

Abstract
Background
Laminins are heterotrimeric complexes, consisting of α, β and γ subunits that form a major component of basement membranes and extracellular matrix. Laminin complexes have different, but often overlapping, distributions and functions.
Methods
Under our clinical protocol, NCT00068224, we have performed extensive clinical and neuropsychiatric phenotyping, neuroimaging and molecular analysis in patients with laminin α1 (LAMA1)-associated lamininopathy. We investigated the consequence of mutations in LAMA1 using patient-derived fibroblasts and neuronal cells derived from neuronal stem cells.
Results
In this paper we describe individuals with biallelic mutations in LAMA1, all of whom had the cerebellar dysplasia, myopia and retinal dystrophy, in addition to obsessive compulsive traits, tics and anxiety. Patient-derived fibroblasts have impaired adhesion, reduced migration, abnormal morphology and increased apoptosis due to impaired activation of Cdc42, a member of the Rho family of GTPases that is involved in cytoskeletal dynamics. LAMA1 knockdown in human neuronal cells also showed abnormal morphology and filopodia formation, supporting the importance of LAMA1 in neuronal migration, and marking these cells potentially useful tools for disease modelling and therapeutic target discovery.
Conclusion
This paper broadens the phenotypes associated with LAMA1 mutations. We demonstrate that LAMA1 deficiency can lead to alteration in cytoskeletal dynamics, which may invariably lead to alteration in dendrite growth and axonal formation. Estimation of disease prevalence based on population studies in LAMA1 reveals a prevalence of 1–20 in 1 000 000.
Trial registration number:
INTRODUCTION
Laminins are heterotrimeric glycoproteins that function within basement membranes and the extracellular matrix to connect cells and maintain the integrity of tissues.1 Twelve human genes are known to encode laminin subunits, nine of which are associated with human disorders2–9 (see online supplementary table S1). Each of the 16 known human laminin complexes (further referred to as laminins) combines one of each of the α5, β4, and γ3 chain laminin subunits (encoded by 12 different genes) to form a cruciform structure. Laminins are named according to their chain composition; for example, laminin-111 is composed of α1, β1 and γ1 chains.10 Laminins perform unique physiological functions within basement membranes1,11,12through their ability to bind to other laminins or extracellular matrix molecules to form extracellular scaffolding and/or through binding to anchor and organise cells within tissues.
It is not surprising that laminin disorders (lamini-nopathies) are highly variable in their clinical presentations, which include epidermolysis bullosa (MIM 600805, MIM 150310, MIM 150292), nephrotic syndrome (MIM 614199), muscular dystrophy with eye abnormalities (MIM 61643), lissencephaly and other cerebral anomalies (MIM 614115) and cardiomyopathy (MIM 600133) (see online supplementary table S1).
Poretti–Boltshauser syndrome (MIM 615960) is a newly described disorder characterised by congenital cerebellar dysplasia, cerebellar vermis hypoplasia and cerebellar cysts in patients with myopia with or without retinal dystrophy.13 Recently, mutations in Laminin α1 (LAMA1) (MIM 150320), which encodes LAMA1, were associated with the same congenital cerebellar dysplastic syndrome.14 How LAMA1 deficiency leads to this phenotype, however, has not been clarified. In mice, Lama1 subunit is essential for proliferation and migration of granule cell precursors in the cerebellum; it is also implicated in the development of the retina, regulation of mesangial cells and matrix and modulation of kidney function.15,16 Here we describe three subjects from two families in whom whole exome sequencing identified biallelic null mutations in LAMA1 that segregated with an expanded phenotype of neuropsychiatric symptoms composed of simple and complex tics, obsessive compulsive traits and anxiety, in addition to the characteristics described in Poretti–Boltshauser syndrome.13,14 We found that LAMA1-deficient patient fibroblasts exhibited impaired activation of the Cdc42 GTPAse that led to abnormal filopodia formation, impaired adhesion and reduced migration. We documented that similar abnormalities were also seen in LAMA1-deficient human neuronal cells. The neuropsychiatric phenotype in our patients encompasses features common to other more common neurological and psychiatric disorders. Our findings underscore the importance of LAMA1 in brain development and the potential role of laminins in the genesis of neuropsychiatric symptoms such as those observed in Tourette’s syndrome.
MATERIAL AND METHODS
Subjects
Subjects were evaluated at the NIH Clinical Center under NIH protocol ‘Clinical and Molecular Investigations into Ciliopathies’ (http://www.clinicaltrials.gov, trial NCT00068224) approved by the National Human Genome Research Institute (NHGRI) Institutional Review Board. Subjects and/or their parents gave written, informed consent.
Imaging
All subjects underwent axial, coronal and sagittal T1-weighted or T2-weighted brain MRI studies at their local hospitals as part of their routine diagnostic evaluation. All available brain MRI data were retrospectively evaluated for infratentorial and supratentorial abnormalities, as previously reported.13 Cerebellar dysplasia was defined as an abnormal cerebellar foliation, fissuration and white matter arborisation. Standard and high-resolution ultrasonographic (HR-USG) studies were performed using 4 and 7 Mhz transducers (AVI Sequoia).
Video ocular motor and neurocognitive evaluations
For video ocular motor assessment, eye tracking was recorded at 100 frames per second via Neuro Kinetics binocular infrared digital video goggles (I-Portal-VOG V.2.4.002). Visual stimuli were presented using a calibrated Neuro Kinetics VNG system (VEST V.7.0). Ocular motor assessment included gaze testing, random saccadic tracking and smooth pursuit testing in both the vertical and horizontal directions, as well as optokinetic testing at 20°, 40° and 60°/s. Neurophysiological evaluations administered by a certified PhD clinical neuropsychologist included Wechsler Adult Intelligence Scale-IV (WAIS-IV); Neuropsychological Assessment Battery: Language Module and Peabody Picture Vocabulary Test-4. Language assessment included auditory comprehension, confrontation naming and oral production as well as word generation and receptive vocabulary.
Sequencing and variant analysis
For exome sequencing, we used the HiSeq2000 (Illumina)17 that employed 101 bp paired-end read sequencing. Image analysis and base-calling were performed using Illumina Genome Analyzer Pipeline software (V.1.13.48.0) with default parameters. Reads were aligned to a human reference sequence (University of California Santa Cruz [UCSC] assembly hg19, NCBI build 37) using a package called Efficient Large-scale Alignment of Nucleotide Databases (Illumina). Genotypes were called at all positions where there were high-quality sequence bases using a Bayesian algorithm called the Most Probable Genotype18 and variants were filtered using the graphical software tool VarSifter (V.1.5).19 The database dbSNP (see URLs) covers the 1.22% of the human genome corresponding to the Consensus Conserved Domain Sequences and more than 1000 non-coding RNAs.20
For dideoxy sequencing, primers were designed to cover all coding exons and flanking intronic regions of LAMA1 (primer sequences available on request). Direct sequencing of the PCR amplification products was carried out using BigDye 3.1 Terminator chemistry (Applied Biosystems) and separated on an ABI 3130xl genetic analyzer (Applied Biosystems). Data were evaluated using Sequencher (V.5.0) software (Gene Codes Corp.).
Fibroblast culture
Fibroblasts were cultured from forearm skin biopsies and grown in high-glucose (4.5 g/L) Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 15% fetal bovine serum (FBS), 2 mM L-glutamine, non-essential amino acid solution and penicillin-streptomycin. For control, normal adult human dermal fibroblasts (ATCC PCS-201–012) were used.
Expression studies
Total RNA was isolated from fibroblasts with TRIZOL reagent (Invitrogen) and from whole blood using PAXgene Blood RNA Kit (Qiagen). RNA was treated with a DNase kit (DNA-free), according to the manufacturer’s protocol (Applied Biosystems). RNA concentration and purity were assessed on a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). First strand cDNA was synthesised using a high-capacity RNA-to-cDNA kit (Applied Biosystems). Human multiple tissue cDNA panels (human Multiple Tissue Panels of cDNA I and II and Human Fetal MT Panel) were purchased from Clontech. Quantitative real-time PCR (qPCR) was performed using a Bio-Rad iQ SYBR Green Supermix and qPCR machine with standard qPCR parameters to analyse the expression of LAMA1, LAMB1, LAMB2 and LAMC1 compared with the control gene ACTB (primer sequences available on request). For analysis of tissue-specific expression, LAMA1 (Hs01074511_m1), Assays-On-Demand primer-probe assays (Applied Biosystems) and a control assay for POL2RA (Hs00172187_m1) were used. PCR amplifications were performed on 100 ng of cDNA using TaqMan Gene Expression Master Mix reagent (Applied Biosystems) and were carried out on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). Results were analysed with the comparative CT method as described.21,22
Immunoblotting
Fibroblasts were grown for 48 h at 1×106 cells per flask in DMEM+10% FBS. Cell lysates, prepared using radioimmuno-precipitation assay buffer (50 mM Tris, pH 7; 150 mM NaCl; 0.1% SDS; 0.5% sodium deoxycholate; 1% Triton x-100; 1 mM EDTA), supplemented with protease inhibitors (complete, Mini, EDTA-free, Roche), were normalised by the bicinchoninic acid protein assay (Biorad) and subjected to immunoblotting. Samples were electrophoresed on a 3%–8% Tris-Acetate gel and blotted onto a 0.2 μm nitrocellulose membrane (Invitrogen). Membranes were blocked with phosphate buffered saline (PBS)+0.1% Tween 20 (PBS-T), supplemented with 5% non-fat milk, washed and incubated with primary rabbit anti-human LAMA1 antibodies (H-300, Santa Cruz Biotechnology), mouse anti-Vinculin Antibody, clone V284 (EMD Millipore), followed by appropriate IRDye 680RD or IRDye 800CW-conjugated secondary antibodies (Li-Cor Biosciences). LAMA1 antibody specificity was verified by doing peptide completion blocking with Laminin-111 (Sigma-Aldrich). In addition, the same findings were seen using LAMA1 antibody from Sigma-Aldrich. Antibodies were diluted in PBS-T, supplemented with 5% non-fat milk.
For all the immunoblots, proteins were detected using the Li-Cor Imaging System (Li-Cor Biosciences). The molecular weight ladder used was the Precision Plus Protein Kaleidoscope Standards (Bio-Rad) or the HiMark Pre-stained Protein Standard (Life Technologies)
Immunofluorescence microscopy
Cells were grown in 4-well chamber slides for 48 h and fixed using 4% paraformaldehyde, permeabilised using 0.1% Triton-X-100, and blocked with donkey serum (1 × PBS) and 0.5% Tween. Phalloidin conjugates were purchased from Invitrogen. After washing, samples were mounted in Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Cells were imaged with a Zeiss 510 META confocal laser-scanning microscope. Optical sections were collected from the xy plane and merged into maximum projection images.
Cell adhesion and migration assays
Cell adhesion was measured as described23 with slight modifications. Culture plates were coated with bovine serum albumin (BSA) (1 mg/mL), laminin 111 (Sigma-Aldrich, 10 μg/mL) and collagen I (Millipore, 10 μg/mL), according to the manufacturer’s instruction. About 0.5×106 fibroblasts (subject and control) were seeded per well using serum-free media (OptiPRO, Invitrogen), and allowed to attach for 90 min at 37°C with 5% CO2. Non-adherent cells were washed off with PBS and the plate was frozen for 30 min. After thawing, cells were lysed with CyQuant dye (Invitrogen) diluted in PBS for 10 min; the fluorescence was read at 520 nm (excitation 480 nm, emission 520 nm). All experiments were performed in triplicate; three independent assays were performed.
For migration assays, fibroblasts were grown to 80% confluence in non-coated chamber slides. At confluence, the medium was changed to serum-free, and a 0.9 mm scratch was made in the confluent layer of cells. Cells were labelled with Calcein AM (Invitrogen), and the migration of the cells into the wound area was imaged at 12, 24, 36, 48 and 60 h using a fluorescent microscope. All experiments were performed in triplicate; six independent assays were performed.
Scanning electron microscopy
Fibroblasts grown on glass coverslips were fixed in 2.5% glutaraldehyde in 0.1 M Cacodylate buffer pH 7.4, post fixed with 1% OsO4 for 1 h and serially dehydrated in ethanol. The samples were critical point-dried (Samdri-795) and placed on carbon adhesive tape coated with 10 nm gold in an EMS 575× sputter coater (Electron Microscopy Sciences). The images were obtained in a Hitachi S3400-N1 SEM (Hitachi High Technologies).
GTPase activation assay
Patient and control fibroblasts were grown to 70% confluency before harvesting. The GTPase activation assay was performed according to the manufacturer’s instructions (Rhoa/Rac1/Cdc42 activation assay; G-LISA: Cytoskeleton, Denver, Colorado, USA). The plates were read immediately by measuring absorbance at 490 nm on a SpectraMax M2 (Molecular Devices) microplate spectrophotometer. Experiments were performed in five replicates; three independent assays were performed.
Production of lentiviral vectors and transduction of cells
cDNA encoding the full-length human LAMA1 (NM_005559.3) was obtained by RT-PCR-mediated cloning using total RNA from testes. LAMA1 was cloned into pENTR11 vector by Seamless cloning, and later subcloned into pLenti6.3/V5-DEST (Invitrogen) using Gateway recombination (Invitrogen). Production of lentiviral constructs was performed by transient co-transfection of 293FT cells (ViraPower Lentiviral Expression Systems, Invitrogen), producing pLenti-LAMA1 and pLenti-empty for control, and concentrated using Lenti-X Concentrator (Clontech) following manufacturer’s protocol to generate high titre virus. Fibroblasts cells (Control, Patients 2 and 3) were transduced with viral preparations (1×108/mL, at an multiplicity of infection or MOI of 10) diluted in fresh culture media supplemented with Polybrene (Sigma, with a final concentration of 8 μg/mL) twice over the duration of 12 h. About 48 h after transduction, generation of cells stably expressing pLenti-LAMA1 or pLenti-empty was initiated by changing the culture media with selection antibiotic (Blasticidin, Invitrogen). After serial dilution and clone selection for stable cell line generation, cells were used for adhesion, migration and apoptosis assays.
Neural stem cell culture
Human neural stem cells (H9-derived, Invitrogen) were dissociated with TrypLE and plated as a homogenous single-cell suspension seeded onto 12-well plates and chamber slides coated with fibronectin. All cell culture media components were purchased from Invitrogen. Cells were cultured in complete medium (KnockOut D-MEM/F-12 with StemPro Neural Supplement, bFGF, EGF, and GlutaMAX-I) until they reached 80% confluence. Differentiation into neurons was initiated by replacing the medium with differentiation medium (Neurobasal medium with B-27 Serum-Free Supplement and GlutaMAX-I). On day 7, siRNA duplexes to LAMA1 gene (SMARTpool, E-012118–00-0050, Dharmacon GE) and non-targeting control (D-001910–02-50) were delivered to cells following the manufacturer’s protocol, and allowed to incubate for 96 h. Knockdown efficiency was assayed using qPCR to measure LAMA1 expression. Rescue experiments with pLenti-LAMA1 were performed using Lenti-X Accelerator (Clontech). Cells were fixed 4 days after knockdown and processed for either immunofluorescence or scanning electron microscopy.
Apoptosis assay
Apoptosis was evaluated using Annexin V staining (Sigma-Aldrich). Quantification of apoptosis was performed by using Apo-ONE Homogeneous Caspase-3/7 Assay purchased from Promega Corporation and performed according to the manufacturer’s instructions. About 24 h after plating, Control and Patient 2 cells were treated with or without 1 nM Staurosporine for 12 and 48 h followed by the addition of caspase-3/7 reagent. Results were read at 485 nm/527 nm fluorescence. Experiments were performed in three replicates; three independent assays were performed.
Statistical analysis
Datasets were analysed using non-parametric test (Mann–Whitney), and p values <0.05 were considered significant. Data are presented as mean±SD.
RESULTS
LAMA1-deficient subjects have myopia, cerebellar cysts and behavioural abnormalities
Under our NIH protocol, NCT00068224, we evaluated three individuals in two families presenting with similar cerebellar and ocular involvement. Patients 1 and 2 were siblings born at term via vaginal delivery without perinatal complications. Their birthweights, lengths and head sizes were normal. Parents were concerned about patient 1’s abnormal eye movements in the first week of life. Ophthalmology evaluation at 3 weeks revealed nystagmus and low vision. An electroretinography (ERG) at 3 months suggested retinal dystrophy and glasses were prescribed for high myopia at 12 months. The boy walked at 27 months and spoke four to five single words at age 2. Patient 2, the older sister of patient 1, had strabismus and nystagmus at 2 months of age. Poor vision with high myopia was diagnosed within the first year. She did not have ptosis or ocular motor apraxia. She walked at 26 months and had a wide-based stance, but speech and language development were on time. She was never able to ride a bicycle, but performed well in school, graduating from college. At the time of his evaluation at the NIH Clinical Center at the age of 21 years, patient 1 (figure 1A) had severe degenerative myopia (~−20D right eye or OD, ~−30D left eye or OS), chorioretinal atrophy (figure 1D, E), nystagmus and anxiety disorder with obsessive-compulsive features and compulsive motor stereotypy (see online supplementary video 1). Pubertal development was normal. Brain MRI showed hypoplasia of the cerebellar vermis with multiple cortical and subcortical cysts (figure 1H–J). Despite having mild motor and speech delays, he graduated from high school. Patient 2 (figure 1B), who was 27 years old, also had severe myopia (~−19D both eyes or OU), chorioretinal atrophy (figure 1F), simple facial motor tics, hypoplasia and dysplasia of the cerebellar vermis and cortical and subcortical cysts (figure 1K–M). The patients had no gross cognitive impairment (table 1). Patients 1 and 2 were previously reported as UW160–3 and UW160–4.14
Figure 1.
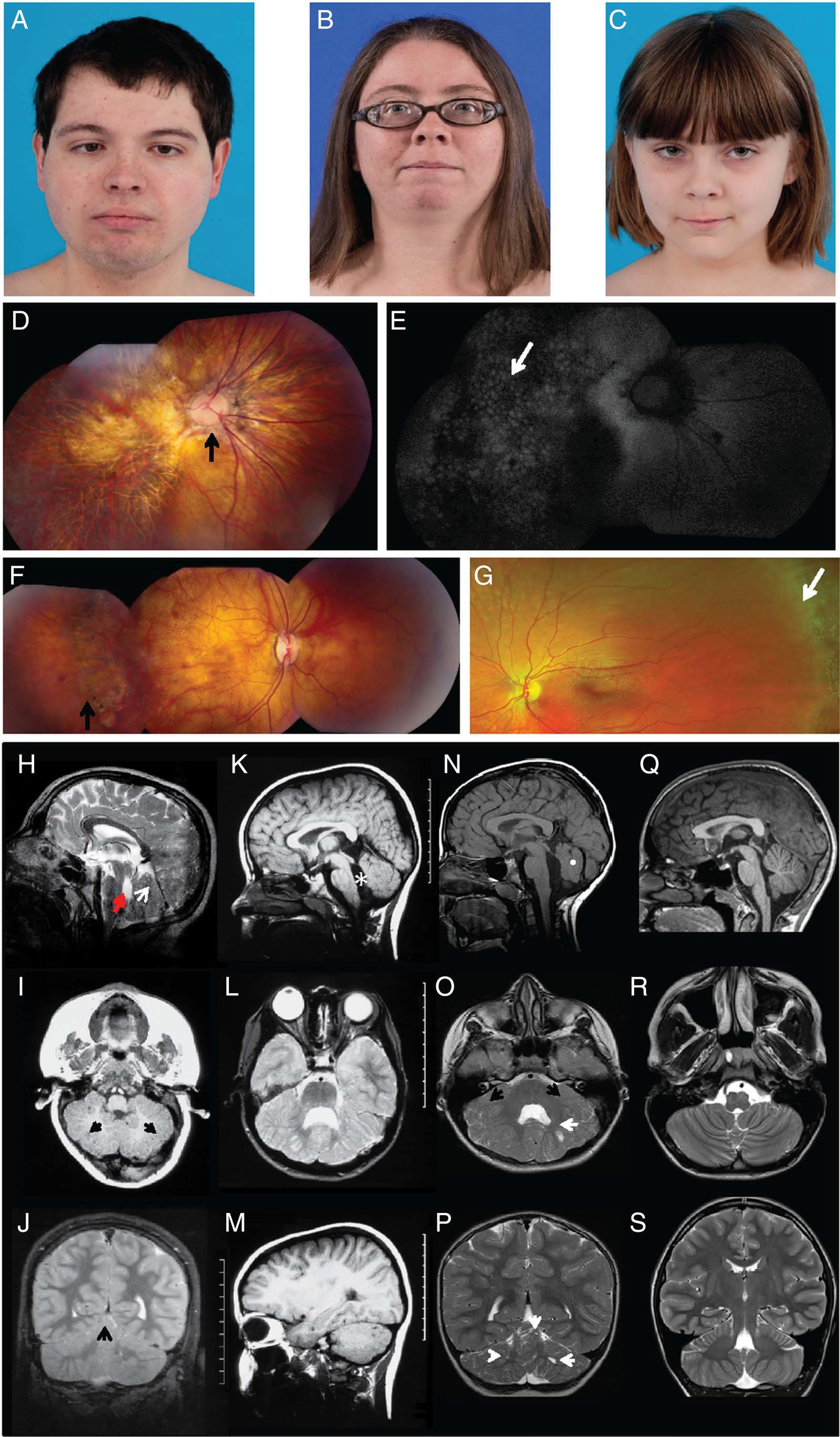
Clinical and neuroimaging features of patients with LAMA1 mutations. (A) Clinical features of patients showing distinct ophthalmologic characteristics. Patients 1 (A), 2 (B) and 3 (C) had unremarkable facial features. Note residual strabismus of patient 1 after corrective surgery. (D) Composite colour fundus photography of patient 1’s right eye demonstrating a dysplastic optic nerve with nasalised retinal vasculature (arrow) and diffuse chorioretinal atrophy involving the macula and midperiphery. (E) Composite fundus autofluorescence of the same eye, showing a ‘cobblestone’ pattern (arrow) indicating chorioretinal atrophy. (F) Composite colour fundus photograph of patient 2, the older sister of patient 1, showing moderate chorioretinal atrophy (arrow). (G) Colour fundus photo of patient 3 showing normal-appearing posterior pole and peripheral lattice degeneration (arrow), despite only mild myopia. (H–S) Brain MRI demonstrates cerebellar dysplasia associated with cysts. Brain MRI findings of patient 1 (H–J), patient 2 (K–M), patient 3 (N–P) and a healthy control (Q–S) are shown. (H) Patient 1 at 7 months of age has multiple cortical/subcortical cysts in the cerebellar vermis (arrow), dysplastic cerebellar vermis and enlarged fourth ventricle (arrowhead) with an abnormal rectangle-like shape (midsagittal-T1). The midbrain is mildly elongated, and the pons is mildly reduced in size. (I) Subcortical cysts (arrows) and dysplasia are also seen in the cerebellar hemispheres (axial-T1). (J) At 10 years 7 months, dysplasia in cerebellar vermis and hemispheres with multiple cortical and subcortical cysts (arrow) remain visible. (K) Patient 2 has hypoplasia and dysplasia of the cerebellar vermis with cortical and subcortical cysts, enlarged fourth ventricle (asterisk), elongated midbrain and pons appearing small (midsagittal-T1). (L) Cerebellar hemispheres are dysplastic and with multiple cortical and subcortical cysts in patient 2 (axial-T2). (M) Cortical and subcortical cysts within the left cerebellar hemisphere of patient 2 (parasagittal-T1) are shown. Images (K–M) were taken at age 6 years and 10 months. (N) Patient 3 has a normal sized but severely dysplastic vermis (white dot) with lack of separation of the vermian lobules (midsagittal-T1). Axial (O) and coronal (P) T2-weighted images of patient 3 show a markedly dysplastic appearance of the cerebellar hemispheres and the vermis with a diffusely abnormal configuration of the cerebellar folia and sulci (black arrows). Multiple small cysts are noted in the cerebellar hemispheres bilaterally (white arrows). Images (N–P) were taken at 4 years 3 months. (Q) A healthy 12-year-old control with normal anatomy of the posterior fossa structures (midsagittal-T1) and a normal triangle-shaped fourth ventricle is shown. Normal morphology of the cerebellum with an ‘onion-like’ foliation/fissuration pattern and white matter arborisation in the cerebellar hemispheres seen in axial (R) and coronal (S) T2 weighted images.
Table 1.
Clinical summary of patients with LAMA1 deficiency
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
|
| |||
| Age (years) | 21 | 26 | 8.5 |
| Sex | M | F | F |
| Brain MRI | Cerebellar dysplasia and cysts | Cerebellar dysplasia and cysts | Cerebellar dysplasia and cysts |
| Cognitive function | WAIS-IV verbal comprehension index normal at 116; PPVT-4 receptive vocabulary score normal at 105*; graduated high school | WAIS-IV Full Scale IQ 89; PPVT-4 receptive vocabulary score normal at 96; graduated college | WISC-IV Full Scale IQ 78; PPVT-4 receptive vocabulary normal at 95; age appropriate classes at school with minimal accommodations to account for ocular motor apraxia and extra time for handwriting |
| Neurological findings | Mildly wide-based gait; fine, rapid nystagmus that worsened in lateral gaze; significantly limited supraduction in both eyes; a 16-prism dioptre esotropia and ocular motor apraxia, especially when initiating voluntary horizontal saccades | Mildly wide-based gait; mild postural tremor and dysmetria of the upper extremities; On video ocular motor assessment, gross abnormalities were observed on all tests with numerous saccadic intrusions documented during gaze holding† | Ocular motor apraxia; mildly wide-based gait; difficulties standing on one foot or in tandem gait |
| Tics | Shoulder shrugging and nose wrinkling | Shoulder shrugging, nose wrinkling and eye rolling | Bilateral arm extensions and flexions, nose wrinkling, mouth pursing‡ |
| Anxiety | Severe | No | Severe |
| Eye findings | Nystagmus and low vision at 3 weeks; retinal dystrophy on ERG at 3 months; strabismus surgery; Currently −20D right eye and −30D left eye; chorioretinal atrophy | Strabismus and nystagmus at 2 months; high myopia diagnosed at 1 year; strabismus surgeries; currently −19D both eyes; chorioretinal atrophy | Ocular motor apraxia diagnosed at 1 year; currently 0.50+0.25 axis 170 right eye and −0.25 left eye; lattice degeneration of peripheral retina |
| Other | Bilateral syndactyly of the second and third toes involving ¾ of toe length | Bilateral syndactyly of second and third toes involving ¼ of toe length on the left and ½ of toe length on the right | Normal toes |
| Muscle bulk and strength | Normal | Normal | Normal |
| Abdominal ultrasonography | Normal | Normal | Normal |
| Kidney and liver-related blood chemistries | Normal | Normal | Normal |
| Serum creatine kinase (U/L) | 151§ | 81§ | 71¶ |
| Echocardiography | Normal | Normal | Normal |
Patient 1 could not complete full cognitive testing due to anxiety.
Online supplementary video 2a for the left eye and online supplementary video 2b for the right eye.
Normal range for adults: 62–386 U/L/.
Normal range for paediatric age group: 0–149 U/L.
F, female; M, male; PPVT-4, Peabody Picture Vocabulary Test-4; WAIS-IV, Wechsler Adult Intelligence Scale-IV.
Patient 3 (figure 1C), an 8.5-year-old female born to non-consanguineous parents Caucasian and Native American descent, had mild myopia associated with peripheral lattice degeneration of retina (figure 1G). She also had a severely dysplastic cerebellar vermis and multiple small cysts in the cerebellar hemispheres (figure 1N–P). She had delayed motor development and ocular motor apraxia (table 1). Significant anxiety, complex tics (see online supplementary video 1) and obsessive-compulsive behaviours began in early childhood. Other clinical features are summarised in table 1.
Patients harbour biallelic LAMA1 variants
Exome sequencing was performed on patients 1 and 2 and their mother. Variants were filtered using the databases of dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), ClinSeq24 and the NIH Undiagnosed Diseases Program.25–27 Only 129 coding sequence variants were found to segregate under a dominant model and two variants under compound heterozygote recessive model; none were homozygous. As the disorder was suspected to be recessive, we focused on the compound heterozygous variants. Both variants were found in LAMA1 (NM_005559.3).
Patients 1 and 2 each had two LAMA1 variants affecting canonical splice sites (NM_005559.3:c.768+1G>A and c.8557–1G>C), inherited from their mother, and a single base deletion in exon 47 (NM_005559.3:c.6701delC; p. Pro2234Leufs*9), presumably inherited from their unavailable father, since it was absent from the mother’s DNA (figure 2A). For patient 3, Sanger sequencing of the LAMA1 gene identified a nonsense mutation in exon 15 (NM_005559.3:c.2160T>A; p.Cys720*) inherited from her mother and a 7 bp deletion in exon 42 (NM_005559.3:c.5985_5991del; p.Ile1996Glufs*7), presumably inherited from her unavailable father (figure 2A). Based on allele frequencies of potential null alleles from public databases, we estimate the prevalence of this disease to be about 1–20 in 1 000 000.
Figure 2.
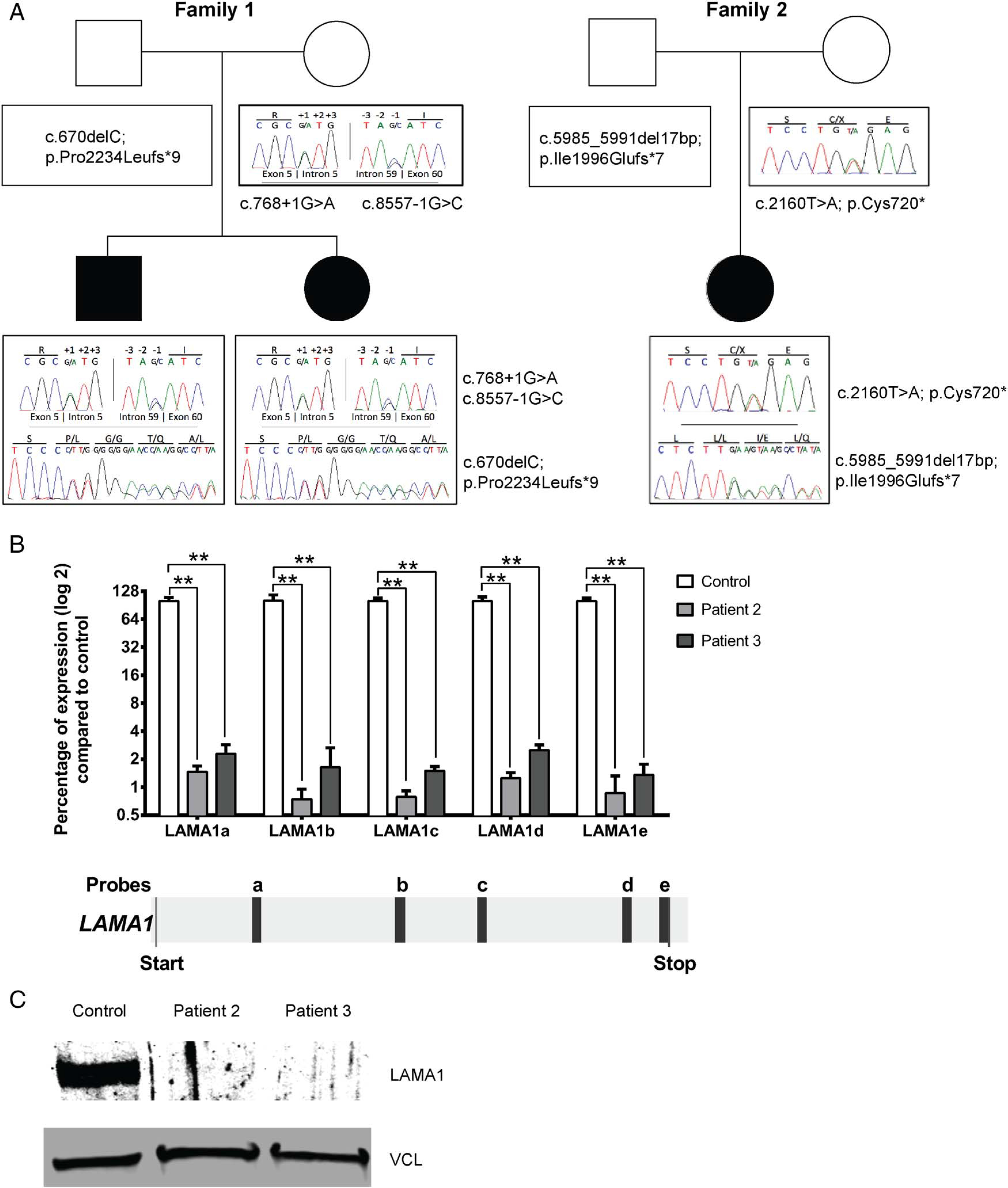
LAMA1 null mutations in patients with lamininopathy. (A) Pedigrees of the two families including chromatograms of their identified LAMA1 variants. (B) Quantitative real-time PCR results for LAMA1 mRNA expression in fibroblast from patient 2 and patient 3 compared with control using five different probes spanning LAMA1 cDNA. Values are percentage expression of LAMA1 in patient cells compared with control cells, normalised to ACTB (error bars represent S.D., n=4 from three independent experiments; **p<0.01, Mann–Whitney test); under the histogram, localisation of the different probes on LAMA1 transcript (NM_005559.3) is shown. (c) Immunoblot of fibroblast lysates of patient 2, patient 3 and control, showing absence of LAMA1 in the patients. Loading was controlled by vinculin (VCL).
mRNA and protein expression are decreased in LAMA1-deficient patients
The LAMA1 gene (Gene ID: 284217) spans ~176 kb, and is divided into 63 exons. Its mRNA is 9657 bp and encodes a 3075 amino acid protein, laminin α1, which is selectively expressed in tissues (see online supplementary figure S1A). LAMA1 mRNA transcript was absent from cultured fibroblasts of patients 2 and 3 (figure 2B), presumably due to nonsense-mediated decay. Indeed, no LAMA1 protein was produced, as confirmed by immunoblotting (figure 2C). In family 1, we could not confirm which splice variant is disease-causing, as no mRNA is produced from the maternal allele; we hypothesise that the c.768+1G>A variant, which occurs earlier in the gene than the c.8557–1G>C variant is more likely deleterious.
As the identified mutations lead to null alleles and no expression of LAMA1 protein (figure 2B), we tested the expression of the laminin subunits that can assemble together with the α1 chain (LAMB1, encoding laminin β1, MIM 150240; LAMB2, encoding laminin β2, MIM 150325 and LAMC1, encoding laminin γ1, MIM 150290). We noticed a significant decrease of the mRNA expression of these three subunits in both patient 2 and patient 3 fibroblasts (see online supplementary figure S1B).
Impaired adhesion and migration of LAMA1-deficient cells
Since LAMA1 is a component of the basement membrane, we evaluated the functional consequences of absent LAMA1 by measuring the adhesion response of trypsinised fibroblasts to different ECM components (collagen-1, laminin-111) (figure 3A). LAMA1-deficient cells showed significantly decreased adhesion to non-coated and coated wells when compared with control fibroblasts (figure 3A). To determine the role of LAMA1 in fibroblasts motility, we used an in vitro monolayer scratch assay. Patient’s LAMA1-deficient fibroblasts demonstrated less migration into the cell-denuded gap than did control fibroblasts (figure 3B). To visualise the effect of LAMA1 deficiency on cell migration, we analysed cell membrane projections (lamellipodia and filopodia) using immunohistochemistry (figure 4A) and scanning electron microscopy (figure 4B). Control fibroblasts on uncoated wells appear flat and stretched, with normal looking slender lamellipodia and scattered filopodia. In contrast, LAMA1-deficient cells from patient 2 appear less stretched out, with short and stubby lamellipodia, and increased microvillus projection on the cell surface (figure 4A). When grown on laminin-111-coated coverslips, control cell membrane projections increased in number and length, reflecting enhanced migration in cells, in addition to increased number of focal adhesion points that appear as small aggregates in the termini of stress fibres. When grown on laminin-111-coated coverslips, patient 2 cell processes increased in number and length with broader lamellipodia, but less visible focal adhesion points, indicating partial rescue in cell architecture (figure 4B).
Figure 3.

LAMA1-deficient cells exhibit impaired adhesion and migration. (A) Equal numbers of control and patient 2 fibroblasts were plated on wells coated with either bovine serum albumin (BSA), collagen-I (Col-I) or Laminin-111 and allowed to attach. OD values represent amounts of DNA in cells that remain attached to the wells after 90 min of incubation. Bar graph gives mean and SD of five replicates from three independent experiments; *p<0.05; **p<0.01 (Mann–Whitney test). (B) Equal numbers of control and patient 2 fibroblasts were seeded on culture wells and allowed to form a monolayer and grow to confluence, after which a 0.9 mm scratch was made. ‘Wound’ closure was monitored by imaging cells stained with Calcein AM after 12, 24, 36, 48 and 60 h. Percentage closure and migration rates were calculated from six experiments. Scale bar represents 100 μm. Bar graph gives mean and SD of three replicates from three independent experiments; *p<0.05; **p<0.01 (Mann–Whitney test).
Figure 4.
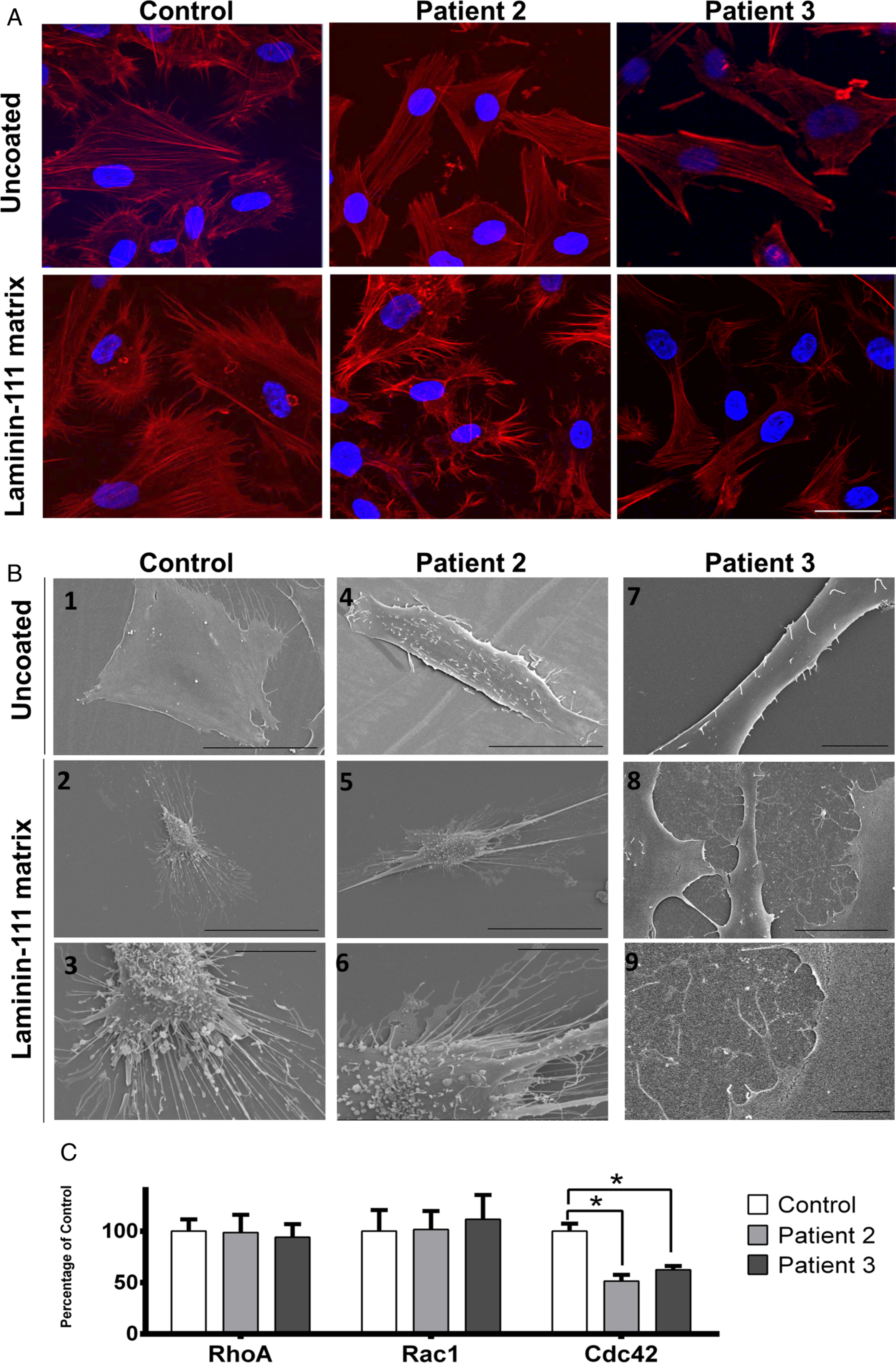
LAMA1 deficiency disrupts cell architecture for focal adhesion in fibroblasts. Equal numbers of cells from control and patient 2 fibroblasts were plated on non-coated or laminin-111-coated coverslips. Laminin-111 is a heterotrimer complex composed of LAMA1, LAMB1 and LAMC1. (A) Cells were imaged after staining with phalloidin (red) that labels actin filaments, and DAPI (blue) that labels the nuclei. On uncoated coverslips, the control cells show intact, highly organised cytoskeletal organisation, and numerous filopodia. In cells of patient 2 and patient 3, there is less complexity in cytoskeletal network, and filopodia structures are remarkably reduced or absent, indicating the importance of LAMA1 in the focal adhesion complex. On laminin-111-coated coverslips, control cells appear to spread more than on non-coated coverslips, while the filopodia length is increased. Cells of patient 2 and patient 3 show increased number of filopodia on laminin-coated coverslips. Scale bar represents 20 μm. (B) On scanning electron microscopy, control cells on uncoated slides appear thoroughly spread and flat with well-formed filopodia structures at the edges, as expected in normal control fibroblasts.45 Patient 2 and patient 3 cells on uncoated slides generally appear ‘lifted’, creating a three-dimensional appearance. The number of filopodia in patient 2 cells are markedly lower as compared with control cells; this number normalised to almost similar to control cells when patient 2 and patient 3 cells were grown on laminin-coated slides. In addition, microvilli protrusions, which are presumably unattached filopodia or lamellipodia, are seen on the surface of the cells. Scale bars in (B) represent 50 μm in B1–2 and B4–5, 10 μm in B3, B6, B7 and B9 and 25 μm in B8. (C) GTPase activation assay of Cdc42, Rac1 and RhoA was performed in control and patient 2 fibroblasts. Results show a significant decrease of the activated form (GTP-bound) of Cdc42 in patient 2 and patient 3 fibroblasts compared with control; no difference was observed for Rac1 and RhoA (error bars represent SD, n=6; *p<0.05, Mann–Whitney test).
Laminin-111 induces neurite outgrowth in mesenchymal stem cells28 and in primary central and peripheral nervous system neurons.29,30 This process requires the activation of the GTPase Cdc42.31 We tested the activation of different GTPases using G-Lisa (figure 4C); Cdc42 was significantly decreased in patients’ fibroblasts compared with control, whereas other GTPases (RhoA and Rac1) were normal. These results confirm the finding of decreased filopodia observed by scanning electron microscopy, since binding of Cdc42 to GTP is implicated in filopodia formation.32
To determine whether LAMA1-associated abnormalities in filopodia formation are also observed in neuronal cells, we knocked down LAMA1 using siRNA in neuronal cells differentiated from neural stem cells. Knockdown efficiency was evaluated using 0.1, 0.2 and 0.6 μM, and we used 0.6 μM for the rest of the study since it gave the most robust knockdown (~80%, see online supplementary figure S2). When non-targeting siRNA was used, neuronal processes appear long and branched, and filopodia structures extend from these processes. In contrast, LAMA1 knockdown resulted in reduced branching of processes (figure 5A) and disappearance of filopodia (figure 5B,C), supporting the importance of LAMA1 in neuronal cell filopodia formation and cell migration. Neuronal cells co-transduced with pLenti-LAMA1 showed preservation of most filopodia as compared with those transduced with pLenti-empty, indicating that the phenotype of impaired filopodia is specific to LAMA1 deficiency.
Figure 5.

Effects of LAMA1 knockdown in neuronal cells (A–C); LAMA1 knockdown in neuronal cells reduced the number of filopodia. Non-targeting (control) siRNA and siRNA to LAMA1 (LAMA1-siRNA) were introduced to neuronal cells differentiated from Neural Stem Cells (NSC). Immunohistochemistry with phalloidin (A) showed that LAMA1-siRNA reduced the number of neurites and branching, in addition to reduction of filopodia (red, phalloidin; blue, DAPI stain for nucleus). Scanning electron microscopy (SEM) (B,C) confirmed that the number of filopodia is reduced when LAMA1 is knocked down (LAMA1-siRNA) as compared with control. Filopodia were partially recovered when wild-type hLAMA1 was reintroduced to cells (LAMA1-siRNA+Rescue). Magnified images of (B) are shown in (C). Scale bars represent 100 μm in (A), 10 μm in (B) and 2 μm in (C). (D) LAMA1 deficiency increases susceptibility to apoptosis. To quantify apoptosis, caspase-3/7 activity was measured at 12 and 30 h after plating control (white bars), patient 2 (light grey bars) and patient 3 cells, with or without the addition of the known apoptosis inducer staurosporine. Results show an increased susceptibility of patient’s cells to apoptosis. Bars represent SD for three replicates of three experiments; *p<0.05; **p<0.01 (Mann–Whitney test).
LAMA1 deficiency increases susceptibility to apoptosis
Since the conditional LAMA1 knockout mouse model shows increased apoptosis,33 we measured apoptosis in LAMA1-deficient patients’ fibroblasts by Annexin V expression and caspase 3/7 activity assays. At baseline, a significantly greater number of LAMA1-deficient cells underwent apoptosis compared with control cells, demonstrated by increased Annexin V expression (data not shown). After treatment with staurosporine, a known apoptosis inducer,34 LAMA1-deficient cells show markedly increased susceptibility to cell death, demonstrated by significantly increased caspase 3/7 activity (figure 5D). Whether apoptosis causes the specific developmental abnormalities of LAMA1-deficient patients remains to be determined.
Wild-type LAMA1 rescues LAMA1-deficient cellular phenotypes
To demonstrate that the above-described cellular phenotypes in fibroblasts are due to LAMA1 deficiency, we introduced full length, wild-type LAMA1 in patients’ fibroblasts by lentiviral transduction. Stable overexpression of pLenti-LAMA1 in patients’ fibroblasts led to an increase in the amount of LAMA1 protein by immunoblot (see online supplementary figure S3A; image of full blots are shown in see online supplementary figure S4), compared with absent LAMA1 protein in patients’ cells without rescue (figure 2C). Patients’ fibroblasts stably expressing LAMA1 showed improvement in cell adhesion (see online supplementary figure S3B), increased migration (see online supplementary figure S3C) and reduced apoptosis (see online supplementary figure S3D) compared with patient fibroblasts cells stably expressing pLenti-empty lentiviral vector. In addition, patient cells transduced with pLenti-LAMA1 exhibited an increase in GTP-bound Cdc42 (see online supplementary figure S3E), indicating that LAMA1 is important in filopodia formation and, consequently, cell adhesion and migration.
DISCUSSION
In this paper, we have shown that LAMA1 deficiency leads to altered cell adhesion and migration. Using immunohistochemistry and ultrastructural studies, we demonstrated that these LAMA1-deficient cells lack filopodia, cytoplasmic projections that are important for the formation of focal adhesions. The role of Rho GTPase family members, including RhoA, Rac1 and Cdc42, in cytoskeletal dynamics has been well described in fibroblast studies. Activation of RhoA, Rac1 and Cdc42 was shown to lead to actin cytoskeletal reorganisation into distinct structures, including stress fibres and focal adhesions, lamellipodia, and filopodia. These cellular structures affect cell–cell and cell–extracellular matrix adhesion, cell motility and neuronal development. Using fibroblasts derived from patients with LAMA1, pull-down assays revealed that there is an impaired activation of Cdc42, which may well explain the lack of filopodia in LAMA1-deficient fibroblasts. Susceptibility of LAMA1-deficient cells to apoptosis is reminiscent of what was seen in the conditional knockout mouse model, and may be related to loss of protection to anoikis that is provided by laminins.
Several findings in our patients are recapitulated in LAMA1-deficient animal models. A conditional LAMA1 knockout mouse (LAMA1CKO)15,16 escaped embryonic lethality, but displayed decreased proliferation and migration of granule cell precursors, marked reduction in numbers of dendritic projections in Purkinje cells and disorganisation of Bergmann glial fibres and end-feet. In LAMA1CKO spheres were markedly hypoplastic and almost complete loss of folia in the anterior part of the cerebellum.15 This is very similar to the cerebellar dysplasia that we observed in all three of our patients.
In mice, laminins are present in many different compartments of the retina, including Bruch’s membrane, the interphotoreceptor matrix, the external limiting membrane, the outer plexiform layer, inner plexiform layer and inner limiting membrane.35 Mice with constitutional ablation of LAMA1 were blind due to a defective inner limiting membrane and abnormal differentiation of the Müller glial cells that support the organisation of the retina, and also due to altered ganglion cell layer, a major neuronal population of the retina.36 Similarly, the zebrafish bashful (baltp82) mutant, with LAMA1 null alleles, displayed abnormal retinal ganglion cell layer and altered optic nerve.37 In humans, severe myopia has been associated with the genomic region MYP2 (myopia 2), in which LAMA1 resides. In fact, rs2089760 polymorphism, located in the promoter region of LAMA1, was found to be significantly enriched in individuals with high myopia.38 In another study,39 SNPs located in LAMA1 were associated with severe myopia. The importance of LAMA1 in the formation of the lens is also corroborated by the loss of the lens in the zebrafish LAMA1 mutant.40,41
Our patients manifested tics, obsessive compulsive features and anxiety. Of note, the conditional LAMA1 knockout mice also display behavioural abnormalities. Interestingly, Online Mendelian Inheritance in Man summarises 19 genes, not including LAMA1 that are associated to tics and anxiety (see online supplementary table S2). Patients with Tourette’s syndrome, a developmental neuropsychiatric disorder characterised by chronic vocal and motor tics are shown to have reduced volumes of the cerebellar hemispheres on MRI.42 Tourette’s syndrome displays familial aggregation and its concordance rates for MZ twins reach 77% compared with 23% for dizygotic twins, suggesting genetic contribution to its aetiology.43 Loss-of-function mutations in SLITRK1 are thought to be associated with Tourette’s syndrome.44 It is interesting that similar to LAMA1, the protein encoded by SLITRK1 promotes dendritic growth. Whether tics, obsessive compulsive traits and anxiety are a primary part of the LAMA1 deficiency phenotype will likely be determined after description of other patients with LAMA1 mutations and targeted testing for LAMA1 mutations in this specific patient cohort. Based on population studies (1000 Genomes, Exome Aggregation Consortium, and NHLBI Exome Sequencing Project; see URLs) and the frequencies of potential null variants (stop gain, frame shift or splice variants) in LAMA1 (see online supplementary table S3), the prevalence of the disease would be about 1–20/1 000 000, indicating that there are undiagnosed patients who may harbour LAMA1 mutations, and these patients might belong to cohort of patients presenting with overlap of obsessive compulsive traits, tics and anxiety, myopia, retinal degeneration and cerebellar abnormalities. It would be interesting to look for LAMA1 mutations in these cohorts of patients.
The cerebellar dysplasia combined with cortical and subcortical cysts, present in our patients, represent uncommon neuroimaging findings. The cerebellar cysts are considered rather specific for α-dystroglycanopathies and have been reported mostly in patients with mutations in FKRP (MIM 606596), POMT2 (MIM 607439), POMGnT1 (MIM 606822) and LARGE (MIM 608840). In our patients, however, the glycosylation of α-dystroglycan, at least in fibroblasts, appears intact (data not shown). Further studies of the relationship between α-dystroglycan and laminin may uncover the reasons for the similar neuroimaging findings in these patients and in our patients with LAMA1 deficiency.
In summary, we describe detailed clinical and molecular functional studies of a lamininopathy associated with LAMA1 mutations. The study of LAMA1-deficient fibroblasts and the use of neuronal stem cell model allowed us to establish the initial steps in understanding what happens with LAMA1 deficiency in the cellular level. Future work such as generation of induced pluripotent stem cells that can be differentiated to several cell types will allow further understanding of the role of LAMA1 in cell signalling and migration, and ultimately, its specific role in human disease.
Supplementary Material
Acknowledgements
The authors thank the Electron Microscopy facility National Heart, Lung, and Blood Institute, NIH, for the use of its critical point dryer, sputter coater and electron microscope. They also thank Deniz Yildirimli, and Courtney M Sinclair (Medical Genetics Branch, National Human Genome Research Institute, NIH) for their technical assistance. They also thank the patients and their families for participating in this study.
Funding This research was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, Maryland, USA.
Footnotes
Competing interests None declared.
Patient consent Obtained.
Ethics approval National Human Genome Research Institute (NHGRI) Institutional Review Board.
URLS
1000 Genomes Project 11_2010 data release project, ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20100804/dbSNP, http://www.ncbi.nlm.nih.gov/snp/
Exome Aggregation Consortium (ExAC), Cambridge, Massachusetts, USA (http://exac.broadinstitute.org) (date accessed October 11, 2015)
NHLBI Exome Sequencing Project EVS (V.0.0.30), http://evs.gs.washington.edu/EVS/ (date accessed October 11, 2015)
REFERENCES
- 1.Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, Yurchenco PD. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol 2005;169:179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliveira J, Santos R, Soares-Silva I, Jorge P, Vieira E, Oliveira ME, Moreira A, Coelho T, Ferreira JC, Fonseca MJ, Barbosa C, Prats J, Ariztegui ML, Martins ML,Moreno T, Heinimann K, Barbot C, Pascual-Pascual SI, Cabral A, Fineza I, Santos M, Bronze-da-Rocha E. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet 2008;74:502–12. [DOI] [PubMed] [Google Scholar]
- 3.Knoll R, Postel R, Wang J, Kratzner R, Hennecke G, Vacaru AM, Vakeel P, Schubert C, Murthy K, Rana BK, Kube D, Knoll G, Schafer K, Hayashi T, Holm T, Kimura A, Schork N, Toliat MR, Nurnberg P, Schultheiss HP, Schaper W, Schaper J, Bos E, Den Hertog J, van Eeden FJ, Peters PJ, Hasenfuss G, Chien KR, Bakkers J. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 2007;116:515–25. [DOI] [PubMed] [Google Scholar]
- 4.Radmanesh F, Caglayan AO, Silhavy JL, Yilmaz C, Cantagrel V, Omar T, Rosti B, Kaymakcalan H, Gabriel S, Li M, Sestan N, Bilguvar K, Dobyns WB, Zaki MS, Gunel M, Gleeson JG. Mutations in LAMB1 cause cobblestone brain malformation without muscular or ocular abnormalities. Am J Hum Genet 2013;92:468–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasselbacher K, Wiggins RC, Matejas V, Hinkes BG, Mucha B, Hoskins BE, Ozaltin F, Nurnberg G, Becker C, Hangan D, Pohl M, Kuwertz-Broking E, Griebel M, Schumacher V, Royer-Pokora B, Bakkaloglu A, Nurnberg P, Zenker M, Hildebrandt F. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int 2006;70:1008–12. [DOI] [PubMed] [Google Scholar]
- 6.Vidal F, Baudoin C, Miquel C, Galliano MF, Christiano AM, Uitto J, Ortonne JP, Meneguzzi G. Cloning of the laminin alpha 3 chain gene (LAMA3) and identification of a homozygous deletion in a patient with Herlitz junctional epidermolysis bullosa. Genomics 1995;30:273–80. [DOI] [PubMed] [Google Scholar]
- 7.Pulkkinen L, Meneguzzi G, McGrath JA, Xu Y, Blanchet-Bardon C, Ortonne JP, Christiano AM, Uitto J. Predominance of the recurrent mutation R635X in the LAMB3 gene in European patients with Herlitz junctional epidermolysis bullosa has implications for mutation detection strategy. J Invest Dermatol 1997;109:232–7. [DOI] [PubMed] [Google Scholar]
- 8.Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E, Uitto J. Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet 2002;110:41–51. [DOI] [PubMed] [Google Scholar]
- 9.Barak T, Kwan KY, Louvi A, Demirbilek V, Saygi S, Tuysuz B, Choi M, Boyaci H, Doerschner K, Zhu Y, Kaymakcalan H, Yilmaz S, Bakircioglu M, Caglayan AO, Ozturk AK, Yasuno K, Brunken WJ, Atalar E, Yalcinkaya C, Dincer A, Bronen RA, Mane S, Ozcelik T, Lifton RP, Sestan N, Bilguvar K, Gunel M. Recessive LAMC3 mutations cause malformations of occipital cortical development. Nat Genet 2011;43:590–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biol 2005;24:326–32. [DOI] [PubMed] [Google Scholar]
- 11.Li S, Edgar D, Fassler R, Wadsworth W, Yurchenco PD. The role of laminin in embryonic cell polarization and tissue organization. Dev Cell 2003;4:613–24. [DOI] [PubMed] [Google Scholar]
- 12.Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha3 isoform. J Cell Biol 1997;137:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poretti A, Hausler M, von Moers A, Baumgartner B, Zerres K, Klein A, Aiello C, Moro F, Zanni G, Santorelli FM, Huisman TA, Weis J, Valente EM, Bertini E, Boltshauser E. Ataxia, intellectual disability, and ocular apraxia with cerebellar cysts: a new disease? Cerebellum 2014;13:79–88. [DOI] [PubMed] [Google Scholar]
- 14.Aldinger KA, Mosca SJ, Tetreault M, Dempsey JC, Ishak GE, Hartley T, Phelps IG, Lamont RE, O’Day DR, Basel D, Gripp KW, Baker L, Stephan MJ, Bernier FP, Boycott KM, Majewski J, University of Washington Center for Mendelian G, Care4Rare C, Parboosingh JS, Innes AM, Doherty D. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Am J Hum Genet 2014;95:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heng C, Lefebvre O, Klein A, Edwards MM, Simon-Assmann P, Orend G, Bagnard D. Functional role of laminin α1 chain during cerebellum development. Cell Adh Migr 2011;5:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichikawa-Tomikawa N, Ogawa J, Douet V, Xu Z, Kamikubo Y, Sakurai T, Kohsaka S, Chiba H, Hattori N, Yamada Y, Arikawa-Hirasawa E. Laminin α1 is essential for mouse cerebellar development. Matrix Biol 2012;31:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O’Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008;456:53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 2010;19:R145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics 2012;28:599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 2009;27:182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak K Comparative Ct method. ABI Prism 7700 Sequence Detection System. 1997. [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 23.Lu W, McCallum L, Irvine AE. A rapid and sensitive method for measuring cell adhesion. J Cell Commun Signal 2009;3:147–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biesecker LG, Mullikin JC, Facio FM, Turner C, Cherukuri PF, Blakesley RW, Bouffard GG, Chines PS, Cruz P, Hansen NF, Teer JK, Maskeri B, Young AC, Manolio TA, Wilson AF, Finkel T, Hwang P, Arai A, Remaley AT, Sachdev V, Shamburek R, Cannon RO, Green ED. The ClinSeq project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res 2009;19:1665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gahl WA, Boerkoel CF, Boehm M. The NIH Undiagnosed Diseases Program: bonding scientists and clinicians. Dis Model Mech 2012;5:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DM, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med 2012;14:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gahl WA, Tifft CJ. The NIH undiagnosed diseases program: lessons learned. JAMA 2011;305:1904–5. [DOI] [PubMed] [Google Scholar]
- 28.Mruthyunjaya S, Manchanda R, Godbole R, Pujari R, Shiras A, Shastry P. Laminin-1 induces neurite outgrowth in human mesenchymal stem cells in serum/differentiation factors-free conditions through activation of FAK-MEK/ERK signaling pathways. Biochem Biophys Res Commun 2010;391:43–8. [DOI] [PubMed] [Google Scholar]
- 29.Luckenbill-Edds L Laminin and the mechanism of neuronal outgrowth. Brain Res Rev 1997;23:1–27. [DOI] [PubMed] [Google Scholar]
- 30.Plantman S, Patarroyo M, Fried K, Domogatskaya A, Tryggvason K, Hammarberg H, Cullheim S. Integrin-laminin interactions controlling neurite outgrowth from adult DRG neurons in vitro. Mol Cell Neurosci 2008;39:50–62. [DOI] [PubMed] [Google Scholar]
- 31.Weston CA, Anova L, Rialas C, Prives JM, Weeks BS. Laminin-1 activates Cdc42 in the mechanism of laminin-1-mediated neurite outgrowth. Exp Cell Res 2000;260:374–8. [DOI] [PubMed] [Google Scholar]
- 32.Fukata M, Nakagawa M, Kaibuchi K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol 2003;15:590–7. [DOI] [PubMed] [Google Scholar]
- 33.Alpy F, Jivkov I, Sorokin L, Klein A, Arnold C, Huss Y, Kedinger M, Simon-Assmann P, Lefebvre O. Generation of a conditionally null allele of the laminin alpha1 gene. Genesis 2005;43:59–70. [DOI] [PubMed] [Google Scholar]
- 34.Belmokhtar CA, Hillion J, Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001;20:3354–62. [DOI] [PubMed] [Google Scholar]
- 35.Libby RT, Champliaud MF, Claudepierre T, Xu Y, Gibbons EP, Koch M, Burgeson RE, Hunter DD, Brunken WJ. Laminin expression in adult and developing retinae: evidence of two novel CNS laminins. J Neurosci 2000;20:6517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards MM, Mammadova-Bach E, Alpy F, Klein A, Hicks WL, Roux M, Simon-Assmann P, Smith RS, Orend G, Wu J, Peachey NS, Naggert JK, Lefebvre O, Nishina PM. Mutations in LAMA1 disrupt retinal vascular development and inner limiting membrane formation. J Biol Chem 2010;285:7697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biehlmaier O, Makhankov Y, Neuhauss SC. Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest Ophthalmol Vis Sci 2007;48:2887–94. [DOI] [PubMed] [Google Scholar]
- 38.Zhao YY, Zhang FJ, Zhu SQ, Duan H, Li Y, Zhou ZJ, Ma WX, Li Wang N. The association of a single nucleotide polymorphism in the promoter region of the LAMA1 gene with susceptibility to Chinese high myopia. Mol Vis 2011;17:1003–10. [PMC free article] [PubMed] [Google Scholar]
- 39.Sasaki S, Ota M, Meguro A, Nishizaki R, Okada E, Mok J, Kimura T, Oka A, Katsuyama Y, Ohno S, Inoko H, Mizuki N. A single nucleotide polymorphism analysis of the LAMA1 gene in Japanese patients with high myopia. Clin Ophthalmol 2007;1:289–95. [PMC free article] [PubMed] [Google Scholar]
- 40.Pathania M, Semina EV, Duncan MK. Lens extrusion from Laminin alpha 1 mutant zebrafish. ScientificWorldJ 2014;2014:524929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zinkevich NS, Bosenko DV, Link BA, Semina EV. laminin alpha 1 gene is essential for normal lens development in zebrafish. BMC Dev Biol 2006;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tobe RH, Bansal R, Xu D, Hao X, Liu J, Sanchez J, Peterson BS. Cerebellar morphology in Tourette syndrome and obsessive-compulsive disorder. Ann Neurol 2010;67:479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price RA, Kidd KK, Cohen DJ, Pauls DL, Leckman JF. A twin study of Tourette syndrome. Arch Gen Psychiat 1985;42:815–20. [DOI] [PubMed] [Google Scholar]
- 44.Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, Davis NR, Ercan-Sencicek AG, Guez DH, Spertus JA, Leckman JF, Dure LSt, Kurlan R, Singer HS, Gilbert DL, Farhi A, Louvi A, Lifton RP, Sestan N, State MW. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science 2005;310:317–20. [DOI] [PubMed] [Google Scholar]
- 45.Grinnell F, Bennett MH. Fibroblast adhesion on collagen substrata in the presence and absence of plasma fibronectin. J Cell Sci 1981;48:19–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.