

Abstract
Liquid chromatography coupled with electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) is a major analytical technique used for nontargeted identification of metabolites in biological fluids. Typically, in LC-ESI-MS/MS based database assisted structure elucidation pipelines, the exact mass of an unknown compound is used to mine a chemical structure database to acquire an initial set of possible candidates. Subsequent matching of the collision induced dissociation (CID) spectrum of the unknown to the CID spectra of candidate structures facilitates identification. However, this approach often fails because of the large numbers of potential candidates (i.e., false positives) for which CID spectra are not available. To overcome this problem, CID fragmentation predication programs have been developed, but these also have limited success if large numbers of isomers with similar CID spectra are present in the candidate set. In this study, we investigated the use of a retention index (RI) predictive model as an orthogonal method to help improve identification rates. The model was used to eliminate candidate structures whose predicted RI values differed significantly from the experimentally determined RI value of the unknown compound. We tested this approach using a set of ninety-one endogenous metabolites and four in silico CID fragmentation algorithms: CFM-ID, CSI:FingerID, Mass Frontier, and MetFrag. Candidate sets obtained from PubChem and the Human Metabolite Database (HMDB) were ranked with and without RI filtering followed by in silico spectral matching. Upon RI filtering, 12 of the ninety-one metabolites were eliminated from their respective candidate sets, i.e., were scored incorrectly as negatives. For the remaining seventy-nine compounds, we show that RI filtering eliminated an average of 58% from PubChem candidate sets. This resulted in an approximately 2-fold improvement in average rankings when using CFM-ID, Mass Frontier, and MetFrag. In addition, RI filtering slightly increased the occurrence of number one rankings for all 4 fragmentation algorithms. However, RI filtering did not significantly improve average rankings when HMDB was used as the candidate database, nor did it significantly improve average rankings when using CSI:FingerID. Overall, we show that the current RI model incorrectly eliminated more true positives (12) than were expected (4–5) on the basis of the filtering method. However, it slightly improved the number of correct first place rankings and improved overall average rankings when using CFM-ID, Mass Frontier, and MetFrag.
Graphical Abstract
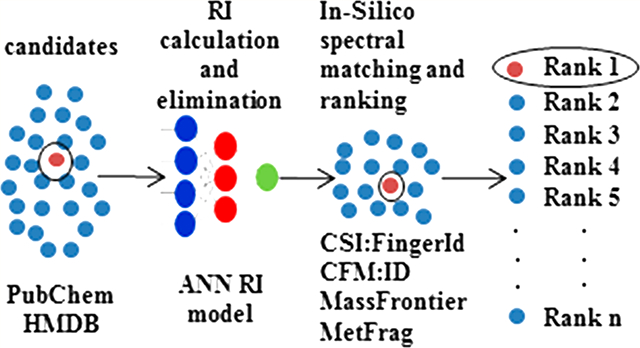
The composition and concentrations of small molecule metabolites (20–1000 Da) in living organisms frequently change over time and represent the biochemical phenotype of an individual. These changes can be due to multiple factors including diet, time of day, environmental exposures, disease, drug exposures, genetic manipulation, gender, and age.1,2 Nontargeted metabolomics is the unbiased quantification and identification of these metabolites in biological samples.3
In nontargeted metabolomics, researchers often utilize liquid chromatography coupled with electrospray ionization mass spectrometry (LC-ESI-MS) as the major analytical technique to separate and accurately measure the precursor masses of thousands of metabolites present in biological samples.4 However, to elucidate the chemical structure of these compounds, tandem mass spectrometry (LC-ESI-MS/MS) is often used.5 In LC-ESI-MS/MS, an experimental collision induced dissociation (CID) spectrum of an isolated precursor ion is used as a fingerprint in matching against a collection of reference CID spectra of known compounds in spectral libraries (e.g., MassBank, Metlin).6–8 Unfortunately, this approach often fails due to the dependency of CID spectral profiles on experimental conditions and lack of coverage of chemical space that pertains to endogenous human metabolites within existing spectral libraries.6,9–11
To overcome this limitation, computational fragmentation software (predictive fragmenters) has been developed with the aim of predicting experimental tandem mass spectral (MS/MS) profiles and the chemical structure of ensuing predictive fragments10,12 Working principles of these predictive fragmenters are described elsewhere;13 thus, only a brief overview is given here.
Commercial predictive fragmenters such as ACD/MS fragmenter (Advanced Chemistry Development Laboratories, www.acdlabs.com) and MassFroniter (Thermo Scientific, www.thermoscientific.com) rely on general ionization fragmentation and rearrangement rules along with fragmentation schemes collected from the literature to predict the chemical structures of energy induced fragment ions generated from precusor ions of specific structural composition (https://tools.thermofisher.com).14 These tools are extremely helpful in aiding manual spectral interpretation, which can be laborintensive.15 However, cost, lack of automated candidate retrieval, and ranking protocols limit their use in high-throughput metabolomics pipelines.16,17 In contrast, predictive fragmenters such as MetFrag and MAGMa attempt to explain ion peaks in an experimental CID spectrum by systematic dissociation of all the bonds in a given molecule. In other words, these predictive fragmenters compute all possible fragments of a molecule and then compare the mass of these fragments with the m/z values of fragments in an experimental CID spectrum. Compounds are ranked by assigning a score which is a function of ion peak intensity and the number of peaks explained in the experimental CID spectrum, bond dissociation energies, and neutral losses to account for rearrangements.10,18,19 Free availability, automated workflows, and faster processing times makes these programs popular among metabolomic researchers.20
Machine learning (ML) is one of the most rapidly growing areas in computer science.21 ML involves the development of computer algorithms that learn from example data or past experience to solve or predict the outcome of an unfamiliar problem.22 Predictive fragmenters such as CFM-ID and CSI:FingerID have been developed on the basis of ML paradigms.20,23 CFM-ID23 utilizes a stochastic, generative Markov model trained using the CID mass spectral profiles of approximately 3500 metabolites randomly chosen from the Metlin database.23 This method allows for the prediction of CID spectral profiles and can also be used to rank candidates based solely on the similarity between predicted and experimental spectra.23 CSI:FingerID is based on fragmentation trees and kernel based support vector machines trained to predict molecular structural features from CID spectra.20,24,25 The predicted set of molecular features (or a fingerprint) is used to rank candidates based on maximum likelihood considerations and Platt probabilities to refine the fingerprint similarity scoring.20 It is important to note that training data used in ML methods have a significant influence on identification quality. Generally, ML based predictive fragmenters outperform other competing predictive fragmenters but have longer processing times.13,20,26
Upon completion of an ESI-LC-MS/MS analysis, measured experimental features such as monoisotopic mass (MIM), retention time, and CID spectra are often utilized by researchers to elucidate the chemical structure of an unknown compound (peak) of interest. In database assisted structure elucidation pipelines, the first step is the acquisition of candidate structures for the unknown by matching the measured MIM to compounds in an all-purpose chemical database such as PubChem or ChemSpider or specialized biological databases such as the Human Metabolite Database (HMDB) or Kyoto Encyclopedia of Genes and Genomes (KEGG).13,27,28 According to the critical assessment of small molecule identification (CASMI) contests,26,29 the use of specialized databases improves the chances of correctly identifying the unknown structure. However, a major disadvantage of this approach is the inherent incompleteness of such databases. If the unknown compound is not contained in the database, it cannot be correctly identified.30 Thus, it can be argued that there are advantages in mining large chemical databases such as PubChem (which currently contains more than 90 million compounds) or ChemSpider (which currently contains more than 59 million compounds) because of their much larger size and the inclusion of more diverse compound classes. Nonetheless, mining such databases may also result in large candidate sets.31,32 For example, searching for 5-hydroxytryptophan (MIM = 220.0848 Da, ±5 ppm) using the MetFrag web interface (http://msbi.ipb-halle.de/MetFragBeta/) yields 8223 candidates from PubChem and 3777 from Chemspider. The same search resulted in 3 candidates from HMDB and 4 candidates from KEGG.
Generating the candidate list from large databases increases the likelihood that the unknown will be included in the candidate list but also dramatically increases the number of false positives. To address this problem, we have developed a software package called MolFind,33 which relies on a set of orthogonal experimental features acquired from LC-ESI-MS/MS experiments. These include Retention Index (RI), Drift Index (DI), and Ecom50 (collision gas normalized energy required to fragment 50% of precursor ions). For each experimental feature, MolFind eliminates false positives by comparing the experimental value of the unknown to a value predicted for each candidate compound using a computational model. A candiate compound is excluded when a predicted value for the candidate is substantially different from the experimental value of the unknown.33 In previous studies, when both RI and Ecom50 filters were applied concurrently with MetFrag, ranking of the correct compound improved from 142 to 102 on average (over 35 sets).33 More importantly, it was suggested that enhancements in the accuracy of the RI and Ecom50 models could lead to removal of 87.2% of candidates on average, attaining a potential increase in an average MetFrag ranking of 15.5.33
The robustness of modeling approaches in predicting experimental features is heavily dependent on the training set. The models used in our earlier study30 were far from optimal, as the Ecom50 model and the RI models were trained using 54 and 400 compounds, respectively, having 99.5% confidence intervals of 2.1 eV and 114 RI units (RIU), respectively.30 Recently, we have improved our RI model by training with a diverse set of synthetic chemicals (1955 in total) covering diverse chemical classes representative of endogenous human metabolites.34 Confirmed endogenous human metabolites were deliberately excluded from the model data set in the previous study. A total of 202 confirmed metabolites were set aside as an independent validation set to test if a model based exclusively on relatively simple synthetic compounds could be used to make predictions for more complex metabolites.34 For the model used in this present study, the 202 independent validation compounds were reintroduced and the model was rebuilt using the same descriptors and learning protocol as the previous study resulting in a model based on 2157 compounds. This was done to take advantage of the available data on confirmed metabolites and expand the model applicability domain.
As an extension to our previous work, the present study investigates the improvement in identification quality by enrichment of candidate sets using the expanded 2157 compoound RI model in conjunction with four different fragmentation algorithms: CFM-ID, CSI-FingerID, Mass Frontier, and MetFrag. Candidate compounds were taken from both a large (PubChem) and small (HMDB) database.
EXPERIMENTAL SECTION
Reagents and Chemicals.
Acetonitrile (HPLC, gradient grade) and methanol (HPLC grade) were purchased from Sigma-Aldrich (St Louis, MO, USA). Water (18.2 MΩ·cm) used for the UPLC mobile phase and sample preparation. Reagent grade water was generated on a Burnstead Nanopure Diamond system (Thermo Scientific, Ward Hill, MA, USA). Heptafluorobutyric acid (HPLC grade) was purchased from Thermo Fisher Scientific Chemicals Inc. (Ward Hill, MA, USA). n-Propionamide, n-butanamide, and n-hexanamide were ordered from Aldrich (St Louis, MO, USA). n-Pentanamide was ordered from MP Biochemicals, LLC (Solon, OH, USA). A series of n-C7–C14 amides were synthesized as described in Supporting Information SI–1. The 91 test compounds and the controls used in the study were purchased from various sources, and the vendor information is summarized in Table S2. HPLC grade formic acid (98–100%) was purchased from EMD Millipore Corporation (Billerica, MA, USA).
Sample Preparation.
Two different approaches were followed for the sample preparation. As the chemicals ordered from IROA technologies were contained in plates of polypropylene wells containing 5 μg of each chemical, 100 μL of solvent (0.1% formic acid in water, 0.05% formic acid in water/methanol (1:1) (v/v), or methanol) based on the XLOGP3 (taken from PubChem) value was added to each well. The plates were covered with sealing tape to prevent evaporation. Dissolution was achieved by shaking wells on an Innova 2100 platform shaker (New Brunswick, CT, USA) for 45 min. Finally, the dissolved chemicals were transferred to 2 mL HPLC vials with micro volume glass inserts (Thermo Fisher Scientific, Ward Hill, MA, USA), sealed with Teflon septum caps, and used directly for UPLC analysis. Stock solutions of all other chemicals were prepared at 1–10 μmol/mL concentrations in the appropriate solvent based on the analyte’s XLOGP3 value as described above. The prepared stock solutions were further diluted at appropriate concentrations and used for the UPLC analysis.
UPLC-ESI-MS/MS.
Retention index values were measured on a Zorbax, SB-C18, 2.1 mm × 150 mm, 1.8 μm column (Agilent Technologies, Santa Clara, CA, USA) using an Acquity UPLC liquid chromatographic system (Waters, Milford, MA, USA). Solvent A was 0.766 mM heptafluoroacetic acid (HFBA) in water, and solvent B was 0.766 mM HFBA in 10% water/acetonitrile (v/v). Compounds were eluted from the column using a solvent program consisting of a 4 min isocratic hold of 2% solvent B followed by a 20 min linear gradient to 100% solvent B and a 5 min isocratic hold at a flow rate of 388 μL/min. The RI model used in this study was trained and validated using retention data for compounds analyzed on an Agilent 1100 capillary HPLC system (Agilent Technologies, Santa Clara, CA, USA). Thus, the protocols used to transfer and validate the LC method to the Acquity UPLC system are summerized in Supporting Information SI–2. The outlet of the UPLC system was connected to the electrospray (ESI) ionization source of a Synapt G2-Si mass spectrometer (Waters, Milford, MA, USA) operating in the positive ion mode. A solution of leucine enkephalin (556.2771 Da, 400 pg/uL) in 0.1% (v/v) formic acid–methanol/water (1:1) was infused as the lock mass reference compound at a flow rate of 5 μL/min. The retention times of the test compounds were measured in duplicate using detection parameters described in Supporting Information SI–2. The mass range of the detector was set to 20–1000 Da. CID was carried out with nitrogen as the collision gas, and the collision energy was varied from 0 to 30 eV (30–60 eV if required) in incremental steps of 2 eV at a scan rate of 12.5 scans/s.
Retention Index Measurements.
RI values were measured on the basis of a method developed by Hall et al.35 At the beginning and the end of each run, 1 μL of a homologues series of n-C3–C14 amides was injected on the described UPLC system. The average retention times (RT) of the individual n-amides were used as the calibration reference for calculating RI values for the test compounds. The RI of each n-amide was defined as 100 times the number of carbon atoms. Owing to the linear relationships between RI vs log of RT in the isocratic part and RT vs RI in the gradient part of the solvent program, RI values of compounds eluted during isocratic and gradient parts were calculated by the following equations, respectively
| (1) |
| (2) |
where Tx is the retention time of the analyte; Tz is the retention time of the n-amides eluting just before the analyte; Tz+1 is the retention time of the n-amides eluting just after the analyte.
Compound Set Diversity.
A set of ninety-one endogenous metabolites, not included in the training set of the RI model, were selected for the study. The set consisted of a variety of chemical classes including alkaloids, amines, benzene and substituted derivatives, benzenoids, carbohydrates and conjugates, carboxylic acids, fatty acyls, flavin nucleotides, imidazopyrimidines, indoles, lipids and lipid like molecules, morphinans, organic acids, organic carbonic acids, organic nitrogen compounds, organic phosphonic acids, organoheterocyclic compounds, pteridines and derivatives, purine nucleosides, pyridines, quinolines, sphingolipids, steroids, stilbenes, and tetrahydroisoquinolines. Four of the compounds had an overall charge of +1 while the rest were neutral. The XLOGP3 values (predicted octanol–water partition coefficient) of the compound set varied from −5.0 to 8.5 (Figure 1) with a standard deviation of 3.4. The MIMs of the compounds varied from 105 to 785 Da with a standard deviation of 99 Da. Structural details for each test compound can be found in Table S3.
Figure 1.

Histogram of XLOGP3 values of the 91 compounds used in the study. Data were extracted from the PubChem database.
Data Analysis.
Retention and mass spectral data were processed using Masslynx version 4.1 (Waters, Milford, MA, USA). The experimental survival yields of precursor ions at each collision energy were calculated using eq 3 on the basis of a code written in Python version 3 (http://www.python.org).
| (3) |
Candidate Structures.
Candidate structures were downloaded from PubChem and HMDB by matching the experimental MIM within a relative mass error of ±15 ppm.36 MIMs were calculated by averaging scans with counts higher than or equal to 25% of the respective precursor ion peak apex in the CID spectrum acquired at an energy which resulted in a precursor survival yield closest to 20%.36 The relative mass error (15 ppm) was calculated at the 3-sigma limit when comparing actual to experimental MIMs. PubChem was queried using an in-house program written in python version 3 using underlying cheminformatics functions of Rdkit (http://rdkit.org/)37 and power user gateway (PUG) service (https://pubchem.ncbi.nlm.nih.gov/pug/). Downloaded candidate sets of compounds were saved in 91 separate structure-data (SD) files. A snapshot of HMDB database was downloaded as an SD file, and http://www.hmdb.ca/downloads was used to acquire candidate compound structures. Resulting candidate sets were saved in 91 separate SD files.
Preprocessing of Candidates.
Compounds in candidate sets that contained33 salts, had disconnections, contained heavy isotopes, had an overall charge (this filter was not applied to the candidate sets of the quaternary ammonium ions), contained only carbon and hydrogen, or were duplicate stereoisomers were removed prior to processing. Compounds with elements other than CHNOPS were also removed.
In Silico RI Prediction and Filtering.
In silico RI prediction of the remaining candidates was carried out using topological descriptors calculated by winMolconn (version 2.1)38 and newly trained parameters. The learning process used to develop the RI model has been described previously.34 Briefly, a 4 × 10 × 10 artificial neural network ensemble model was built on RI data for 2157 compounds (1955 commercial synthetic compounds and 202 confirmed human endogenous metabolites) measured according to the protocol described above. The model was trained according to the learning method described in the previous publication34 with RPROP40 back propagation on a network architecture of 47 input neurons and one hidden layer of 23 neurons.
The significant difference between the model used for this study and the previous publication is that confirmed human endogenous metabolites were deliberately excluded from the model data set in the previous publication. This was done in part to determine if a model based exclusively on relatively simple synthetic compounds could be used to make reasonable predictions for more complex human metabolites. To facilitate this, 202 confirmed human metabolites were set aside as an independent validation set. For this study, the independent validation set was reintroduced to the model data set to. After the additional data was added, the model was rebuilt using the same descriptors and learning method as the previous study. The metabolite data was reintroduced because the goal of this present study was to test RI model performance in the context of the MolFind algorithm, not to test if a model based on synthetic compounds could be used to predict the RI of metabolites. The addition of the confirmed human metabolites makes use of all available data so as to achieve the maximal applicability domain.
RI predictions were made for compounds in the PubChem and HMDB candidate lists corresponding to each of the 91 unknowns. Candidates with predicted RI values that deviated more than a threshold value were eliminated. The threshold windows were chosen on the basis of an algorithm that utilized the experimental RI value of the “unknown” and the similarity of each candidate to model data. Similarity was evaluated using a partial molecular fingerprint encoded in a bit key. The filter windows can be found in Table 4. The bit key similarity approach and algorithm for derivation of the filter windows are discussed in Supporting Information SI–4. The resulting RI-filtered candidate sets were saved separately.
Table 4.
Sensitivity and Specificity Results Using Four Different Approaches (SE; Bit Key; Range; Bit Key and Range) for Setting the RI Model Filter Windows
| “unknown” RIa | similarb | SEc | bit keyd | rangee | bit key and rangef |
|---|---|---|---|---|---|
|
| |||||
| <300 | HBK | <359 | <359 | <359 | <359 |
| <300 | LBK | <359 | <383 | <359 | <383 |
| 300–850 | HBK | ±105 | ±105 | ±97 | ±97 |
| 300–850 | LBK | ±105 | ±157 | ±97 | ±146 |
| 851–1400 | HBK | ±105 | ±105 | ±124 | ±124 |
| 851–1400 | LBK | ±105 | ±157 | ±124 | ±186 |
| >1400 | HBK | >1262 | >1262 | >1262 | >1262 |
| >1400 | LBK | >1262 | >1239 | >126 | >1239 |
| sensitivityg | 80% | 85% | 80% | 87% | |
| specificityh | 60% | 56% | 62% | 59% | |
Range where experimental RI value of the “unknown” occurs.
Whether a predicted compound is similar to model data (HBK) or not (LBK).
Filter windows for SE (standard error) method.
Filter windows for bit key method.
Filter windows for range method.
Filter windows for bit key and range method.
True positive rate for each filter window method.
True negative rate for each filter window method.
In Silico Predictive Fragmenters.
CFM-ID, CSI:FingerID, Mass Frontier, and MetFrag were used without modifications to rank candidate sets resulting from pre- and RI filtering. MAGMa19 was not used in the study as it does not support processing compounds with an overall charge of +1 (adduct type [M+]). Relative and absolute mass errors of 15.0 ppm and 0.01 Da were used to annotate fragments against peaks in respective experimental CID spectra with relative ion intensities closest to 20% survival yield (eq 3).
For single energy CFM-ID (version 2.0), the pretrained model params_se_cfm, having parameter file param_output.-log, was used (https://sourceforge.net/p/cfm-id/wiki/Home/). The experimental CID spectrum was repeated in all three energies (low, medium, high) such that CFM-ID assigned an average Jaccard score (default method) in ranking candidates.23
CSI:FingerID (version 3.5) was run with instrument mode set to “qtof” and setting all the other parameters to defaults. Candidates with molecular formulas having tree scores lower than the cutoff of <75% of the highest scoring tree were eliminated. The remaining candidates were processed with FingerID.
Mass Frontier (version 7.0.5.9) was run in batch mode using general fragmentation rules. Parameter settings were as follows : ionization method = protonation, maximum number of reaction steps = 5, reaction limit = 10 000, and mass range = 20–1000 Da. As Mass Frontier lacked functionality in automated candidate ranking, a python program was written to calculate number of peaks matched in the CID spectrum by Mass Frontier generated fragments (within either 15 ppm or 0.01 Da). Candidates were ranked considering the number of peaks matched in the experimental CID spectrum.
The command line version of MetFrag (version 2.3.1) was downloaded and used http://c-ruttkies.github.io/MetFrag/. None of the pre- or postprocessing filters (MetFragPreProcessingCandidateFilter and MetFragPostProcessingCandidateFilter) were applied. Default values for all other parameters were used.27
It is important to note that the objective of the study was to investigate the performance of the RI model in eliminating false positives contained in candidate sets and the subsequent improvement in predictive fragmenter performance. Our intent was not to evaluate or benchmark the 4 predictive fragmenters that we used (as done in previous CASMI contests).26 Researchers are encouraged to use this study as a guide to test the RI model with any predictive fragmenter of their choice.
RESULTS AND DISCUSSION
Reduction in Candidate Set Sizes Upon RI Filtering.
The aim of this study was to determine whether we could improve identification rankings by eliminating false positives using an RI filter prior to CID spectral matching. CFM-ID, CSI:FingerID, Mass Frontier, and MetFrag were used for spectral matching using data sets downloaded from PubChem and HMDB for 91 endogenous metabolites treated as “unknowns”. As with all approaches such as this, we face the problem of false negatives; i.e., the RI model will incorrectly eliminate a certain percentage of correct candidates. In this case, 12 of the ninety-one compounds evaluated were eliminated from their respective candidate sets after RI filtering. A sensitivity (true positive rate) of 95% was anticipated given the method used to set the filter ranges. This would equate to 4–5 expected false negative predictions. The lower sensitivity observed (79/91 = 87%) was unexpected as the new RI model was anticipated to outperform its predecessor which also had a sensitivity of approximately 87%.33 The model comes close to the expected sensitivity for compounds with RI values <900 (92% sensitivity, with six false negatives, Figure 2).
Figure 2.

Predicted and observed RI for 91 unknowns. True positive predictions are shown as diamonds; false negative predictions are shown as triangles. Model sensitivity is close to the expected 95% for RI < 900 (92% sensitivity) and RI > 1120 (91% sensitivity) but poor in the intervening range. Sensitivity is 100% for unknowns with RI values <300 and >1400. A detailed version of this figure is included in Supporting Information SI–4 showing separate results for each of the 8 filter range categories.
Nevertheless, for the remaining 79 compounds, the RI filter eliminated 58% of false positives from candidate sets acquired from the PubChem database (Figure 3). This was an improvement over the previous model,33 which eliminated approximately 21% of compounds. Likewise, approximately 32% of compounds were removed from candidate sets acquired from HMDB (Figure 3). There were only 22 of the 79 “unknowns” where the final number of compounds remaining in the PubChem set after RI filtering was <175. A complete list of these results is given in Tables S4, S5, and S6.
Figure 3.

Average number of compounds after RI filtering of candidate sets acquired from PubChem (a) and HMDB (b). n = 79 compounds. Figure summarizes data in Tables S4 and S5.
Predictive Fragmenters Performance with and without RI Filtering.
The performance of each predictive fragmenter was evaluated using the method described by Allen et al.23 CSI:FingerID scored 53 correct number one ranks (the known compound ranked as the best match after predictive fragmenter evaluation) when ranked using candidate sets acquired from PubChem database. After the RI filter, the number of correct number one ranks increased to 56. Mass Frontier scored 13 correct number one ranks which improved to 16 after RI filtering. Both CFM-ID and MetFrag had three correct number one ranked sets which increased to seven and six, respectively, after RI filtering. CSI:FingerID and Mass Frontier both scored 66 correct number one ranks with candidate sets acquired from the HMDB database. Upon applications of the RI filter, the number of correct number one rankings increased to 68 and 71, respectively. CFM-ID achieved 62 and 64 correct number one ranks from the HMDB database, while for MetFrag the number of correct number one ranks were 54 and 59 before and after the RI filter. Although these improvements in number one rankings are small, when taken together, they are statistically significant (paired t test; p < 0.001). Thus, our results suggest that RI filtering slightly improved a predictive fragmenter’s ability to rank an unknown as the correct number one ranked candidate. Interestingly, 37 of the 53 compounds that were correctly ranked number one by CSI:FingerID were compounds included in the algorithm’s original training set.20 This likely causes a significant performance skew in favor of CSI:FingerID and is consistent with the results of the 2016 CASMI contest where ML methods were shown to perform better when unknowns were included in their training sets.26 In contrast, for CFM-ID, only 18 of the 79 compounds studied were in its training set.
In addition to an evaluation of our RI model based on correct number one rankings, we also compared the results based on overall average rankings. The fragmentation tree filter used in CSI:FingerID eliminated glycocholic acid from its candidate set.20 Thus, Table 1 summarizes overall average rankings of the remaining 78 candidate sets. As seen (Table 1), following RI filtering, the performance of CFM-ID, Mass Frontier, and MetFrag was improved by approximately 2-fold (1.7-, 1.8-, and 1.9-fold, respectively; p < 0.05) when candidate sets acquired from PubChem database were used. This is consistent with the approximate 2-fold reduction in candidates following RI filtering (Figure 3a). There was no significant improvement in the performance of CSI:FingerID upon RI filtering using candidate sets from PubChem, and the RI filter provided no significant improvement for any of the fragmenters when using candidate sets from HMDB. It is important to note that the SD values in Table 1 are relatively large suggesting that the average ranking results are markedly dependent on the structural characteristics of the compounds in each candidate list.
Table 1.
Average Predictive Fragmenter Performance with and without RI Filtering Using Candidate Sets Acquired from PubChem and HMDB Databasesa
| fragmenter | database | without RI filter |
with RI filter |
||
|---|---|---|---|---|---|
| average ranking | S.D.b | average ranking | S.D.b | ||
|
| |||||
| CSI:FingerID | PubChem | 18.6 | 93.5 | 13.9 | 71.6 |
| CSI:FingerID | HMDB | 1.2 | 0.7 | 1.2 | 0.6 |
| CFM-ID | PubChemc | 149.2 | 282.1 | 87.3 | 205.7 |
| CFM-ID | HMDB | 1.3 | 0.8 | 1.2 | 0.6 |
| MetFrag | PubChemc | 345.2 | 677.3 | 182.6 | 453.7 |
| MetFrag | HMDB | 1.6 | 1.1 | 1.4 | 0.8 |
| Mass Frontier | PubChemc | 403.1 | 929.7 | 219.5 | 563.3 |
| Mass Frontier | HMDB | 1.3 | 0.8 | 1.2 | 0.6 |
Numbers indicate average ranking using 78 “unknown” compounds (i.e., n = 78 replicate rankings) for each predictive fragmenter and each database.
Standard deviation.
With RI filter significantly different from without RI filter at p < 0.05.
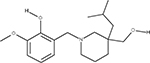
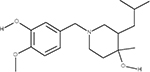
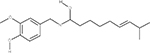
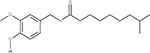
In our previous work,33 we used multiple orthologous experimental features that can be measured by LC-MS (such as RI) in order to eliminate false positive from candidate sets. The RI model used in this study was based on quantitative structure property relationships (QSPR) using a set of 2D structural descriptors.30 Matching the experimental and predicted RI values (considering the error of the model) enabled us to eliminate a large percentage of interfering false positives (Table 2). To better understand how RI filtering improves predictive fragmenter ranking, let us consider dihydrocapsaicin as an example of where the method performed well (Table 2) using MetFrag and candidates taken from PubChem. Dihydrocapsaicin was ranked as the fourth best match by MetFrag (Table 2). Moreover, for compounds a and b, MetFrag successfully annotated five peaks in the experimental CID spectrum of dihydrocapsaicin, while only detecting four peaks for compounds c and d. Thus, for this example, CID peak matching alone did not result in the correct ranking of dihydrocapsaicin (d) using MetFrag. However, the predicted RI values of compounds a, b, and c were sufficiently different from the experimental value so that they were excluded from the candidate set, permitting the ranking of d to become number one.
Table 2.
Chemical Structures of Top Four Candidates of Dihydrocapsaicin along with Predictive Fragmenter Rankings and Predicted and Experimental RI Valuesa
Fragmentation Tree Filter.
CSI:FingerID, developed by Dührkop et al., uses fragmentation trees (FT) to eliminate false positives contained in candidate sets prior to FingerID ranking.20 The theoretical and implementation details of FT are given elsewhere; however, a brief description is provided here.24 The first step in the CSI:FingerID algorithm is to generate all possible candidate molecular formulas of the “unknown” (based on precursor mass) and use them to generate fragmentation trees. Each tree is assigned a score based on how well each explains the experimental CID spectrum. Next, candidate molecular formulas having tree scores less than 75% of the top scored tree are excluded. Finally, the remaining formulas are used to eliminate false positives (i.e., candidate structures having molecular formulas other than the >75% scored trees) from the candidate set (e.g., acquired from PubChem). Therefore, we investigated the effect of tree filtering on the performance of CFM-ID, Mass Frontier, and MetFrag (Table 3), in addition to CSI:FingerID. The FT filter eliminated on average approximately 28% (1710 compounds) of false positives in candidate sets acquired from PubChem. It eliminated glycocholic acid from its respective candidate set (false negative). As shown in Table 3, the average performance of CFM-ID, MetFrag, and Mass Frontier improved following FT filtering. However, overall average performance was lower when compared to the RI filter (Table 1). Similar to the RI filter, applying the FT filter on HMDB candidate sets resulted in no increased performance for any of the predictive fragmenters studied.
Table 3.
Predictive Fragmenter Performance with FT Filtering Using Candidate Sets Acquired from PubChem and HMDBa
| fragmenter | database | without FT filter |
with FT filter |
||
|---|---|---|---|---|---|
| average ranking | S.D.b | average ranking | S.D.b | ||
|
| |||||
| CSI:FingerId | PubChem | 13.9 | 71.6 | ||
| CSI:FingerId | HMDB | 1.6 | 1.1 | ||
| CFM-ID | PubChemc | 149.2 | 282.1 | 126.0 | 256.3 |
| CFM-ID | HMDB | 1.3 | 0.8 | 1.3 | 0.8 |
| MetFrag | PubChemc | 345.2 | 677.3 | 217.9 | 594.9 |
| MetFrag | HMDB | 1.6 | 1.1 | 1.6 | 1.1 |
| Mass Frontier | PubChemc | 403.1 | 929.7 | 350.9 | 832.8 |
| Mass Frontier | HMDB | 1.3 | 0.8 | 1.3 | 0.7 |
n = 78 compounds.
Standard deviation.
With FT filter significantly different from without FT filter at p < 0.05.
Evaluation of RI Model Filter Windows.
As mentioned, 12 of the ninety-one compounds were eliminated from their respective candidate sets upon RI filter enrichment. We identify this as a shortcoming of this technique in that only 4–5 false eliminations were anticipated on the basis of the method used to set the filter window. The magnitude of the filter window for each candidate was based on the structural similarity of each candidate to the RI model data and the experimental RI of the “unknown”. A compound was defined as “similar” (high bit key (HBK)) when three or more compounds in the model data had the same bit key value as the compound being predicted (bit key match). This means that at least three compounds in the model data have the same combination of heteroatomic structure features as the compound being predicted. Compounds with two or fewer data set compounds with a matching bit key were considered “not similar” (low bit key (LBK)). The bit key similarity metric is described in detail in Supporting Information SI–4. A filter window of ±2SE (validation standard error) was used when a candidate was similar to model data, and a window of ±3SE was used when a candidate was not similar. Further, an SE value of 48.5 RIU was used when the compound’s RI value was 300–850 (data range where the model performed best), and a larger SE value of 62 RIU was used when the compound’s RI was 851–1400 (data range where the model preformed least well). This method (termed the “bit key and range” approach) and the metrics used to assess similarity are discussed in Supporting Information SI–4. This method was suggested by the independent validation results of a previous study.34
According to the similarity metrics, only four of the 12 eliminated compounds had low structural similaritiy to the model data (Table S6). This raises the question as to whether using the described approach to set the filter window gives optimal results. In order to evaluate this, additional statistics were calculated using three other approaches for setting the filter window: SE (standard error), bit key, and range. The SE method used a filter window of ±2SE based on the validation SE of the entire data set (52.4 RIU) and used the 95% cutoff values where the “unknown” RI was outside the RI range of the reference standards (<RI 300 or >RI 1400). The bit key method used a filter window of ±2SE (52.4 RIU) when a candidate was similar to the model’s data and a filter window of ±3SE (78.6 RI Units) when a candidate was not similar. For “unknown” RI values <300 or >1400, the bit key method used the 95% cutoff when the candidate was similar and the 98.5% cutoff when the candidate was not similar. The range method used a filter window of ±2SE (48.5 RIU) when the “unknown” RI was 300–850 and a filter window of ±2SE (62.0 RIU) when the “unknown” RI was 851–1400. The range method used the 95% cutoff values where the “unknown” RI was <300 or >1400. Table 4 gives the filter window values used for all 4 approaches along with the sensitivity and specificity for each.
The highest sensitivity is found for the combined bit key and range approach at 87%. In contrast, the SE and range approaches have sensitivities of only 80%. As expected, the use of wider filter windows when screening some compounds results in a loss of specificity for the bit key and range approach. Both the SE and range approaches removed more false positives. The differences, however, are small at 1–3%, and since the elimination of an unknown from the candidate list results in failure, it is likely necessary to accept the reduced specificity in favor of greater sensitivity.
An analysis of Figure 2 indicates that, by far, sensitivity is worse where the RI of the unknown falls between 950 and 1120 RIU. Sensitivity is only 38% with false negative classifications for 5 out of the 8 compounds in this range. All 5 false negative compounds are significantly under-predicted. It may be of interest that 4 out the 5 compounds are lipid like with a hydrocarbon tail of at least 15 carbons. All 4 of these lipids are also predicted to have a charge of +1 at the pH of the mobile phase (pH 2.5). It is unclear if these structural characteristics are responsible for the poor prediction. The sample size in this RI range is small, but the poor performance of the mode in this range suggests that this approach would not be recommended in cases where the RI of the unknown falls in the range of 900–1150.
An analysis of Figure 2 also indicates that there are several close misses in the “unknowns” that were eliminated from their respective candidate lists. Two “unknowns” are within 20 RIU of being retained and 3 more are within 45. Boundary effects are anticipated when cutoff values are used and cannot be entirely avoided. Even so, given the failure of the method when the unknown is eliminated from its candidate list, some consideration of larger filter windows should be made. Larger filter windows will likely reduce the specifcity of the RI filter, but the results of this study suggest that the reduction could be small. An increase in the width of the filter range by 1/2SE increases the sensitivity to 90% while decreasing the specificity to 53%. Ultimately, the study showed that the RI model improved CID spectral rankings, but the overall level of improvement is modest. Further increases in model accuracy could be made by increasing the number and diversity of the training set to account for the current 3:1 skew toward the lower RI range, including additional relevant descriptors (such as 3D structural information) and QSAR-feature engineering. However, it is important to note that inclusion of 3D descriptors was avoided in the current RI model, as obtaining an accurate global minimum of flexible compounds (e.g., lipids) in a computationally efficient way remains problematic in the field of computational chemistry.39 Our results would suggest that, as candidate lists increase in size, the number of structurally similar compounds also likely increases. Thus, distinguishing among these extremely similar structures, either by CID prediction algorithms or by RI models, becomes increasingly difficult. Clearly, additional orthologous structure identification methods would help in these situations. We are in the process of upgrading our existing Ecom50 model which we hope to use as an additional filter to further enrich acquired candidate sets. From such enrichment, we foresee a potential increase in predictive fragmenter performance due to further reduction in candidate sizes.33 Additionally, the inclusion of an Ecom50 filter would allow us to use broader RI filter windows enabling us to overcome some of the limitations shown here. In addition, future improvements in the sensitivity of NMR and/or IR spectroscopy methods would dramatically enhance our ability to identify unknown compounds in nontargeted metabolomics applications.
CONCLUSIONS
We show that RI based enrichment of large candidate sets prior to in silico predictive fragmenter ranking slightly improved the performance of three of the four predictive fragmenters studied. We have developed an approach termed “bit key and range” to define the filter windows, but in spite of this, the model eliminated 12 of the 91 “unknowns” from their respective candidate sets where only 4–5 eliminations were expected. In the remaining PubChem candidate sets, approximately 58% of the false positives were eliminated on average and overall average rankings were improved 2-fold. Further improvements in the RI model (e.g., 3D descriptors, additional training compounds) would increase its reliability in LC-ESI-MS/MS based database assisted structure elucidation pipelines.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge contributions from Sebastian Böcker and Kai Dührkop in processing candidate sets through CSI:FingerID. We acknowledge contributions from Ming-Hui Chen for technical help and discussion in performing bootstrap statistics. We thank Jair Roberson for preparing stock solutions, Matthew Chambers of Proteowizard for technical help in using Msconvert, and ChemAxon for granting a free license to use their neutralize plugin. This work was supported by NIH grant GM087714.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b03118.
Synthesis of C7–C14 n-amides and preparation of C3–C14 n-amide RI reference standard; modification of original HPLC system for RI analysis on Acquity UPLC system; synapt G2-Si system parameters; method to attempt the reduction of false negative RI predictions by including similarity and RI value of compounds being screened in the magnitude of the RI filter range; supporting tables (PDF)
REFERENCES
- (1).Griffiths WJ Metabolomics, metabonomics and metabolite profiling; Royal Society of Chemistry: London, 2008; Vol. 9. [Google Scholar]
- (2).Fiehn O Plant Mol. Biol. 2002, 48 (1–2), 155–171. [PubMed] [Google Scholar]
- (3).Lämmerhofer M; Weckwerth W Metabolomics in practice: successful strategies to generate and analyze metabolic data; John Wiley & Sons: New York, 2013. [Google Scholar]
- (4).Plumb R; Castro-Perez J; Granger J; Beattie I; Joncour K; Wright A Rapid Commun. Mass Spectrom. 2004, 18 (19), 2331–2337. [DOI] [PubMed] [Google Scholar]
- (5).Schymanski EL; Jeon J; Gulde R; Fenner K; Ruff M; Singer HP; Hollender J Identifying small molecules via high resolution mass spectrometry: communicating confidence; ACS Publications: Washington, DC, 2014. [DOI] [PubMed] [Google Scholar]
- (6).Milman BL; Zhurkovich IK TrAC, Trends Anal. Chem. 2016, 80, 636–640. [Google Scholar]
- (7).Horai H; Arita M; Kanaya S; Nihei Y; Ikeda T; Suwa K; Ojima Y; Tanaka K; Tanaka S; Aoshima K; et al. J. Mass Spectrom. 2010, 45 (7), 703–714. [DOI] [PubMed] [Google Scholar]
- (8).Smith CA; O’Maille G; Want EJ; Qin C; Trauger SA; Brandon TR; Custodio DE; Abagyan R; Siuzdak G Ther. Drug Monit. 2005, 27 (6), 747–751. [DOI] [PubMed] [Google Scholar]
- (9).Dubey R; Hill DW; Lai S; Chen M-H; Grant DF Metabolomics 2015, 11 (3), 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wolf S; Schmidt S; Müller-Hannemann M; Neumann S BMC Bioinf. 2010, 11 (1), 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Johnson SR; Lange BM Front. Bioeng. Biotechnol. 2015, 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Scheubert K; Hufsky F; Böcker SJ Cheminf. 2013, 5 (1), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hufsky F; Böcker S Mass Spectrom. Rev. 2017, 36, 624–633. [DOI] [PubMed] [Google Scholar]
- (14).Meringer M MOLGEN-MSF; molgen group: Munich, 2009. [Google Scholar]
- (15).Zhang MY; Pace N; Kerns EH; Kleintop T; Kagan N; Sakuma TJ Mass Spectrom. 2005, 40 (8), 1017–1029. [DOI] [PubMed] [Google Scholar]
- (16).Zhou J; Weber RJ; Allwood JW; Mistrik R; Zhu Z; Ji Z; Chen S; Dunn WB; He S; Viant MR Bioinformatics 2014, 30 (4), 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hufsky F; Scheubert K; Böcker S TrAC, Trends Anal. Chem. 2014, 53, 41–48. [Google Scholar]
- (18).Wang Y; Kora G; Bowen BP; Pan C Anal. Chem. 2014, 86 (19), 9496–9503. [DOI] [PubMed] [Google Scholar]
- (19).Ridder L; van der Hooft JJ; Verhoeven S Mass Spectrom. 2014, 3 (Special_Issue_2), S0033–S0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dührkop K; Shen H; Meusel M; Rousu J; Böcker S Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (41), 12580–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jordan MI; Mitchell TM Science 2015, 349 (6245), 255–260. [DOI] [PubMed] [Google Scholar]
- (22).Alpaydin E Introduction to machine learning; MIT press: Cambridge, MA, 2014. [Google Scholar]
- (23).Allen F; Greiner R; Wishart D Metabolomics 2015, 11 (1), 98–110. [Google Scholar]
- (24).Shen H; Dührkop K; Böcker S; Rousu J Bioinformatics 2014, 30 (12), i157–i164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Heinonen M; Shen H; Zamboni N; Rousu J Bioinformatics 2012, 28 (18), 2333–2341. [DOI] [PubMed] [Google Scholar]
- (26).Schymanski EL; Ruttkies C; Krauss M; Brouard C; Kind T; Dührkop K; Allen F; Vaniya A; Verdegem D; Böcker S; et al. J. Cheminf. 2017, 9 (1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ruttkies C; Schymanski EL; Wolf S; Hollender J; Neumann SJ Cheminf. 2016, 8 (1), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Allen F; Pon A; Wilson M; Greiner R; Wishart D Nucleic Acids Res. 2014, 42 (W1), W94–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nishioka T; Kasama T; Kinumi T; Makabe H; Matsuda F; Miura D; Miyashita M; Nakamura T; Tanaka K; Yamamoto A Mass Spectrom. 2014, 3 (Special_Issue_2), S0039–S0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hall LM; Hall LH; Kertesz TM; Hill DW; Sharp TR; Oblak EZ; Dong YW; Wishart DS; Chen M-H; Grant DF J. Chem. Inf. Model. 2012, 52 (5), 1222–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pence HE; Williams A ChemSpider: an online chemical information resource; ACS Publications: Washington, DC, 2010. [Google Scholar]
- (32).Bolton EE; Wang Y; Thiessen PA; Bryant SH Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- (33).Menikarachchi LC; Cawley S; Hill DW; Hall LM; Hall L; Lai S; Wilder J; Grant DF Anal. Chem. 2012, 84 (21), 9388–9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hall LM; Hill DW; Bugden K; Cawley S; Hall LH; Chen M-H; Grant DF J. Chem. Inf. Model. 2018, 58 (3), 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hall LM; Hill DW; Menikarachchi LC; Chen M-H; Hall LH; Grant DF Bioanalysis 2015, 7 (8), 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hill DW; Kertesz TM; Fontaine D; Friedman R; Grant DF Anal. Chem. 2008, 80 (14), 5574–5582. [DOI] [PubMed] [Google Scholar]
- (37).Landrum G RDKit: Open-Source Cheminformatics Software; SourceForge: 2014. [Google Scholar]
- (38).Hall L; Kellogg G; Haney DM-Z Google Scholar; Hall Associates Consulting: Quincy, MA, 1991. [Google Scholar]
- (39).Bolton EE; Chen J; Kim S; Han L; He S; Shi W; Simonyan V; Sun Y; Thiessen PA; Wang J; et al. J. Cheminf. 2011, 3 (1), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Riedmiller M; Braun H A Direct Adaptive Method for Faster Backpropagation Learning: The RPROP Algorithm. Proceedings of 1993 IEEE International Conference on Neural Networks (ICNN ′93), San Francisco, CA, USA, 28 March to 1 April, 1993; pp 586–591. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.