

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that broadly affect cellular and physiological function in all multicellular organisms. Here, the role of miRNAs in neuroinflammation is considered. miRNAs are 21-23 oligonucleotide RNAs that regulate translation of specific RNAs by binding to complementary regulatory RNA sequences, thereby causing mRNA degradation or sequestration. More than 5000 miRNAs likely exist in humans, and each miRNA binds an average of 200 RNAs. Specific immunomodulatory miRNAs can regulate a set of RNAs in a coordinated manner, suggesting that effective miRNA-based therapeutic manipulations for neuroinflammatory conditions may be revealed. For instance, miRNAs that preferentially inhibit translation of many cellular anti-inflammatory proteins could drive a pro-inflammatory response. Key pro-inflammatory (miR-155, miR-27b, miR-326), anti-inflammatory (miR-124, miR-146a, miR-21, miR-223), and mixed immunomodulatory (let-7 family) miRNAs regulate neuroinflammation in various pathologies, including spinal cord injury, multiple sclerosis, ischemic stroke, and Alzheimer’s disease. miRNAs represent a newly-revealed layer of physiological complexity, the therapeutic benefits of which remain to be fully explored and exploited. In this review, we discuss the role of miRNAs in neuroinflammatory regulation, and how controlling miRNAs could alter cellular machinery to improve neuroinflammatory dynamics.
Keywords: central nervous system, traumatic brain injury, inflammation, immune response, neuropathology
Introduction
In 1993, a new class of small RNA molecules was discovered in Caenorhabditis elegans (Lee and others, 1993; Wightman and others, 1993), though their key regulatory functions remained undefined until 2000 (Pasquinelli and others, 2000; Reinhart and others, 2000). These small non-protein coding RNAs, called microRNAs (miRNAs), regulate innumerable cellular processes by causing degradation or sequestration of specific RNAs, thereby preventing protein translation (Dong and others, 2013). The observation that miRNAs have been conserved throughout evolution highlights their importance; over 5000 miRNAs likely exist in humans (Londin and others, 2015), miRNAs target 30-80% of protein-coding genes (Friedman and others, 2009; Lu and Clark, 2012), and each miRNA regulates translation of hundreds of distinct mRNAs (Krek and others, 2005). Because microRNAs (and other non-coding RNAs) were only recently discovered, concepts related to our biological systems must be revised to include this additional layer of physiological regulation. Understanding how specific miRNAs regulate key cellular processes could reveal powerful new endogenous combinatorial therapeutic targets. Here, we consider how miRNAs influence neuroinflammatory dynamics.
Although inflammation in the nervous system can be beneficial, it often can worsen pathology (Popovich and others, 1999; Kigerl and others, 2009; Hooten and others, 2015; Grace and others, 2016). In models of central nervous system (CNS) trauma, neuroinflammation can exacerbate injury by causing secondary damage (Faulkner and others, 2004; Beck and others, 2010; David and Kroner, 2011; Kigerl and others, 2016). Inflammation also can modify pathology caused by stress, sickness, or neurodegenerative disease (e.g., multiple sclerosis (MS), Alzheimer’s disease (AD), and Parkinson’s disease) (Gagne and Power, 2010; Pisanu and others, 2014; Hoppmann and others, 2015). Therapies that harness the beneficial aspects of inflammation, while restricting its ability to cause pathology could improve neurological function (Stirling and others, 2004; Peng and others, 2009; Tentillier and others, 2016). miRNAs are emerging as novel therapeutic targets for various human diseases, including those that affect the CNS (Kabaria and others, 2015; Kamphuis and others, 2015; Christopher and others, 2016; Fonken and others, 2016a; Gaudet and others, 2016b; Huang and others, 2016a; Severin and others, 2016).
In this review, we discuss recent advances in dynamic miRNA regulation of neuroinflammation. First, the contribution of inflammation to various neuropathologies is considered. Next, we provide a brief overview of miRNA production and function. In the following section, specific pro-inflammatory (miR-155, miR-27b, miR-326), anti-inflammatory (miR-124, miR-146a, miR-21, miR-223), and mixed immunomodulatory (let-7 family) miRNA are considered in the context of immunity and neuroinflammation. Finally, we discuss strategies for interrogating the biological role of miRNAs, ideas for research design, and future directions.
Inflammation contributes to neuropathologies
Neuroinflammation is an adaptive response to tissue injury/infection that can also cause and worsen pathology in neurological disorders (Fig. 1). In the healthy adult CNS, tissue-resident microglia and astrocytes maintain a quiescent yet vigilant state (Nimmerjahn and others, 2005). These cells respond to infection or injury by transiently increasing inflammatory actions to isolate and remove the causative agents (Fonken and others, 2015; Norden and others, 2015; Frank and others, 2016). Microglia are the main antigen-presenting cells of the CNS; once activated, they increase expression of major histocompatibility complex class II (MHC II) molecules, which are necessary to present antigens to adaptive immune cells (Frank and others, 2007). Activated microglia and astrocytes also release cytokines, chemokines, and other factors that help mount an inflammatory response and subsequently restore CNS homeostasis. Indeed, pro-inflammatory factors in the CNS can be beneficial. For example, the detection and propagation of an immune signal throughout the CNS causes a suite of behavioral and physiological modifications, collectively known as the sickness response, which are critical to host defense (Dantzer and others, 2008). Behavioral changes associated with CNS inflammation include reduced food and water intake, increased sleep, decreased exploration and social behavior, hyperalgesia, and global changes in mood and cognition (Dantzer and others, 2008). Overall, the sickness response represents a shift in an organism’s motivational state and is considered highly adaptive. After successfully removing pathogens or recovering from minor injury, the inflammatory response may resolve. However, neuroinflammation has a “dark side”; pathological neuroinflammation occurs when the inflammatory response is exaggerated or persists long-term such as after CNS injury. As an immune-privileged tissue (Engelhardt and others, 2017), healthy CNS parenchyma is not typically exposed to peripheral immune cells or robust inflammatory responses and is therefore vulnerable to neuroinflammatory-elicited damage and autoimmune disease.
Figure 1. Inflammatory dynamics follow a stereotyped pattern in neuroinflammatory disorders.

Upper portion describes neuroinflammatory events, whereas lower portion depicts immune cell responses over time. In healthy tissue, quiescent microglia and astrocytes sample the local CNS microenvironment for signs of infection or damage. Neuroinflammation can be initiated by trauma (brain or spinal cord injury), ischemia, infection, aging, or unknown causes. Microglia and astrocytes detect the immune stimulus and respond by becoming activated, proliferating, and releasing cytokines/chemokines. These cytokines/chemokines cause peripheral immune cells (which usually have limited CNS access) to translocate into CNS parenchyma. Initially, neutrophils and macrophages (and likely dendritic cells) – cells of the innate immune system – enter the CNS compartment. Macrophages in particular persist in the CNS for long periods and maintain a potentially cytotoxic pro-inflammatory phenotype. Since the blood-brain barrier (BBB) becomes more permeable and new antigens become exposed with CNS damage (e.g., inner portions of myelin), CNS antigens may be newly presented to naïve cells of the adaptive immune system (acute-mid-stage neuroinflammation). Many of these antigens may not have been presented outside the CNS before, so CNS antigen-specific adaptive immune cells may exist and undergo clonal expansion during neuroinflammation. At mid-stage to chronic neuroinflammation, these potentially autoreactive T cells and B cells translocate into the CNS parenchyma. A strong adaptive response to non-resolving neuroinflammation could exacerbate pathology. The course of neuroinflammatory dynamics is similar in traumatic, ischemic, autoimmune, and neurodegenerative disorders.
Uncontrolled neuroinflammation can have pathological effects. For instance, traumatic spinal cord injury (SCI) causes prolonged pro-inflammatory responses that fail to resolve or effectively repair the spinal cord. Instead, prolonged SCI-elicited neuroinflammation is likely neurotoxic and causes secondary damage (Kigerl and others, 2009). Below, we discuss evidence that hyperinflammatory CNS responses worsen pathology in several neurological disorders, including neurotrauma, ischemic injury, MS, and AD/aging.
Traumatic spinal cord and brain injury
Experimental models of traumatic SCI are useful for understanding neuroinflammatory dynamics, because the initial injury occurs at a discrete time and post-injury inflammatory cascades can be assessed systematically. SCI damages the blood-spinal cord barrier and creates a gradient of chemotactic inflammatory factors that promotes the infiltration of peripheral immune cells to the lesion epicenter and peri-lesional zone (Popovich and others, 1996; Figley and others, 2014). Thus, SCI causes activation of resident CNS cells (astrocytes, microglia, ependymal cells), but also involves peripheral immune cells that are not typically exposed to CNS antigens. After SCI, the responding peripheral immune cells appear in waves similar to what is observed following peripheral trauma (Donnelly and Popovich, 2008). Neutrophils respond to injury by producing chemokines that recruit blood monocytes, which differentiate into lesion-localized macrophages (Schnell and others, 1999; Kigerl and others, 2006; Gaudet and others, 2015). In the periphery, macrophages respond efficiently to environmental cues and participate in the efficient and rapid phagocytic removal of tissue debris, culminating in a switch (within 7-10 d) to a reparative and/or resolving anti-inflammatory cell type (Gaudet and others, 2011). In contrast, in the CNS, macrophages maintain and even exaggerate their pro-inflammatory phenotype into chronic stages (Kigerl and others, 2009; Kroner and others, 2014), which may amplify signaling and activation of adaptive immune cells (T cells and B cells, which typically have no CNS access) to further drive pro-inflammatory cascades (Sroga and others, 2003; Fleming and others, 2006; Kigerl and others, 2006; Rieckmann and others, 2017). Thus, feed-forward inflammatory cascades caused in part by inefficient phagocytosis of debris, the accumulation of damage-associated molecular patterns (DAMPs) and ineffective immune cell clearance (e.g., phagocytosis of apoptotic cells) likely contribute to chronic, non-resolving neuroinflammation after SCI.
Dampening or shifting the post-SCI inflammatory response could improve neuroprotection and neurological function. Ablating peripheral macrophages reduced SCI pathology (Popovich and others, 1999), and macrophages in the spinal cord cause axon dieback (Busch and others, 2009; Evans and others, 2014) and neurotoxicity (Kigerl and others, 2009). Pathologic B cell and T cell responses also alter post-SCI outcomes (Jones and others, 2002; Ankeny and others, 2009). Astrocytes form a scar that limits the spread of immune cell infiltrates beyond the site of primary trauma (Faulkner and others, 2004), but also restricts axon plasticity and regeneration (McKeon and others, 1991; Alilain and others, 2011; Bartus and others, 2014). Thus, modifying post-SCI neuroinflammatory responses to improve neuroprotection, remyelination, and axon plasticity could benefit neurologic recovery.
Although reducing the inflammatory response to traumatic SCI could boost neuroprotection, there are also beneficial aspects of the pro-inflammatory response. Microglia activated by the pro-inflammatory cytokine interferon-γ (IFN-γ) promote adult neural progenitor cells to differentiate into neurons (Butovsky and others, 2006). In the injured peripheral nerve, CD11b+ myeloid cells are required for myelin clearance, growth factor induction, angiogenesis, and effective axon regeneration (Barrette and others, 2008), suggesting that optimizing CNS microglial/macrophage responses could improve spinal cord repair (e.g., McPhail and others, 2004). In the spinal cord, axons from transplanted fluorescent neurons extend towards foci of activated macrophages, but these axons are destroyed upon closely approaching or touching activated macrophages (Gensel and others, 2009). In demyelinating models, activating microglia can improve remyelination and oligodendrocyte progenitor cell proliferation (Olah and others, 2012; Plemel and others, 2014; Doring and others, 2015). Therefore, there are aspects of the pro-inflammatory response that, if modulated specifically and cautiously, could help improve tissue repair.
Traumatic brain injury (TBI)-elicited neuroinflammation is similar to that caused by SCI (Johnson and others, 2013; Corps and others, 2015; Gyoneva and Ransohoff, 2015). After TBI, microglia develop a mixed pro- and anti-inflammatory phenotype that polarizes towards pro-inflammatory activation over time (Kumar and others, 2016). Limiting microglial hyperactivation by deleting NOX2, an enzyme that produces cytotoxic superoxide, reduced post-TBI neuropathology (Dohi and others, 2010). After TBI in humans, microglial/macrophage activation and neuropathology often persist for years post-injury (Johnson and others, 2013). Although individuals with TBI often achieve apparent complete cognitive and functional recovery, these patients are more susceptible to neuropsychiatric disorders (e.g., depression, cognitive impairment, and neurodegenerative disease) (Witcher and others, 2015). As with other neuroinflammatory conditions, TBI-elicited inflammation has both detrimental and beneficial aspects (Russo and McGavern, 2016); thus, one must optimize the timing, duration, and intensity of any immunomodulatory treatment.
Ischemic brain injury
The brain consumes 20% of whole-body oxygen intake (Raichle and Gusnard, 2002); thus, loss of cerebral blood flow (via vessel blockade or hypoperfusion) causes devastating neuropathology. Ischemic stroke, which is the most prevalent stroke subtype, is caused by obstruction of a brain-supplying artery that prevents oxygen delivery (Roger and others, 2012; Gesuete and others, 2014). The only current treatments for ischemic stroke are fibrinolytic therapy (Shobha and others, 2011), which degrades the blood clot to restore blood flow, and clot removal by surgery. Ideally, these procedures are initiated within three hours of stroke onset. Unfortunately, there is nearly complete cell death in the ischemic core (Morrison and Filosa, 2013), and reperfusion after a period of ischemia can drive pathological neuroinflammation.
Cerebral ischemia-elicited neuroinflammation has a similar course and function to that observed after SCI and TBI (Yilmaz and Granger, 2010). Ischemia activates brain-resident cells, including microglia, astrocytes, and endothelia. Increased production of endothelial cell adhesion molecules and inflammatory cytokines/chemokines, combined with increased blood-brain barrier permeability, enable ischemia-induced peripheral immune cell infiltration and activation. Within the first 4 h after ischemia-reperfusion, neutrophils are the major immune cell infiltrate (Yilmaz and others, 2006). Other cell types (monocytes/macrophages and T cells) become more prevalent at/after 24 h post-ischemia.
Dampening the responses of several immune cell types has often proven protective in cerebral ischemia-reperfusion models. After middle cerebral artery occlusion (MCAO), rats treated with an anti-neutrophil antibody (i.p., 24 h prior and immediately after) showed reduced infarct size (Matsuo and others, 1994). Oxygen-glucose deprivation causes microglia/macrophages to develop an initially more balanced inflammatory response that eventually biases towards a pro-inflammatory phenotype, which could exacerbate injury (Hu and others, 2012). For instance, HMGB1, which is a chromatin binding protein that is normally expressed intracellularly, is released by stressed or necrotic cells into the extracellular space. Extracellular HMGB1 acts as a DAMP that can bind to immune receptors on microglia/macrophages to drive inflammation. Blocking HMGB1 and other DAMPs can reduce post-MCAO microglial/macrophage activation and infarct size (Kim and others, 2006; Shichita and others, 2017). T cells can also exacerbate ischemic injury: MCAO mice lacking CD4+ or CD8+ T cells exhibit reduced inflammation and ameliorated neuropathology (Yilmaz and others, 2006). Neuroinflammatory activation also occurs in humans after ischemic stroke (Denes and others, 2010), and identifying effective neuroprotective therapies is a top priority (Chamorro and others, 2016). Although there are some protective roles of neuroinflammation prior to (preconditioning) and after ischemia (Karelina and others, 2009; Yilmaz and Granger, 2010), it is most clear that neuroinflammation exacerbates pathology after cerebral ischemia.
Multiple sclerosis
MS is a neurodegenerative demyelinating disorder that often first presents in 20- to 40-year olds. There are four types of MS: relapsing-remitting MS (RRMS), the most common form, which consists of transient MS symptoms with periods of remission; secondary progressive MS, during which MS clinical signs steadily progress after an initial period of RRMS; primary progressive MS, which presents as immediate progression of MS clinical signs; and progressive relapsing MS, during which relapsing and progressive MS occur concomitantly (Mahad and others, 2015). Although the exact etiology of MS remains unknown, neuroinflammatory processes appear to contribute to MS pathology. Chronic MS-related pathological neuroinflammation eventually evolves into a more neurodegenerative form at later stages. Different aspects of MS can be explored using specific experimental models: for instance, inflammatory pathology in MS is modeled using experimental autoimmune encephalomyelitis (EAE), which can be induced by immunization with CNS antigens (e.g., myelin-oligodendrocyte glycoprotein); the cellular and molecular determinants of demyelination and remyelination can be assessed using focal intraparenchymal injection of lysolecithin or ethidium bromide or by adding cuprizone in rodent chow (Ransohoff, 2012).
As mentioned, although the molecular mechanisms that initiate MS pathology remain elusive, neuroinflammation likely causes or exacerbates MS pathology. Focally activated microglia demyelinate axons and help recruit monocytes, T cells, and B cells (Berard and others, 2010; Rawji and Yong, 2013; Hemmer and others, 2015). T and B cells expand clonally and worsen inflammation (Prineas and Graham, 1981; Skulina and others, 2004; Obermeier and others, 2011). Various T cell subsets influence MS/EAE outcomes: CD8+ cytotoxic T cells, Tbet+ Th1 cells, GATA3+ Th2 cells, and ROR-γt+ Th17 cells can contribute to EAE pathology (Sie and others, 2014; Grifka-Walk and others, 2015; Sinha and others, 2015) (although these cells can also have reparative properties). In contrast, FOXP3+ regulatory T (Treg) cells, which maintain tolerance to self-antigens and could thereby reduce neuroinflammation, likely have impaired regulatory activity during EAE (Nyirenda and others, 2015). B cells also influence EAE outcomes through their ability to secrete antibodies, present antigens, and release cytokines. B cells may limit EAE onset, but worsen EAE progression by augmenting T cell activation and infiltration (Matsushita and others, 2008; Pierson and others, 2014). The early protective actions of B cells are carried out by a rare IL-10-producing regulatory B cell subset that can reduce neuroinflammation. Unfortunately, the benefit of this small endogenous B cell population is likely overwhelmed by inflammatory changes as the disease progresses (Matsushita and others, 2008; Shen and others, 2014; Zhang and others, 2015).
The inflammatory basis for MS is highlighted by recent clinical developments. Indeed, a humanized antibody (ocrelizumab) for treating RRMS acts by binding to CD20 to deplete B cells (Barun and Bar-Or, 2012; Sorensen and Blinkenberg, 2016); B cell depletion showed efficacy in the EAE model (Matsushita and others, 2008; Weber and others, 2010) and ocrelizumab has recently been approved by the U.S. Food and Drug Administration as a therapy for MS. In phase 3 trials for RRMS, ocrelizumab reduced relapse rate by 46% compared to a gold standard treatment, IFN-β1a (Hauser and others, 2017). Remarkably, ocrelizumab also effectively reduced symptoms of primary progressive MS, which currently has no standard treatment (possibly due to a more neurodegenerative, less inflammatory basis compared to RRMS) (Calabresi, 2017; Montalban and others, 2017).
Aging and Alzheimer’s disease
Neuroinflammation also contributes to age-related cognitive decline, AD, and other neurodegenerative conditions (Gomez-Nicola and Perry, 2015). In the brains of aging animals and humans, inflammatory stimuli elicit an exaggerated response from microglia (Streit and others, 2004; Barrientos and others, 2009; Henry and others, 2009; Frank and others, 2010). Aged microglia are considered to be in a “primed” state; they express increased basal levels of immune receptors and host defense genes and are hypersensitive to immune stimulation (Sierra and others, 2007; Hickman and others, 2013; Fonken and others, 2016b). For instance, aged rats injected with Escherichia coli have exaggerated and prolonged neuroinflammatory responses that correlate with a prolonged sickness response and related cognitive deficits. Although hippocampal microglia from aged rats do not exhibit elevated cytokines at baseline, they have increased expression of pattern recognition receptors and danger signals. Targeting HMGB1 with its competitive antagonist, Box-A, prevents age-related exaggerated neuroinflammatory and behavioral responses to infection (Fonken and others, 2016c).
Similarly, neuroinflammation likely contributes to AD (Heneka and others, 2015). In AD, misfolded and aggregated proteins (amyloid β oligomers) accumulate in the extracellular space; these complexes trigger inflammatory signaling in microglia by activating pattern recognition receptors such as toll-like receptors (TLRs) (Heneka and others, 2015). As described above in the context of CNS injury, microglia in AD brain are activated; however they are inefficient phagocytes and do not efficiently clear amyloid β oligomers (Heneka and others, 2015). Because these immune-reactive proteins are constantly deposited in AD brain and persist in the extracellular space indefinitely, the aggregates are associated with chronic neuroinflammation (Lue and others, 2001; Laske and others, 2010). This likely worsens the neurodegeneration and impaired cognition that is characteristic of AD (Tarkowski and others, 2003; Jin and others, 2008; Holmes and others, 2009). Neuroinflammation also contributes to other chronic neurodegenerative disorders including amyotrophic lateral sclerosis, prion disease, and possibly Parkinson’s and Huntington’s diseases (see Gomez-Nicola and Perry, 2015)).
microRNA production and function
Transcription of miRNAs is usually mediated by RNA polymerase II and its associated transcription factors (Cai and others, 2004; Lee and others, 2004). As with production of protein-coding mRNAs, transcription of different miRNAs can be elicited by the same transcription factor. For instance, the canonical pro-inflammatory transcription factor NFκB increases expression of both pro-inflammatory (miR-155) and anti-inflammatory (miR-124, miR-146a) miRNAs (Ma and others, 2014; Doxaki and others, 2015), illustrating how miRNA induction can both elicit inflammation and provide negative feedback regulation of the inflammatory response (see below for more detail). Transcription of a miRNA gene produces miRNA primary transcripts (pri-miRNAs), which are several kilobases long and contain local stem-loop structures (Ha and Kim, 2014).
Canonical miRNA biogenesis occurs in a multi-step process that involves the key processing proteins Drosha, DGCR8, and Dicer (Fig. 2) (Kim and others, 2009; Daugaard and Hansen, 2017). miRNAs also can be produced via alternative pathways that generate mature miRNAs independent of Drosha/DGCR8 and/or Dicer (see Daugaard and Hansen, 2017).
Figure 2. An overview of microRNA processing and function.

Most miRNA primary transcripts (pri-miRNAs) are transcribed by RNA polymerase II. The pri-miRNA is loaded into the microprocessor complex, which consists of the proteins Drosha and DGCR8. Drosha cleaves the pri-miRNA to create a small hairpin RNA, the 70-90 nucleotide pre-miRNA. The pre-miRNA is bound by Exportin-5 (linked to Ran-GTP); then, the Exportin-5-miRNA complex is translocated to the cytosol. The pre-miRNA is then loaded into a protein complex including Dicer and Argonaute (Ago). Dicing the pre-miRNA results in a ~22 nucleotide long miRNA duplex. The miRNA duplex consists of the miR-5p (e.g., miR-155-5p), which is the sequence that was closest to the 5’ end of the pri-miRNA; and the complementary miR-3p (e.g., miR-155-3p), which is the sequence that was closest to the 3’ end of the pri-miRNA. One of the strands (usually the more stable strand, which is often the −5p strand) is loaded into the Ago in the microRNA-induced silencing complex (miRISC). In animals, the miRNA seed sequence (5-7 nucleotides) binds with partial complementarity to sequences in the 3’ untranslated region of target mRNAs. The miRISC complex then directs these target mRNAs for degradation or translational repression. Bold text indicates miRNA species. Please see text for further detail and citations.
For canonical miRNA biogenesis, the RNase type II protein, Drosha, along with cofactor DGCR8, form a microprocessor complex that recognizes specific motifs in the pri-miRNA (Lee and others, 2003; Gregory and others, 2004; Han and others, 2004; Kwon and others, 2016). Ultimately, the microprocessor complex defines the mature miRNA sequence to be used by cleaving at the stem of the hairpin structure then releasing a small RNA hairpin called the pre-miRNA (Lee and others, 2002). Next, the pre-miRNA is exported to the cytoplasm by the nuclear transport receptor Exportin-5. Exportin-5 binds cooperatively to the pre-miRNA and a cofactor, GTP-bound Ran; once in the cytosol, GTP is hydrolyzed and the pre-miRNA cargo is released (Bohnsack and others, 2004; Lund and others, 2004).
Next, the protein Dicer acts with cofactors to cleave the pre-miRNA into ~22 nucleotide miRNA duplexes (double-stranded RNA). The ~22 nucleotide RNA duplex is incorporated into an Argonaute protein (four members, Ago1-4, in humans) to create the miRNA-induced silencing complex (miRISC) (Chendrimada and others, 2005; Iwasaki and others, 2010). Based on relative stability, one strand of the duplex is released; the other remains in the miRISC to participate in miRNA-mediated inhibition of translation (Kwak and Tomari, 2012). Although both strands are active and have knockdown potential, generally one strand is more prevalent and biologically relevant than the other strand due to differing stability/half-lives of each strand (Meijer and others, 2014).
The miRNA in the miRISC helps target the complex to specific RNAs and dampens their expression through translational repression, mRNA decay, and mRNA deadenylation (Bartel, 2009; Eichhorn and others, 2014). In animals, miRNAs identify their target RNAs by partial complementary binding between the miRNA seed sequence – a sequence at positions 2-7 from the 5’ end – and regions in the 3’ untranslated region of the target RNA (Ameres and Zamore, 2013; Daugaard and Hansen, 2017). Although partial complementarity is one key mechanism of target recognition, nucleotides downstream of the seed sequence can also modulate RNA target recognition and other mechanisms of miRNA-mediated inhibition have been identified. It is clear that partial complementarity would enable an individual miRNA to modulate expression of hundreds of mRNAs, and at least one miRNA binding site exists in 30-80% of protein-coding genes (Friedman and others, 2009; Lu and Clark, 2012). Thus, miRNA-based regulation likely has roles in nearly all biological processes and pathologies (Bartel, 2009; Mendell and Olson, 2012; Cloonan, 2015).
miRNA nomenclature is evolving; details on nomenclature are reviewed elsewhere (Griffiths-Jones and others, 2006; Budak and others, 2016). Briefly, miRNAs derived from a single duplex are distinguished by −5p and −3p suffixes (e.g., miR-124-5p for 5’ arm; miR-124-3p for 3’ arm), and a lettered suffix represents closely-related mature miRNAs derived from distinct precursors or loci (e.g., miR-146a, miR-146b) (Budak and others, 2016).
Mature miRNAs can have wide-ranging effects on the function and translation of other RNAs (Fig. 3a). As mentioned, each individual miRNA binds on average 200 mRNAs. Conversely, mRNAs can be bound by multiple miRNAs (Krek and others, 2005; Tsang and others, 2010). miRNAs often act by binding the 3’ untranslated region of target mRNAs, but they can also target sequences in the 5’ region or protein-coding domain (Ameres and Zamore, 2013). Although individual miRNAs reduce translation/function of target RNAs, they generally do not completely shut down target RNA function. Instead, they act to dampen partially the function of these RNAs, although miRNAs acting in concert can have more profound effects (Cech and Steitz, 2014). Further, miRNAs can target other classes of non-coding RNA, including circular RNAs and long non-coding RNAs (Jeggari and others, 2012; Millan, 2017). Thus, miRNAs fine-tune output of the transcriptome.
Figure 3. MicroRNAs dynamically regulate post-transcriptional function and translation of RNAs, both within the producing cell and between cells (intercellular communication).
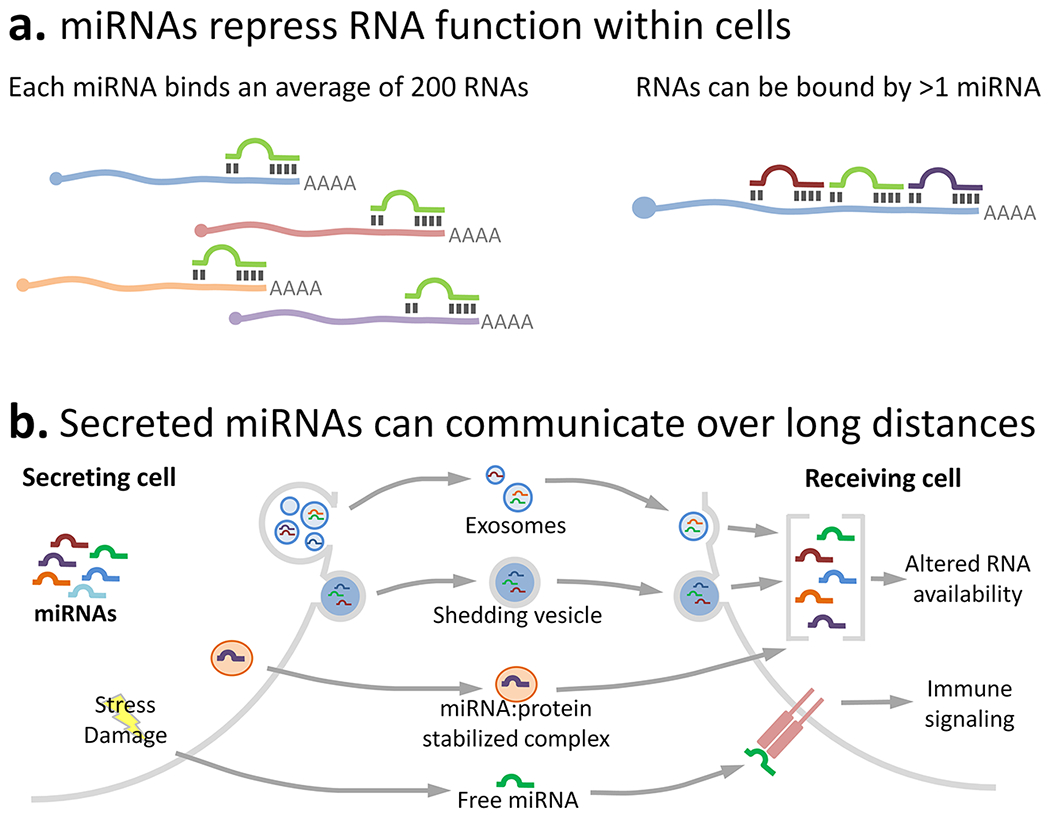
(a) MicroRNAs bind RNA sequences complementary to the miRNA’s 6-7 oligonucleotide “seed sequence”. miRNAs typically bind RNAs in the 3’ untranslated region, although they can bind in the 5’ region or in coding areas. Once bound, miRNAs cause RNA degradation or sequestration. The complexity of miRNA functions is highlighted by the fact that each miRNA binds an average of 200 RNAs (left), and individual RNAs can be bound by multiple miRNAs (right). (b) miRNAs can also be secreted/released to act over short or long distances in a paracrine manner. miRNAs from the secreting cell can be sorted and packaged into microvesicles (exosomes or shedding vesicles), which are then received by another cell by endocytosis or membrane fusion (miRNAs can also be packaged in synaptic vesicles; not shown). miRNAs can also be released in association with a protective protein (e.g., Ago2 or high-density lipoprotein); miRNA:protein complexes are stabilized for long periods in the extracellular space. Receiving cells can take up these secreted or released miRNAs, which regulate RNA availability and protein translation in that cell. In addition, cell stress, damage, or necrosis causes release of free miRNAs that bind to immune receptors to initiate inflammatory changes (e.g., let-7b binds to the immune receptor TLR7). In this manner, miRNAs act as short- or long-range communication cues that affect cell physiology throughout the body during health and pathology.
In addition to their autocrine regulatory roles, miRNAs can be released to alter function of other cells nearby or throughout the body (Chen and others, 2012; Hulsmans and Holvoet, 2013) (Fig. 3b). miRNAs exist in the extracellular space in small vesicles (exosomes, shedding microvesicles, and synaptic vesicles) (Valadi and others, 2007; Mitchell and others, 2008; Li and others, 2015) or protected in extracellular protein-miRNA complexes (e.g., with Ago2 or high-density lipoprotein) (Arroyo and others, 2011; Vickers and others, 2011). The miRNA repertoire in microvesicles can be significantly different from that in their parent cell, suggesting that specific miRNAs are selectively packaged into vesicles (Diehl and others, 2012). These extracellular miRNAs likely affect health and pathology. For instance, inactivated endothelial cells secrete miRNA-containing exosomes to suppress monocyte activation (Njock and others, 2015), and circulating adipose-derived exosomal miRNAs regulate target mRNAs in distant tissues to control metabolic function (Thomou and others, 2017). Extracellular miRNAs may also act as DAMPs; e.g., the miRNA let-7b can bind to TLR7 to elicit neurodegeneration (Lehmann and others, 2012). Therefore, release of miRNAs during neuropathology could shape the inflammatory response, including modulation of cell phenotype, response intensity, and the balance between toxic or reparative effector functions.
microRNAs: role in CNS neuroinflammation
miRNAs (and other non-coding RNAs) were discovered only recently. As such, their functional roles in the context of neuroinflammation remain largely unknown (Fig. 4). Still, it is increasingly clear that miRNAs control inflammation initiation and maintenance (e.g., Liu and Abraham, 2013). Here, we consider the roles of specific pro- and anti-inflammatory miRNAs in neuroinflammation (Fig. 5).
Figure 4. MicroRNAs remain understudied.

(a) The number of articles (as of April 2017) for each miRNA from miR-1 to miR-100. Many miRNAs have very few related published papers. (b) Published articles about miRNAs highlighted in this review, in the context of inflammation (black bar), the nervous system (light grey), and total papers (dark grey). Many of the top immunomodulatory miRNAs are under-studied, highlighting that the miRNA-neuroinflammation field is in its infancy.
Figure 5. Key miRNAs with regulatory roles in inflammatory cells.

Immunomodulatory miRNAs, their functions, and validated RNA targets for each cell type are summarized. Please see text for citations. --, no RNAs identified; N/A, not applicable.
Pro-inflammatory miRNAs in neuroinflammatory disorders
miR-155
miR-155-5p is a pro-inflammatory miRNA that is uniquely positioned: it has been widely studied in inflammation, and its published potent pro-inflammatory actions across immune cell types are unparalleled by any other miRNA. miR-155’s role in inflammation was first identified in 2005 when its expression was found to be elevated in human B cell lymphoma (Eis and others, 2005; Kluiver and others, 2005). Subsequent studies showed that miR-155 is required for typical B cell function and cytokine production (Thai and others, 2007). Similarly, miR-155 is critical for effective responses of macrophages and T cells. In macrophages, miR-155 is upregulated by TLR ligands and by the pro-inflammatory cytokine IFN-γ (O’Connell and others, 2007; Tili and others, 2007). Key validated anti-inflammatory RNA targets of miR-155-5p include the inositol phosphatase Ship1 (O’Connell and others, 2009), the transcription factor Cebpb (Worm and others, 2009), the STAT1 suppressor Socs1 (Lu and others, 2009a; Cardoso and others, 2012), and the anti-inflammatory receptor IL-13Rα1 (Martinez-Nunez and others, 2011). Thus, induction of miR-155 may release an endogenous anti-inflammatory “brake”, resulting in increased inflammation. Indeed, using microarrays, our group found that activation of inflammatory signaling in miR-155 KO macrophages was significantly blunted (Jablonski and others, 2016). In macrophages from miR-155 knockout mice stimulated with IFN-γ + LPS, i.e., stimuli that elicit consistently strong inflammatory cascades in macrophages, 66% fewer genes were up- or down-regulated (vs. media-treated; WT: 1989 genes, KO: 671 genes) via these activating stimuli (Jablonski and others, 2016). These data suggest that miR-155 critically regulates inflammatory signaling in macrophages.
miR-155 is a key pro-inflammatory miRNA, so removing or inhibiting miR-155 should improve damaging aspects of neuroinflammation. Our group found that miR-155 deletion was neuroprotective and improved histological and functional outcome measures in an experimental SCI model (Gaudet and others, 2016b). In a novel co-culture model, miR-155 KO macrophages improved growth and survival of wild-type neurons, particularly under inflammatory conditions. Interestingly, miR-155 KO neurons also had improved intrinsic growth capacity, suggesting that miR-155 inhibition could also affect axon growth and plasticity independent of its effects on macrophages. In vivo, miR-155 KO mice with SCI showed enhanced neuroprotection and axon regeneration, and expedited locomotor recovery (Gaudet and others, 2016b). In separate studies, our group reported that miR-155 deletion reduced pathology in other diseases or disorders exacerbated by inflammation: miR-155 KO mice had reduced anxiety- and depressive-like symptoms (Fonken and others, 2016a) and reduced diet-induced obesity (Gaudet and others, 2016b).
A pro-inflammatory role for miR-155 has been observed in several other neuropathologies. In MS patients, miR-155 is robustly upregulated in brain lesions (Junker et al., 2009) and serum (Paraboschi and others, 2011). In a rodent MS model (EAE), miR-155 deletion (O’Connell and others, 2010) or miR-155-5p inhibition (even after disease onset; Murugaiyan and others, 2011; Zhang and others, 2014) are neuroprotective and attenuate neurologic impairment. In EAE using miR-155 KO or inhibitor-treated mice, induction of cytotoxic T cells, regulatory Th17 cells, and dendritic cell-induced T cell activation are reduced (O’Connell and others, 2010; Murugaiyan and others, 2011; Zhang and others, 2014). Endothelial miR-155 regulates blood-brain barrier permeability to worsen EAE (Lopez-Ramirez and others, 2014). Further, a recent study suggests the involvement of miR-155-3p in EAE pathology (Mycko and others, 2015). After ischemic stroke in mice, miR-155-5p inhibition (intravenous, beginning 48 h post-occlusion) reduced pro-inflammatory processes (Pena-Philippides and others, 2016) and improved neuroprotection, brain perfusion, and functional recovery (Caballero-Garrido and others, 2015). miR-155 upregulation may be pathological in a mouse model of AD (Guedes and others, 2014). Finally, in a mouse model of amyotrophic lateral sclerosis (male and female SOD1G93A mice), miR-155 deletion/inhibition improved survival, likely by reducing the inflammatory potential of microglia (Koval and others, 2013; Butovsky and others, 2015).
Overall, miR-155 (miR-155-5p in particular) is a critical pro-inflammatory miRNA that is commonly upregulated in inflammatory and neurological disorders. In fact, no other miRNA has been identified that has such profound pro-inflammatory effects. Therefore, miR-155 inhibition or removal – perhaps in combination with an anti-inflammatory miRNA or other reparative factor – could be explored as a therapy for various neurological disorders.
Other pro-inflammatory miRNAs
Although miR-155 is the most studied pro-inflammatory miRNA, other miRNAs also are known to be pro-inflammatory. miR-27b targets an anti-inflammatory transcriptional activator, PPAR-γ; in human macrophages, this interaction blocks the induction of an anti-inflammatory phenotype. Inhibiting miR-27b also limits inflammatory signalling. For example, miR-27b inhibition reduces the ability of LPS to increase macrophage production of inflammatory cytokines including IL-6 and TNF-α (Jennewein and others, 2010); this likely occurs by de-repression of PPAR-γ, which normally dampens pro-inflammatory network activation (Lee and others, 2012; see also Zhou and others, 2012). PPAR-γ expression is dysregulated in SCI (McTigue and others, 2007), MS (Klotz and others, 2005), and AD (Sastre and others, 2006), suggesting the possibility that PPAR-γ levels in neurologic disorders could be altered by miR-27b. Accordingly, miR-27b expression increases in several neuroinflammatory disorders; miR-27b is upregulated in CD4+ T cells of MS patients (Guerau-de-Arellano and others, 2011) and in the brain of Alzheimer’s patients (Cogswell and others, 2008). Therefore, inhibiting miR-27b could be a viable strategy for ameliorating neuroinflammation.
miR-326 has been implicated in MS pathology. miR-326 expression in leukocytes correlated with disease severity in MS patients and in mice with EAE (Du and others, 2009; Honardoost and others, 2014). miR-326 drives differentiation of IL-17-producing Th17 cells, which worsen MS pathology. Conversely, silencing miR-326 reduced EAE pathology (Duand others, 2009). The role of miR-326 in other inflammatory and neurologic disorders remains unclear.
With the exception of miR-155, which has broad pro-inflammatory effects in an array of immune cell types (including microglia (Cardoso and others, 2012) and astrocytes (Tarassishin and others, 2011)), there are few other examples of key pro-inflammatory miRNAs in the literature. Future studies will help clarify whether miR-155 is the major pro-inflammatory miRNA, or whether there are additional miRNAs with similarly potent pro-inflammatory activities.
Anti-inflammatory miRNAs in neuroinflammatory disorders
miR-124
miR-124 is expressed most robustly in the nervous system and has predominantly anti-inflammatory effects (Sempere and others, 2004). miR-124 can have anti-inflammatory actions in macrophages via the cholinergic anti-inflammatory pathway and the vagus nerve. The vagus nerve acts on splenic T cells, which produce acetylcholine that binds the α7-nicotonic acetylcholine receptor on macrophages to promote anti-inflammatory polarization (Rosas-Ballina and others, 2011). Within these macrophages, miR-124 drives anti-inflammatory polarization by reducing Stat3 (and downstream IL-6) and TNF-α converting enzyme (and downstream TNF-α). In macrophages, miR-124 is upregulated in response to anti-inflammatory cytokines IL-4 and IL-13 and is necessary for regulating the expression of genes associated with the anti-inflammatory macrophage phenotype (i.e., increased CD206 and Ym1; decreased CD86, iNOS, and TNF) (Veremeyko and others, 2013). These anti-inflammatory effects of miR-124 could be via translational repression of the transcription factor Cebpa and/or the cytokine receptor IL6R (Hatziapostolou and others, 2011; Ponomarev and others, 2011). Interestingly, miR-124 is also upregulated in macrophages by pro-inflammatory stimuli (MyD88-dependent), and acts as a brake on inflammation (Maand others, 2014). miR-124 gain-of-function also improved survival in a model of sepsis (Sun and others, 2013b). Thus, miR-124 has a critical anti-inflammatory role in macrophages. Overexpressing miR-124 in T cells caused them to develop an effector phenotype that was protective in a mouse glioma model (i.e., T cells activated by miR-124 help clear glioma) (Wei and others, 2013); however, this miR-124-elicited effector T cell response could be damaging in neuroinflammatory conditions.
miR-124 also appears to have protective effects in EAE. In adult mice miR-124 was expressed in microglia, but not peripheral monocytes or macrophages (Ponomarev and others, 2011). miR-124 overexpression in microglia reduced induction of pro-inflammatory TNF-α and nitric oxide (Louw and others, 2016). Similarly, overexpressing miR-124 in macrophages transformed them into more quiescent cells, likely by targeting C/ebp-α (Ponomarev and others, 2011). During EAE, microglial miR-124 was downregulated. Peripheral miR-124 administration, either prior to or after EAE onset, deactivated macrophages and T cells, and improved neurologic outcomes (Ponomarev and others, 2011).
In rodent models of stroke, most studies show a neuroprotective role for miR-124. miR-124 delivery, particularly at acute post-injury times, was neuroprotective and polarized CNS macrophages towards an anti-inflammatory phenotype (Doeppner and others, 2013; Sun and others, 2013a; Hamzei Taj and others, 2016). However, miR-124 also may have a detrimental role in stroke pathology; cerebral miR-124 knockdown in rats (24 h prior to occlusion) reduced infarct size and boosted neurologic outcomes (Zhu and others, 2014). It is possible that different experimental models or timing of miR-124 delivery could explain these divergent results. Regardless, serum exosome concentrations of miR-124 have been identified as a biomarker that predicts the incidence and severity of acute ischemic stroke (Ji and others, 2016).
miR-124 may also benefit other neurologic disorders. In AD, miR-124 expression may be downregulated in hippocampus of humans with AD (Lukiw, 2007) and this coincides with an increase in the potentially damaging BACE1 protein. Reducing BACE1 activity could dampen Aβ secretion, and miR-124 appears to downregulate BACE1 expression (Fang and others, 2012). Similarly, miR-124 overexpression was neuroprotective in a Drosophila model of AD (Kong and others, 2015). In mouse peripheral neuropathic pain models, intrathecal infusion of miR-124 relieved hypersensitivity (Willemen and others, 2012). Future studies will further reveal potential anti-inflammatory effects of miR-124 and its neuroprotective actions in the context of neurotrauma, ALS, and AD.
miR-146a and miR-146b
miR-146a acts as a negative regulator of inflammation. miR-146a is induced by NFκB activation and feeds back on this pathway by inhibiting translation of IRAK1 and TRAF6 mRNAs. Accordingly, miR-146a is upregulated in various neurological conditions, suggesting that cells are compensating for pathological inflammation and attempting to restore homeostasis. For instance, in MS patients, miR-146a and miR-146b were upregulated in peripheral blood mononuclear cells compared to controls (Fenoglio and others, 2011). This may represent a compensatory anti-inflammatory response; miR-146a expressed by brain endothelia reduces NFκB activation and T-cell adhesion (by targeting NFκB pathway activators RhoA, Nfat5, IRAK1, and TRAF6; (Wu and others, 2015), which could limit immune cell infiltration and neuroinflammation during pathology.
miR-146a has been studied in other preclinical models involving neuroinflammation. In a rat model of stroke, miR-146a was found to increase oligodendrogenesis by targeting IRAK1 (Liu and others, 2017). miR-146a is also protective after ischemia-reperfusion injury in other tissues including myocardium (Wang and others, 2013), liver (Jiang and others, 2014), and intestine (Chassin and others, 2012) (all through reducing IRAK1 and/or TRAF6 translation). In temporal lobe epilepsy, miR-146a is increased in astrocytes (Aronica and others, 2010). In a spared-nerve injury mouse model of neuropathic pain, overexpressing miR-146a-5p inhibited TRAF6-JNK-CCL2 signaling in astrocytes to limit neuropathic pain (Lu and others, 2015).
NFκB directly induces both anti-inflammatory miR-146a and pro-inflammatory miR-155, so the expression of these miRNAs is often considered in parallel. It is interesting that NFκB-dependent transcription involves activation of two miRNAs with such divergent roles (Fig. 6).
Figure 6. miRNAs both amplify and dampen inflammatory signaling pathways, such as the NFκB pathway.

The NFκB signaling pathway is shown as an example of inflammatory signaling. TLR activation by binding of DAMPs, pathogen-associated molecular patterns, or extracellular RNA/DNA activates MyD88-dependent intracellular signaling, resulting in nuclear translocation of NFκB transcription factors p65 and p50. NFκBs upregulate both pro- and anti-inflammatory miRNAs. Pro-inflammatory miRNAs, such as miR-155 (left side), reduce mRNA availability of factors that inhibit activation of this inflammatory pathway. These pro-inflammatory miRNA pathways can amplify pro-inflammatory signaling cascades. Anti-inflammatory miRNAs (right side) reduce mRNA availability of inflammatory receptors, signaling mediators, and cytokines. These anti-inflammatory miRNAs provide a negative feedback mechanism and act as a “brake” on inflammation. Blue, signaling mediators; grey, inhibitory binding partner; beige-gold, inhibitor of signaling; red, pro-inflammatory cytokines; turquoise, miRNAs.
miR-21
miR-21 is another anti-inflammatory miRNA that could effectively modulate neuroinflammation. miR-21 is upregulated in activated immune cells, including neutrophils, dendritic cells, monocytes/macrophages, and T cells (see Sheedy, 2015). In mouse bone marrow-derived macrophages and human blood monocytes, miR-21 is induced by LPS downstream of TLR4-MyD88-NFκB signaling. miR-21 directly targets Pdcd4; PDCD4 participates in pro-inflammatory signaling by increasing IL-6 and decreasing IL-10 (by preventing a Twist2-c-Maf-IL-10 transcriptional cascade) (Sheedy and others, 2010; van den Bosch and others, 2014). The importance of PDCD4 is highlighted by the fact that more Pdcd4 KO mice survive a potentially lethal LPS dose (a model of sepsis) (Sheedyand others, 2010). Conversely, survival times are decreased in miR-21 KO mice challenged with LPS-induced peritonitis (Barnett and others, 2016). There is some evidence for a pro-inflammatory role of miR-21 in macrophages. miR-21 KO macrophages are better able to adopt an anti-inflammatory phenotype; this may be due to de-repressed expression of the miR-21 target Stat3 (Wang and others, 2015).
miR-21 has several additional targets that are relevant to neuroinflammation. miR-21 targets Smad7, thereby de-repressing TGF-β signaling (Barnettand others, 2016). miR-21 targets Spry1 to boost MAP kinase signaling (Thum and others, 2008). miR-21 also affects differentiation of other immune cells, including T cells and dendritic cells. miR-21 directly targets IL-12p35, a subunit of the cytokine IL-12 (Lu and others, 2009b). IL-12 drives Th1 cell differentiation and production of the Th1 cytokine IFN-γ. Dendritic cells that are deficient in miR-21 express higher IL-12 levels, and miR-21 deficiency enhances Th1 and Th17 cell responses (Lu and others, 2011). Others have shown that miR-21 is required for T cells to develop a Th17 phenotype (Murugaiyan and others, 2015).
Reducing or blocking miR-21 is beneficial in EAE. In mouse T cells, miR-21 increased in Th17 cells and promoted their differentiation by targeting Smad7. Adoptively transferring Th17 cells that were polarized in the presence of miR-21 inhibitor, or systemically inhibiting miR-21 (using anti-miR-21) prior to disease onset, ameliorated EAE neurologic symptoms (Murugaiyan and others, 2015). miR-21 expression also associates with disease progression in human MS. In CD4+ T cells, miR-21 was upregulated in cells from patients with relapse-remitting MS (Fenoglioand others, 2011), but was downregulated in cells from patients with secondary progressive MS (Sanders and others, 2016). Thus, miR-21 has detrimental roles in EAE by driving Th17 cell differentiation; however, miR-21 likely has divergent roles in MS that vary by cell type and disease type/progression.
In contrast with its role in EAE, miR-21 has anti-inflammatory functions in other diseases with prominent neuroinflammatory cascades. Bhalala and colleagues (2012) overexpressed miR-21 or a miR-21 sponge (a synthetic RNA that contains several complementary binding sites to the seed region of a miRNA of interest; a dominant-negative method) specifically in astrocytes. They found that overexpressed miR-21 reduced astrocyte hypertrophy in the traumatically injured spinal cord. Conversely, a miR-21 sponge boosted SCI-induced astrocyte hypertrophy – but also increased axon sprouting into a glial scar that normally blocks axon growth. In another SCI study, intrathecal mini-pump administration of a miR-21 antagomir in rats exacerbated intraspinal pathology and limited spontaneous recovery of function (Hu and others, 2013). After TBI, miR-21 is a biomarker for severe injury (Di Pietro and others, 2017) and overexpressing miR-21 improves blood-brain barrier maintenance, angiogenesis, and neuroprotection, as well as functional recovery (Ge and others, 2014; Ge and others, 2015). miR-21 overexpression also indirectly (via reduced microglial toxicity) and directly protected cultured cortical neurons from apoptosis caused by oxygen and glucose deprivation (Buller and others, 2010; Zhang and others, 2012), suggesting that increasing miR-21 could be therapeutic after stroke. In the context of aging, miR-21 upregulation is a biomarker of aging (Olivieri and others, 2012) and in mouse neurons, treatment with Aβ protein reduces miR-21 (Schonrock and others, 2010). Although these preliminary data on miR-21 in aging are promising, the function of miR-21 in aging, AD, and other neurodegenerative diseases is not well characterized.
Overall, miR-21 has predominantly anti-inflammatory and neuroprotective effects that could benefit neurologic diseases with toxic neuroinflammatory cascades. However, it is clear that miR-21 also can have detrimental effects in MS and other inflammatory conditions, highlighting the importance of understanding how potential miRNA therapeutics can affect the phenotype of different cell types in a specific neuroinflammatory disorder and disease stage.
miR-223
miR-223 has anti-inflammatory properties in peripheral immune cells. In macrophages, miR-223 drives typical anti-inflammatory macrophage phenotype (Zhuang and others, 2012; Ying and others, 2015; Deiuliis and others, 2016). miR-223 in macrophages limits translation of Nlrp3 mRNA, which encodes a key component of the NLRP3 inflammasome (Bauernfeind and others, 2012; Haneklaus and others, 2012). miR-223 may also reduce inflammatory signaling in neutrophils (Li and others, 2017) and dendritic cells (Zhou and others, 2015). In T cells, miR-223 was upregulated in patients with rheumatoid arthritis and miR-223 impaired activation of a protective IGF-1/IL-10 axis (Lu and others, 2014).
miR-223 is under-studied in the nervous system; however, existing data indicate that miR-223 both positively and negatively affects neuroinflammatory cascades. miR-223 reduced neurotoxicity after global ischemia and excitotoxic injury by enhancing the degradation of mRNA encoding glutamate receptors (Harraz and others, 2012). A miR-223 antagonist may reduce SCI pathology by improving neuroprotection and angiogenesis (Liu and others, 2015), although more studies are required. In EAE, miR-223 deletion in mice reduced dendritic cell activation of Th17 (but not Th1) cell differentiation, improved myeloid-derived suppressor cell activity, and enhanced neurologic function (Ifergan and others, 2016; Cantoni and others, 2017). miR-223 was significantly upregulated in CD4+ T cells from patients in the relapse-remitting phase of MS, suggesting a possible role in positively regulating pathogenic cascade that contributes to relapse-remitting MS (Hosseini and others, 2016). Reduced serum miR-223 may be a hallmark of AD (Jia and Liu, 2016), although the function of miR-223 in AD and other neurodegenerative disorders remains unclear.
miRNAs with pro- and anti-inflammatory actions: the Let-7 family
The Lethal-7 (let-7) miRNA is conserved across species (from C. elegans to humans) (Reinhartand others, 2000), and was the first miRNA to be identified in humans (Pasquinelli and others, 2000). In humans and mice, nine mature let-7 miRNAs exist and each has distinct nucleotide sequences, but all contain highly conserved seed regions (Lee and others, 2016). Let-7 family members generally elicit cell differentiation and are tumour suppressors (Lee and others, 2016).
Let-7 miRNAs modulate inflammation. Increasing let-7 expression in macrophages promotes differentiation into an anti-inflammatory phenotype, likely by reducing expression of the transcription factor C/ebp-δ (Banerjee and others, 2013). Other key mRNA targets of let-7 include the inflammatory cytokine IL6 (Schulte and others, 2011), and the highly conserved pattern recognition receptor, Tlr4 (Teng and others, 2013). Let-7 provides negative feedback to limit inflammatory activation; however, it is downregulated by NFκB activation (Schulte and others, 2011). NFκB drives transcription of the RNA-binding protein Lin28, which inhibits let-7 (Iliopoulos and others, 2010). IL-6 translation is therefore disinhibited; IL-6 signaling can activate STAT3-dependent NFκB transcription, thereby closing a positive inflammatory feed-forward loop that amplifies inflammation. In dendritic cells activated by LPS, let-7 inhibits Socs1, which promotes dendritic cell maturation and their ability to drive T cell proliferation (Zhang and others, 2011; Kim and others, 2013). let-7 may also limit self-renewal of memory T cells (Almanza and others, 2010), suppress CD4+ T cell activation, promote T cell anergy (Marcais and others, 2014), and inhibit Th17 cell differentiation (Zhang and others, 2013). In the CNS, let-7 limits microglial activation (Cho and others, 2015) and promotes differentiation of cultured glial progenitor cells into astrocytes (Shenoy and others, 2015).
In neuroinflammatory disorders, let-7 has some protective roles. After ischemic stroke, overexpression of let-7 reduced post-stroke neurotoxicity and improved neurologic outcomes, an effect that might be caused by let-7-mediated reduction of caspase-3. These effects were also associated with reduced microglial activation (Ni and others, 2015). After T10 transection SCI, let-7 was increased in the lumbar spinal cord (Liu and others, 2010), although the biological effects of let-7 induction remain undefined.
Paradoxically, let-7 can also contribute to neuropathology. During insult or in neurodegenerative disease, let-7 can be released by dying neurons into the extracellular space, where it acts as a DAMP (Lehmann and others, 2012; Coleman and others, 2017). Extracellular let-7 can act as a ligand for TLR7, an endolysosome-localized receptor that binds to extracellular-derived single-stranded RNA (which is found at low levels under healthy conditions) (Kawai and Akira, 2010). When bound, let-7:TLR7 elicits microglia and macrophage activation and propagates neurotoxicity. The increased expression of TLR7 and binding by let-7 indicates tissue damage or infection and immune cell activation. In fact, elevated let-7 has been proposed as a biomarker in MS (Gandhi and others, 2013), stroke (Huang and others, 2016b), and AD (Lehmann and others, 2012). In EAE, let-7 was found to drive pathogenic Th1 and Th17 cell differentiation to worsen disease by targeting IL-10 mRNA (Guan and others, 2013). In a newt model of tail/spinal cord regeneration, let-7 is downregulated; application of a let-7 mimic prevents tail regeneration, likely by reducing the effectiveness of the ependymal response to amputation (Lepp and Carlone, 2015).
Thus, it appears that let-7 acts within immune cells to promote both anti- and pro-inflammatory actions, whereas extracellular let-7 may worsen neuroinflammatory conditions. The let-7 miRNA family remains underexplored in the context of several neurological conditions. Future studies could reveal whether the role of let-7 as a DAMP is specific to a selected set of miRNAs (e.g., miR-21 also binds TLR7; Yelamanchili and others, 2015), or whether all/most miRNAs released during cell stress act as DAMPs and similarly activate immune cells.
Future directions: Manipulating immunomodulatory microRNAs to improve CNS and PNS neuroinflammatory pathologies
Several strategies can be used to reveal the functional importance of miRNAs. Deletion of essential miRNA machinery components Dicer, Drosha, or DGCR8 has been used to establish broad functional roles of miRNAs in health and disease. For instance, Dicer conditional deletion in developing parvalbumin-expressing dorsal root ganglion neurons prevents maintenance of proprioceptive cell fate and peripheral connectivity (O’Toole and others, 2017); deletion of Dicer in forebrain neurons predisposes mice to neurodegeneration in adulthood (Hebert and others, 2010); and Dicer-deficient T cells show reduced differentiation capacity and preferentially differentiate into inflammatory Th1 cells (Muljo and others, 2005). These results indicate that miRNAs play a critical role in cell development, cell fate, and cell survival. However, there are caveats to strategies relying on deletion of these miRNA regulators: Dicer, Drosha, and DGCR8 have functions that are independent of regulating miRNA processing, so deleting these key genes will have pleiotropic effects that limit physiologic and therapeutic relevance (i.e., removing all miRNAs, even in a single cell type, could have wide-ranging effects that preclude therapeutic relevance) (Macias and others, 2013). For example, Dicer-null embryonic stem cells lack both miRNAs and small interfering RNAs, Drosha cleaves and thereby destabilizes mRNAs and long non-coding RNAs, and DGCR8 also influences levels of other small RNAs. Regardless, complementing these studies with microarrays and follow-up research on newly-identified functional miRNAs can be powerful. For example, Dicer conditional deletion in mature mouse oligodendrocytes caused demyelination, neuroinflammation, and shortened lifespan; using miRNA microanalysis combined with target prediction analyses of 3’ UTRs, miR-219 and its target RNA ELOVL7 were identified as novel mediators controlling oligodendrocyte physiology (Shin and others, 2009). There are several in silico target prediction algorithms that can reveal potential miRNA-target interactions and networks (Steinkraus and others, 2016).
Once a miRNA of interest is recognized, then specific gain- and loss-of-function experiments can be completed. In particular, several unique loss-of-function strategies can be used to identify key biological effects of miRNAs. In addition to typical mutagenesis (KO of specific miRNA; though there can exist miRNA redundancy), more therapeutically relevant competitive miRNA inhibitors (e.g., antimiRs, antagomirs, LNA-based antimiRs, and sponges) or miRNA response element blockers (i.e., a blocker that spans the miRNA binding site on a specific RNA to physically prevent miRNA binding) can be used to test the necessity and downstream mechanisms of action for a given miRNA (see Steinkraus and others, 2016). Thus, coordinating bioinformatic predictive approaches with robust biological readouts and functional assays will facilitate discovery of relevant new miRNA targets.
In studying miRNAs, one must take into account several considerations. First, a miRNA labeled as a “biomarker” of inflammation suggests its potential involvement in the disease process, but the effects of the miRNA could be pro- or anti-inflammatory and could be indicative of an ongoing pathological response or, conversely, an attempt to restore homeostasis (e.g., miR-155, miR-146, and miR-21 are all upregulated by LPS stimulation, yet have divergent roles). Second, there remain innumerable under-studied miRNAs; examining existing microarray databases, profiling-based strategies, and other data could provide clues regarding undiscovered immunomodulatory miRNAs. Third, of the studied miRNAs, it is likely that most have other as-yet unidentified roles. This is expected, since ongoing research is often biased by previous findings. For instance, our group found that miR-155 – which had known roles in cancer and inflammation – also regulated axon growth (Gaudet and others, 2016b), susceptibility to obesity (Gaudet and others, 2016a), and anxiety- and depressive-like symptoms (Fonken and others, 2016a). Fourth, miRNAs can have hundreds of “predicted” targets; however, it is important to determine whether these miRNA-RNA interactions are valid and have biological relevance. Conversely, it could be useful to work “backwards” to identify potential immunomodulatory miRNAs; i.e., one could identify an inflammatory mRNA that they would like to downregulate, then use prediction algorithms to determine putative miRNAs that would bind to that inflammatory mRNAs. Finally, it is important to consider the double-edged sword of modulating miRNAs: by binding several mRNAs, they may act as intrinsic combinatorial therapies, but such co-regulation could also have unintended consequences. For instance, miR-21 has beneficial effects in several neuroinflammatory disorders (SCI, TBI, ischemia, and aging), but miR-21 also worsens EAE (Murugaiyan and others, 2015), binds TLR7 to act as a DAMP (Yelamanchili and others, 2015), and is an oncogene (Medina and others, 2010).
Conclusions
Thousands of published papers discuss the roles of miRNAs in the nervous system, yet microRNA research is still in its infancy. New genomic loci for non-coding RNAs continue to be discovered, increasing the complexity in our models of post-transcriptional regulatory networks. Further, evidence that miRNAs can be delivered between cells – even over long distances – suggests that these small RNAs can communicate physiologic status and alter function of cells throughout the body.
Given that miRNAs have important intra- and intercellular roles, it is clear that they could control aspects of neuroinflammation. Indeed, as discussed above, miRNAs with roles in pro-inflammatory (miR-155, miR-27b, miR-326), anti-inflammatory (miR-124, miR-146a, miR-21, miR-223), and mixed immunomodulatory (let-7 family) responses regulate neuroinflammation in rodent models of neurologic trauma and disease. Undoubtedly, other miRNAs exist that have immunomodulatory function, but remain to be revealed and/or tested in the nervous system. It is remarkable that the miniscule seven-nucleotide miRNA-RNA interaction can have such wide-ranging cellular and physiologic functions.
Identifying miRNA-based strategies that improve neurological disorders could be useful, since miRNAs target multiple RNAs and act as intrinsic combinatorial modulators. Therapeutic modulation of miRNAs in CNS disorders is in clinical trials for glioblastoma (Christopher and others, 2016), underscoring the clinical potential of these small molecules. Thus, revealing therapeutically-relevant immunomodulatory miRNAs could lead to novel therapies that dampen neuroinflammation and improve outcomes in neurological disorders.
Grant support:
Grant support was provided by the Paralyzed Veterans of America (LRW: #3004), the United States’ Department of Defense (LRW: W81XWH-13-1-0277/SC120066), the Craig H. Neilsen Foundation (SFM), the Wings for Life Foundation (LRW/ADG), NIH R01 DE921966 (LRW) and the Ray W. Poppleton endowment (PGP).
Abbreviations:
- AD
Alzheimer’s disease
- BBB
blood-brain barrier
- CNS
central nervous system
- DAMPs
damage-associated molecular patterns
- EAE
experimental autoimmune encephalomyelitis
- IFN
interferon
- IL
interleukin
- i.p.
intraperitoneal
- LPS
lipopolysaccharide
- MAP
mitogen-activated protein
- MCAO
middle cerebral artery occlusion
- MHC
major histocompatibility complex
- miRNA
microRNA
- MS
multiple sclerosis
- PNS
peripheral nervous system
- RRMS
relapsing-remitting multiple sclerosis
- SCI
spinal cord injury
- TBI
traumatic brain injury
- Th
T helper
- TLR
toll-like receptor
- Treg
regulatory T cell
References
- Alilain WJ, Horn KP, Hu H, Dick TE, Silver J. 2011. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 475(7355):196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almanza G, Fernandez A, Volinia S, Cortez-Gonzalez X, Croce CM, Zanetti M. 2010. Selected microRNAs define cell fate determination of murine central memory CD8 T cells. PLoS One. 5(6):e11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Zamore PD. 2013. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 14(8):475–488. [DOI] [PubMed] [Google Scholar]
- Ankeny DP, Guan Z, Popovich PG. 2009. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J Clin Invest. 119(10):2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E and others.2010. Expression pattern of miR-146a, an inflammation-associated microRNA, in experimental and human temporal lobe epilepsy. Eur J Neurosci. 31(6):1100–1107. [DOI] [PubMed] [Google Scholar]
- Arroyo JD and others.2011. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 108(12):5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S and others.2013. MicroRNA let-7c regulates macrophage polarization. J Immunol. 190(12):6542–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett RE and others.2016. Anti-inflammatory effects of miR-21 in the macrophage response to peritonitis. J Leukoc Biol. 99(2):361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrette B and others.2008. Requirement of myeloid cells for axon regeneration. J Neurosci. 28(38):9363–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Watkins LR, Rudy JW, Maier SF. 2009. Characterization of the sickness response in young and aging rats following E. coli infection. Brain Behav Immun. 23(4):450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell. 136(2):215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus K and others.2014. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J Neurosci. 34(14):4822–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barun B, Bar-Or A. 2012. Treatment of multiple sclerosis with anti-CD20 antibodies. Clin Immunol. 142(1):31–37. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. 2012. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol. 189(8):4175–4181. [DOI] [PubMed] [Google Scholar]
- Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. 2010. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain. 133(Pt 2):433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berard JL, Kerr BJ, Johnson HM, David S. 2010. Differential expression of SOCS1 in macrophages in relapsing-remitting and chronic EAE and its role in disease severity. Glia. 58(15):1816–1826. [DOI] [PubMed] [Google Scholar]
- Bhalala OG and others.2012. microRNA-21 regulates astrocytic response following spinal cord injury. J Neurosci. 32(50):17935–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack MT, Czaplinski K, Gorlich D. 2004. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. Rna. 10(2):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budak H, Bulut R, Kantar M, Alptekin B. 2016. MicroRNA nomenclature and the need for a revised naming prescription. Brief Funct Genomics. 15(1):65–71. [DOI] [PubMed] [Google Scholar]
- Buller B and others.2010. MicroRNA-21 protects neurons from ischemic death. Febs j. 277(20):4299–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Horn KP, Silver DJ, Silver J. 2009. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 29(32):9967–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O and others.2006. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 31(1):149–160. [DOI] [PubMed] [Google Scholar]
- Butovsky O and others.2015. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 77(1):75–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero-Garrido E and others.2015. In Vivo Inhibition of miR-155 Promotes Recovery after Experimental Mouse Stroke. J Neurosci. 35(36):12446–12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Hagedorn CH, Cullen BR. 2004. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna. 10(12):1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi PA. 2017. B-Cell Depletion - A Frontier in Monoclonal Antibodies for Multiple Sclerosis. N Engl J Med. 376(3):280–282. [DOI] [PubMed] [Google Scholar]
- Cantoni C and others.2017. Mir-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 133(1):61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. 2012. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 135(1):73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, Steitz JA. 2014. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 157(1):77–94. [DOI] [PubMed] [Google Scholar]
- Chamorro A, Dirnagl U, Urra X, Planas AM. 2016. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15(8):869–881. [DOI] [PubMed] [Google Scholar]
- Chassin C and others.2012. MicroRNA-146a-mediated downregulation of IRAK1 protects mouse and human small intestine against ischemia/reperfusion injury. EMBO Mol Med. 4(12):1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liang H, Zhang J, Zen K, Zhang CY. 2012. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 22(3):125–132. [DOI] [PubMed] [Google Scholar]
- Chendrimada TP and others.2005. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 436(7051):740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KJ, Song J, Oh Y, Lee JE. 2015. MicroRNA-Let-7a regulates the function of microglia in inflammation. Mol Cell Neurosci. 68(167–176. [DOI] [PubMed] [Google Scholar]
- Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. 2016. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 7(2):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N 2015. Re-thinking miRNA-mRNA interactions: intertwining issues confound target discovery. Bioessays. 37(4):379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogswell JP and others.2008. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 14(1):27–41. [DOI] [PubMed] [Google Scholar]
- Coleman LG Jr., Zou J, Crews FT. 2017. Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J Neuroinflammation. 14(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corps KN, Roth TL, McGavern DB. 2015. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 72(3):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 9(1):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard I, Hansen TB. 2017. Biogenesis and Function of Ago-Associated RNAs. Trends Genet. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A. 2011. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 12(7):388–399. [DOI] [PubMed] [Google Scholar]
- Deiuliis JA and others.2016. Visceral Adipose MicroRNA 223 Is Upregulated in Human and Murine Obesity and Modulates the Inflammatory Phenotype of Macrophages. PLoS One. 11(11):e0165962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Thornton P, Rothwell NJ, Allan SM. 2010. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 24(5):708–723. [DOI] [PubMed] [Google Scholar]
- Di Pietro V and others.2017. MicroRNAs as Novel Biomarkers for the Diagnosis and Prognosis of Mild and Severe Traumatic Brain Injury. J Neurotrauma. [DOI] [PubMed] [Google Scholar]
- Diehl P and others.2012. Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovasc Res. 93(4):633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeppner TR and others.2013. MicroRNA-124 protects against focal cerebral ischemia via mechanisms involving Usp14-dependent REST degradation. Acta Neuropathol. 126(2):251–265. [DOI] [PubMed] [Google Scholar]
- Dohi K and others.2010. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation. 7(41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X. 2013. MicroRNA: function, detection, and bioanalysis. Chem Rev. 113(8):6207–6233. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. 2008. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 209(2):378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring A and others.2015. Stimulation of monocytes, macrophages, and microglia by amphotericin B and macrophage colony-stimulating factor promotes remyelination. J Neurosci. 35(3):1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxaki C, Kampranis SC, Eliopoulos AG, Spilianakis C, Tsatsanis C. 2015. Coordinated Regulation of miR-155 and miR-146a Genes during Induction of Endotoxin Tolerance in Macrophages. J Immunol. 195(12):5750–5761. [DOI] [PubMed] [Google Scholar]
- Du C and others.2009. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 10(12):1252–1259. [DOI] [PubMed] [Google Scholar]
- Eichhorn SW and others.2014. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell. 56(1):104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eis PS and others.2005. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 102(10):3627–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, Vajkoczy P, Weller RO. 2017. The movers and shapers in immune privilege of the CNS. Nat Immunol. 18(2):123–131. [DOI] [PubMed] [Google Scholar]
- Evans TA and others.2014. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol. 254(109-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M and others.2012. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol Lett. 209(1):94–105. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. 2004. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 24(9):2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenoglio C and others.2011. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci Lett. 504(1):9–12. [DOI] [PubMed] [Google Scholar]
- Figley SA, Khosravi R, Legasto JM, Tseng YF, Fehlings MG. 2014. Characterization of vascular disruption and blood-spinal cord barrier permeability following traumatic spinal cord injury. J Neurotrauma. 31(6):541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JC and others.2006. The cellular inflammatory response in human spinal cords after injury. Brain. 129(Pt 12):3249–3269. [DOI] [PubMed] [Google Scholar]
- Fonken LK, Gaudet AD, Gaier KR, Nelson RJ, Popovich PG. 2016a. MicroRNA-155 deletion reduces anxiety- and depressive-like behaviors in mice. Psychoneuroendocrinology. 63(362-369. [DOI] [PubMed] [Google Scholar]
- Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF. 2015. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun. 45(171-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Kitt MM, Gaudet AD, Barrientos RM, Watkins LR, Maier SF. 2016b. Diminished circadian rhythms in hippocampal microglia may contribute to age-related neuroinflammatory sensitization. Neurobiol Aging. 47(102-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK and others.2016c. The Alarmin HMGB1 Mediates Age-Induced Neuroinflammatory Priming. J Neurosci. 36(30):7946–7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Watkins LR, Maier SF. 2010. Aging sensitizes rapidly isolated hippocampal microglia to LPS ex vivo. J Neuroimmunol. 226(1-2):181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. 2007. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 21(1):47–59. [DOI] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF. 2016. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav Immun. 55(215-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19(1):92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne JJ, Power MC. 2010. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 74(12):995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R and others.2013. Circulating microRNAs as biomarkers for disease staging in multiple sclerosis. Ann Neurol. 73(6):729–740. [DOI] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG, Ramer MS. 2011. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. 8(110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Sweet DR, Polinski NK, Guan Z, Popovich PG. 2015. Galectin-1 in injured rat spinal cord: implications for macrophage phagocytosis and neural repair. Mol Cell Neurosci. 64(84-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD and others.2016a. miR-155 Deletion in Female Mice Prevents Diet-Induced Obesity. Sci Rep. 6(22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD and others.2016b. miR-155 Deletion in Mice Overcomes Neuron-Intrinsic and Neuron-Extrinsic Barriers to Spinal Cord Repair. J Neurosci. 36(32):8516–8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X and others.2015. MiR-21 alleviates secondary blood-brain barrier damage after traumatic brain injury in rats. Brain Res. 1603(150-157. [DOI] [PubMed] [Google Scholar]
- Ge XT and others.2014. miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci Rep. 4(6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG. 2009. Macrophages promote axon regeneration with concurrent neurotoxicity. J Neurosci. 29(12):3956–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesuete R, Kohama SG, Stenzel-Poore MP. 2014. Toll-like receptors and ischemic brain injury. J Neuropathol Exp Neurol. 73(5):378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nicola D, Perry VH. 2015. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 21(2):169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM and others.2016. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 113(24):E3441–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI and others.2004. The Microprocessor complex mediates the genesis of microRNAs. Nature. 432(7014):235–240. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. 2006. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34(Database issue):D140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifka-Walk HM, Giles DA, Segal BM. 2015. IL-12-polarized Th1 cells produce GM-CSF and induce EAE independent of IL-23. Eur J Immunol. 45(10):2780–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H and others.2013. MicroRNA let-7e is associated with the pathogenesis of experimental autoimmune encephalomyelitis. Eur J Immunol. 43(1):104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes JR, Custodia CM, Silva RJ, de Almeida LP, Pedroso de Lima MC, Cardoso AL. 2014. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum Mol Genet. 23(23):6286–6301. [DOI] [PubMed] [Google Scholar]
- Guerau-de-Arellano M and others.2011. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain. 134(Pt 12):3578–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S, Ransohoff RM. 2015. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci. 36(7):471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 15(8):509–524. [DOI] [PubMed] [Google Scholar]
- Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M. 2016. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials. 91(151-165. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. 2004. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18(24):3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneklaus M and others.2012. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J Immunol. 189(8):3795–3799. [DOI] [PubMed] [Google Scholar]
- Harraz MM, Eacker SM, Wang X, Dawson TM, Dawson VL. 2012. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc Natl Acad Sci U S A. 109(46):18962–18967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziapostolou M and others.2011. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 147(6):1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SL and others.2017. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med. 376(3):221–234. [DOI] [PubMed] [Google Scholar]
- Hebert SS and others.2010. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum Mol Genet. 19(20):3959–3969. [DOI] [PubMed] [Google Scholar]
- Hemmer B, Kerschensteiner M, Korn T. 2015. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 14(4):406–419. [DOI] [PubMed] [Google Scholar]
- Heneka MT and others.2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14(4):388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. 2009. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 23(3):309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE and others.2013. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 16(12):1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C and others.2009. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 73(10):768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honardoost MA, Kiani-Esfahani A, Ghaedi K, Etemadifar M, Salehi M. 2014. miR-326 and miR-26a, two potential markers for diagnosis of relapse and remission phases in patient with relapsing-remitting multiple sclerosis. Gene. 544(2):128–133. [DOI] [PubMed] [Google Scholar]
- Hooten KG, Beers DR, Zhao W, Appel SH. 2015. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics. 12(2):364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppmann N and others.2015. New candidates for CD4 T cell pathogenicity in experimental neuroinflammation and multiple sclerosis. Brain. 138(Pt 4):902–917. [DOI] [PubMed] [Google Scholar]
- Hosseini A and others.2016. Upregulation of CD4+T-Cell Derived MiR-223 in The Relapsing Phase of Multiple Sclerosis Patients. Cell J. 18(3):371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JZ, Huang JH, Zeng L, Wang G, Cao M, Lu HB. 2013. Anti-apoptotic effect of microRNA-21 after contusion spinal cord injury in rats. J Neurotrauma. 30(15):1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X and others.2012. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 43(11):3063–3070. [DOI] [PubMed] [Google Scholar]
- Huang Q and others.2016a. MicroRNAs associated with the pathogenesis of multiple sclerosis. J Neuroimmunol. 295-296(148-161. [DOI] [PubMed] [Google Scholar]
- Huang S and others.2016b. Identification of Blood Let-7e-5p as a Biomarker for Ischemic Stroke. PLoS One. 11(10):e0163951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmans M, Holvoet P. 2013. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc Res. 100(1):7–18. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Chen S, Zhang B, Miller SD. 2016. Cutting Edge: MicroRNA-223 Regulates Myeloid Dendritic Cell-Driven Th17 Responses in Experimental Autoimmune Encephalomyelitis. J Immunol. 196(4):1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. 2010. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 39(4):493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S and others.2010. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol Cell. 39(2):292–299. [DOI] [PubMed] [Google Scholar]
- Jablonski KA, Gaudet AD, Amici SA, Popovich PG, Guerau-de-Arellano M. 2016. Control of the Inflammatory Macrophage Transcriptional Signature by miR-155. PLoS One. 11(7):e0159724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggari A, Marks DS, Larsson E. 2012. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 28(15):2062–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein C, von Knethen A, Schmid T, Brune B. 2010. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA destabilization. J Biol Chem. 285(16):11846–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q and others.2016. Increased Brain-Specific MiR-9 and MiR-124 in the Serum Exosomes of Acute Ischemic Stroke Patients. PLoS One. 11(9):e0163645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia LH, Liu YN. 2016. Downregulated serum miR-223 servers as biomarker in Alzheimer’s disease. Cell Biochem Funct. 34(4):233–237. [DOI] [PubMed] [Google Scholar]
- Jiang W and others.2014. miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS One. 9(7):e101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. 2008. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 5(23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. 2013. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 136(Pt 1):28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TB and others.2002. Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: implications for autoimmune vaccine therapy. J Neurosci. 22(7):2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E. 2015. Inhibition of miR-34b and miR-34c enhances alpha-synuclein expression in Parkinson’s disease. FEBS Lett. 589(3):319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis WW, Derada Troletti C, Reijerkerk A, Romero IA, de Vries HE. 2015. The blood-brain barrier in multiple sclerosis: microRNAs as key regulators. CNS Neurol Disord Drug Targets. 14(2):157–167. [DOI] [PubMed] [Google Scholar]
- Karelina K, Norman GJ, Zhang N, Morris JS, Peng H, DeVries AC. 2009. Social isolation alters neuroinflammatory response to stroke. Proc Natl Acad Sci U S A. 106(14):5895–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 11(5):373–384. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. 2006. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 494(4):578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. 2009. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 29(43):13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Hall JC, Wang L, Mo X, Yu Z, Popovich PG. 2016. Gut dysbiosis impairs recovery after spinal cord injury. J Exp Med. 213(12):2603–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB and others.2006. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 26(24):6413–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Gregersen PK, Diamond B. 2013. Regulation of dendritic cell activation by microRNA let-7c and BLIMP1. J Clin Invest. 123(2):823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 10(2):126–139. [DOI] [PubMed] [Google Scholar]
- Klotz L and others.2005. Proinflammatory stimulation and pioglitazone treatment regulate peroxisome proliferator-activated receptor gamma levels in peripheral blood mononuclear cells from healthy controls and multiple sclerosis patients. J Immunol. 175(8):4948–4955. [DOI] [PubMed] [Google Scholar]
- Kluiver J and others.2005. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 207(2):243–249. [DOI] [PubMed] [Google Scholar]
- Kong Y, Wu J, Zhang D, Wan C, Yuan L. 2015. The Role of miR-124 in Drosophila Alzheimer’s Disease Model by Targeting Delta in Notch Signaling Pathway. Curr Mol Med. 15(10):980–989. [DOI] [PubMed] [Google Scholar]
- Koval ED and others.2013. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 22(20):4127–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A and others.2005. Combinatorial microRNA target predictions. Nat Genet. 37(5):495–500. [DOI] [PubMed] [Google Scholar]
- Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. 2014. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 83(5):1098–1116. [DOI] [PubMed] [Google Scholar]
- Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. 2016. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J Neurotrauma. 33(19):1732–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak PB, Tomari Y. 2012. The N domain of Argonaute drives duplex unwinding during RISC assembly. Nat Struct Mol Biol. 19(2):145–151. [DOI] [PubMed] [Google Scholar]
- Kwon SC and others.2016. Structure of Human DROSHA. Cell. 164(1-2):81–90. [DOI] [PubMed] [Google Scholar]
- Laske C and others.2010. Macrophage colony-stimulating factor (M-CSF) in plasma and CSF of patients with mild cognitive impairment and Alzheimer’s disease. Curr Alzheimer Res. 7(5):409–414. [DOI] [PubMed] [Google Scholar]
- Lee H, Han S, Kwon CS, Lee D. 2016. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell. 7(2):100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Drakaki A, Iliopoulos D, Struhl K. 2012. MiR-27b targets PPARgamma to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene. 31(33):3818–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 75(5):843–854. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. 2002. MicroRNA maturation: stepwise processing and subcellular localization. Embo j. 21(17):4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y and others.2004. MicroRNA genes are transcribed by RNA polymerase II. Embo j. 23(20):4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y and others.2003. The nuclear RNase III Drosha initiates microRNA processing. Nature. 425(6956):415–419. [DOI] [PubMed] [Google Scholar]
- Lehmann SM and others.2012. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 15(6):827–835. [DOI] [PubMed] [Google Scholar]
- Lepp AC, Carlone RL. 2015. MicroRNA dysregulation in response to RARbeta2 inhibition reveals a negative feedback loop between MicroRNAs 1, 133a, and RARbeta2 during tail and spinal cord regeneration in the adult newt. Dev Dyn. 244(12):1519–1537. [DOI] [PubMed] [Google Scholar]
- Li H, Wu C, Aramayo R, Sachs MS, Harlow ML. 2015. Synaptic vesicles contain small ribonucleic acids (sRNAs) including transfer RNA fragments (trfRNA) and microRNAs (miRNA). Sci Rep. 5(14918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M and others.2017. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47phox-oxidative stress pathway in neutrophils. Gut. 66(4):705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Huang Y, Jia C, Li Y, Liang F, Fu Q. 2015. Administration of antagomir-223 inhibits apoptosis, promotes angiogenesis and functional recovery in rats with spinal cord injury. Cell Mol Neurobiol. 35(4):483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Abraham E. 2013. MicroRNAs in immune response and macrophage polarization. Arterioscler Thromb Vasc Biol. 33(2):170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Keeler BE, Zhukareva V, Houle JD. 2010. Cycling exercise affects the expression of apoptosis-associated microRNAs after spinal cord injury in rats. Exp Neurol. 226(1):200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS and others.2017. MicroRNA-146a Promotes Oligodendrogenesis in Stroke. Mol Neurobiol. 54(1):227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londin E and others.2015. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci U S A. 112(10):E1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Ramirez MA and others.2014. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. Faseb j. 28(6):2551–2565. [DOI] [PubMed] [Google Scholar]
- Louw AM and others.2016. Chitosan polyplex mediated delivery of miRNA-124 reduces activation of microglial cells in vitro and in rat models of spinal cord injury. Nanomedicine. 12(3):643–653. [DOI] [PubMed] [Google Scholar]
- Lu J, Clark AG. 2012. Impact of microRNA regulation on variation in human gene expression. Genome Res. 22(7):1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF and others.2009a. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 30(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu MC, Yu CL, Chen HC, Yu HC, Huang HB, Lai NS. 2014. Increased miR-223 expression in T cells from patients with rheumatoid arthritis leads to decreased insulin-like growth factor-1-mediated interleukin-10 production. Clin Exp Immunol. 177(3):641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TX, Munitz A, Rothenberg ME. 2009b. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 182(8):4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TX and others.2011. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol. 187(6):3362–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cao DL, Jiang BC, Yang T, Gao YJ. 2015. MicroRNA-146a-5p attenuates neuropathic pain via suppressing TRAF6 signaling in the spinal cord. Brain Behav Immun. 49(119-129. [DOI] [PubMed] [Google Scholar]
- Lue LF and others.2001. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 35(1):72–79. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. 2007. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 18(3):297–300. [DOI] [PubMed] [Google Scholar]
- Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. 2004. Nuclear export of microRNA precursors. Science. 303(5654):95–98. [DOI] [PubMed] [Google Scholar]
- Ma C and others.2014. microRNA-124 negatively regulates TLR signaling in alveolar macrophages in response to mycobacterial infection. Mol Immunol. 62(1):150–158. [DOI] [PubMed] [Google Scholar]
- Macias S, Cordiner RA, Caceres JF. 2013. Cellular functions of the microprocessor. Biochem Soc Trans. 41(4):838–843. [DOI] [PubMed] [Google Scholar]
- Mahad DH, Trapp BD, Lassmann H. 2015. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 14(2):183–193. [DOI] [PubMed] [Google Scholar]
- Marcais A and others.2014. microRNA-mediated regulation of mTOR complex components facilitates discrimination between activation and anergy in CD4 T cells. J Exp Med. 211(11):2281–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. 2011. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. 286(3):1786–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y and others.1994. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 25(7):1469–1475. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. 2008. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 118(10):3420–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. 1991. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 11(11):3398–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail LT, Stirling DP, Tetzlaff W, Kwiecien JM, Ramer MS. 2004. The contribution of activated phagocytes and myelin degeneration to axonal retraction/dieback following spinal cord injury. Eur J Neurosci. 20(8):1984–1994. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi R, Wei P, Lash AT. 2007. The PPAR gamma agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Exp Neurol. 205(2):396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina PP, Nolde M, Slack FJ. 2010. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 467(7311):86–90. [DOI] [PubMed] [Google Scholar]
- Meijer HA, Smith EM, Bushell M. 2014. Regulation of miRNA strand selection: follow the leader? Biochem Soc Trans. 42(4):1135–1140. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. 2012. MicroRNAs in stress signaling and human disease. Cell. 148(6):1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. 2017. Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer’s disease: an integrative review. Prog Neurobiol. [DOI] [PubMed] [Google Scholar]
- Mitchell PS and others.2008. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 105(30):10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalban X and others.2017. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med. 376(3):209–220. [DOI] [PubMed] [Google Scholar]
- Morrison HW, Filosa JA. 2013. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 10(4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. 2005. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 202(2):261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. 2011. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 187(5):2213–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugaiyan G and others.2015. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. 125(3):1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycko MP, Cichalewska M, Cwiklinska H, Selmaj KW. 2015. miR-155-3p Drives the Development of Autoimmune Demyelination by Regulation of Heat Shock Protein 40. J Neurosci. 35(50):16504–16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J and others.2015. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun. 49(75-85. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. 2005. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308(5726):1314–1318. [DOI] [PubMed] [Google Scholar]
- Njock MS and others.2015. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood. 125(20):3202–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, Godbout JP. 2015. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 96(Pt A):29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyirenda MH and others.2015. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J Immunol. 194(12):5761–5774. [DOI] [PubMed] [Google Scholar]
- O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. 2009. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 106(17):7113–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 104(5):1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM and others.2010. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 33(4):607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole SM and others.2017. Dicer maintains the identity and function of proprioceptive sensory neurons. J Neurophysiol. 117(3):1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier B and others.2011. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J Neuroimmunol. 233(1-2):245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M and others.2012. Identification of a microglia phenotype supportive of remyelination. Glia. 60(2):306–321. [DOI] [PubMed] [Google Scholar]
- Olivieri F and others.2012. Age-related differences in the expression of circulating microRNAs: miR-21 as a new circulating marker of inflammaging. Mech Ageing Dev. 133(11-12):675–685. [DOI] [PubMed] [Google Scholar]
- Paraboschi EM and others.2011. Genetic association and altered gene expression of mir-155 in multiple sclerosis patients. Int J Mol Sci. 12(12):8695–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE and others.2000. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 408(6808):86–89. [DOI] [PubMed] [Google Scholar]
- Pena-Philippides JC, Caballero-Garrido E, Lordkipanidze T, Roitbak T. 2016. In vivo inhibition of miR-155 significantly alters post-stroke inflammatory response. J Neuroinflammation. 13(1):287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W and others.2009. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A. 106(30):12489–12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson ER, Stromnes IM, Goverman JM. 2014. B cells promote induction of experimental autoimmune encephalomyelitis by facilitating reactivation of T cells in the central nervous system. J Immunol. 192(3):929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanu A and others.2014. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis. 71(280-291. [DOI] [PubMed] [Google Scholar]
- Plemel JR, Wee Yong V, Stirling DP. 2014. Immune modulatory therapies for spinal cord injury--past, present and future. Exp Neurol. 258(91-104. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. 2011. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 17(1):64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Horner PJ, Mullin BB, Stokes BT. 1996. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol. 142(2):258–275. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. 1999. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 158(2):351–365. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Graham JS. 1981. Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol. 10(2):149–158. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Gusnard DA. 2002. Appraising the brain’s energy budget. Proc Natl Acad Sci U S A. 99(16):10237–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. 2012. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 15(8):1074–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawji KS, Yong VW. 2013. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin Dev Immunol. 2013(948976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ and others.2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 403(6772):901–906. [DOI] [PubMed] [Google Scholar]
- Rieckmann JC and others.2017. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. [DOI] [PubMed] [Google Scholar]
- Roger VL and others.2012. Executive summary: heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 125(1):188–197. [DOI] [PubMed] [Google Scholar]
- Rosas-Ballina M and others.2011. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 334(6052):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, McGavern DB. 2016. Inflammatory neuroprotection following traumatic brain injury. Science. 353(6301):783–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KA and others.2016. Next-generation sequencing reveals broad down-regulation of microRNAs in secondary progressive multiple sclerosis CD4+ T cells. Clin Epigenetics. 8(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M and others.2006. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 103(2):443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. 1999. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci. 11(10):3648–3658. [DOI] [PubMed] [Google Scholar]
- Schonrock N and others.2010. Neuronal microRNA deregulation in response to Alzheimer’s disease amyloid-beta. PLoS One. 5(6):e11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte LN, Eulalio A, Mollenkopf HJ, Reinhardt R, Vogel J. 2011. Analysis of the host microRNA response to Salmonella uncovers the control of major cytokines by the let-7 family. Embo j. 30(10):1977–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. 2004. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 5(3):R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin ME and others.2016. MicroRNAs targeting TGFbeta signalling underlie the regulatory T cell defect in multiple sclerosis. Brain. 139(Pt 6):1747–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ. 2015. Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response. Front Immunol. 6(19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ and others.2010. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 11(2):141–147. [DOI] [PubMed] [Google Scholar]
- Shen P and others.2014. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 507(7492):366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy A, Danial M, Blelloch RH. 2015. Let-7 and miR-125 cooperate to prime progenitors for astrogliogenesis. Embo j. 34(9):1180–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T and others.2017. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med. [DOI] [PubMed] [Google Scholar]
- Shin D, Shin JY, McManus MT, Ptacek LJ, Fu YH. 2009. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 66(6):843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shobha N, Buchan AM, Hill MD. 2011. Thrombolysis at 3-4.5 hours after acute ischemic stroke onset--evidence from the Canadian Alteplase for Stroke Effectiveness Study (CASES) registry. Cerebrovasc Dis. 31(3):223–228. [DOI] [PubMed] [Google Scholar]
- Sie C, Korn T, Mitsdoerffer M. 2014. Th17 cells in central nervous system autoimmunity. Exp Neurol. 262Pt A(18-27. [DOI] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. 2007. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 55(4):412–424. [DOI] [PubMed] [Google Scholar]
- Sinha S, Boyden AW, Itani FR, Crawford MP, Karandikar NJ. 2015. CD8(+) T-Cells as Immune Regulators of Multiple Sclerosis. Front Immunol. 6(619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulina C and others.2004. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci U S A. 101(8):2428–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen PS, Blinkenberg M. 2016. The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neurol Disord. 9(1):44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. 2003. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol. 462(2):223–240. [DOI] [PubMed] [Google Scholar]
- Steinkraus BR, Toegel M, Fulga TA. 2016. Tiny giants of gene regulation: experimental strategies for microRNA functional studies. Wiley Interdiscip Rev Dev Biol. 5(3):311–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP and others.2004. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 24(9):2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. 2004. Dystrophic microglia in the aging human brain. Glia. 45(2):208–212. [DOI] [PubMed] [Google Scholar]
- Sun Y and others.2013a. MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther. 19(10):813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y and others.2013b. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 23(11):1270–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassishin L, Loudig O, Bauman A, Shafit-Zagardo B, Suh HS, Lee SC. 2011. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155*. Glia. 59(12):1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Andreasen N, Tarkowski A, Blennow K. 2003. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 74(9):1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng GG, Wang WH, Dai Y, Wang SJ, Chu YX, Li J. 2013. Let-7b is involved in the inflammation and immune responses associated with Helicobacter pylori infection by targeting Toll-like receptor 4. PLoS One. 8(2):e56709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentillier N and others.2016. Anti-Inflammatory Modulation of Microglia via CD163-Targeted Glucocorticoids Protects Dopaminergic Neurons in the 6-OHDA Parkinson’s Disease Model. J Neurosci. 36(36):9375–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai TH and others.2007. Regulation of the germinal center response by microRNA-155. Science. 316(5824):604–608. [DOI] [PubMed] [Google Scholar]
- Thomou T and others.2017. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 542(7642):450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T and others.2008. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 456(7224):980–984. [DOI] [PubMed] [Google Scholar]
- Tili E and others.2007. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 179(8):5082–5089. [DOI] [PubMed] [Google Scholar]
- Tsang JS, Ebert MS, van Oudenaarden A. 2010. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol Cell. 38(1):140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 9(6):654–659. [DOI] [PubMed] [Google Scholar]
- van den Bosch MW, Palsson-Mcdermott E, Johnson DS, O’Neill LA. 2014. LPS induces the degradation of programmed cell death protein 4 (PDCD4) to release Twist2, activating c-Maf transcription to promote interleukin-10 production. J Biol Chem. 289(33):22980–22990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED. 2013. IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS One. 8(12):e81774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. 2011. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 13(4):423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X and others.2013. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc Res. 97(3):432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z and others.2015. MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E2-mediated M2 generation. PLoS One. 10(2):e0115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MS and others.2010. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 68(3):369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J and others.2013. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 73(13):3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. 1993. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 75(5):855–862. [DOI] [PubMed] [Google Scholar]
- Willemen HL, Huo XJ, Mao-Ying QL, Zijlstra J, Heijnen CJ, Kavelaars A. 2012. MicroRNA-124 as a novel treatment for persistent hyperalgesia. J Neuroinflammation. 9(143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher KG, Eiferman DS, Godbout JP. 2015. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 38(10):609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worm J and others.2009. Silencing of microRNA-155 in mice during acute inflammatory response leads to derepression of c/ebp Beta and down-regulation of G-CSF. Nucleic Acids Res. 37(17):5784–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D and others.2015. Brain endothelial miR-146a negatively modulates T-cell adhesion through repressing multiple targets to inhibit NF-kappaB activation. J Cereb Blood Flow Metab. 35(3):412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamanchili SV and others.2015. MiR-21 in Extracellular Vesicles Leads to Neurotoxicity via TLR7 Signaling in SIV Neurological Disease. PLoS Pathog. 11(7):e1005032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN. 2010. Leukocyte recruitment and ischemic brain injury. Neuromolecular Med. 12(2):193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. 2006. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 113(17):2105–2112. [DOI] [PubMed] [Google Scholar]
- Ying W and others.2015. MicroRNA-223 is a crucial mediator of PPARgamma-regulated alternative macrophage activation. J Clin Invest. 125(11):4149–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lapato A, Bodhankar S, Vandenbark AA, Offner H. 2015. Treatment with IL-10 producing B cells in combination with E2 ameliorates EAE severity and decreases CNS inflammation in B cell-deficient mice. Metab Brain Dis. 30(5):1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cheng Y, Cui W, Li M, Li B, Guo L. 2014. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 266(1-2):56–63. [DOI] [PubMed] [Google Scholar]
- Zhang L, Dong LY, Li YJ, Hong Z, Wei WS. 2012. miR-21 represses FasL in microglia and protects against microglia-mediated neuronal cell death following hypoxia/ischemia. Glia. 60(12):1888–1895. [DOI] [PubMed] [Google Scholar]
- Zhang M and others.2011. Inhibition of microRNA let-7i depresses maturation and functional state of dendritic cells in response to lipopolysaccharide stimulation via targeting suppressor of cytokine signaling 1. J Immunol. 187(4):1674–1683. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Zhong M, Zhang M, Suo Q, Lv K. 2013. MicroRNA let-7a ameliorates con A-induced hepatitis by inhibiting IL-6-dependent Th17 cell differentiation. J Clin Immunol. 33(3):630–639. [DOI] [PubMed] [Google Scholar]
- Zhou H and others.2015. MicroRNA-223 Regulates the Differentiation and Function of Intestinal Dendritic Cells and Macrophages by Targeting C/EBPbeta. Cell Rep. 13(6):1149–1160. [DOI] [PubMed] [Google Scholar]
- Zhou R, Gong AY, Eischeid AN, Chen XM. 2012. miR-27b targets KSRP to coordinate TLR4-mediated epithelial defense against Cryptosporidium parvum infection. PLoS Pathog. 8(5):e1002702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L. 2014. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci. 52(1):148–155. [DOI] [PubMed] [Google Scholar]
- Zhuang G and others.2012. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation. 125(23):2892–2903. [DOI] [PubMed] [Google Scholar]