

Summary
Cells counter DNA damage through repair or apoptosis, yet a direct mechanism for this choice has remained elusive. When facing interstrand crosslinks (ICL), the ICL-repair protein FANCI heterodimerizes with FANCD2 to initiate ICL excision. We found that FANCI alternatively interacts with a pro-apoptotic factor, PIDD1, to enable PIDDosome (PIDD1-RAIDD-caspase-2) formation and apoptotic death. FANCI switches from FANCD2/repair to PIDD1/apoptosis signaling in the event of ICL repair failure. Specifically, removing key endonucleases downstream of FANCI/FANCD2, increasing ICL levels, or allowing damaged cells into mitosis (when repair is suppressed) all suffice for switching. Reciprocally, apoptosis-committed FANCI reverts from PIDD1 to FANCD2 after a failed attempt to assemble the PIDDosome. Monoubiquitination and deubiquitination at FANCI K523 impact interactor selection. These data unveil a repair-or-apoptosis switch in eukaryotes. Beyond ensuring the removal of unrepaired genomes, the switch’s bidirectionality reveals that damaged cells can offset apoptotic defects via de novo attempts at lesion repair.
Keywords: DNA repair, apoptosis, fate choice, molecular switch, FANCI, FANCD2, PIDD1, PIDDosome, DNA interstrand crosslink, monoubiquitination, deubiquitination, repair failure
Graphical Abstract
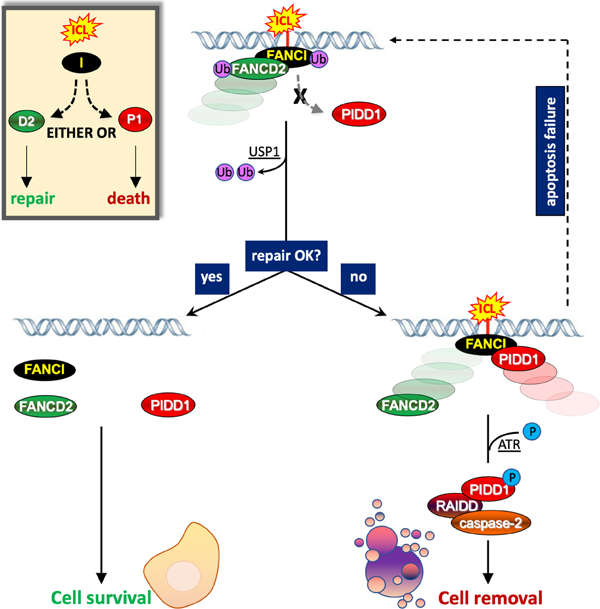
eTOC Blurb:
Shah et al. describe a mechanism by which cells decide their fate, repair or self-removal, after DNA damage. The mechanism relies on the ability of a DNA repair effector, FANCI, to either engage an apoptotic device or repair activity in response to interstrand crosslinks.
Introduction
Damage to genomic DNA is a major threat to the viability of organisms and that of their offspring (Jackson and Bartek, 2009). In long lived, cancer-prone animals such as mammals, DNA lesions are either resolved by dedicated repair factories, or removed altogether by DNA damage-induced apoptosis (DDIA) or senescence pathways (Campisi and d’Adda di Fagagna, 2007; Ciccia and Elledge, 2010; Norbury and Zhivotovsky, 2004; Roos et al., 2016). DNA repair and DDIA are both critical to tumor suppression and tumor responses to radiotherapy and genotoxic chemotherapies (Helleday et al., 2008; Igney and Krammer, 2002). Less understood is how cells make the decision to repair (and thus survive) the lesions, or commit to apoptosis. Mechanistic knowledge of this choice is key for predicting and overcoming treatment resistance in patients (Carvajal and Manfredi, 2013; Helleday et al., 2008; Vousden and Lu, 2002).
Current views of the repair vs. apoptosis decision mainly derive from studies of the DNA damage-inducible transcription factor p53. In a widely held model, DNA lesions first trigger a DNA damage response, which in turn stabilizes p53, which in turn transactivates pro-repair or pro-apoptotic targets, which in turn repair the DNA or kill the cell (Aylon and Oren, 2007; Krenning et al., 2019; Vousden and Lu, 2002). Beyond the intricate nature of the process, how p53 selects its targets is unclear. Dynamic p53 pulses resulting from low vs. severe/sustained levels of DNA injury have been implicated, alongside differing p53 affinities for pro-repair vs. pro-apoptotic promoters, among others (Aylon and Oren, 2007; Carvajal and Manfredi, 2013; Krenning et al., 2019; Purvis et al., 2012; Vousden and Lu, 2002; Zhang et al., 2009).
In contrast, a direct mechanism enabling damaged cells to choose between repair and apoptosis—possibly involving a DNA repair factor with an added, intrinsic cell-killing activity (De Zio et al., 2013)—has yet to be conclusively identified. While the homologous recombination (HR) repair protein BRCA1 physically interacts with p53 to stimulate its transcriptional activity (Evers and Jonkers, 2006), this action is geared toward p53 targets involved in cell cycle control and DNA repair, not pro-apoptotic targets (MacLachlan et al., 2002). In a 2011 study (De Zio et al., 2011), Cecconi and colleagues showed that the double strand break (DSB) sensor Ku, a critical player in non-homologous end joining (NHEJ) repair, represses APAF1 in response to DNA damage, thus possibly impacting caspase-9 activation within the apoptosome (Cyt c-Apaf1-caspase-9). Interestingly, as cells presumably accumulated DNA damage over time, Ku vacated the APAF1 promoter, correlating with apoptosis induction (De Zio et al., 2011). In another study (Stoyanova et al., 2009), the nucleotide excision repair (NER) component XPE/DDB2 contributed to Cul4A-mediated degradation of p53 after low-dose UV irradiation. Reminiscent of the Ku-Apaf1 study, XPE/DDB2 action was relieved at greater UV doses, again correlating with apoptosis (Stoyanova et al., 2009). These studies did not directly address whether or how Ku and XPE/DDB2 switched from pro-survival to pro-apoptotic functionalities. However, they lent credence to the concept that DNA repair factors might exist that can additionally trigger apoptosis should DNA lesions accumulate in damaged cells (De Zio et al., 2013).
The activation platform for caspase-2 (C2), the PIDDosome (or C2-PIDDosome) (Tinel and Tschopp, 2004), has been implicated in both p53-dependent and p53-independent DDIA, although the biologic contexts in which it is mobilized remain unclear (Janssens and Tinel, 2012; Sladky et al., 2017). The PIDDosome comprises the death domain (DD) scaffold protein PIDD1 (p53-induced protein with DD; LRDD), the caspase adaptor RAIDD (RIP-associated ICH-1/CED-3-homologous protein with DD; CRADD), and the C2 protease (Tinel and Tschopp, 2004). Five PIDD1 monomers nucleate the complex via DD:DD interactions with seven RAIDD molecules, which in turn support seven pro-C2 monomers for activation by homodimerization and autoproteolytic cleavage (Park et al., 2007), a process termed induced-proximity (Bouchier-Hayes et al., 2009). Double-strand DNA breaks (DSBs), as induced by IR (Ando et al., 2012) or topoisomerase inhibitors (Ando et al., 2017), trigger PIDDosome formation via ATM-mediated phosphorylation of T788 within the PIDD1 DD, which enables RAIDD recruitment and C2 activation (Ando et al., 2012). PIDDosome signaling has been consistently linked to genomic instability in mitosis, as caused by DSBs (Thompson et al., 2015), aneuploidy (Dawar et al., 2017), or supernumerary centrosomes (Fava et al., 2017; Sladky et al., 2020). As such, checkpoint kinase 1 inhibitors (Chk1i) such as Gö6976 (Chen et al., 2009; Kohn et al., 2003; Sidi et al., 2008), which eliminate the G2/M checkpoint, markedly enhance PIDDosome signaling in damaged cells (Ando et al., 2012; Ando et al., 2017; Ho et al., 2009; Manzl et al., 2013; Thompson et al., 2015).
Results
ICLs trigger PIDDosome signaling
While searching for DNA lesions other than DSBs that trigger PIDDosome assembly in combination with Chk1i, we found that ICLs, as induced by mitomycin C (MMC), bendamustine or cisplatin, are potent inducers of PIDDosome signaling (Figures 1 and S1). This was evidenced by PIDD1- and RAIDD-dependent but caspase-3-independent induction of C2 cleavage into its mature 19 kDa product in response to ICL+Chk1i (Figures 1A and S1A–F). Effective PIDDosome formation was verified in endogenous PIDDosome assembly assays (see Figure 3 below) and by C2 bimolecular fluorescence complementation (C2 BiFC) in control, PIDD1–/– and RAIDD–/– cells stably expressing a bipartite reporter of C2 dimerization (C2 Pro-BiFC, Figure 1B–C; see Methods for details) (Ando et al., 2017; Bouchier-Hayes et al., 2009). PIDDosome formation led to a marked decline in cell survival at 5 days post-treatment (Figure 1D).
Figure 1. ICLs trigger PIDDosome signaling.
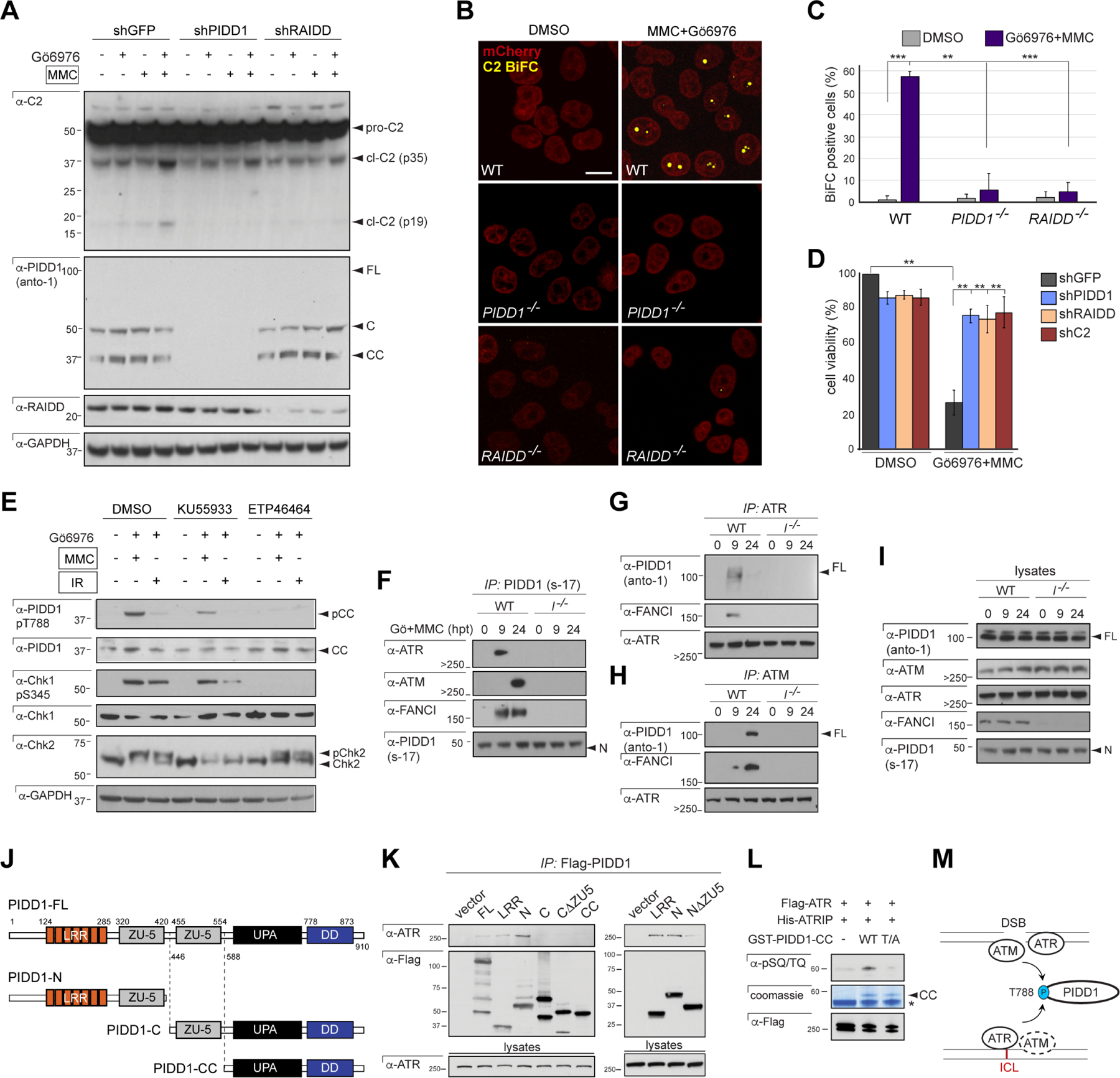
(A) HeLa cells stably expressing the indicated shRNAs, treated with or without Gö6976 and MMC (1 μM each) and harvested 24hrs post-MMC were analyzed by western blot. pro-C2, procaspase-2; p35, intermediate cleavage fragment; p19, mature product. FL, full-length; C, large C-terminal autocleavage product; CC, short, distal, C-terminal autocleavage product (see J).
(B) HeLa cells of indicated genotypes stably expressing C2 Pro-BiFC and mCherry were treated with Gö6976 and MMC (5μM each) plus VD-OPH (20 μM) and fixed at 24 hours. Confocal images (5 μm sections) show C2 BiFC (yellow) and mCherry (red) expression. Scale bar, 20 μm.
(C) Quantification of images as in (B) over 3 independent experiments, with data expressed as means +/− SD. **p < 0.01, two-tailed Student’s t-test.
(D) HeLa cells stably expressing the indicated shRNAs were treated with or without Gö6976 and MMC (1 μM each). Cells were stained with the vital dye alamarBlue 72 hr post-treatment. Data are means +/− SD of 3 independent experiments. **p < 0.01, two-tailed Student’s t-test.
(E) HeLa cells initially treated with ATM inhibitor (KU55933) and ATR inhibitor (ETP46464), 10 μM each, were treated with or without Gö6976 and MMC (1 μM each) or IR (10 Gy) after 1 hr. Cells were harvested 24hrs post-treatment and analyzed by western blot.
(F-I) HCT116 cells of indicated FANCI genotypes and stably expressing shp53 were treated with Gö6976 and MMC (1 μM each) and harvested at 0 hrs, 9 hrs and 24 hrs post-MMC. Lysates (I) were immunoprecipitated with s-17 antibody to the PIDD1 N-terminus (F) or monoclonal anti-ATR (G) and anti-ATM (H) antibodies and analyzed by western blot.
(J) Schematic diagram of full-length (FL) PIDD1 (910 a.a.) and autoproteolytic cleavage products PIDD1-N, PIDD1-C and PIDD1-CC. LRR, leucine-rich repeats; ZU-5, ZO-1 and Unc5-like domain; UPA, uncharacterized protein domain in UNC5, PIDD and Ankyrin, implicated in PIDD1 oligomerization (Janssens and Tinel, 2012); DD, death domain.
(K) HeLa cells transfected with the indicated Flag-tagged PIDD deletion constructs were harvested 24hrs post-transfection. Flag IPs were analyzed by western blot.
(L) Recombinant GST-PIDD1-CC proteins (WT and T788A phosphomutant, T/A) were incubated with Flag-ATR and His-ATRIP for an in vitro kinase assay. Reactions were analyzed by Coomassie staining and western blot. α-pSQ/TQ, polyclonal antibody to phosphorylated SQ or TQ motifs, such as T788Q789 in PIDD1 (Ando et al., 2012). *, non-specific band.
(M) Schematic of PIDD1 phosphorylation by ATR and ATM in response to ICLs and DSBs. ATM plays a delayed role in the ICL response (dotted).
Figure 3. FANCI acts directly to enable ATR/ATM-mediated PIDD1 phosphorylation and PIDDosome formation.
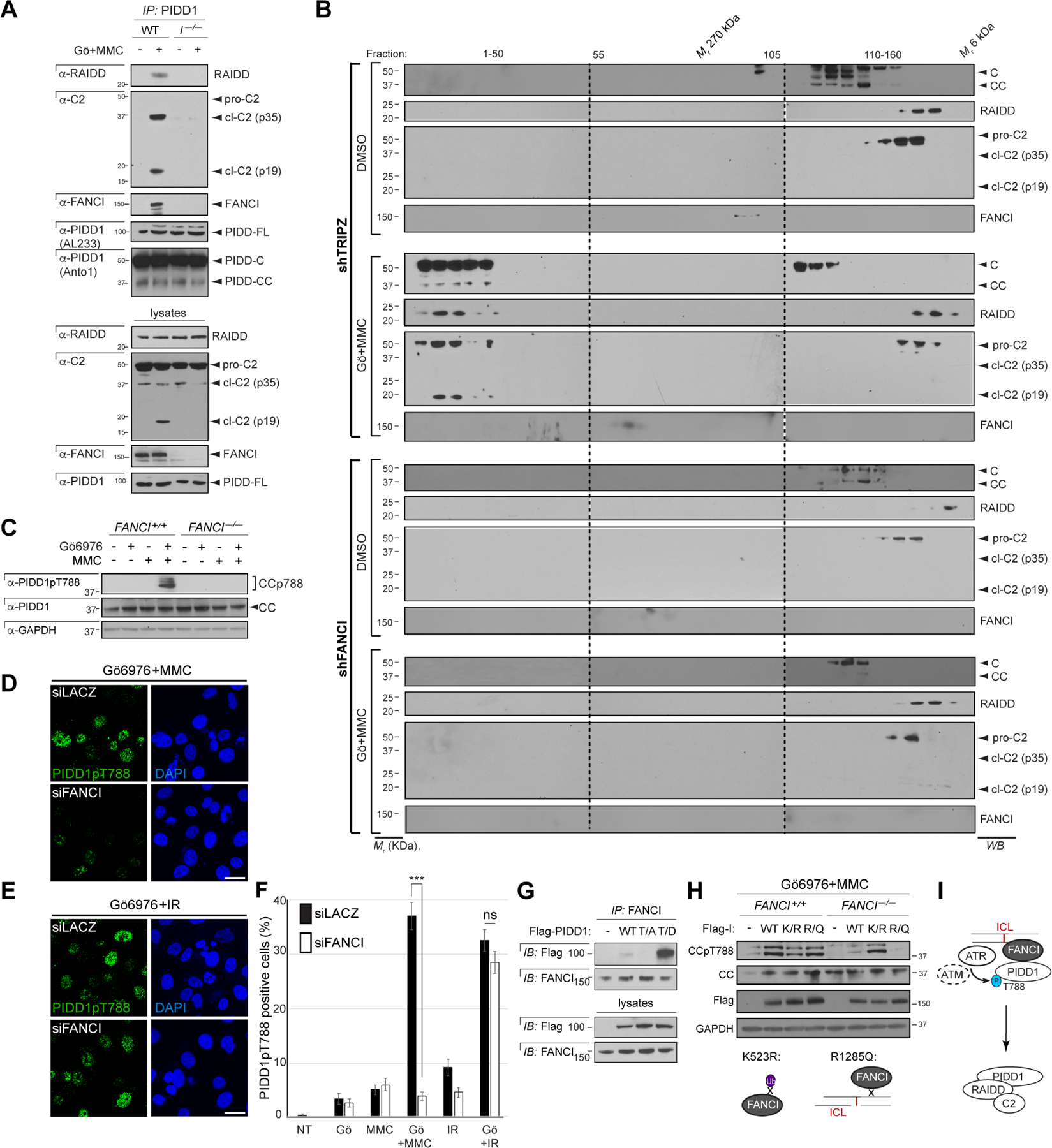
(A) HCT116 cells of indicated FANCI genotypes and stably expressing shp53 were treated with Gö6976 and MMC (1 μM each), harvested 24 hrs post-MMC and monoclonal PIDD1 IPs were analyzed by western blot. I–/–, FANCI–/–.
(B) HeLa cells stably expressing the indicated shRNAs were treated with or without Gö6976 and MMC (1 μM each), lysed 24 hrs after treatment and run on a S400 HiPrep 16/60 Sephacryl column (1 ml/min). An aliquot of each fraction was concentrated and analyzed by western blot.
(C) HCT116 cells of indicated FANCI genotypes and stably expressing shp53 were treated with Gö6976 and MMC (1 μM each), harvested at 24 hrs post-MMC and analyzed by western blot.
(P) (D) PC3 cells grown on coverslips and transfected with indicated siRNAs for 48hrs were treated with Gö6976 and MMC (1 μM each). Cells were fixed after 10hrs of treatment, stained using indicated antibodies and visualized by confocal microscopy (0.8 μm sections). Scale bar, 40 μm.
(E) As in (D) but substituting IR (10 Gy) for MMC. Scale bar, 40 μm.
(F) Quantification of images as in (D-E) over three independent experiments. Data shown as means +/− SEM. ***p<0.001 and ns, non-significant, two-tailed Student’s t test.
(G) HeLa cells transfected with the indicated C-terminally Flag-tagged PIDD1 constructs were lysed at 24 hpt and FANCI IPs were analyzed by western blot. WT, wild-type; T/A, T788A; T/D, T788D.
(H) HCT116 cells of indicated FANCI genotypes and stably expressing shp53 were transfected with indicated FANCI cDNAs and treated with Gö6976 and MMC (1 mM each) after 24 hrs. Cells were harvested 24hrs post-treatment and analyzed by western blot. Bottom, schematics of K523R (mono-Ub-deficient) and R1285Q (DNA binding-deficient).
(I) Model for FANCI-mediated PIDDosome signaling (see text).
ICLs, like DSBs, also strongly induced PIDD1 phosphorylation on T788 (Figure 1E), which marks the initiating step in PIDDosome assembly (Ando et al., 2012). While ATM was necessary for this event, ATR was also clearly required (Figure 1E), which is not the case after IR (Ando et al., 2012). Endogenous co-immunoprecipitation (co-IP) showed that ATR bound PIDD1 earlier than ATM (Figure 1F–I). These differential timings coincided with respectively early and late requirements for ATR and ATM in ICL-induced PIDD1 T788 phosphorylation (Figure S1G–H‘). These dynamics were consistent with the timings of ATR and ATM activation in the ICL response (Lopez-Martinez et al., 2016).
Transiently transfected PIDD1 deletion constructs mapped the ATR/PIDD1 interface to the PIDD1 N-terminus (PIDD1-N fragment), with the leucine-rich repeat (LRR) and ZO-1 and Unc5-like (ZU-5) domains both required for the interaction (Figure 1K). The LRR domain was sufficient for ATR binding (Figure 1 K), similar to that previously reported for ATM (Ando et al., 2012). ATR, like ATM (Ando et al., 2012), phosphorylated a recombinant GST-PIDD1-CC fragment on T788 in vitro when incubated with its obligate interacting partner ATRIP (ATR-interacting protein) (Figure 1L) (Liu et al., 2011). Finally, the nuclear phosphochaperone NPM1, which provides a scaffold for PIDDosome formation in response to DSBs (Ando et al., 2017), was also required for PIDDosome signaling in response to ICLs (Figure S1I). Thus, ICLs trigger PIDDosome signaling via a mechanism related to, but partially distinct from, that in response to DSBs (Figure 1M).
FANCI interacts with PIDD1 in response to ICLs
ICLs are detected and resolved through the Fanconi Anemia (FA) pathway of DNA repair, whose constituents have been largely identified as genes mutated in FA patients (Lopez-Martinez et al., 2016; Niraj et al., 2019) (see Figure 4A below). Interestingly, of the ~30 FA and FA-associated proteins thus far identified, one, FANCI (FA Complementation Group I), was identified by Tschopp and colleagues as a candidate PIDD1-binding protein (Logette et al., 2011) (Figure 2A). FANCI is one of the FA proteins earliest detected at ICLs and is absolutely essential for ICL repair (Castella et al., 2015; Dorsman et al., 2007; Lopez-Martinez et al., 2016; Niraj et al., 2019; Sims et al., 2007; Smogorzewska et al., 2007).
Figure 4. ICL-induced PIDDosome signaling does not strictly require FA pathway function.
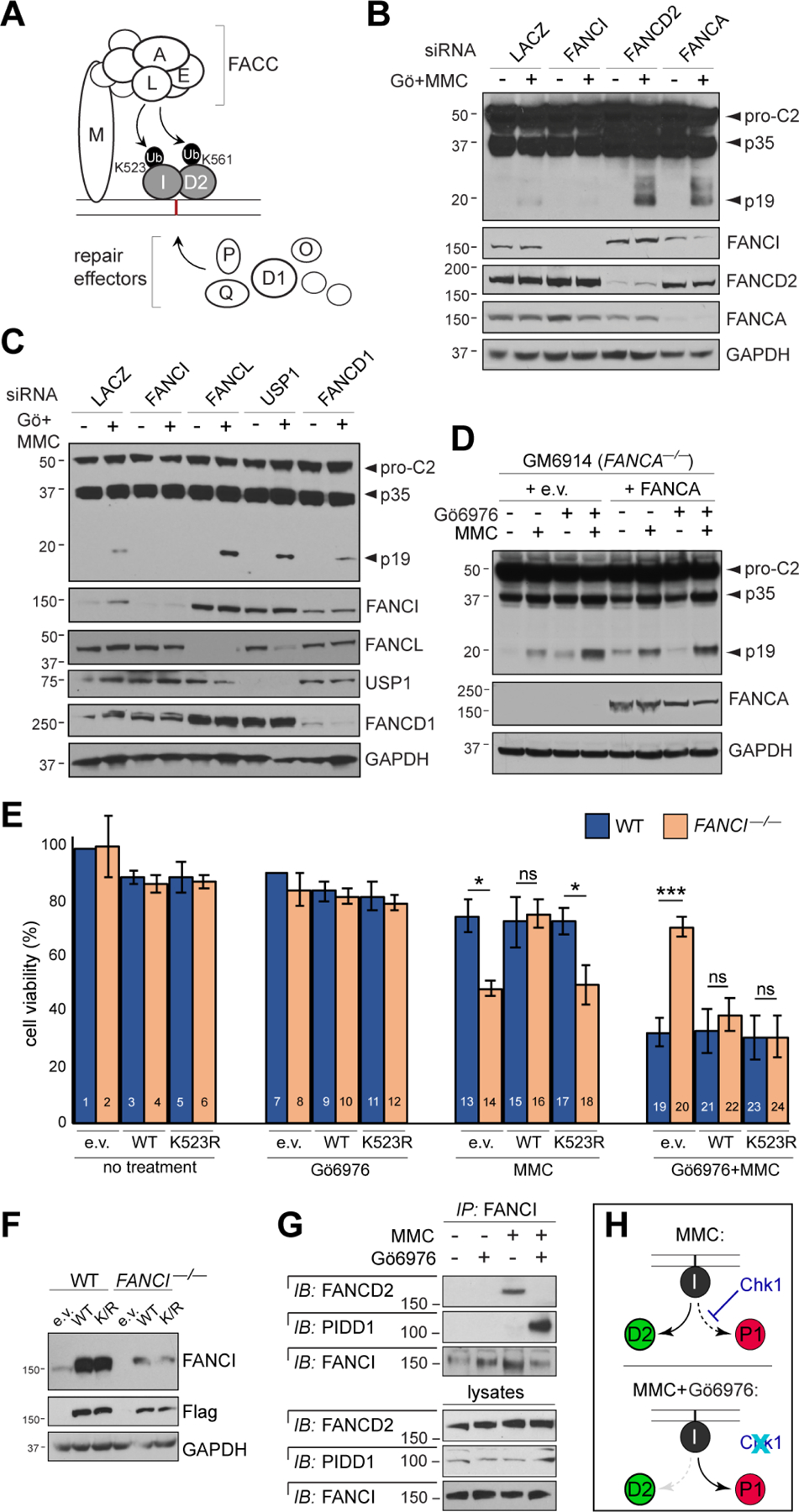
(A) Simplified diagram of the FA pathway. FACC, Fanconi core complex.
(B-C) HeLa cells transfected with indicated siRNAs were treated with or without Gö6976 and MMC (1 μM each), harvested 24hrs post-treatment and analyzed by western blot.
(D) Patient-derived GM6914 FANCA–/– fibroblasts reconstituted with empty vector or WT FANCA were treated with Gö6976 and MMC (1 μM each), harvested 24 hrs post-MMC and analyzed by western blot.
(E) HCT116 cells of indicated FANCI genotypes and stably expressing shp53 were reconstituted with empty vector (e.v) or indicated Flag-FANCI constructs and treated with or without Gö6976 (1 μM) and MMC (0.1 μM). AlamarBlue was added 72 hrs post-treatment. Data are means +/− SD of 3 independent experiments. *p < 0.05; **p < 0.01 ***p < 0.001, two-tailed Student’s t-test.
(F) Western blot from the experiment in (E) showing equal, near-physiologic expression of Flag-FANCI constructs.
(G) HeLa cells synchronized as in Table S1A were treated with or without Gö6976 and MMC (1 μM each) and harvested after 9hrs. FANCI IPs were analyzed by western blot.
(H) Schematic highlighting the effects of Chk1 (top) and Chk1i (bottom) on binding partner choice by FANCI in response to ICLs.
Figure 2. FANCI interacts with PIDD1 and is essential for ICL-induced PIDDosome signaling.
(A) Venn diagram featuring FANCI as the sole ICL-repair protein identified in a mass spectrometry analysis of Flag-PIDD1 immunoprecipitates (Logette et al., 2011).
(B) HeLa cells treated with or without Gö6976 and MMC (1 μM each) were harvested after 24 hrs and monoclonal PIDD1 IPs were analyzed by western blot. *non-specific band.
(C) HeLa cells treated with or without Gö6976 and MMC (1 μM each) were harvested after 24hrs and monoclonal FANCI IPs were analyzed by western blot.
(D) HeLa cells grown on coverslips and synchronized as in Table S1A were treated with or without Gö6976 and MMC (1 μM each), fixed 9 hrs post-MMC, stained with the indicated antibodies and visualized by confocal microscopy (0.8 μm sections are shown). Arrowheads mark colocalization areas. Scale bar, 20 μm.
(E) Schematic diagram of FANCI (1328 a.a) highlighting the FANCD2 binding domain on its C-terminal tail (aa 1001–1328). ARM, Armadillo domain, α-α superhelix folds involved in protein-protein interactions. Leu, leucine-rich region. EDGE, EDGE motif.
(F) Schematic diagram of PIDD1-FL (910 a.a.)
(G) HeLa cells transfected with indicated Flag-FANCI constructs were harvested after 24 hrs and Flag IPs were analyzed by western blot.
(H) HeLa cells transfected with the indicated Flag-PIDD1 constructs were harvested after 24 hrs and FANCI IPs were analyzed by western blot.
(I) HCT116 cells of indicated FANCI genotypes and stably expressing shp53 were treated with Gö6976 and MMC (1 μM each) and harvested 24 hrs post-MMC. Lysates were analyzed by western blot.
(J) Patient-derived FA-I 010191 fibroblasts reconstituted with FANCI or empty vector (e.v.) were treated and analyzed as in (I).
(K) HeLa cells stably expressing the indicated shRNAs were treated with or without Gö6976 (1 μM), MMC (1 μM) or IR (10 Gy) and analyzed as in (I).
(L) HeLa cells stably expressing the indicated shRNAs were treated with Gö6976 (0.1 μM) and MMC (0.05 μM). 14 days post-treatment, cells were fixed and stained with crystal violet.
(M) Colonies from (M) of greater than 50 cells were counted. Data from 3 independent experiments expressed as means +/− SEM. *p < 0.05; **p < 0.01 ***p < 0.001, two-tailed Student’s t-test.
(N) p53–/– zebrafish embryos of indicated fanci genotypes were treated with or without Gö6976 (1 μM) at 17 hrs post fertilization (hpf) and MMC (30 μM) 1 hr later and stained with the cell death marker acridine orange (AO) at 24 hpf. Boxed spinal-cord areas are magnified. Scale bar, 0.2 mm. As in human cells (L), zebrafish FANCI is required for death induction in embryos treated with MMC+Chk1i.
(O) Quantification of spinal cord AO stains as shown in (N). Data were collected from 2 independent experiments with at least 5 embryos scored per condition in each and reported as means +/− SEM. **p < 0.01, two-tailed Student’s t-test.
In reciprocal co-IP and immunofluorescence (IF) assays, we found that FANCI and PIDD1 indeed physically associate at the endogenous level, specifically in cells instructed to assemble the PIDDosome by means of MMC+Chk1i (Figures 2B–D and S2A–C). In the IF assay, 43% of cells (n=40) showed colocalization between FANCI and PIDD1pT788, with 54.8% of PIDD1pT788 foci (n=110) co-occupied by FANCI (Figure 2D and Figure S2B–C). PIDD1 did not otherwise associate with the FANCI paralog FANCD2, while FANCI did not detectably associate with the PIDD1-related protein FADD (Figures 2B–C and S2A). The FANCI-PIDD1 interaction occurred specifically in response to ICLs and not DSBs (Figure S2A).
Deletion constructs (Logette et al., 2011; Yuan et al., 2009) mapped the PIDD1-FANCI interface to the FANCI C-terminal tail [also known to bind FANCD2 (Wang et al., 2020; Yuan et al., 2009); Figure 2E,G] and the PIDD1 distal ZU-5 domain and UPA domain [implicated in PIDD1 oligomerization (Janssens and Tinel, 2012); Figure 2F–H]. FANCI binding to these distal domains was consistent with its ability to pull down PIDD1-FL as well as both C-terminal autoproteolytic fragments, PIDD1-C and PIDD1-CC (Figure 1J), in the endogenous setting (Figure 2C). Collectively, these data confirmed a physical association between a DNA repair protein, FANCI, and an apoptotic effector, PIDD1, specifically in ICL-bearing cells fated to die via PIDDosome signaling.
FANCI is essential for PIDDosome formation in response to ICLs
We next investigated the significance of the FANCI-PIDD1 interaction. Genetic ablation of FANCI via siRNA, shRNA, or CRISPR/Cas9 targeting in human cell lines and zebrafish embryos, or in patient-derived FA-I-010191 fibroblasts carrying the FANCI R1299X mutation (Colnaghi et al., 2011), all demonstrated an essential role for FANCI in ICL-induced PIDDosome activation. This was demonstrated in cells/embryos treated with MMC+Chk1i using: (i) the C2 cleavage assay (Figures 2I–K, 3A and S2D–F, note the block or reduction of p19 maturation in FANCI-deficient cells); (ii) the C2 BiFC fluorescent reporter assay (Figure S2G–H, note the absence of C2 dimerization in FANCI-depleted cells); (iii) apoptosis, cell viability and/or clonogenic assays in vitro (Figures 2L–M and S2I–M) and in vivo (Figure 2N–O); and importantly, (iv), endogenous PIDDosome assembly assays by means of monoclonal PIDD1 pulldowns (Figure 3A, note the failure of RAIDD and C2 to associate with PIDD1 in FANCI–/– cells) and size exclusion chromatography (Figure 3B, note the failure of all three platform subunits to mobilize to higher molecular weight fractions in shFANCI cells). The requirement for FANCI was yet again specific to ICLs, with FANCI playing no role in DSB-induced PIDDosome signaling (Figures 2K and S2A). These experiments identified FANCI as a novel regulator of PIDDosome signaling, exerting an essential and direct role in platform formation in response to ICLs.
FANCI acts to enable PIDD1 phosphorylation by ATR and ATM
Having uncovered an essential role for FANCI in ICL-induced PIDDosome signaling, we investigated the underlying mechanism. Because FANCI was required for the PIDD1-RAIDD interaction itself (Figure 3A) and colocalized with PIDD1pT788 foci (Figures 2D and S2B,C), we asked whether FANCI exerted its role in the initiating step in PIDDosome formation, i.e., ICL-induced PIDD1 phosphorylation by ATR/ATM. Indeed, FANCI was essential for this event (Figures 3C,D,F and S3A–B) and, again, was so specifically in response to ICLs (Figures 3D,E,F and S3B). Mechanistically, FANCI acted to enable PIDD1 recruitment to ATR and ATM in the early (9 hr) and late (24 hr) stages of the ICL response, respectively (Figure 1F–I). FANCI was essential in this respect, as deletion of FANCI completely abolished PIDD1 recruitment to, and phosphorylation by, ATR and ATM (Figures 1F–I and 3C). FANCI was not otherwise necessary for ICL-induced, ATR-mediated phosphorylation of Chk1 on S345 or ATM/ATR-mediated phosphorylation events in general, as detected with an anti-phospho-SQ/TQ antibody (Figure S3C).
Pulldown assays with transiently transfected phosphomutant (PIDD1T788A) and phosphomimetic (PIDD1T788D) PIDD1 variants (Ando et al., 2012) indicated that the FANCI-PIDD1 interaction only strengthens with phosphorylation (Figure 3G). Furthermore, whereas wild-type (WT) FANCI restored PIDD1 phosphorylation in FANCI–/– cells, a FANCI variant deprived of DNA binding, FANCIR1285Q (Yuan et al., 2009), failed to do so (Figure 3H). In fact, this mutant, as well as a FANCI variant (FANCIK294E/K339E; Castella et al., 2015) which blocks DNA binding without affecting the PIDD1 binding domain, abolished the FANCI-PIDD1 interaction altogether (Figure S3D–E). Overall, the data suggested a model for FANCI-mediated PIDDosome control whereby ICL-bound FANCI mobilizes PIDD1 to ATR and ATM molecules located in the vicinity, resulting in the phosphorylation of PIDD1 and PIDDosome formation (Figure 3I).
FA repair factors other than FANCI are not necessary for ICL-induced PIDDosome signaling
In the FA pathway, FANCI promotes DNA repair in conjunction with its paralog FANCD2, with which it heterodimerizes to form the FANCI/FANCD2 (I/D2) complex (Figure 4A). Recent studies indicate that I/D2 arranges in a DNA clamp analogous to a PCNA ring (Alcon et al., 2020; Tan et al., 2020; Wang et al., 2020). When monoubiquitylated by the Fanconi Core Complex (FACC), I/D2 recruits downstream effectors involved in ICL unhooking and subsequent strand repair via combinations of HR, translesion synthesis (TLS) and NER (Figure 4A) (Lopez-Martinez et al., 2016; Niraj et al., 2019).
Surprisingly, we found that FANCD2 was not necessary for ICL-induced PIDDosome signaling (Figures 4B and S4A), and neither were the FACC (FANCA, FANCE, FANCL, FANCM) or critical repair effectors downstream of I/D2 (FANCD1/BRCA2, FANCO/RAD51C, FANCP/SLX4) (Figures 4B–D and S4A). Further consistent with PIDDosome signaling representing a new function of FANCI, a monoubiquitination-deficient FANCI variant, FANCIK523R (Smogorzewska et al., 2007), retained full PIDDosome-inducing activity while otherwise devoid of repair functionality. This was evidenced by the ability of FANCIK523R to restore PIDD1 phosphorylation on T788 (Figure 3H) and cell death induction in FANCI–/– cells treated with MMC+Chk1i, while simultaneously unable to rescue the cells’ hypersensitivity to MMC alone (Figure 4E,F). Taken together, these results indicated that FA signaling, including through I/D2 itself, is not strictly necessary for ICL-induced PIDDosome signaling, in contrast to FANCI monomers.
If anything, FANCD2 acted to suppress PIDDosome activation, as evidenced by enhanced PIDD1 T788 phosphorylation and C2 cleavage in FANCD2-depleted cells (Figure 4B). Such gain-of PIDD1 phosphorylation mirrored that seen in FANCI–/– cells reconstituted with FANCIK523R (Figure 3H), a mutation which disrupts proper I/D2 signaling (Chen et al., 2015; Smogorzewska et al., 2007). Furthermore, whereas FANCI bound FANCD2 in response to MMC alone, as expected, the interaction was notably lost in cells co-treated with Chk1i, in which FANCI associated with PIDD1 instead (Figures 4G and S4B).
Exclusive recruitments of FANCD2 and PIDD1 by FANCI enable a binary fate switch
The above-observed, binary-like nature of binding partner selection by FANCI (Figure 4H); its recruitment of FANCD2 and PIDD1 via the same domain (Figures 2G and 5A); and FANCD2 seemingly acting to suppress PIDDosome signaling (Figure 4B) altogether suggested that FANCD2 and PIDD1 might compete for access to FANCI. Indeed, increasing levels of FANCD2 interfered with the FANCI-PIDD1 interaction (Figure 5B), whereas depletion of FANCD2 enhanced the interaction (Figure 5C, right). Reciprocally, excess and depletion of PIDD1 respectively acted to displace and stimulate the FANCI-FANCD2 interaction (Figures 5B, and 5C, left). Notably, removal of either binding partner was sufficient to force recruitment of the other in its place: Depletion of FANCD2 enabled PIDD1 recruitment after MMC alone (Figure 5C, right), while depletion of PIDD1 allowed for I/D2 formation after MMC+Chk1i (Figure 5C, left). These ectopic recruitments provided first evidence that binding partner choice at the FANCI C-terminus is actively regulated, with FANCI appearing to behave as a binary fate switch which engages its pro-repair and pro-apoptotic partner in a mutually exclusive manner.
Figure 5. Exclusive recruitments of FANCD2 and PIDD1 by FANCI enable a binary fate switch in response to ICLs.
(A) Schematic diagram of FANCI highlighting the overlapping FANCD2- and PIDD1- binding domains (D2BD and P1BD, respectively; see Figure 2G–H).
(B) HeLa cells transfected with fixed or increasing amounts of expression vectors encoding HA-FANCI, Flag-PIDD1-FL and GFP-FANCD2, as indicated. Lysates were harvested 24 hrs post transfection and HA IPs were analyzed by western blot.
(C) HeLa cells stably expressing the indicated shRNAs or transfected with indicated siRNAs were synchronized as in Table S1B, treated with or without Gö6976 and MMC (1 μM each) and harvested at 9 hrs post-treatment. FANCI IPs were analyzed by western blot
(D) HeLa cells synchronized as in Table S1A were treated with or without Gö6976 and MMC (1μM each), harvested at indicated time points post-treatment, stained with phospho-Histone H3 antibody and analyzed by flow cytometry.
(E) FANCI IPs from HeLa cells as in (D) were analyzed by western blot. hpt, hours post-treatment; int, interphase; M, mitosis.
(F) HeLa cells transfected with indicated siRNAs and treated with or without MMC (1 μM) were harvested at 24 hrs. FANCI IPs were analyzed by western blot
(G) HeLa cells synchronized as in Table S1A were treated with or without RO-3306 (10 μM) or nocodazole (200 ng/ml) 1 hr after release. Cells were then treated with Gö6976 (1 μM) and MMC (1 μM) and harvested 9 hrs later. FANCI IPs were analyzed by western blot.
(H) Schematic summarizing the results in panels D-G.
Should FANCI mount a biochemical switch for repair vs. apoptosis in response to ICLs, how would fate choice be decided? The ability of Chk1 to block switching from FANCD2 to PIDD1 (Figure 4H) provided an entry point into this question. Chk1 exerts a number of functions in genome maintenance of which the DNA damage-induced G2/M checkpoint is conserved from yeast to humans (Dai and Grant, 2010; Kastan and Bartek, 2004). An involvement of the G2/M checkpoint seemed likely in light of recent studies tying PIDDosome formation to mitotic progression in multiple settings (Dawar et al., 2017; Fava et al., 2017; Sladky et al., 2017; Sladky et al., 2020; Thompson et al., 2015).
As expected, Chk1i abrogated an MMC-induced G2/M checkpoint, thus forcing mitotic entry of damaged cells (Figure 5D). PIDD1 recruitment to FANCI and FANCI-mediated PIDDosome activation both coincided with this event (Figures 5E and S5A). Depletion of the G2/M checkpoint effector Wee1 (O’Connell et al., 1997) was also sufficient for FANCI to switch from FANCD2 to PIDD1 and trigger PIDDosome signaling (Figures 5F and S5B–C). Conversely, denying mitotic entry or progression by means of RO-3306 (a CDK inhibitor) and nocodazole, respectively, blocked PIDD1 recruitment to FANCI (Figures 5G and S5D). Removal of caspase-3 had no effect, ruling out that the observed switch to PIDD1 merely resulted from apoptosis induction (Figure S5E). Taken together, these data indicated that Chk1 controls FANCI switch activity via its G2/M checkpoint function, and that FANCI switches from FANCD2 to PIDD1 should ICL-bearing cells enter mitosis (Figure 5H).
FANCI switches from FANCD2 to PIDD1 in response to repair failure
It has long been observed that DNA repair machineries are shut off during mitosis (Hustedt and Durocher, 2016), presumably to prevent telomere fusions (Orthwein et al., 2014). This applies to FA repair, which is actively suppressed during mitosis via several mechanisms (Kee et al., 2009). We thus hypothesized that FANCI switching from FANCD2 to PIDD1 might reflect a more general mechanism via which damaged cells transduce defective DNA repair into commitment to cell death.
If this were the case, then mimicking repair failure by introducing excess lesions might be sufficient for FANCI to switch from FANCD2 to PIDD1 in cells with intact Chk1. Indeed, whereas FANCI interacted with FANCD2 at lower doses of MMC (1–10 μM), a greater dose (20 μM) led to PIDD1 recruitment instead, followed by PIDDosome activation (Figures 6A–C, and 7A below).
Figure 6. FANCI mounts a bidirectional fate switch that transduces failure of DNA repair into apoptosis, and vice versa.
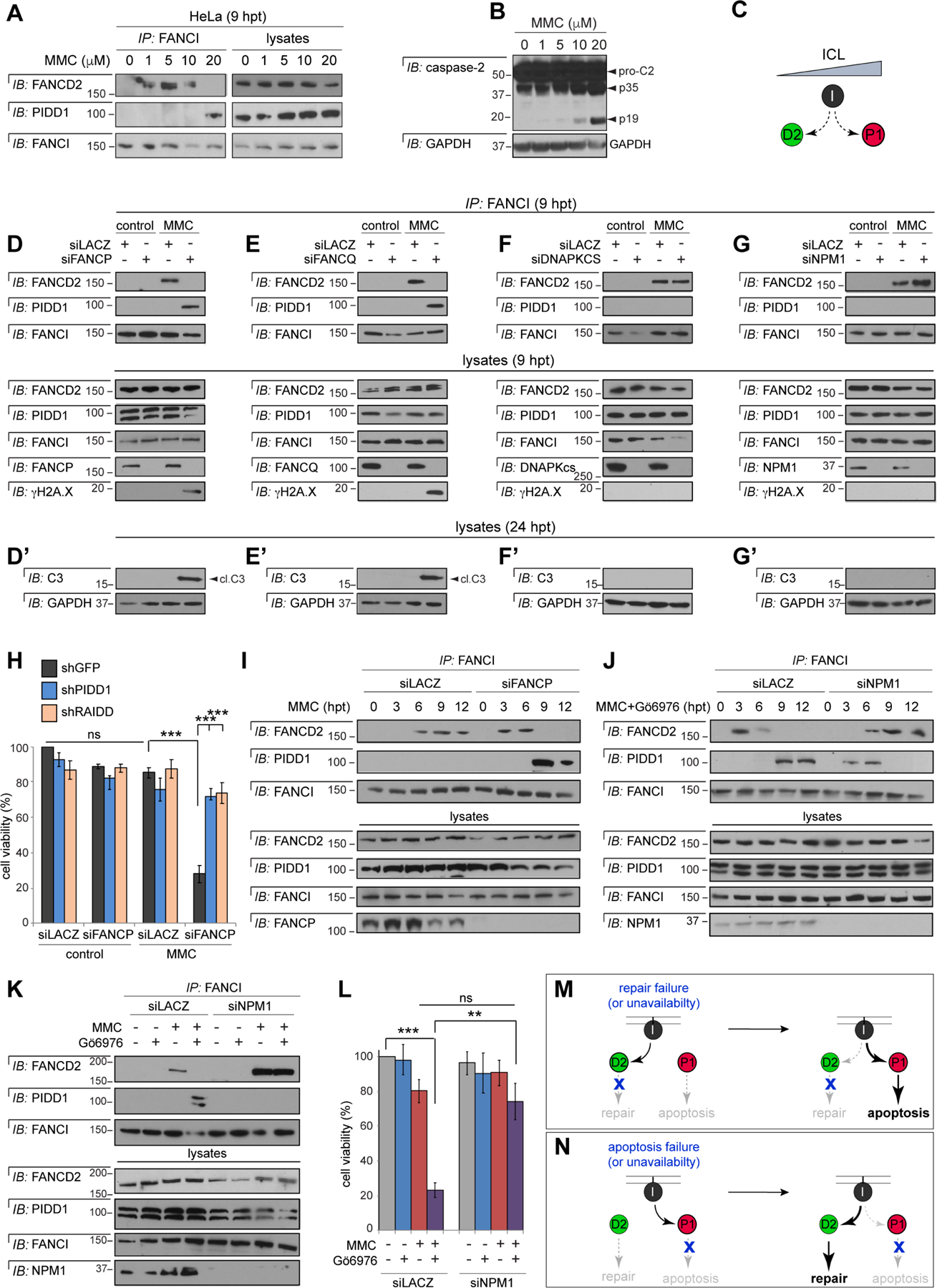
(A) HeLa cells treated with indicated doses of MMC were harvested at 9 hrs. FANCI IPs were analyzed by western blot.
(B) HeLa cells treated with indicated doses of MMC were harvested at 24 hrs and analyzed by western blot.
(C) Model derived from the results in panels A-B: FANCI switches from FANCD2 to PIDD1 with increasing ICL levels.
(D-G) HeLa cells synchronized as in Table S1B were transfected with indicated siRNAs, treated with or without MMC (1 μM) and harvested 9 hrs post-MMC. FANCI IPs were analyzed by western blot.
(D’ – G’) As above but harvested 24 hrs post-MMC and whole-cell lysates analyzed by western blot. cl.C3, cleaved caspase-3.
(H) HeLa cells expressing the indicated shRNAs were transfected with indicated siRNAs, treated with or without MMC (0.1 μM) and stained with alamarBlue 72 hrs post-treatment. Data are means +/− SD of 3 independent experiments. n.s: non-significant, ***p < 0.001, two-tailed Student’s t-test.
(I-J) HeLa cells synchronized as in Table S1B were transfected with indicated siRNAs and treated with MMC (1 μM) (I) or Gö6976 and MMC (1 μM each) (J) and harvested at indicated time points post-MMC. FANCI IPs were analyzed by western blot.
(K) HeLa cells synchronized as in Table S1B were transfected with indicated siRNAs, treated with or without Gö6976 and MMC (1 μM each) and harvested 9 hrs post-MMC. FANCI IPs were analyzed by western blot.
(L) HeLa cells transfected with indicated siRNAs were treated with or without Gö6976 (1 μM) and MMC (0.1 μM). Alamarblue was added 72 hrs post-treatment. Data are means +/− SEM of 3 independent experiments. **p < 0.01 ***p < 0.001, two-tailed Student’s t-test.
(M-N) Schematic diagrams summarizing the results in panels D-I (M) and J-L (N).
Figure 7. Monoubiquitination status impacts binding partner selection by FANCI.
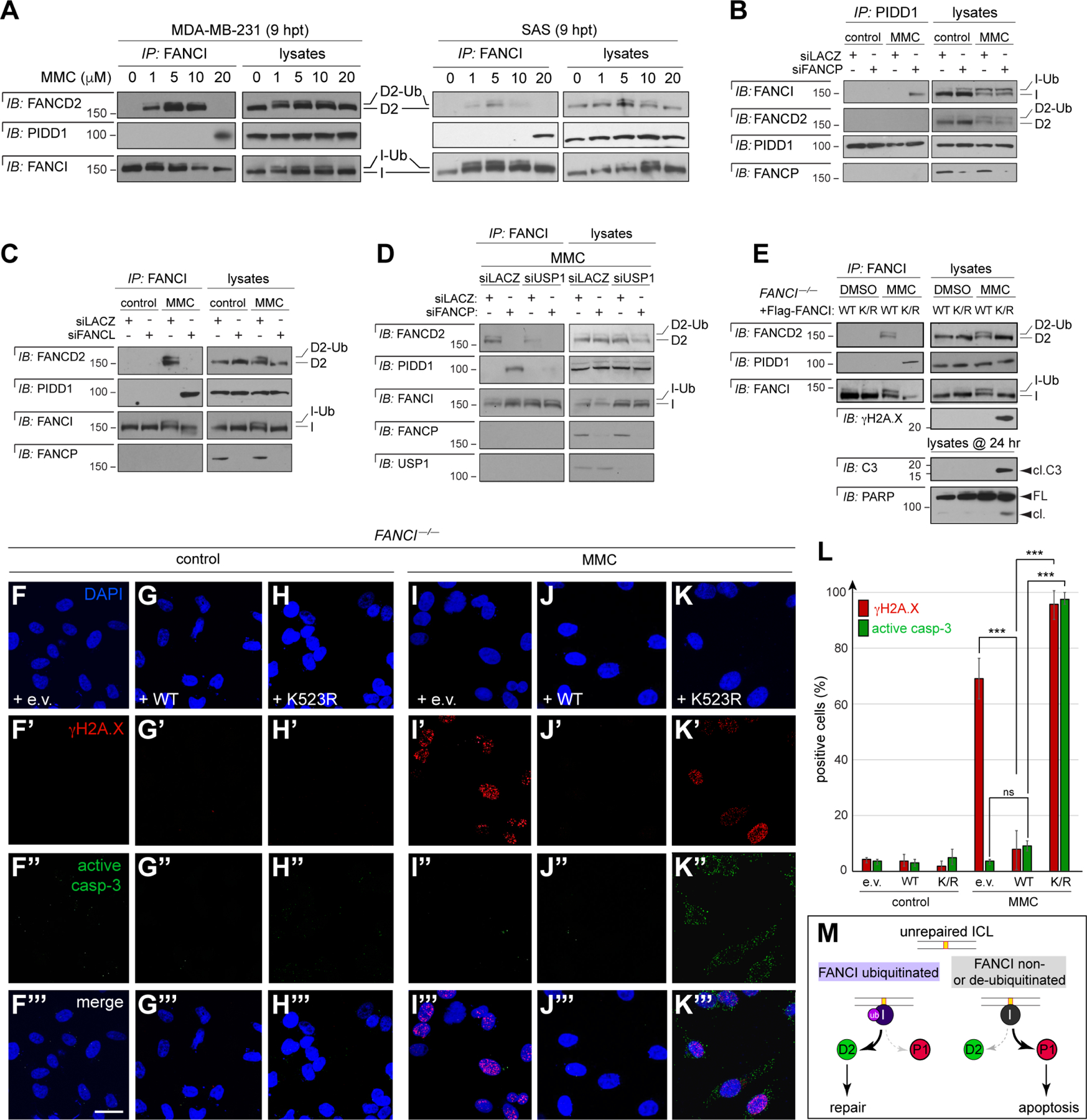
(A) MDA-MB-231 and SAS cells treated with indicated doses of MMC and harvested at 9 hrs post-treatment. FANCI IPs were analyzed by western blot. Ub, monoubiquitinated form.
(B-D) HeLa cells synchronized as in Table S1B were transfected with indicated siRNAs and treated with MMC (1 μM), harvested at 7.5 hrs post-treatment, immunoprecipitated with monoclonal PIDD1 (B-C) and FANCI (D) antibodies and analyzed by western blot.
(E) FANCI–/– HCT116 cells transfected with sip53 were synchronized and reconstituted with indicated Flag-FANCI cDNAs as in Table S1C, treated with MMC (1 μM) and harvested 9 hrs later. FANCI IPs were analyzed by western blot. Cleaved C3 (cl.C3) and PARP (cl.) were assessed in 24 hr lysates.
(F-K’’’) FANCI–/– HCT116 cells stably expressing shp53 were grown on cover slips, synchronized, reconstituted with indicated Flag-FANCI cDNAs, treated with or without MMC (1 μM) and harvested 19 hrs post-MMC. Cells were stained with DAPI (blue, F-K), anti-γH2A.X (red, F’-K’) and anti-active caspase-3 (green, F’’-K’’) and imaged by confocal microscopy (0.6 μm sections). Scale bar, 50 μm.
(L) Quantification of γH2A.X and/or active caspase-3 positive cells, as shown in (F-K’’’), over three independent experiments. Data expressed as means +/−SD, ***p < 0.001, two-tailed Student’s t-test.
(M) Model for the regulation of FANCI switch function by K523 monoubiquitination.
To directly test whether FANCI switches from FANCD2 to PIDD1 as a response to repair failure, we disrupted ICL repair downstream of intact FANCI and FANCD2 in cells with repairable levels of ICLs (1 μM MMC). We targeted the endonucleases FANCP (SLX4) (Kim et al., 2011) and FANCQ (ERCC4/XPF) (Bogliolo et al., 2013) because they are essential for ICL unhooking (Hoogenboom et al., 2019; Klein Douwel et al., 2014), a signaling bottleneck in the FA pathway immediately downstream of I/D2 (Figure 4A) (Lopez-Martinez et al., 2016; Niraj et al., 2019). As expected, silencing of either endonuclease led to repair failure in MMC-treated cells (Figure 6D–E, γH2A.X blots, and Figure S6A–B). Strikingly, in such cells with otherwise repairable levels of ICLs, silencing of FANCP or FANCQ was sufficient for FANCI to switch from commitment to FANCD2/repair to commitment to PIDD1/apoptosis (Figure 6D–E’). Knockdown of the NHEJ repair factor DNAPKcs, which is not involved in ICL repair, had no effect (Figure 6F–F’), while knockdown of the PIDDosome scaffold NPM1 not only failed to induce switching but enhanced FANCD2 binding (Figure 6G–G’).
The switch from FANCD2 to PIDD1 and resulting commitment to apoptosis we observed in FANCP-depleted cells was biologically significant because it resulted in a marked, PIDDosome-mediated decrease in cell survival at 5 days post-treatment (Figure 6H). Crucially, in these cells lacking repair activity downstream of intact FANCI and FANCD2, FANCI first bound FANCD2—until ~6 hours post MMC treatment, coinciding with the would-be expected timing of repair completion at this dose (Nijman et al., 2005)—only after which did it mobilize PIDD1 (Figure 6I). Thus, FANCI indeed functions as a biochemical switch, engaging the PIDDosome only in response to a prior but failed attempt at repair.
The FANCI switch is bidirectional, reverting to repair upon apoptosis failure
If FANCI can trigger apoptosis in response to repair failure, it might also be able to drive repair—or revert to repair—should apoptosis fail. We reasoned this might occur in cells instructed to engage the PIDDosome but in which formation of the complex is blocked downstream of intact FANCI and PIDD1. We took advantage of the fact that ICL-induced PIDDosome signaling, like DSB-induced PIDDosome signaling (Ando et al., 2017), requires the phospho-chaperone NPM1 (Figure S1I). Depleting NPM1 from cells treated with MMC+Chk1i was sufficient for FANCI to switch from PIDD1 to FANCD2 (Figure 6K), coinciding with a reversal from commitment to cell death to that of survival (Figure 6L). Remarkably, in these cells instructed to die but unable to assemble the PIDDosome, FANCI first bound pro-apoptotic PIDD1—until 6–9 hours post MMC+Chk1i, coinciding with the time necessary for FANCI to coordinate platform formation (Figure S5A)— only after which did it revert to pro-repair FANCD2 (Figure 6J). Thus, reminiscent of the FANCI response to repair failure (Figure 6M), FANCI reverted to ICL repair only in response to a prior but failed attempt at apoptosis (Figure 6N). This bidirectionality validates FANCI as a molecular switch that controls a cell’s decision to repair, or succumb to, ICLs.
Monoubiquitination status at K523 impacts FANCI switch function
Mutually exclusive recruitments of FANCD2 and PIDD1 to FANCI provided a biochemical basis for FANCI switch function (Figure 5B–C), yet did not address the mechanisms controlling binding partner choice. Several observations suggested that monoubiquitination of FANCI at K523 might be involved. First, depletions of FACC components such as FANCA or FANCL enhanced FANCI-mediated PIDDosome signaling (Figure 4B–D). Second, FANCIK523R not only restored PIDDosome signaling in FANCI–/– cells (Figure 4E) but in fact enhanced ATR/ATM-mediated PIDD1 phosphorylation (Figure 3H). Third, a cell cycle stage when FANCI is actively deubiquitinated, that of mitosis (Kee et al., 2009), was highly permissive for the FANCI-PIDD1 interaction (Figures 5E–H and S7A). Fourth, we observed a similar correlation between deubiquination and the FANCI-PIDD1 interaction when we ran MMC dose-response experiments, such as initially shown in Figure 6A, on low-percent acrylamide gels. Indeed, cells treated with greater doses of MMC, in which the FANCI-PIDD1 interaction specifically occurred, exhibited markedly reduced levels of monoubiquitinated FANCI (Figure 7A). Thus, K523 status might regulate FANCI switch function, whereby monoubiquitination favors FANCD2 while deubiquitination or aberrant lack thereof enables PIDD1 recruitment.
In this model, PIDD1 would be expected to show greater affinity for non-ubiquitinated versus mono-ubiquitinated FANCI. To test this, we depleted FANCP to trigger the FANCI-PIDD1 interaction via repair failure (as in Figure 6D) all the while leaving FACC-mediated FANCI monoubiquitination intact. In this context where both mono- and non-ubiquitinated forms were present, PIDD1 specifically interacted with the non-ubiquitinated form (Figure 7B).
Further consistent with the model, depletion of the E3 ubiquitin ligase itself, FANCL, stimulated the FANCI-PIDD1 interaction, whereas depletion of the FANCI deubiquitinase, USP1, had the opposite effect (Figures 7C,D). The latter could be revealed only early in the ICL response (7.5 hpt), with USP1 depletion having no effect at later stages (Figure S7C). Indeed, USP1 knockdown delayed but did not abolish FANCI deubiquitination, as previously observed with similar timing (Nijman et al., 2005).
A final prediction was that the FANCI K523R mutation alone might be sufficient for PIDD1 recruitment in place of FANCD2. We reasoned that when transfected into MMC-treated FANCI–/– cells, this mutant, which compromises FA repair (Chen et al., 2015; Smogorzewska et al., 2007), would dictate a situation where unprocessed ICLs are solely sensed by non-ubiquitinable FANCI. Indeed, FANCIK523R was sufficient to force the switch to PIDD1 in this context (Figure 7E) and for cells to switch from commitment to DNA repair to commitment to apoptosis (Figure 7E–K’’’). Specifically, while WT FANCI suppressed γH2A.X immunoreactivity in FANCI–/– cells (I’ vs. J’), FANCIK523R failed to restore repair (I’ vs. K’) and induced caspase-3 activation and PARP cleavage instead (I’’ vs. K’’, and panel E, γH2A.X, C3 and PARP blots). Together, the data indicated that fate choice by FANCI is regulated by ubiquitination at K523. Whereas monoubiquitination favors I/D2-mediated ICL repair, it’s non- or deubiquitination in the face of persistent ICLs—reflective of unavailability or failure of lesion repair—signal PIDD1 recruitment and removal of the damaged cell (Figure 7M, and 5H and 6M).
Discussion
We describe a direct mechanism via which cells can decide between repair or self-removal after DNA damage. At its core, this mechanism relies on the ability of a repair protein, FANCI, to not only physically engage an apoptotic device, but do so in manner exclusive with its repair activity.
The data thus far indicate that damaged cells can exploit the FANCI switch to counter genomic instability resulting from DNA repair failure (Figure 6M). Surprisingly, such a direct mechanism dedicated to the removal repair-failing cells had not been previously described. Repair failure results in the accumulation of DNA damage and, as such, has long been assumed to constitute a prime trigger for p53-mediated DDIA (Ciccia and Elledge, 2010; De Zio et al., 2013; Evers and Jonkers, 2006; Matt and Hofmann, 2016; Roos et al., 2016). Central to this assumption is that loss of Tp53 partially rescues widespread apoptosis in Brca1–/– and Brca2–/– mice (Evers and Jonkers, 2006; Hakem et al., 1997; Ludwig et al., 1997). However, inactivation of the p53 target and G1/S checkpoint effector, p21, also rescued embryonic lethality in BRCA-deficient mice (Evers and Jonkers, 2006). BRCA proteins also have repair-independent functions whose deficiencies might have accounted for apoptosis induction (Evers and Jonkers, 2006). More mechanistic insights were generally needed to tie repair dysfunction to apoptosis induction. Our characterization of FANCI as a DNA repair factor with an added, intrinsic cell-killing activity fills this gap in the context of the ICL response, with potentially additional factors playing similar roles in response to other lesions.
We note that p53, when present, can also ultimately ensure the removal of such repair-failing cells (Chen et al., 2009; Jacquemont et al., 2012) in a manner independent of the FANCI-PIDDosome axis (Figure S2L, boxed in red). The mechanism is unclear but may involve apoptotic mediators such as the caspase-9-activating apoptosome or caspase-8-activating DISC platform, both of which participate in p53-dependent DDIA (Norbury and Zhivotovsky, 2004; Roos and Kaina, 2006), or even non-apoptotic cell death mediators, as previously suggested (Chen et al., 2009). In the absence of p53, however, the FANCI-PIDDosome axis becomes indispensable for preventing the survival of cells with unrepaired ICL (Figure S2L–M, boxed in blue), with potentially important implications for cancer therapeutics (see closing section).
The decision by FANCI to kill a cell should it fail to resolve ICLs, while desirable from the perspective of tumor suppression, is no less a radical choice. Indeed, the selection of PIDD1 by FANCI is tightly regulated, at minimum by FANCI monoubiquitination and, indirectly, by Chk1 and Wee1. This might ensure that FANCI kills a repair-failing cell only in dire situations, such as if it violates the G2/M checkpoint to carry the ICL into mitosis (Figure 5H). In this particular instance, the switch to apoptosis might be justified by a “what’s the alternative?” dilemma, as reversal to FANCD2 or utilization of compensatory repair pathways are not available options during cell division (Hustedt and Durocher, 2016; Kee et al., 2009; Orthwein et al., 2014). Interestingly, FANCI’s role as a Chk1-regulated repair vs. apoptosis switch gives rise to a rare occurrence of a synthetic viable interaction (Hartwell et al., 1997; Kaelin, 2005), as captured by the clonogenic assay shown in Figure 2L–M: reduced FANCI and Chk1 activities are individually lethal in the presence of ICLs while their simultaneous silencing restores viability.
We identified DNA-binding by, and monoubiquitination of, FANCI as first determinants of FANCI switch function. While DNA binding is not necessary for the FANCI-FANCD2 interaction, it is required for the FANCI-PIDD1 interaction. Conversely, FANCI monoubiquitination acts to inhibit the FANCI-PIDD1 interaction. Cryo-EM studies have revealed that FANCI monoubiquitination participates, along with that of FANCD2, in a conformational change in I/D2 that seals it as a clamp around DNA and exposes completely different surfaces of either monomer (Alcon et al., 2020; Li et al., 2020; Wang et al., 2020). This change, together with I/D2 heterodimerization itself (Figures 5B–C), likely prevent access of PIDD1 to FANCI. Because non-ubiquitinated FANCI cannot constitutively bind PIDD1 and only does so after a failed repair attempt, we propose that changes associated with I/D2 dissociation, including deubiquitination and possibly additional modifications, ultimately combine to signal PIDD1 engagement. Indeed, the timing of PIDD1 recruitment to FANCI (~7.5 hours post injury) is consistent with that of physiologic FANCI deubiquitination (Nijman et al., 2005; Taniguchi et al., 2002).
Unexpectedly, in addition to allowing for the removal of repair-failing cells, the dual signaling properties of FANCI enable the reciprocal conversion of an apoptotic defect into a de novo attempt at lesion repair (Figure 6N). To our knowledge, this is the first reported instance of cells attempting to offset DDIA failure through re-engagement of lesion repair. Whether a similar phenomenon occurs in cells exposed to lesions other than ICLs, and whether it impacts the efficacy of genotoxic cancer treatments or that of DNA repair inhibitors (Helleday et al., 2008), are key questions for the future. The phenomenon’s relevance to other processes involving—or deleteriously impacted by—DDIA, including aging, immunity and neurodegenerative disease (Jackson and Bartek, 2009), will also be of interest. Overall, the FANCI repair/apoptosis switch can thus far be viewed as a dual failsafe which ensures that dysfunction in either genome-surveillance pathway is offset by activation of the other (Figure 6M–N).
Our discovery of a FANCI function that does not strictly require FANCD2 adds to a growing number of studies, including in mouse knockout models (Dubois et al., 2019; Houghtaling et al., 2003; Yang et al., 2019), which describe independent roles for these proteins in replication stress, meiotic recombination, limb development and ribosome biogenesis, among other processes (Castella et al., 2015; Chen et al., 2015; Dubois et al., 2019; Sondalle et al., 2019; Thompson et al., 2017). A particularly intriguing finding in the context of our results is that whereas FANCD2 promotes forks recovery and cell survival in response to replication stress, FANCI appears to play an opposite role in this setting (Thompson et al., 2017). Although mechanisms other than an active cell death-inducing role for FANCI were proposed, it is possible that the FANCI-PIDDosome axis we identified in response to repair failure also acts to ensure the death of cells that fail to resolve replication stress. Understanding the non-overlapping biologic roles of FANCD2 and FANCI should illuminate differences in the clinical manifestations of patients in either complementation group (Boisvert and Howlett, 2014; Savage et al., 2016).
Despite C2 being the closest relative to the C. elegans caspase CED-3, the biological significance of the PIDDosome has remained elusive since its discovery in 2004 (Janssens and Tinel, 2012; Sladky et al., 2017; Tinel and Tschopp, 2004). In addition to FANCI, other DNA repair factors have been reported to physically associate with PIDD1. These include PCNA (Logette et al., 2011) and DNA-PKcs (Lin et al., 2018) in the TLS and NHEJ pathways, respectively. However, PIDD1 appears to promote repair in these situations, in contrast to its role as an apoptosis executioner when engaged by FANCI. When taken with converging evidence linking PIDDosome formation to various forms of genomic instability (Ando et al., 2012; Ando et al., 2017; Burigotto et al., 2021; Dawar et al., 2017; Evans et al., 2021; Fava et al., 2017; Sladky et al., 2020; Thompson et al., 2015; Tsabar et al., 2020), the platform is emerging as a genome surveillance device involved in repair quality control, ploidy control, and other processes.
To avoid possible contributions of p53-dependent DDIA pathways to C2 cleavage and apoptosis readouts (Inoue et al., 2009; Manzl et al., 2009), the experiments herein were performed on TP53 mutant, depleted or null backgrounds. Thus, the FANCI repair/apoptosis switch should be operational in the more than half of human tumors with mutant p53 (Olivier et al., 2002), which typically fail to respond to traditional DNA-damaging therapies (Igney and Krammer, 2002; Liu et al., 2019). Future development of compounds that modulate FANCI activity to toggle the switch in the desired position, as defined by genetic landscape, might define novel therapeutic strategies as single agents or in combinations with DNA-crosslinking chemotherapies.
Limitations of the Study
While our analyses of fanci–/– zebrafish provide in vivo support for our findings, the current lack of antibodies directed to zebrafish Fanci, Fancd2 and Pidd1 precluded us from assessing FANCI switch function biochemically in this physiologic setting. Additionally, the relevance of FANCI switch activity to bone marrow failure observed in FA patients, while supported by the stimulatory effects of FANCA, FANCD2 or FANCL deficiencies on the FANCI-PIDD1 interaction, remains to be addressed by crossing Pidd1 null alleles into appropriate mouse FA models.
STAR Methods
RESOURCES AVAILABILITY
Lead Contact
Further information and request for resources should be directed to and will be fulfilled by the Lead Contact, Dr. Samuel Sidi (samuel.sidi@mssm.edu).
Materials Availability
Unique reagents generated in this study are available from the lead contact upon request.
Data and Code Availability
No large datasets or new code were generated as part of this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Adult zebrafish were maintained on a 14:10 hour light:dark cycle at 28°C in accordance with the regulations and policies of the Mount Sinai Institutional Animal Care and Use Committee (IACUC). CRISPR/Cas9-generated tp53–/–;fanci+/+ and tp53–/–;fanci–/– zebrafish lines (Ramanagoudr-Bhojappa et al., 2018) were kept and maintained as described (Ramanagoudr-Bhojappa et al., 2018; Sidi et al., 2008). All experiments involved up to 1-day old embryos and were performed in accordance with the regulations and policies of the Mount Sinai IACUC.
METHODS DETAILS
Cell Culture and Reagents
To circumvent potential ICL-dependent, PIDD1 transactivation by p53 (Lin et al., 2000) or contributions from p53-dependent DDIA pathways to C2 cleavage and apoptosis and cell viability assays (Inoue et al., 2009; Manzl et al., 2009) performed herein, all experiments were performed on p53 mutant or deficient backgrounds. HeLa (cervical, p53-defective via HPV-E6), PC3 (prostate, TP53 null), CAL27 (HNSCC, TP53 H183L), SAS (HNSCC, TP53 E336*), MDA-MB-231 (breast, TP53 R280K) and isogenic FANCI+/+ and FANCI–/– and TP53+/+ and TP53–/– HCT116 (colon) cancer cell lines (Bunz et al., 1998) were cultured in DMEM medium (Life Technologies) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma- Aldrich) and 1% penicillin/streptomycin (P/S) (Life Technologies). T98G (GBM, TP53 M237I) cells were cultured in EMEM medium (Life Technologies) supplemented with 10% FBS and 1% P/S. SV40-transformed (p53 degraded), patient-derived FA-I F010191 (R1299X) and GM6914 (FANCA–/–) fibroblasts were cultured as described (Colnaghi et al., 2011; Naf et al., 1998). The isogenic pair of FANCI+/+ and FANCI–/– HCT116 cell lines (Thompson et al., 2017) was kindly provided by Dr. Alexandra Sobeck. The pair was stably transfected with doxycycline-inducible TP53 shRNA (see RNAi) and cultured in McCoy’s medium (Corning 10–050-CV) and substituted with 5% FBS, 1% Glutamine and 1% P/S. HeLa.C2-Pro-BiFC cells of WT, PIDD–/– and RAIDD–/– genotypes (Ando et. al. JCB, 2017) were cultured in DMEM medium (Life Technologies) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma- Aldrich) and 1% penicillin/streptomycin (P/S) (Life Technologies). All other cell lines were obtained as previously described (Liu et al., 2019). Gö6976, MMC, nocodazole, RO-3306, ETP46464, cisplatin, Staurosporine and doxycycline were purchased from Sigma-Aldrich. KU-55933 was purchased from TOCRIS. Bendamustine was a gift from Dr. Joshua Brody.
RNAi
siRNA transfections were performed using X-tremeGENE siRNA transfection reagent (Roche) and 20nM siRNA according to the manufacturer’s instructions. Cells were treated with Gö6976 +/− MMC at 48 hrs post-transfection (but see Alamarblue). Previously validated siRNAs were siLACZ (Sidi et al., 2008), siPIDD1 and siRAIDD (RAIDD-2) (Ando et al., 2012; Janssens et al., 2005; Tinel and Tschopp, 2004), and siNPM1 (Ando et al., 2017) (Qiagen). siRNAs to FANCA (MacKay et al., 2010), BRCA2 (FANCD1), FANCD2 (Vinciguerra et al., 2010), FANCE, FANCI (Sims et al., 2007), FANCL, FANCM, RAD51C (FANCO), SLX4 (FANCP), ERCC4 (FANCQ), USP1, CHEK1 (Sidi et al., 2008), WEE1 and CASP3 were purchased from Qiagen. HeLa cells stably expressing shGFP, shPIDD1, shRAIDD and shCASP2 have been previously described (Ando et al., 2012). Doxycycline-inducible shTRIPZ, shFANCI and shp53 cells, generated as previously described (Ando et al., 2012; Ando et al., 2017), were treated with doxycycline (1μg/ml) for 48 hrs before treatment. See Supplemental Table S4 for siRNA and shRNA sequences.
Expression Vectors and DNA Transfections
Plasmid DNA was transfected into HeLa cells using X-tremeGENE HP (Roche) according to the manufacturer’s instructions. Transfected cells were treated with or without Gö6976 at 24 hrs post-transfection and MMC was added one hour later. C-terminally Flag-tagged PIDD1-FL, PIDD1-N, PIDD1-C, PIDD1-CC, LRR, and CDZU5, cloned in pcDNA5/FRT (Logette et al., 2011; Tinel et al., 2007; Tinel and Tschopp, 2004), were kind gifts from Dr Emanuelle Logette. FANCI deletion constructs and DNA-binding mutant (R1285Q) (Yuan et al., 2009) were kind gifts from Dr. Yanbin Zhang. FANCI WT and mono-ubiquitination mutant K523R (Smogorzewska et al., 2007) were kind gifts from Dr. Agata Smogorzewska. MYC-FANCI WT and K294E/K339E (KKEE) (Castella et al., 2015) were kind gifts from Dr. Toshiyasu Taniguchi. The T788A and T788D mutations were introduced into Flag-PIDD-FL using QuikChange II XL Site-Directed Mutagenesis kit (STRATAGENE) according to the manufacturer’s instructions (Ando et al., 2012). GFP-FANCD2 (Liang et al., 2016) was a kind gift from Dr. Martin A. Cohn. Flag-ATR and His-ATRIP (Liu et al., 2011) were kind gifts from Dr Lee Zou. Unless mentioned otherwise, all cDNA constructs were transfected at a concentration of 1 mg/ml. FANCIR1285Q was transfected at 0.25 mg/ml. HA-FANCI was generated from HA-Flag-FANCI (Smogorzewska et al., 2007) using a Q5® Site-Directed Mutagenesis Kit (NEB, E0554S) with the primers FANCI_dflag_F (5’)TACCCATACGATGTTCCAG(3’) and FANCI_dflag_R (5’)CATGGTGGCAGATCAACC(3’) to delete the Flag sequence. FANCI deletion constructs were subcloned from pFastBac-HT-B into pCMV-3Tag1 for expression in mammalian cells. Briefly, pFastBac-HT-B was digested using BamHI and SalI, while pCMV-3Tag1 was digested with BamHI and XhoI restriction enzymes (New England Biolabs) to generate compatible cohesive ends, followed by purification and ligation (NEB Quick Ligase). The ligated FANCI constructs in pCMV-3Tag1 vector were transformed into One Shot™ TOP10 Chemically Competent E. coli cells (Thermo Fisher Scientific) and presence of insert was confirmed by restriction digestion using BamHI and either XbaI or ApaI restriction digestion.
Caspase-2 Bimolecular Fluorescence Complementation (C2 BiFC) Imaging
Bimolecular fluorescence complementation (BiFC) uses nonfluorescent fragments (“split Venus”) of the yellow fluorescent protein Venus that can associate to reform the fluorescent complex when fused to interacting proteins (Shyu et al., 2006). When the C2 prodomain is fused to each half of split Venus, recruitment of C2 to the PIDDosome and the resulting induced proximity leads to enforced association of the two Venus halves, culminating in the C2 BiFC signal. Thus Venus fluorescence acts as a terminal readout for PIDDosome assembly (Bouchier-Hayes et al., 2009). HeLa.C2 Pro-BiFC cells (Ando et al., 2017) harbor a bicistronic construct in which C2 Pro-VC and C2 Pro-VN, separated by the viral 2A self-cleaving peptide, are translated from a single mRNA transcript. This ensures that C2 Pro-VC and C2 Pro-VN are expressed at equal levels. The sensitivity of the endogenous C2 BiFC reporter is sufficient to detect C2 induced proximity in the nucleolus, a major but not unique site for PIDDosome formation (Ando et al., 2017). HeLa.C2 Pro-BiFC cells of parental, PIDD–/– and RAIDD–/– genotypes (1 x 105 cells) (Ando et al., 2017) were seeded directly on coverslips, treated with qVD-OPH (20 μM) and Gö6976 (5 μM) and MMC (5μM) and harvested 24 hours post-treatment. Cells expressing the BiFC components were identified by fluorescence of the linked mCherry protein in stable cell lines. Venus channel image data was analyzed to determine the cells positive for C2 BiFC. More than 100 cells were counted over three independent experiments.
Confocal Microscopy
Parental, PIDD–/– and RAIDD–/– HeLa.C2 Pro-BiFC cells (see above), and HeLa, PC3 and FANCI–/– HCT116 cells (5 × 104) were seeded directly onto coverslips, fixed in 1% paraformaldehyde, permeabilized in 0.5% Triton-X100, and stained as described (Thompson et al., 2015). Confocal microscopy was performed using a Leica TCS SP5 II Confocal over an inverted microscope. Images were acquired using LAS software. See Supplemental Table S3 for a table of antibodies used and specific staining methods.
Zebrafish Maintenance and Embryonic Assays
tp53–/–;fanci+/+ and tp53–/–;fanci–/– zebrafish lines (Ramanagoudr-Bhojappa et al., 2018) were genotyped with primers fanci_F (5’)TTTATCTGCAGTGTGTTCGG(3’), fanci_R (5’)CTACCTTAAAGCTCTCCAGG(3’), tp53_F (5’)TTGCAGTATTCACCGGACC(3’), and tp53_R (5’)TGTCTGTACTATCTCCATCC(3’). Embryos derived from tp53–/–;fanci+/+ and tp53–/–;fanci–/– incrosses were incubated with DMSO or Gö6976 (1 μM) at 17 hours post fertilization (hpf) and with MMC (30 μM) at 18 hpf. 24 hpf embryos were stained with 10 μg/ml acridine orange as previously described (Liu et al., 2019; Sidi et al., 2008). Briefly, embryos were labeled live with AO at 10 μg/mL in egg water for 20 min and washed three times. Embryos were imaged with a Nikon SMZ 1500 fluorescence microscope and analyzed using ImageJ as previously described (Liu et al., 2019; Sidi et al., 2008). The study is compliant with all relevant ethical regulations regarding zebrafish research.
In Vitro Kinase Assays
The ATR kinase assay was performed as previously described (Liu et al., 2011) but utilizing GST-PIDD1-CC (Ando et al., 2012) as substrate. Briefly, HEK293T cells were transfected with Flag-ATR and His-ATRIP expression plasmids (Liu et al., 2011) using X-tremeGENE HP (Roche). 48 hours after transfection, cells were extracted in 0.1% NP-40 lysis buffer (0.1% NP-40, 50 mM Tris-HCl [pH 7.5], 250 mM NaCl, 5 mM EDTA) supplemented with protease and phosphatase inhibitors (Roche). Lysates (10 mg) were immunoprecipitated with 20 μL protein A beads (Bio-Rad) coupled to anti-FLAG M2 antibody (Sigma). Recombinant GST-PIDD1-CC was expressed in DH5a (Invitrogen) and purified with glutathione beads (GE Healthcare). Flag-ATR and GST-PIDD1-CC beads were mixed together and resuspended in kinase reaction buffer with 200 μM ATP (Cell Signaling) for 30 min at 30°C. Flag-ATR and His-ATRIP were generous gifts from Dr Lee Zou.
Western blotting and Antibodies
Cells seeded at a density of 1x106 in 10 cm plates were grown to 60–70% confluence for treatment with DMSO or Gö6976 (1 μM final). After 1 hour, cells were treated with MMC (1 or 0.1 μM final) or IR (10Gy). Cells were harvested at various time points and lysed using 1% NP-40 buffer (Boston BioProducts) with protease and phosphatase inhibitors. Lysates (180 – 200ug) were incubated at 70°C for 10 minutes after adding NuPAGE LDS Sample Buffer (4X) (Life Technologies) and 5% 2-Mercaptoethanol (Sigma Aldrich). For Figures 1–6, S1–S6 and S7B, samples were run on 4–12% or 10% Bis-Tris gels using 1X MES or 1X MOPS buffer (Life Technologies) for 1 hour at 170 V. For Figures 7, S7A and S7C, samples were run on a 3–8% Tris-Acetate (TA) gel using 1X TA buffer (Life Technologies) for 4 hrs at 135 V. After electrophoresis, samples were transferred on a nitro-cellulose membrane (Thermo Fisher Scientific) at 94 V for 100 minutes. Membranes were then blocked with 5% Bovine serum albumin (BSA, Sigma Aldrich) in TBS with 0.1% Tween and probed with primary antibodies at 4°C overnight. Membranes were then rinsed with TBS-Tween (5x5 min) and probed with specific HRP-linked secondary antibody in 5% milk (in TBS-Tween) for 1 hr at room temperature. Membranes were washed as described earlier and placed in SuperSignal West Pico Chemiluminescent Substrate or SuperSignal West Dura Extended Duration Substrate (Pierce Biotechnology). The membrane was then developed with photographic film. A full list of antibodies and protocols can be found in Supplementary Table S2.
Co-Immunoprecipitation
Lysates for immunoprecipitation (IP) were prepared in 1 or 0.1% NP-40 buffer (1 or 0.1% NP-40, 50 mM Tris-HCl [pH 8.0], 150–250 mM NaCl, 5mM EDTA, 1mM phenylmethylsulfonyl fluoride, protease inhibitors cocktail [Complete Mini, Roche] and phosphatase inhibitor cocktail [PhosSTOP, Roche]). For endogenous IPs, whole-cell lysates (1–5 mg) were mixed with Protein-G magnetic beads (Invitrogen, 20 µl of a 50% slurry) and α-PIDD1 (Anto-1), α-PIDD1 (AL233), α-PIDD1 (s-17), α-ATM (D2E2), α-ATR (E1S3S) or α-FANCI (A-7) antibody (5 µg, 1 ml final volume) for 10 min to 3 hours at room temp on a rotating wheel. Beads were then washed three times with PBS-Tween20 (0.02%), resolved by SDS-PAGE, and probed with primary antibodies detected with the corresponding secondary antibodies or mouse TrueBlot HRP-conjugated secondary antibodies [eBioscience]). For α-Flag or α-HA IPs, whole-cell lysates (0.15–2 mg) were mixed with 20 µL beads (50% slurry) and mouse α-Flag (M2) antibody (3 µg) or α–HA (3ug) in 1% NP-40 buffer (500 µL final volume) for 10 min on a rotating wheel. Beads were then washed three times with PBS-T, resolved by SDS-PAGE and analyzed by western blot.
Size Exclusion Chromatography (Gel Filtration)
SEC was performed using an AKTApurifier FPLC System according to the manufacturer’s instructions (GE). Cells were washed twice with PBS and resuspended in hypotonic buffer (20 mM Hepes- KOH, 10 mM KCl, 1 mM MgCl2, 1mM EDTA, 1 mM EGTA, 1 mM DTT, pH 7.5) supplemented with protease inhibitors (Complete Mini, Roche) and phosphatase inhibitors (PhosSTOP, Roche). Resuspended cells were subjected to three rounds of freeze thawing in liquid nitrogen. Debris was removed by centrifugation at 10,000×g for 20 min at 4°C, followed by filtration at 0.2 μm. The column was equilibrated with gel filtration buffer (150 mM NaCl, 20 mM Hepes- KOH, 10 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, pH 7.5). Whole cell lysates (5 mg) were applied to a S400 (HiPrep 16/60 Sephacryl) gel filtration column (Amersham Biosciences). Samples were eluted at 1 ml/min collected at every 0.75mL and monitored with an online detector at 280 nm. Every fifth sample was then concentrated using Amicon®Ultracel® 3K centrifugal filter units (Millipore) and analyzed by western blot.
Cell Viability Assays
AlamarBlue-based cell viability assays were performed as described (Liu et al., 2019) with several modifications. Cells were seeded into 96-well plates at a density of 400 cells/well. After 16 hours, cells were treated with DMSO (1 μM), Gö6976 (1 μM), and an hour later MMC (0.1 μM) was added. 3 days post-treatment, cells were incubated with alamarBlue (Thermo Fisher) at a final concentration of 10%. Absorbance was measured at a wavelength of 570 nm with a 600nm reference wavelength. Relative fluorescence (RFU) was calculated using cell free wells as a control reference and percent survival was calculated compared to DMSO-treated, non-treated controls. Reverse transfections were performed for siRNAs while seeding the cells. shRNA lines were treated with doxycycline 24 hours prior to seeding and fresh doxycycline (1 μg/mL) was also added while seeding. cDNA transfections were performed 24 hrs post-seeding.
Clonogenic Assays
Colony survival assays were performed as previously described (Liu et al., 2019) with several modifications. Single-cell suspensions were seeded into 6-well plates (10000 cells/well) and doxycycline (1 μg/mL) was added. After 48 hours, cells were treated with Gö6976 (0.1 μM) and MMC (0.05 μM). After being cultured for 14 days, plates were rinsed with PBS and fixed / stained by 0.5% crystal violet (Sigma-Aldrich) in methanol for 30 min at room temperature. Colonies consisting of at least 50 cells were scored. Clonogenic assays performed on shRNA transfected lines required refreshing media with 1 μg/mL doxycycline every 48 hours.
TUNEL Assay
TUNEL assays were performed on cells were harvested 24hrs post treatment using the APO-BRDU kit (BD Biosciences) and analyzed by flow cytometry as described previously (Thompson et al., 2015).
Cell Synchronization
Cells were seeded at a density of 1 x 106 / plate for western blotting and 3 x 10 6 for co-immunoprecipitation. See Supplemental Table S1 for detailed synchronization protocols. Thymidine was purchased from Sigma Aldrich.
Flow Cytometry
Cells were treated as stated, collected by trypsinization and fixed in ice cold 70% Ethanol overnight at −20°C. Cells were washed once in PBS and permeabilized for 5 mins in cold PBS containing 0.25% Triton-X-100. Following a further wash in wash buffer (PBS + 0.1% azide + 1% FBS) cells were incubated for 2 hours at 37°C with 1:100 pHH3 antibody (Cell signaling) in wash buffer. Cells were washed twice and incubated with alexa-488 labelled anti-rabbit secondary antibody (Invitrogen) for 30 mins at room temperature in the dark. Following two more washes, cells were incubated in propidium iodide + RNase A for 30 minutes in the dark prior to analysis on FACS Canto (BD biosciences).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was analyzed by two-tailed Student’s t-test using Microsoft Excel. Data are represented as means ± SD or SEM as indicated and p-values <0.05 were considered statistically significant. P-values, as indicated in the figures or figure legends, were coded as follows: *p < 0.05, **p < 0.005, ***p < 0.001, ns indicated no significance. Definitions of analytical units (n) are given in the figure legends and text. The figures were prepared with Adobe Photoshop CC and Adobe Illustrator CC. Graphs were generated using Microsoft Excel.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-Caspase2 mAb (clone 11B4) | Millipore Sigma | Cat# MAB3507, RRID:AB_94894 |
| Mouse anti-PIDD (Anto-1) mAb | Novus Biological | Cat# ALX-804–837, RRID:AB_2297204 |
| Rabbit anti-PIDD (AL233) pAb | Adipogen | Cat# AG-25B-0015, RRID:AB_2490450 |
| Goat anti-PIDD (S-17) pAb | Santacruz biotech | Cat# sc-32161, RRID:AB_2281483 |
| Mouse anti-RAIDD mAb (clone 4B12) | MBL | Cat# M056–3, RRID:AB_592601 |
| Rabbit anti-GAPDH mAb (14C10) | Cell Signaling Technology | Cat# 5014, RRID:AB_10693448 |
| Mouse anti-Chk1(G-4) mAb | Santacruz Biotech | Cat# sc-8408, RRID:AB_627257 |
| Mouse anti-Chk2 (clone 7) mAB | EMD Millipore | Cat# 05–649, RRID:AB_2244941 |
| Rabbit anti-p-chk1 (s345) mAb | Cell Signaling Technology | Cat# 2348, RRID:AB_331212 |
| Rabbit anti-phospho-(Ser/Thr) ATM/ATR Substrates (phospho-SQ/TQ) | Cell Signaling Technology | Cat# 2851, RRID:AB_330318 |
| Mouse anti-FANCI(A-7) mAb | Santacruz Biotech | Cat# sc-271316, RRID:AB_10612555 |
| Rabbit anti-FANCI | FARF | N/A |
| Mouse anti-FANCD2(FI17) mAb | Santacruz Biotech | Cat# sc-20022, RRID:AB_2278211 |
| Rabbit anti-FANCA | FARF | N/A |
| Mouse anti-FANCD1(BRCA2) (3D12) mAB | Santacruz Biotech | Cat# sc-293185 |
| Rabbit anti-FANCE pAb | Novus Biologicals | Cat# 21310002, RRID:AB_2102047 |
| Mouse anti-FANCL (c-4) mAb | Santacruz Biotech | Cat# sc-137076, RRID:AB_2262590 |
| Rabbit anti-FANCM | FARF | N/A |
| Rabbit anti-FANCP | FARF | N/A |
| Mouse anti-FANCO(RAD51C) (F-11) mAb | Santacruz Biotech | Cat# sc-390697 |
| Rabbit anti-USP1 | FARF | N/A |
| Rabbit anti-ATM(D2E2) mAb | Cell Signaling Technology | Cat# 2873, RRID:AB_2062659 |
| Rabbit anti-ATR pAb | Cell Signaling Technology | Cat# 2790, RRID:AB_2227860 |
| Rabbit anti-NPM1 pAb | Cell Signaling Technology | Cat# 3542, RRID:AB_2155178 |
| Mouse anti-Wee1 (B-11) mAb | Santacruz Biotech | Cat# sc-5285, RRID:AB_628447 |
| Mouse anti-FLAG-M2 mAb | Sigma aldrich | Cat# F3165, RRID:AB_259529 |
| Rabbit anti-FLAG (DYKDDDDK) pAb | Cell Signaling Technology | Cat# 14793, RRID:AB_2572291 |
| Mouse anti-eGFP(F56–6A1.2.3) mAb | Thermofisher Scientific | Cat# MA1–952, RRID:AB_889471 |
| Rabbit anti-HA(SG77) pAb | Thermofisher Scientific | Cat# 71–5500, RRID:AB_2533988 |
| Rabbit anti-FADD | Cell Signaling Technology | Cat# 2782, RRID:AB_2100484 |
| Rabbit anti-phospho-PIDD1 (pT788) | Ando et al., 2012 | N/A |
| Mouse anti-ϒ-H2AX(ser139) (clone JBW301) | EMD Millipore | Cat# 05–636, RRID:AB_309864 |
| Mouse anti-PRKDC (G-4) | Santacruz Biotech | Cat# sc-5282, RRID:AB_2172848 |
| Mouse anti-MYC-tag(9B11) mAb | Cell Signaling Technology | Cat# 2276, RRID:AB_331783 |
| Rabbit anti-Active Caspase3 | BD Pharmingen | Cat# 559565, RRID:AB_397274 |
| Rabbit anti-ATR (E1S3S) mAb | Cell Signaling Technology | Cat# 13934, RRID:AB_2798347 |
| Rabbit anti-PARP (9542) | Cell Signaling Technology | Cat# 9542, RRID:AB_2160739 |
| Mouse anti-Caspase3 (31A1067) mAb | SantaCruz Biotech | Cat# sc-56053, RRID:AB_781826 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11008, RRID:AB_143165 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21422, RRID:AB_2535844 |
| Anti-goat IgG, HRP-linked Antibody | Santacruz Biotech | Cat# sc-2354, RRID:AB_628490 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076, RRID:AB_330924 |
| Anti-rat IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7077, RRID:AB_10694715 |
| Bacterial and virus strains | ||
| One Shot™ TOP10 Chemically Competent E. coli cells | Thermo Fisher Scientific | Cat# C404003 |
| Human TRIPZ lentiviral inducible shRNAmir individual clone | GE Dharmacon | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Sigma Aldrich | Cat# D2650–100ML ; CAS Number 67–68-5 |
| Mitomycin C | Sigma Aldrich | Cat# M4287–2MG CAS# 50 07 2 |
| Go6976 | Sigma Aldrich | 365250–1MG CAS 136194–77-9 |
| KU55933 | Tocris | Cat. No. 3544 CAS# 587871–26-9 |
| ETP 46464 | Sigma Aldrich | Cat# SML1321 CAS Number: 1345675–02-6 |
| Acridine Orange | Sigma Aldrich | Cat# A9231 CAS Number 65–61-2 |
| QVD-OPH | Selleckchem | Cat# S7311 CAS# 1135695–98-5 |
| Nocodazole | Sigma Aldrich | Cat# M1404 CAS Number: 31430–18-9 |
| Ro3306 | Sigma Aldrich | Cat# SML0569 CAS Number: 872573–93-8 |
| Cisplatin | Sigma Aldrich | Cat# PHR1624 CAS Number: 15663–27-1 |
| Bendamustine | Dr. Joshua Brody | N/A |
| Staurosporine | LC Laboratories | Cat# S-9300 CAS# 62996–74-1 |
| Crystal Violet | Sigma Aldrich | Cat# C6158 CAS# 548–62-9 |
| Doxycycline | Sigma Aldrich | Cat# 9891 CAS Number 24390–14-5 |
| Methanol | VWR Scientific | Cat# BDH1135–4LP CAS# 67–561 |
| Protease Inhibitor Complete Mini | Roche | Cat# 11836170001 |
| Phosphatase inhibitor PHOSSTOP | Roche | Cat# 04906837001 |
| Benzonase | Millipore-Sigma | Cat# 71205–25KUN |
| NP-40 lysis buffer | Boston Bioproducts | Catalog Number: BP-119 |
| Tris-Buffered Saline | Boston Bioproducts | Catalog Number: BM-300 |
| PBS | Boston Bioproducts | Cat# |
| Tween | Thermofisher Scientific | Cat# P7949 CAS# 9005–64-5 |
| Fetal Bovine Serum | Millipore Sigma | Cat# F0926–500ML Batch Number: 20J481 |
| Penicillin/Streptomycin | Life technologies | Cat# 15140–122 |
| DMEM | Life technologies | Cat# 11995–065 |
| EMEM | ATCC | Cat# 30–2003 |
| McCoy’s Media | Corning | Cat# 10–050-CV |
| Bovine Serum albumin | NEB | Cat# B9000S |
| Thymidine | Sigma aldrich | Cat# 89270 |
| Critical commercial assays | ||
| AlamarBlue Cell Viability assay | Thermofisher. | Cat# DAL1025 |
| TUNEL assay | BDBioSciences | Cat# 556405 |
| X-tremeGene HP DNA transfection kit | Roche | Cat# 6366236001 |
| X-tremeGene siRNA transfection kit | Roche | Cat# 4476115001 |
| QuickChange II XL Site-Directed Mutagenesis Kit | Stratagene | Cat. #200524 |
| Q5 Site-Directed Mutagenesis Kit | New England BioLabs | Cat# E0554S |
| Experimental models: Cell lines | ||
| HeLa (cervical, p53-defective via HPV-E6), | Ando et. al, 2012 | N/A |
| PC3 (prostate, TP53 null) | Lab of Dr. William Oh | N/A |
| CAL27 (HNSCC, TP53 H183L) | ATCC | Cat# CRL-2095 |
| SAS (HNSCC, TP53 E336*) | HSRRB/ JCRB | Cat# JCRB0260 |
| MDA-MB-231 (breast, TP53 R280K) | Lab of Dr. Doris Germain | N/A |
| Isogenic FANCI+/+ and FANCI–/– HCT116 (colon) cancer cell lines |
Lab of Dr. Alexandra Sobeck | Thompson et. al, 2017 |
| Isogenic FANCI+/+ and FANCI–/– HCT116 (colon) cancer cell lines transduced with shp53 | This paper | N/A |
| Isogenic TP53+/+ and TP53–/– HCT116 (colon) cancer cell lines |
A. Thomas Look Lab | Bunz et al., 1998 |
| T98G (GBM, TP53 M237I) | ATCC | Cat#CRL1690 |
| SV40-transformed (p53 degraded), patient-derived FA-I F010191 (R1299X) |
Lab of Dr. Tony Huang | Colnaghi et al., 2011; |
| GM6914 (FANCA–/–) fibroblasts | Lab of Dr. Alan D. Andrea | Naf et al., 1998 |
| HeLa.C2-Pro-BiFC cells of Wild type, PIDD–/– and RAIDD–/– genotypes | Lab of Dr. Lisa Bouchier-Hayes | Ando et. al., 2017 |
| Experimental models: Organisms/strains | ||
| tp53–/–;fanci+/+ and tp53–/–;fanci–/– zebrafish lines | Ramanagoudr-Bhojappa et al., 2018 | N/A |
| Oligonucleotides | ||
| siRNA, shRNA and primer sequences are listed in Table S4 | Table S4 | Table S4 |
| Recombinant DNA | ||
| FLAG-PIDD1 deletion constructs (PCDNA5/FRT-PIDD-3’FLAG) |
Logette et. al., 2011, Tinel and Tschopp et. al., 2004 | N/A |
| FLAG-FANCI deletion constructs (pFastBac-HT-B) | Yuan et. al., 2009 | N/A |
| FLAG FANCI deletion constructs (pCMV-3Tag1) (subcloned from pFastBac-HT-B) |
This paper | N/A |
| FLAG- tagged DNA binding mutant (R1285Q) | Yuan et. al., 2009 | N/A |
| FLAG- tagged DNA binding mutant (R1285Q) (subcloned from pFastBac-HT-B into pCMV-3Tag1) | This paper | N/A |
| HA-FLAG-FANCI WT and K523R (AS177 PHAGE CMV IP N-HA-FLAG) |
Smogorzewska et. al., 2007 | N/A |
| HA-FANCI WT (AS177 PHAGE CMV IP N-HA-FLAG) Generated from HA-FLAG-FANCI WT |
This paper | N/A |
| MYC-FANCI WT and K294E/K339E (KKEE) (pLentiX1-puro) |
Castella et. al., 2015 | N/A |
| FLAG-PIDD1 WT, T788A and T788D (PCDNA5/FRT-PIDD1–3’FLAG) |
Ando et. al., 2012 | N/A |
| GFP-FANCD2 (pOZ-FH-N) |
Liang et. al., 2016 | N/A |
| Flag-ATR | Liu et. al., 2011 | N/A |
| His-ATRIP | Liu et. al., 2011 | N/A |
| Software and algorithms | ||
| Leica Application Suite (LAS) | Leica |
https://www.leica-microsystems.com/products/microscope-software/p/leica-application-suite/ RRID:SCR_016555 |
| FIJI | ImageJ |
https://imagej.net/Fiji/ RRID:SCR_002285 |
| MS Excel | Microsoft |
https://www.microsoft.com/en-us/microsoft-365/excel RRID:SCR_016137 |
Highlights:
FANCI binds either pro-repair FANCD2 or pro-apoptotic PIDD1 in response to ICLs
Interactor selection by FANCI is binary and regulated by deubiquitination
Repair failure triggers PIDD1 recruitment, PIDDosome formation and apoptotic death
Apoptosis failure triggers reversal to FANCD2 and de novo attempts at ICL repair
Acknowledgements:
We thank R. Baer, R. Cagan, R. Krauss, M. O’Connell and S. Sarti for helpful discussions, P. Liu and Y. Gupta for technical assistance, C. Franco and D. Carles for zebrafish care, and A. Smogorzewska, L. Zou, Y. Zhang, A. Sobeck, T. Taniguchi, M. Cohn and J. Brody for critical reagents. Y.L. was supported by a National Cancer Center postdoctoral fellowship award. R.R-B. and S.C.C. acknowledge the support from the Intramural Research Program of the National Genome Research Institute of NIH. T.T.H. was supported by grants from NIH/NIGMS (GM107257) and V Foundation for BRCA Research. A.D.D. was supported by NIH/NHLBI (R37HL052725). A.K.A. was supported by NIH/NIGMS (R35GM131780). R.T. was supported by a Royal Society Dorothy Hodgkin fellowship (DH160106) and Royal Society grants RGF/R1/181008 and RGF/EA/180015. S.S. was supported by grants from NIH/NCI (R01CA178162), NIH/NIGMS (RO1GM135301), Searle Scholars Program (11-SSP-196), Pershing Square Sohn Cancer Research Alliance and New York Community Trust (P16-000373), Claudia Adams Barr Award (PS# 9617093) and JJR Foundation (Young Cancer Research Scientist Award).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References
- Alcon P, Shakeel S, Chen ZA, Rappsilber J, Patel KJ, and Passmore LA (2020). FANCD2-FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat Struct Mol Biol 27, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT, Wang J, Bouchier-Hayes L, and Sidi S (2012b). PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol Cell 47, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Parsons MJ, Shah RB, Charendoff CI, Paris SL, Liu PH, Fassio SR, Rohrman BA, Thompson R, Oberst A, et al. (2017). NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J Cell Biol 216, 1795–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, and Oren M (2007). Living with p53, dying of p53. Cell 130, 597–600. [DOI] [PubMed] [Google Scholar]
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillon J, Ramirez MJ, Pujol R, et al. (2013). Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 92, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert RA, and Howlett NG (2014). The Fanconi anemia ID2 complex: dueling saxes at the crossroads. Cell Cycle 13, 2999–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchier-Hayes L, Oberst A, McStay GP, Connell S, Tait SW, Dillon CP, Flanagan JM, Beere HM, and Green DR (2009). Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell 35, 830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, and Vogelstein B (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A, et al. (2021). Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. EMBO J 40, e104844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, and d’Adda di Fagagna F (2007). Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8, 729–740. [DOI] [PubMed] [Google Scholar]
- Carvajal LA, and Manfredi JJ (2013). Another fork in the road--life or death decisions by the tumour suppressor p53. EMBO Rep 14, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castella M, Jacquemont C, Thompson EL, Yeo JE, Cheung RS, Huang JW, Sobeck A, Hendrickson EA, and Taniguchi T (2015). FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet 11, e1005563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Kennedy RD, Sidi S, Look AT, and D’Andrea A (2009). CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol Cancer 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, Rothenberg E, Jallepalli PV, and Huang TT (2015). ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 58, 323–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnaghi L, Jones MJ, Cotto-Rios XM, Schindler D, Hanenberg H, and Huang TT (2011). Patient-derived C-terminal mutation of FANCI causes protein mislocalization and reveals putative EDGE motif function in DNA repair. Blood 117, 2247–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, and Grant S (2010). New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 16, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawar S, Lim Y, Puccini J, White M, Thomas P, Bouchier-Hayes L, Green DR, Dorstyn L, and Kumar S (2017). Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 36, 2704–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zio D, Bordi M, Tino E, Lanzuolo C, Ferraro E, Mora E, Ciccosanti F, Fimia GM, Orlando V, and Cecconi F (2011). The DNA repair complex Ku70/86 modulates Apaf1 expression upon DNA damage. Cell Death Differ 18, 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zio D, Cianfanelli V, and Cecconi F (2013). New insights into the link between DNA damage and apoptosis. Antioxid Redox Signal 19, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, et al. (2007). Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol 29, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois EL, Guitton-Sert L, Beliveau M, Parmar K, Chagraoui J, Vignard J, Pauty J, Caron MC, Coulombe Y, Buisson R, et al. (2019). A Fanci knockout mouse model reveals common and distinct functions for FANCI and FANCD2. Nucleic Acids Res 47, 7532–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans LT, Anglen T, Scott P, Lukasik K, Loncarek J, and Holland AJ (2021). ANKRD26 recruits PIDD1 to centriolar distal appendages to activate the PIDDosome following centrosome amplification. EMBO J 40, e105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers B, and Jonkers J (2006). Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene 25, 5885–5897. [DOI] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al. (2017). The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Elia A, Potter J, and Mak TW (1997). Partial rescue of Brca1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat Genet 16, 298–302. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, and Sharma RA (2008). DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8, 193–204. [DOI] [PubMed] [Google Scholar]
- Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, and Kumar S (2009). A tumor suppressor function for caspase-2. Proc Natl Acad Sci U S A 106, 5336–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom WS, Boonen R, and Knipscheer P (2019). The role of SLX4 and its associated nucleases in DNA interstrand crosslink repair. Nucleic Acids Res 47, 2377–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, and Grompe M (2003). Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev 17, 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, and Durocher D (2016). The control of DNA repair by the cell cycle. Nat Cell Biol 19, 1–9. [DOI] [PubMed] [Google Scholar]
- Igney FH, and Krammer PH (2002). Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2, 277–288. [DOI] [PubMed] [Google Scholar]
- Inoue S, Browne G, Melino G, and Cohen GM (2009). Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ 16, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont C, Simon JA, D’Andrea AD, and Taniguchi T (2012). Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol Cancer 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, and Tinel A (2012). The PIDDosome, DNA-damage-induced apoptosis and beyond. Cell Death Differ 19, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Tinel A, Lippens S, and Tschopp J (2005). PIDD mediates NF-kappaB activation in response to DNA damage. Cell 123, 1079–1092. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr. (2005). The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5, 689–698. [DOI] [PubMed] [Google Scholar]
- Kastan MB, and Bartek J (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Kee Y, Kim JM, and D’Andrea AD (2009). Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev 23, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, and Smogorzewska A (2011). Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 43, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, and Knipscheer P (2014). XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn EA, Yoo CJ, and Eastman A (2003). The protein kinase C inhibitor Go6976 is a potent inhibitor of DNA damage-induced S and G2 cell cycle checkpoints. Cancer Res 63, 31–35. [PubMed] [Google Scholar]
- Krenning L, van den Berg J, and Medema RH (2019). Life or Death after a Break: What Determines the Choice? Mol Cell 76, 346–358. [DOI] [PubMed] [Google Scholar]
- Li L, Tan W, and Deans AJ (2020). Structural insight into FANCI-FANCD2 monoubiquitination. Essays Biochem 64, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Li Z, Lopez-Martinez D, Nicholson WV, Venien-Bryan C, and Cohn MA (2016). The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat Commun 7, 12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Ma W, and Benchimol S (2000). Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet 26, 122–127. [DOI] [PubMed] [Google Scholar]
- Lin YF, Shih HY, Shang ZF, Kuo CT, Guo J, Du C, Lee H, and Chen BPC (2018). PIDD mediates the association of DNA-PKcs and ATR at stalled replication forks to facilitate the ATR signaling pathway. Nucleic Acids Res 46, 1847–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PH, Shah RB, Li Y, Arora A, Ung PM, Raman R, Gorbatenko A, Kozono S, Zhou XZ, Brechin V, et al. (2019). An IRAK1-PIN1 signalling axis drives intrinsic tumour resistance to radiation therapy. Nat Cell Biol 21, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, and Zou L (2011). ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logette E, Schuepbach-Mallepell S, Eckert MJ, Leo XH, Jaccard B, Manzl C, Tardivel A, Villunger A, Quadroni M, Gaide O, et al. (2011). PIDD orchestrates translesion DNA synthesis in response to UV irradiation. Cell Death Differ 18, 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Martinez D, Liang CC, and Cohn MA (2016). Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci 73, 3097–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, and Efstratiadis A (1997). Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 11, 1226–1241. [DOI] [PubMed] [Google Scholar]
- MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, et al. (2010). Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLachlan TK, Takimoto R, and El-Deiry WS (2002). BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol Cell Biol 22, 4280–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, Fava LL, Krumschnabel G, Peintner L, Tanzer MC, Soratroi C, Bock FJ, Schuler F, Luef B, Geley S, et al. (2013). Death of p53-defective cells triggered by forced mitotic entry in the presence of DNA damage is not uniquely dependent on Caspase-2 or the PIDDosome. Cell Death Dis 4, e942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F, Logette E, Tschopp J, and Villunger A (2009). Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol 185, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt S, and Hofmann TG (2016). The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci 73, 2829–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naf D, Kupfer GM, Suliman A, Lambert K, and D’Andrea AD (1998). Functional activity of the fanconi anemia protein FAA requires FAC binding and nuclear localization. Mol Cell Biol 18, 5952–5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, and Bernards R (2005). The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 17, 331–339. [DOI] [PubMed] [Google Scholar]
- Niraj J, Farkkila A, and D’Andrea AD (2019). The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol 3, 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ, and Zhivotovsky B (2004). DNA damage-induced apoptosis. Oncogene 23, 2797–2808. [DOI] [PubMed] [Google Scholar]
- O’Connell MJ, Raleigh JM, Verkade HM, and Nurse P (1997). Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J 16, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, and Hainaut P (2002). The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 19, 607–614. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, and Durocher D (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. [DOI] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, and Wu H (2007). Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, and Lahav G (2012). p53 dynamics control cell fate. Science 336, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanagoudr-Bhojappa R, Carrington B, Ramaswami M, Bishop K, Robbins GM, Jones M, Harper U, Frederickson SC, Kimble DC, Sood R, et al. (2018). Multiplexed CRISPR/Cas9-mediated knockout of 19 Fanconi anemia pathway genes in zebrafish revealed their roles in growth, sexual development and fertility. PLoS Genet 14, e1007821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos WP, and Kaina B (2006). DNA damage-induced cell death by apoptosis. Trends Mol Med 12, 440–450. [DOI] [PubMed] [Google Scholar]
- Roos WP, Thomas AD, and Kaina B (2016). DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 16, 20–33. [DOI] [PubMed] [Google Scholar]
- Savage SA, Ballew BJ, Giri N, Laboratory, N.D.C.G.R., Chandrasekharappa SC, Ameziane N, de Winter J, Alter BP, and Group, N.D.C.S.W. (2016). Novel FANCI mutations in Fanconi anemia with VACTERL association. Am J Med Genet A 170A, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu YJ, Liu H, Deng X, and Hu CD (2006). Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 40, 61–66. [DOI] [PubMed] [Google Scholar]
- Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, et al. (2008). Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 133, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. (2007). FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 14, 564–567. [DOI] [PubMed] [Google Scholar]
- Sladky V, Schuler F, Fava LL, and Villunger A (2017). The resurrection of the PIDDosome - emerging roles in the DNA-damage response and centrosome surveillance. J Cell Sci 130, 3779–3787. [DOI] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Soratroi C, Heppke J, Eichin F, Rocamora-Reverte L, Szabo TG, Bongiovanni L, Westendorp B, Moreno E, et al. (2020). E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev Cell 52, 335–349 e337. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondalle SB, Longerich S, Ogawa LM, Sung P, and Baserga SJ (2019). Fanconi anemia protein FANCI functions in ribosome biogenesis. Proc Natl Acad Sci U S A 116, 2561–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanova T, Roy N, Kopanja D, Bagchi S, and Raychaudhuri P (2009). DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci U S A 106, 10690–10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, van Twest S, Leis A, Bythell-Douglas R, Murphy VJ, Sharp M, Parker MW, Crismani W, and Deans AJ (2020). Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, and D’Andrea AD (2002). S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100, 2414–2420. [DOI] [PubMed] [Google Scholar]
- Thompson EL, Yeo JE, Lee EA, Kan Y, Raghunandan M, Wiek C, Hanenberg H, Scharer OD, Hendrickson EA, and Sobeck A (2017). FANCI and FANCD2 have common as well as independent functions during the cellular replication stress response. Nucleic Acids Res [DOI] [PMC free article] [PubMed]
- Thompson R, Shah RB, Liu PH, Gupta YK, Ando K, Aggarwal AK, and Sidi S (2015). An Inhibitor of PIDDosome Formation. Mol Cell 58, 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, and Tschopp J (2007). Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. Embo J 26, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, and Tschopp J (2004). The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846. [DOI] [PubMed] [Google Scholar]
- Tsabar M, Mock CS, Venkatachalam V, Reyes J, Karhohs KW, Oliver TG, Regev A, Jambhekar A, and Lahav G (2020). A Switch in p53 Dynamics Marks Cells That Escape from DSB-Induced Cell Cycle Arrest. Cell Rep 33, 108392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinciguerra P, Godinho SA, Parmar K, Pellman D, and D’Andrea AD (2010). Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest 120, 3834–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, and Lu X (2002). Live or let die: the cell’s response to p53. Nat Rev Cancer 2, 594–604. [DOI] [PubMed] [Google Scholar]
- Wang R, Wang S, Dhar A, Peralta C, and Pavletich NP (2020). DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature 580, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Xie H, Zhong Y, Li D, Ke X, Ying H, Yu B, and Zhang T (2019). Severe Fanconi Anemia phenotypes in Fancd2 depletion mice. Biochem Biophys Res Commun 514, 713–719. [DOI] [PubMed] [Google Scholar]
- Yuan F, El Hokayem J, Zhou W, and Zhang Y (2009). FANCI protein binds to DNA and interacts with FANCD2 to recognize branched structures. J Biol Chem 284, 24443–24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XP, Liu F, Cheng Z, and Wang W (2009). Cell fate decision mediated by p53 pulses. Proc Natl Acad Sci U S A 106, 12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No large datasets or new code were generated as part of this study.