; Juan R. R. S.; Masuda J. D.; Stephan D. W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]; g Frey G. D.; Lavallo V.; Donnadieu B.; Schoeller W. W.; Bertrand G. Facile Splitting of Hydrogen and Ammonia by Nucleophilic Activation at a Single Carbon Center. Science 2007, 316, 439–441. 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]; h Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S. Synthesis, Structure and Reaction Chemistry of a Nucleophilic Aluminyl Anion. Nature 2018, 557, 92–95. 10.1038/s41586-018-0037-y. [DOI] [PubMed] [Google Scholar]; i Légaré M.-A.; Bélanger-Chabot G.; Dewhurst R. D.; Welz E.; Krummenacher I.; Engels B.; Braunschweig H. Nitrogen Fixation and Reduction at Boron. Science 2018, 359, 896–900. 10.1126/science.aaq1684. [DOI] [PubMed] [Google Scholar]; j Rösch B.; Gentner T. X.; Langer J.; Färber C.; Eyselein J.; Zhao L.; Ding C.; Frenking G.; Harder S. Dinitrogen Complexation and Reduction at Low-valent Calcium. Science 2021, 371, 1125–1128. 10.1126/science.abf2374. [DOI] [PubMed] [Google Scholar]
- a Ruffell K.; Ball L. T. Organobismuth Redox Manifolds: Versatile Tools for Synthesis. Trends in Chemistry 2020, 2, 867–869. 10.1016/j.trechm.2020.07.008. [DOI] [Google Scholar]; b Lipshultz J. M.; Li G.; Radosevich A. T. Main Group Redox Catalysis of Organopnictogens: Vertical Periodic Trends and Emerging Opportunities in Group 15. J. Am. Chem. Soc. 2021, 143, 1699–1721. 10.1021/jacs.0c12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Brien C. J.; Tellez J. L.; Nixon Z. S.; Kang L. J.; Carter A. L.; Kunkel S. R.; Przeworski K. C.; Chass G. A. Recycling the Waste: The Development of a Catalytic Wittig Reaction. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]; b van Kalkeren H. A.; Leenders S. H. A. M.; Hommersom C. R. A.; Rutjes F. P. J. T.; van Delft F. L. In Situ Phosphine Oxide Reduction: A Catalytic Appel Reaction. Chem. - Eur. J. 2011, 17, 11290–11295. 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]; c Dunn N. L.; Ha M.; Radosevich A. T. Main Group Redox Catalysis: Reversible PIII/PV Redox Cycling at a Phosphorus Platform. J. Am. Chem. Soc. 2012, 134, 11330–11333. 10.1021/ja302963p. [DOI] [PubMed] [Google Scholar]; d van Kalkeren H. A.; Bruins J. J.; Rutjes F. P. J. T.; van Delft F. L. Organophosphorus-Catalysed Staudinger Reduction. Adv. Synth. Catal. 2012, 354, 1417–1421. 10.1002/adsc.201100967. [DOI] [Google Scholar]; e Buonomo J. A.; Aldrich C. C. Mitsunobu Reactions Catalytic in Phosphine and a Fully Catalytic System. Angew. Chem., Int. Ed. 2015, 54, 13041–13044. 10.1002/anie.201506263. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhao W.; Yan P. K.; Radosevich A. T. A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PV=O Redox Cycling. J. Am. Chem. Soc. 2015, 137, 616–619. 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]; g Nykaza T. V.; Cooper J. C.; Li G.; Mahieu N.; Ramirez A.; Luzung M. R.; Radosevich A. T. Intermolecular Reductive C–N Cross Coupling of Nitroarenes and Boronic Acids by PIII/PV=O Catalysis. J. Am. Chem. Soc. 2018, 140, 15200–15205. 10.1021/jacs.8b10769. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Longwitz L.; Werner T. Reduction of Activated Alkenes by PIII/PV Redox Cycling Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2760–2763. 10.1002/anie.201912991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Planas O.; Wang F.; Leutzsch M.; Cornella J. Fluorination of Arylboronic Esters Enabled by Bismuth Redox Catalysis. Science 2020, 367, 313–317. 10.1126/science.aaz2258. [DOI] [PubMed] [Google Scholar]; b Planas O.; Peciukenas V.; Cornella J. Bismuth-Catalyzed Oxidative Coupling of Arylboronic Acids with Triflate and Nonaflate Salts. J. Am. Chem. Soc. 2020, 142, 11382–11387. 10.1021/jacs.0c05343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang F.; Planas O.; Cornella J. Bi(I)-Catalyzed Transfer-Hydrogenation with Ammonia-Borane. J. Am. Chem. Soc. 2019, 141, 4235–4240. 10.1021/jacs.9b00594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pang Y.; Leutzsch M.; Nothling N.; Cornella J. Catalytic Activation of N2O at a Low-Valent Bismuth Redox Platform. J. Am. Chem. Soc. 2020, 142, 19473–19479. 10.1021/jacs.0c10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schwamm R. J.; Lein M.; Coles M. P.; Fitchett C. M. Catalytic Oxidative Coupling Promoted by Bismuth TEMPOxide Complexes. Chem. Commun. 2018, 54, 916–919. 10.1039/C7CC08402A. [DOI] [PubMed] [Google Scholar]; b Ramler J.; Krummenacher I.; Lichtenberg C. Well-defined, Molecular Bismuth Compounds: Catalysts in Photochemically-induced Radical Dehydrocoupling Reactions. Chem. - Eur. J. 2020, 26, 14551–14555. 10.1002/chem.202002219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuehnel M. F.; Lentz D.; Braun T. Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem., Int. Ed. 2013, 52, 3328–3348. 10.1002/anie.201205260. [DOI] [PubMed] [Google Scholar]; b Whittlesey M. K.; Peris E. Catalytic Hydrodefluorination with Late Transition Metal Complexes. ACS Catal. 2014, 4, 3152–3159. 10.1021/cs500887p. [DOI] [Google Scholar]; c Hu J.-Y.; Zhang J.-L.. Hydrodefluorination Reactions Catalyzed by Transition-Metal Complexes. In Organometallic Fluorine Chemistry; Braun T., Hughes R. P., Eds.; Springer International Publishing: Cham, 2015; pp 143–196. [Google Scholar]
- a Aizenberg M.; Milstein D. Catalytic Activation of Carbon-Fluorine Bonds by a Soluble Transition Metal Complex. Science 1994, 265, 359–361. 10.1126/science.265.5170.359. [DOI] [PubMed] [Google Scholar]; b Vela J.; Smith J. M.; Yu Y.; Ketterer N. A.; Flaschenriem C. J.; Lachicotte R. J.; Holland P. L. Synthesis and Reactivity of Low-Coordinate Iron(II) Fluoride Complexes and Their Use in the Catalytic Hydrodefluorination of Fluorocarbons. J. Am. Chem. Soc. 2005, 127, 7857–7870. 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]; c Reade S. P.; Mahon M. F.; Whittlesey M. K. Catalytic Hydrodefluorination of Aromatic Fluorocarbons by Ruthenium N-Heterocyclic Carbene Complexes. J. Am. Chem. Soc. 2009, 131, 1847–1861. 10.1021/ja806545e. [DOI] [PubMed] [Google Scholar]; d Yow S.; Gates S. J.; White A. J. P.; Crimmin M. R. Zirconocene Dichloride Catalyzed Hydrodefluorination of C–F bonds. Angew. Chem., Int. Ed. 2012, 51, 12559–12563. 10.1002/anie.201207036. [DOI] [PubMed] [Google Scholar]; e Lv H.; Cai Y.-B.; Zhang J.-L. Copper-Catalyzed Hydrodefluorination of Fluoroarenes by Copper Hydride Intermediates. Angew. Chem., Int. Ed. 2013, 52, 3203–3207. 10.1002/anie.201208364. [DOI] [PubMed] [Google Scholar]; f Cybulski M. K.; McKay D.; Macgregor S. A.; Mahon M. F.; Whittlesey M. K. Room Temperature Regioselective Catalytic Hydrodefluorination of Fluoroarenes with trans-[Ru(NHC)4H2] through a Concerted Nucleophilic Ru–H Attack Pathway. Angew. Chem., Int. Ed. 2017, 56, 1515–1519. 10.1002/anie.201610820. [DOI] [PubMed] [Google Scholar]
- a Braun T.; Izundu J.; Steffen A.; Neumann B.; Stammler H.-G. Reactivity of a Palladium Fluoro Complex towards Silanes and Bu3SnCH=CH2: Catalytic Derivatisation of Pentafluoropyridine Based on Carbon–fluorine Bond Activation Reactions. Dalton Trans. 2006, 5118–5123. 10.1039/B608410A. [DOI] [PubMed] [Google Scholar]; b Breyer D.; Braun T.; Penner A. Isolation and Reactivity of Palladium Hydrido Complexes: Intermediates in the Hydrodefluorination of Pentafluoropyridine. Dalton Trans. 2010, 39, 7513–7520. 10.1039/c0dt00086h. [DOI] [PubMed] [Google Scholar]; c Fischer P.; Götz K.; Eichhorn A.; Radius U. Decisive Steps of the Hydrodefluorination of Fluoroaromatics using [Ni(NHC)2]. Organometallics 2012, 31, 1374–1383. 10.1021/om2009815. [DOI] [Google Scholar]; d Lv H.; Zhan J.-H.; Cai Y.-B.; Yu Y.; Wang B.; Zhang J.-L. π–π Interaction Assisted Hydrodefluorination of Perfluoroarenes by Gold Hydride: A Case of Synergistic Effect on C–F Bond Activation. J. Am. Chem. Soc. 2012, 134, 16216–16227. 10.1021/ja305204y. [DOI] [PubMed] [Google Scholar]; e Zhan J.-H.; Lv H.; Yu Y.; Zhang J.-L. Catalytic C-F Bond Activation of Perfluoroarenes by Tricoordinated Gold(I) Complexes. Adv. Synth. Catal. 2012, 354, 1529–1541. 10.1002/adsc.201100843. [DOI] [Google Scholar]; f Chen Z.; He C.-Y.; Yin Z.; Chen L.; He Y.; Zhang X. Palladium-catalyzed Ortho-selective C–F Activation of Polyfluoroarenes with Triethylsilane: A Facile Access to Partially Fluorinated Aromatics. Angew. Chem., Int. Ed. 2013, 52, 5813–5817. 10.1002/anie.201300400. [DOI] [PubMed] [Google Scholar]; g Li J.; Zheng T.; Sun H.; Li X. Selectively Catalytic Hydrodefluorination of Perfluoroarenes by Co(PMe3)4 with Sodium Formate as Reducing Agent and Mechanism Study. Dalton Trans. 2013, 42, 13048–13053. 10.1039/c3dt50409c. [DOI] [PubMed] [Google Scholar]; h Gianetti T. L.; Bergman R. G.; Arnold J. Carbon–fluorine Bond Cleavage in Fluoroarenes via a Niobium(III) Imido Complex: from Stoichiometric to Catalytic Hydrodefluorination. Chem. Sci. 2014, 5, 2517–2524. 10.1039/c4sc00006d. [DOI] [Google Scholar]; i Nakai H.; Jeong K.; Matsumoto T.; Ogo S. Catalytic C–F Bond Hydrogenolysis of Fluoroaromatics by [(η5-C5Me5)RhI(2,2′-bipyridine)]. Organometallics 2014, 33, 4349–4352. 10.1021/om500647h. [DOI] [Google Scholar]
- a Senaweera S. M.; Singh A.; Weaver J. D. Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics. J. Am. Chem. Soc. 2014, 136, 3002–3005. 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]; b Lu J.; Khetrapal N. S.; Johnson J. A.; Zeng X. C.; Zhang J. “π-Hole−π” Interaction Promoted Photocatalytic Hydrodefluorination via Inner-Sphere Electron Transfer. J. Am. Chem. Soc. 2016, 138, 15805–15808. 10.1021/jacs.6b08620. [DOI] [PubMed] [Google Scholar]
- Kikushima K.; Grellier M.; Ohashi M.; Ogoshi S. Transition-Metal-Free Catalytic Hydrodefluorination of Polyfluoroarenes by Concerted Nucleophilic Aromatic Substitution with a Hydrosilicate. Angew. Chem., Int. Ed. 2017, 56, 16191–16196. 10.1002/anie.201708003. [DOI] [PubMed] [Google Scholar]
- Stahl T.; Klare H. F. T.; Oestreich M. Main-Group Lewis Acids for C–F Bond Activation. ACS Catal. 2013, 3, 1578–1587. 10.1021/cs4003244. [DOI] [Google Scholar]
- a Jana A.; Samuel P. P.; Tavčar G.; Roesky H. W.; Schulzke C. Selective Aromatic C–F and C–H Bond Activation with Silylenes of Different Coordinate Silicon. J. Am. Chem. Soc. 2010, 132, 10164–10170. 10.1021/ja103988d. [DOI] [PubMed] [Google Scholar]; b Samuel P. P.; Singh A. P.; Sarish S. P.; Matussek J.; Objartel I.; Roesky H. W.; Stalke D. Oxidative Addition Versus Substitution Reactions of Group 14 Dialkylamino Metalylenes with Pentafluoropyridine. Inorg. Chem. 2013, 52, 1544–1549. 10.1021/ic302344a. [DOI] [PubMed] [Google Scholar]; c Chu T.; Boyko Y.; Korobkov I.; Nikonov G. I. Transition Metal-like Oxidative Addition of C–F and C–O Bonds to an Aluminum(I) Center. Organometallics 2015, 34, 5363–5365. 10.1021/acs.organomet.5b00793. [DOI] [Google Scholar]; d Crimmin M. R.; Butler M. J.; White A. J. P. Oxidative Addition of Carbon–fluorine and Carbon–oxygen Bonds to Al(I). Chem. Commun. 2015, 51, 15994–15996. 10.1039/C5CC07140B. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Styra S.; Melaimi M.; Moore C. E.; Rheingold A. L.; Augenstein T.; Breher F.; Bertrand G. Crystalline Cyclic (Alkyl)(amino)carbene-tetrafluoropyridyl Radical. Chem. - Eur. J. 2015, 21, 8441–8446. 10.1002/chem.201500740. [DOI] [PubMed] [Google Scholar]; f Bakewell C.; White A. J. P.; Crimmin M. R. Addition of Carbon–Fluorine Bonds to a Mg(I)–Mg(I) Bond: An Equivalent of Grignard Formation in Solution. J. Am. Chem. Soc. 2016, 138, 12763–12766. 10.1021/jacs.6b08104. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kysliak O.; Görls H.; Kretschmer R. Cooperative Bond Activation by a Bimetallic Main-Group Complex. J. Am. Chem. Soc. 2021, 143, 142–148. 10.1021/jacs.0c12166. [DOI] [PubMed] [Google Scholar]
- Lim S.; Radosevich A. T. Round-Trip Oxidative Addition, Ligand Metathesis, and Reductive Elimination in a PIII/PV Synthetic Cycle. J. Am. Chem. Soc. 2020, 142, 16188–16193. 10.1021/jacs.0c07580. [DOI] [PubMed] [Google Scholar]
- a Šimon P.; de Proft F.; Jambor R.; Růžička A.; Dostál L. Monomeric Organoantimony(I) and Organobismuth(I) Compounds Stabilized by an NCN Chelating Ligand: Syntheses and Structures. Angew. Chem., Int. Ed. 2010, 49, 5468–5471. 10.1002/anie.201002209. [DOI] [PubMed] [Google Scholar]; b Vránová I.; Alonso M.; Lo R.; Sedlák R.; Jambor R.; Růžička A.; Proft F. D.; Hobza P.; Dostál L. From Dibismuthenes to Three- and Two-Coordinated Bismuthinidenes by Fine Ligand Tuning: Evidence for Aromatic BiC3N Rings through a Combined Experimental and Theoretical Study. Chem. - Eur. J. 2015, 21, 16917–16928. 10.1002/chem.201502724. [DOI] [PubMed] [Google Scholar]
- a Denmark S. E.; Stavenger R. A.; Faucher A.-M.; Edwards J. P. Cyclopropanation with Diazomethane and Bis(oxazoline)palladium(II) Complexes. J. Org. Chem. 1997, 62, 3375–3389. 10.1021/jo970044z. [DOI] [PubMed] [Google Scholar]; b Motoyama Y.; Makihara N.; Mikami Y.; Aoki K.; Nishiyama H. Chiral Bis(oxazolinyl)phenyl Rhodium and Palladium Complexes. Chem. Lett. 1997, 26, 951–952. 10.1246/cl.1997.951. [DOI] [Google Scholar]; c Motoyama Y.; Okano M.; Narusawa H.; Makihara N.; Aoki K.; Nishiyama H. Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes: Synthesis, Structure, Enantioselective Allylation of Aldehydes, and Mechanistic Studies. Organometallics 2001, 20, 1580–1591. 10.1021/om010018y. [DOI] [Google Scholar]; d Stol M.; Snelders D. J. M.; Godbole M. D.; Havenith R. W. A.; Haddleton D.; Clarkson G.; Lutz M.; Spek A. L.; van Klink G. P. M.; van Koten G. 2,6-Bis(oxazolinyl)phenylnickel(II) Bromide and 2,6-Bis(ketimine)phenylnickel(II) Bromide: Synthesis, Structural Features, and Redox Properties. Organometallics 2007, 26, 3985–3994. 10.1021/om061055y. [DOI] [Google Scholar]; e Bugarin A.; Connell B. T. Chiral Nickel(II) and Palladium(II) NCN-Pincer Complexes Based on Substituted Benzene: Synthesis, Structure, and Lewis Acidity. Organometallics 2008, 27, 4357–4369. 10.1021/om8004367. [DOI] [Google Scholar]; f Bugarin A.; Connell B. T. A Highly Active and Selective Palladium Pincer Catalyst for the Formation of α-Aryl Ketones via Cross-coupling. Chem. Commun. 2011, 47, 7218–7220. 10.1039/c1cc12071a. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for details.
- These compounds have two independent molecules in the asymmetric units and the averaged bond distances are given in the text.
- Wang G.; Freeman L. A.; Dickie D. A.; Mokrai R.; Benkő Z.; Gilliard R. J. Jr Isolation of Cyclic(Alkyl)(Amino) Carbene–Bismuthinidene Mediated by a Beryllium(0) Complex. Chem. - Eur. J. 2019, 25, 4335–4339. 10.1002/chem.201900458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ojima I.; Kwon H. B. Remarkable Effects of a Pentafluorophenyl Group on the Stereoselective Reactions of a Chiral Iron Acyl Complex. J. Am. Chem. Soc. 1988, 110, 5617–5621. 10.1021/ja00225a004. [DOI] [Google Scholar]; b Hirai T.; Hamasaki A.; Nakamura A.; Tokunaga M. Enhancement of Reaction Efficiency by Functionalized Alcohols on Gold(I)-Catalyzed Intermolecular Hydroalkoxylation of Unactivated Olefins. Org. Lett. 2009, 11, 5510–5513. 10.1021/ol9023166. [DOI] [PubMed] [Google Scholar]; c Korenaga T.; Abe K.; Ko A.; Maenishi R.; Sakai T. Ligand Electronic Effect on Reductive Elimination of Biphenyl from cis-[Pt(Ph)2(diphosphine)] Complexes Bearing Electron-Poor Diphosphine: Correlation Study between Experimental and Theoretical Results. Organometallics 2010, 29, 4025–4035. 10.1021/om100073j. [DOI] [Google Scholar]; d Crisenza G. E. M.; Sokolova O. O.; Bower J. F. Branch-Selective Alkene Hydroarylation by Cooperative Destabilization: Iridium-Catalyzed ortho-Alkylation of Acetanilides. Angew. Chem., Int. Ed. 2015, 54, 14866–14870. 10.1002/anie.201506581. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fernández D. F.; Gulías M.; Mascareñas J. L.; López F. Iridium(I)-Catalyzed Intramolecular Hydrocarbonation of Alkenes: Efficient Access to Cyclic Systems Bearing Quaternary Stereocenters. Angew. Chem., Int. Ed. 2017, 56, 9541–9545. 10.1002/anie.201705105. [DOI] [PubMed] [Google Scholar]; f Lee Y. H.; Morandi B. Palladium-Catalyzed Intermolecular Aryliodination of Internal Alkynes. Angew. Chem., Int. Ed. 2019, 58, 6444–6448. 10.1002/anie.201812396. [DOI] [PubMed] [Google Scholar]
- Tyrra W.; Aboulkacem S.; Hoge B.; Wiebe W.; Pantenburg I. Silver Compounds in Synthetic Chemistry: Part 4. 4-Tetrafluoropyridyl Silver(I), AgC5F4N in Redox Transmetallations—Possibilities and Limitations in Reactions with Group 15 Elements. J. Fluorine Chem. 2006, 127, 213–217. 10.1016/j.jfluchem.2005.10.009. [DOI] [Google Scholar]
- Schaub T.; Fischer P.; Steffen A.; Braun T.; Radius U.; Mix A. C–F Activation of Fluorinated Arenes using NHC-Stabilized Nickel(0) Complexes: Selectivity and Mechanistic Investigations. J. Am. Chem. Soc. 2008, 130, 9304–9317. 10.1021/ja074640e. [DOI] [PubMed] [Google Scholar]
- Pyykkö P.; Atsumi M. Molecular Single-Bond Covalent Radii for Elements 1–118. Chem. - Eur. J. 2009, 15, 186–197. 10.1002/chem.200800987. [DOI] [PubMed] [Google Scholar]
- Hejda M.; Jirásko R.; Růžička A.; Jambor R.; Dostál L. Probing the Limits of Oxidative Addition of C(sp3)–X Bonds toward Selected N,C,N-Chelated Bismuth(I) Compounds. Organometallics 2020, 39, 4320–4328. 10.1021/acs.organomet.0c00418. [DOI] [Google Scholar]
- a Tyrra W.; Naumann D. On Pentavalent Perfluoroorgano Bismuth Compounds. Can. J. Chem. 1989, 67, 1949–1951. 10.1139/v89-303. [DOI] [Google Scholar]; b Olaru M.; Nema M. G.; Soran A.; Breunig H. J.; Silvestru C. Mixed Triorganobismuthines RAr2Bi [Ar = C6F5, 2,4,6-(C6F5)3C6H2] and Hypervalent Racemic Bi-chiral Diorganobismuth(III) Bromides RArBiBr (Ar = C6F5, Mes, Ph) with the Ligand R = 2-(Me2NCH2)C6H4. Influences of the Organic Substituent. Dalton Trans. 2016, 45, 9419–9428. 10.1039/C5DT05074J. [DOI] [PubMed] [Google Scholar]
- Solyntjes S.; Bader J.; Neumann B.; Stammler H.-G.; Ignat’ev N.; Hoge B. Pentafluoroethyl Bismuth Compounds. Chem. - Eur. J. 2017, 23, 1557–1567. 10.1002/chem.201604910. [DOI] [PubMed] [Google Scholar]
- Aldridge S.; Downs A. J. Hydrides of the Main-Group Metals: New Variations on an Old Theme. Chem. Rev. 2001, 101, 3305–3366. 10.1021/cr960151d. [DOI] [PubMed] [Google Scholar]
- Amberger E. Hydride des Wismuts. Chem. Ber. 1961, 94, 1447–1452. 10.1002/cber.19610940607. [DOI] [Google Scholar]
- Balázs G.; Breunig H. J.; Lork E. Synthesis and Characterization of R2SbH, R2BiH, and R2Bi–BiR2 [R = (Me3Si)2CH]. Organometallics 2002, 21, 2584–2586. 10.1021/om020202z. [DOI] [Google Scholar]
- Hardman N. J.; Twamley B.; Power P. P. (2,6-Mes2H3C6)2BiH, a Stable, Molecular Hydride of a Main Group Element of the Sixth Period, and Its Conversion to the Dibismuthene (2,6-Mes2H3C6)BiBi(2,6-Mes2C6H3). Angew. Chem., Int. Ed. 2000, 39, 2771–2773. . [DOI] [PubMed] [Google Scholar]
- Olaru M.; Duvinage D.; Lork E.; Mebs S.; Beckmann J. Heavy Carbene Analogues: Donor-Free Bismuthenium and Stibenium Ions. Angew. Chem., Int. Ed. 2018, 57, 10080–10084. 10.1002/anie.201803160. [DOI] [PubMed] [Google Scholar]
- Ashe A. J.; Ludwig E. G.; Oleksyszyn J. Preparation and Properties of Dibismuthines. Organometallics 1983, 2, 1859–1866. 10.1021/om50006a027. [DOI] [Google Scholar]
- Vícha J.; Novotný J.; Komorovsky S.; Straka M.; Kaupp M.; Marek R. Relativistic Heavy-Neighbor-Atom Effects on NMR Shifts: Concepts and Trends Across the Periodic Table. Chem. Rev. 2020, 120, 7065–7103. 10.1021/acs.chemrev.9b00785. [DOI] [PubMed] [Google Scholar]
- A few reports on the spectroscopic identification of organobismuth hydrides (R2BH) are known in the literature. Ashe (ref (33)) and Breunig (ref (30)) reported chemical shifts of 2.10–3.24 ppm for the hydrides of (alkyl)2BiH however, the hydride of Power’s Ar2BiH appears at 19.39 ppm (ref (31)). See ref (34) for more details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.