

Abstract
The suprachiasmatic nucleus (SCN) of the hypothalamus functions as the master circadian clock. The phasing of the SCN oscillator is locked to the daily solar cycle, and an intracellular signaling cassette from the small GTPase Ras to the p44/42 mitogen-activated protein kinase (ERK/MAPK) pathway is central to this entrainment process. Here, we analyzed the expression and function of SynGAP—a GTPase-activating protein that serves as a negative regulator of Ras signaling—within the murine SCN. Using a combination of immunohistochemical and Western blotting approaches, we show that SynGAP is broadly expressed throughout the SCN. In addition, temporal profiling assays revealed that SynGAP expression is regulated over the circadian cycle, with peak expression occurring during the circadian night. Further, time-of-day-gated expression of SynGAP was not observed in clock arrhythmic BMAL1 null mice, indicating that the daily oscillation in SynGAP is driven by the inherent circadian timing mechanism. We also show that SynGAP phosphorylation at serine 1138—an event that has been found to modulate its functional efficacy—is regulated by clock time and is responsive to photic input. Finally, circadian phenotypic analysis of Syngap1 heterozygous mice revealed enhanced locomotor activity, increased sensitivity to light-evoked clock entrainment, and elevated levels of light-evoked MAPK activity, which is consistent with the role of SynGAP as a negative regulator of MAPK signaling. These findings reveal that SynGAP functions as a modulator of SCN clock entrainment, an effect that may contribute to sleep and circadian abnormalities observed in patients with SYNGAP1 gene mutations.
Keywords: C57/Bl6, circadian, ERK/MAPK, suprachiasmatic nucleus, SynGAP
1 |. INTRODUCTION
The suprachiasmatic nucleus (SCN) of the hypothalamus functions as the master circadian (24 hr) pacemaker (Hastings et al., 2018; Welsh et al., 2010). Synaptic and paracrine output from the SCN serves as a phasing cue to ancillary oscillator populations throughout the body, thus ensuring that precise, clock-gated, cellular- and systems-level biochemistry and physiology is achieved (Kriegsfeld & Silver, 2006; Silver & Kriegsfeld, 2014).
The phasing of the SCN oscillator is tightly regulated by photic input signals from the retina, and this entraining effect allows clock-gated physiology and behavior to be properly segregated across the 24 hr light/dark cycle (Fernandez et al., 2016; Golombek & Rosenstein, 2010; Hattar et al., 2006; Tanaka et al., 1993, 1997).
At a molecular level, the circadian timing system arises from a well-characterized set of interlocking and autoregulatory processes formed from a transcriptional-translational feedback loop. This feedback loop is centered on the rhythmic (24 hr) expression of period and cryptochrome classes of genes, where the transcriptional drive for this rhythm is mediated by a heterodimeric basic helix-loop-helix transcription factor formed by CLOCK and BMAL1, and autoregulatory transcriptional inhibition is achieved via the association of PERIOD/CRYPTOCHROME heterodimers with the CLOCK/BMAL1 transcriptional complex (Cermakian & Sassone-Corsi, 2000; Meng et al., 2008; Sangoram et al., 1998; Zheng & Sehgal, 2012).
The periodicity and phasing of this core molecular clock feedback loop is modulated by numerous kinase signaling pathways. For example, via its phosphorylation-mediated degradation of PERIOD, Casein Kinase 1δ/ε plays a key role in regulating clock periodicity (Gallego & Virshup, 2007; Lee et al., 2009; Meng et al., 2008). Further, via its ability to couple light to the induction of period gene expression, the p44/42 mitogen-activated protein kinase (ERK/MAPK) pathway is a key conduit by which photic input sets the phasing of the SCN oscillator (Butcher et al., 2002; Coogan & Piggins, 2003; Goldsmith & Bell-Pedersen, 2013; Obrietan et al., 1998). Focusing on ERK/MAPK signaling, complementary studies examining ERK modulators (e.g., scaffolds and phosphatases), as well as studies examining signaling via the small GTPase Ras (an upstream regulator of ERK/MAPK activity), have provided important insights into the molecular events that regulate the ERK/MAPK cascade in the SCN (Cheng et al., 2006; Doi et al., 2007; Serchov & Heumann, 2017; Wheaton et al., 2018). However, there is still much to learn about the second messenger pathways that affect the RAS/ERK/MAPK signaling cassette. To this end, we examined the expression and function of the GTPase SynGAP (Synaptic Ras GTPase-activating protein) in the SCN.
SynGAP is one of the most abundant postsynaptic density proteins, where it functions as a negative regulator of Ras and Rap1 signaling and serves as a component of the NMDA receptor signaling complex (Chen et al., 1998; Gamache et al., 2020; Kim et al., 1998; Rumbaugh et al., 2006; Walkup et al., 2018). Along these lines, NMDA receptor activity has been shown to trigger the CaMKII-mediated phosphorylation of SynGAP, which leads to an increase in its Ras GAP activity; this, in turn, increases the rate of the inactivation of Ras at the synapse (Oh et al., 2004; Walkup et al., 2015). Ras activity has profound effects on synaptic physiology, including increased AMPA receptor insertion into the active zone, spine remodeling, and enhanced LTP (Arendt et al., 2004; Manabe et al., 2000; Qin et al., 2005; Zhu et al., 2002). Notably, many of these effects of Ras are mediated by its downstream target: the ERK/MAPK cascade. These findings, coupled with the well-established role of ERK/MAPK signaling in the SCN, raise the possibility that SynGAP could serve as an important signaling intermediate that regulates ERK/MAPK activity, and in turn, the sensitivity of the SCN clock to light.
Here we show that SynGAP expression is gated by the SCN clock and that its phosphorylation state is regulated by photic input. Further, we utilized Syngap1 WT and heterozygous mice to examine the functional contribution of SynGAP to clock physiology and ERK/MAPK activation in the SCN. We show that Syngap1 heterozygosity alters circadian-gated locomotor activity and entrainment capacity and potentiates the activation of the ERK/MAPK pathway after nighttime light exposure. These findings suggest that SynGAP may function upstream of the ERK/MAPK cellular signaling cassette to modulate light-mediated entrainment of the master circadian clock.
2 |. MATERIALS AND METHODS
2.1 |. Mice
All procedures were approved by the Institutional Animal Care and Use Committee at The Ohio State University. In total, ~220 mice (male and female—age and sex-matched between groups) were used. In order to minimize the number of animals, whenever possible, animals were utilized for more than one experiment (e.g., they were used for behavioral wheel running assays and then were sacrificed for tissue collection). Experiments were carried out in C57/Bl6 WT animals (Figures 1–3) derived from our in-house breeding colony, or from animals derived from the Syngap1lx-st mouse line (referred to here as Syngap1+/−, Jax Stock No. 029304: Clement et al., 2012; Figures 4–6). Syngap1+/− mice (males or females) were crossed to Syngap1+/+ mice (males or females) in order to generate Syngap1+/− and Syngap1+/+ experimental animals. Syngap1+/− mice were genotyped using the methods described previously (Clement et al., 2012; Creson et al., 2019; Ozkan et al., 2014). BMAL1−/− mice were generated from breeder animals that were acquired from Jackson Laboratories (Jax Stock No. 009,100); genotyping was performed as previously described (Bunger et al., 2000). Animals were kept on a standard 12 hr/12 hr light-dark (LD) cycle—unless otherwise noted and were fed ad libitum.
FIGURE 1.
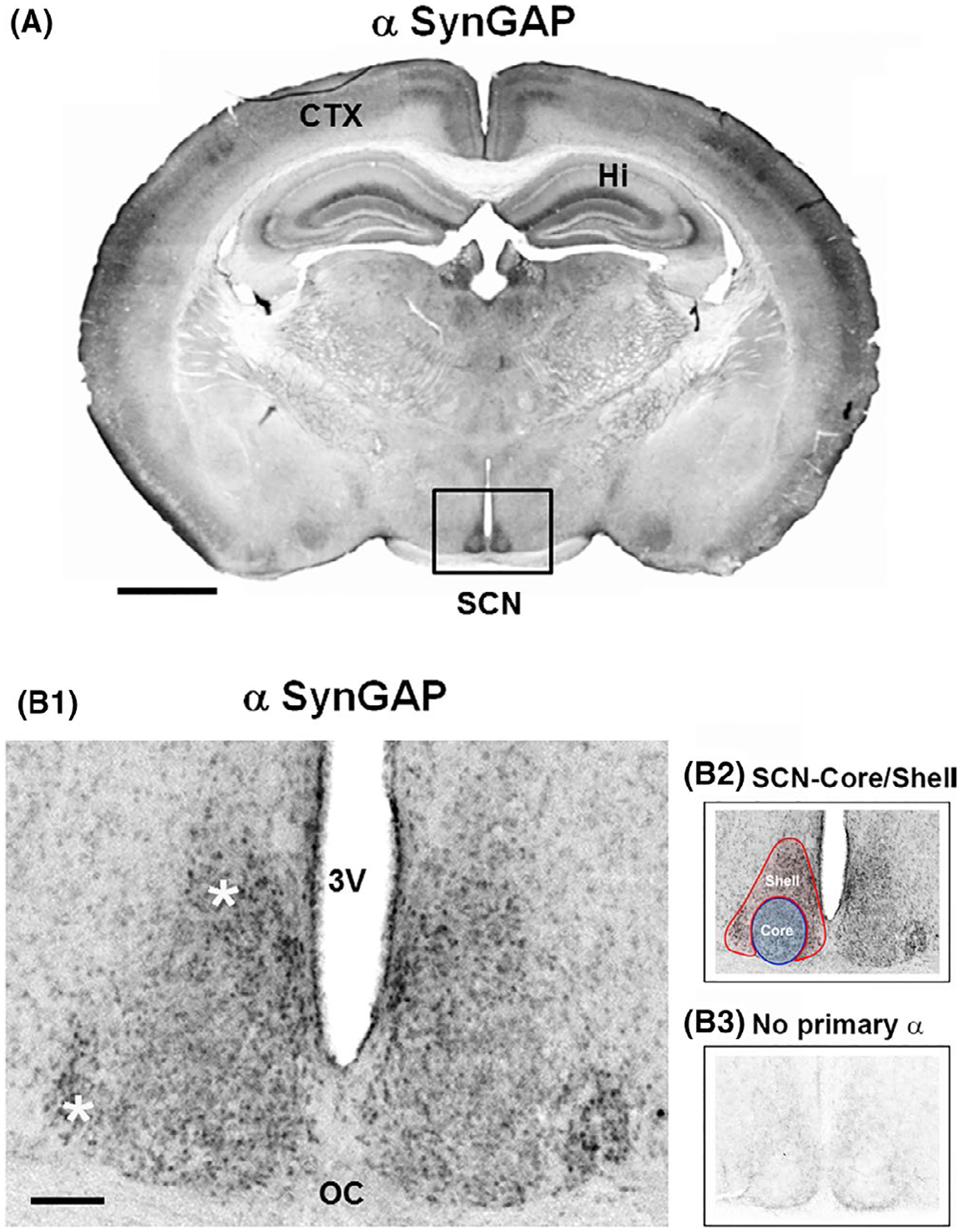
Expression of SynGAP in the SCN. (A) Coronal mouse brain section immunolabeled for SynGAP. Marked immunoreactivity is observed within the hippocampus (Hi), cortex (CTX), and SCN (boxed in black). Scale bar = 1 mm. (B1) High-magnification image of SynGAP expression in the central SCN. Note that SynGAP is observed throughout the SCN, with strong expression in the dorsal and lateral regions (white asterisks). 3V: third ventricle; OC: optic chiasm. Scale bar = 200 μm. (B2) The neuroanatomical demarcation of the SCN shell (red) and core (blue) is denoted. (B3) As an immunolabeling control, SCN tissue was processed without incubation with the primary antibody
FIGURE 3.
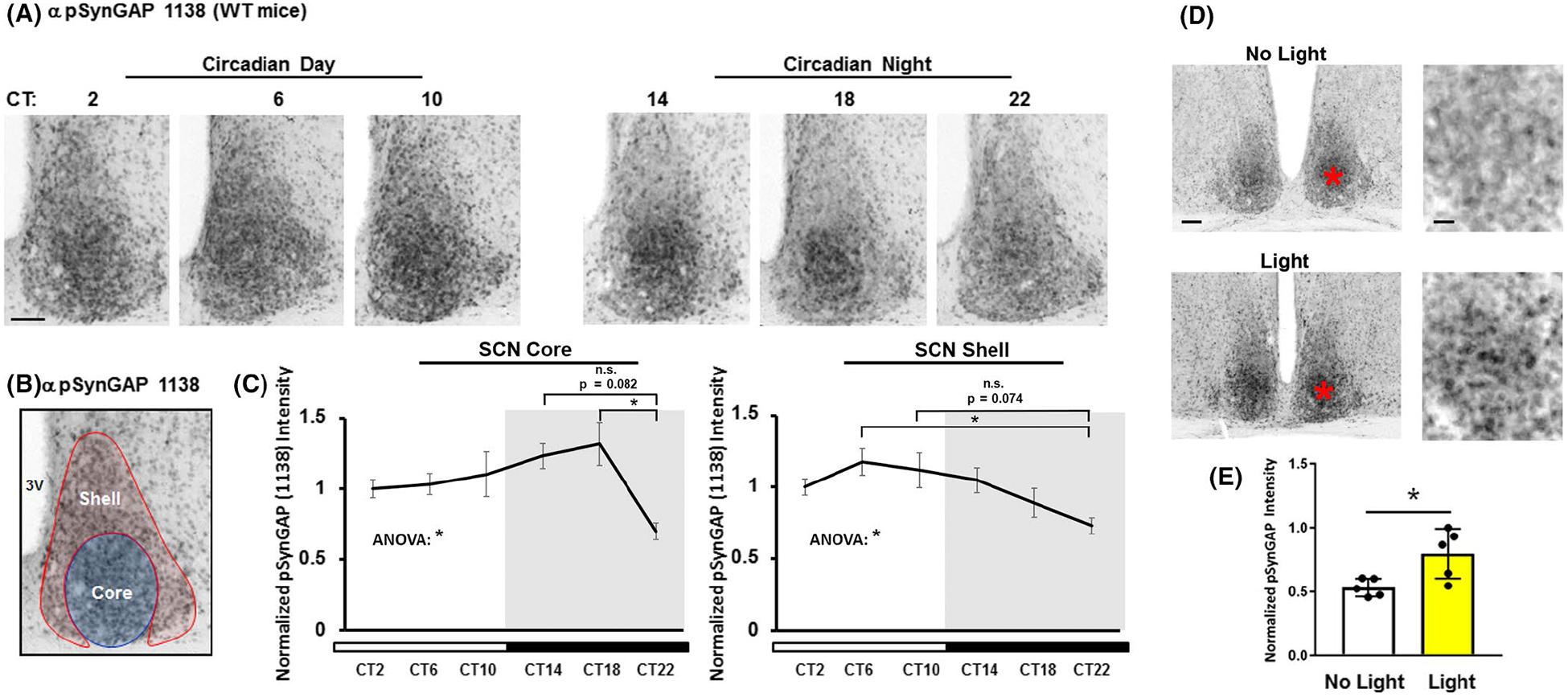
SynGAP phosphorylation at serine 1138 (pSynGAP) in the SCN across the circadian day and after a nighttime light pulse. (A) Immunolabeling-based profiling of pSynGAP over the circadian cycle: tissue was collected at four hour intervals beginning at CT2. Scale bar = 100 μm. (B) As a reference, the neuroanatomical demarcation of the SCN shell (red) and core (blue) is denoted. (C) pSynGAP expression as a function of circadian time is profiled in the SCN core (left panel) and shell (right panel). Data were normalized to the CT2 mean intensity values, which were set equal to 1. N = 5–6 animals per timepoint; data were analyzed using one-way ANOVA followed by post-hoc tests. *: p < .05; n.s. = not significant. (D) Representative immunolabeling for pSynGAP after a 15 minute (~100 lux) light pulse (or no pulse) at CT15. Note that the asterisks in the low magnification images (left panels) approximate the locations of the magnified regions in the right panels. Scale bar = 200 μm for low magnification images and 100 μm for high magnification images. (E) Graphical representation of relative pSynGAP intensity under the two conditions. Data were normalized to the light-pulsed animal with the highest pSynGAP expression, which was set equal to 1. N = 5 animals per timepoint; data were analyzed using the Student’s t-test. *: p < .05.
FIGURE 4.
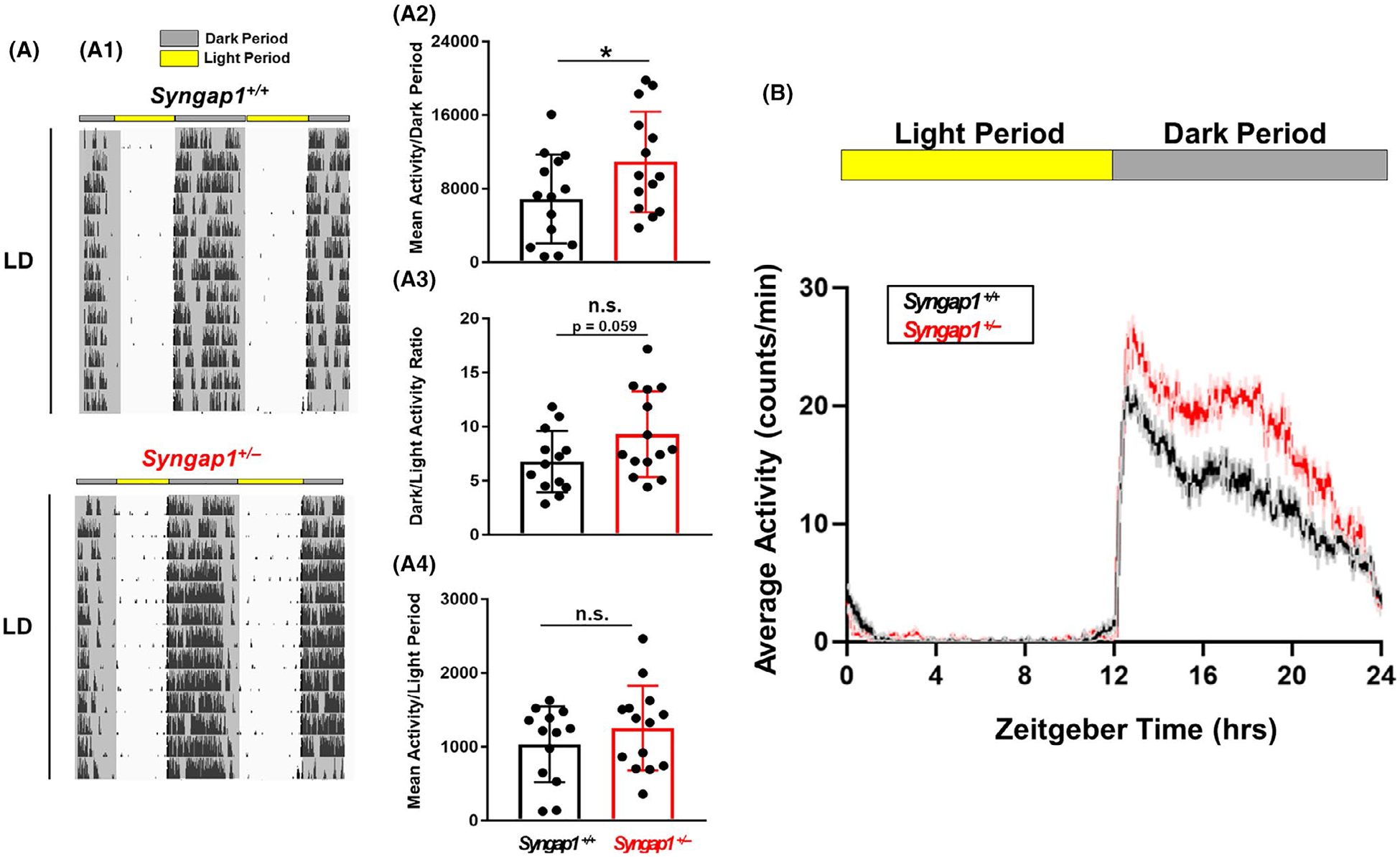
Light-gated locomotor activity in Syngap1 +/− mice. (A: A1) Representative double-plotted actograms of wheel-running activity from a Syngap1+/+ and Syngap1+/−animal. Throughout the profiling, mice were maintained on a 12:12 hr light/dark (LD) cycle. (A2) Daily activity (wheel rotations) during the ‘Dark Period’ is presented as the mean ± SEM value for each genotype; mean dark period activity for each animal is also presented as a scatter plot. (A3) The mean ± SEM dark/light period activity ratio for each genotype is plotted, as is the mean daily ratio for each animal. (A4) Daily activity (wheel rotations) during the ‘Light Period’ are presented as the mean ± SEM value for each genotype; mean daily light period for each animal is also presented as a scatter plot. Data presented in A were averaged from 13 to 14 animals per genotype. *: p < .05; n.s. = not significant; assessed via Student’s t-tests. (B) Mean daily activity profiles of Syngap1 +/+ and Syngap1 +/− mice over a 24 hr LD cycle. Plotted data were averaged from 14 to 15 mice per genotype
FIGURE 6.
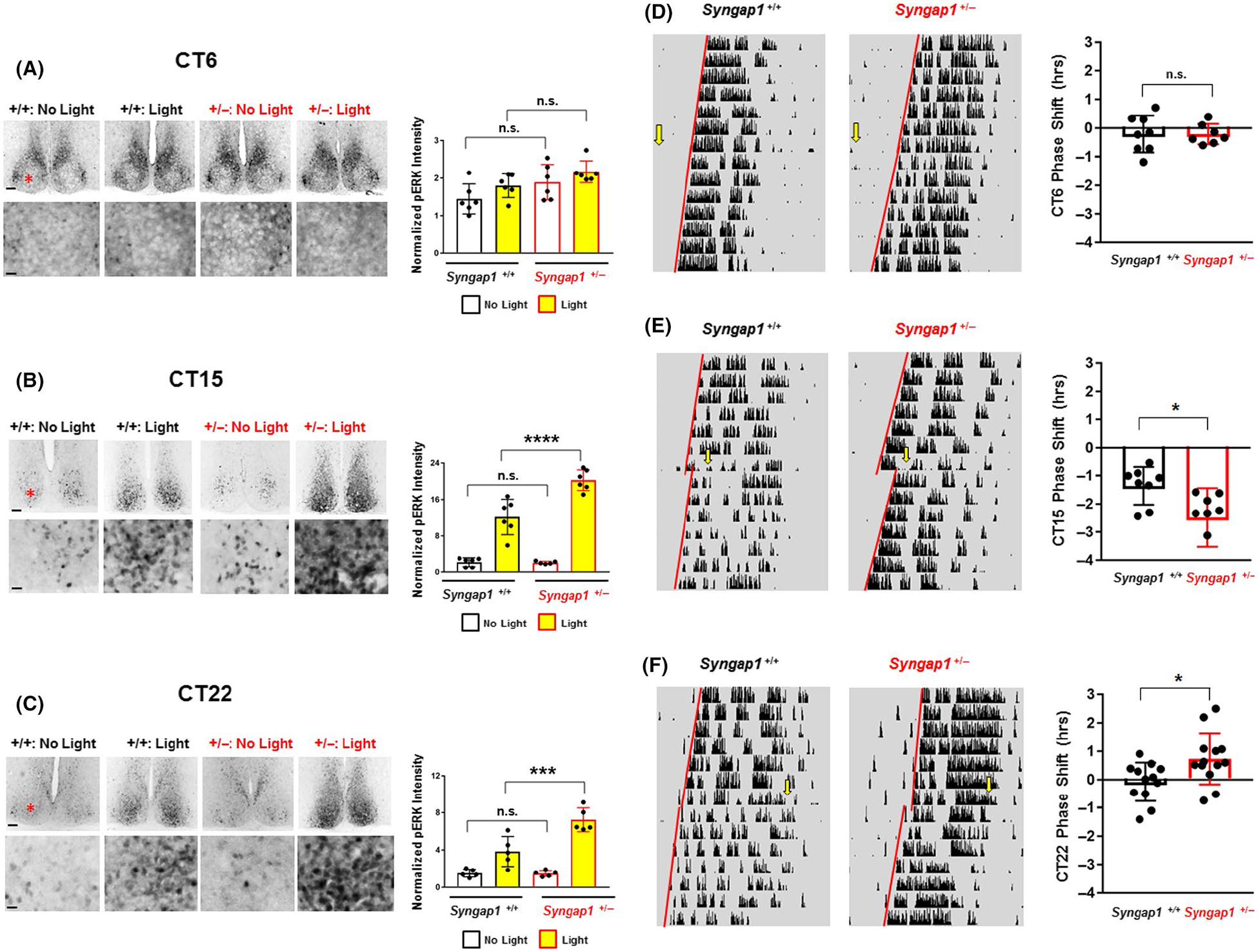
Syngap1 regulates light-evoked ERK/MAPK activation and clock entrainment. (A-C: left panels) Representative low (top panels) and high (bottom panels) magnification images of pERK immunostaining in Syngap1 +/+ and Syngap1 +/− mice exposed to light (15 min; ~40 lux) or not exposed to light during the subjective day (CT6) (A), early subjective night (CT 15) (B) and late subjective night (CT 22) (C). Note that the red asterisk in the top panel approximates the location of the region depicted in the bottom panel. Scale bar = 200 μm for low magnification images and 100 μm for high magnification images. (A–C: right panels) Quantitative analysis of pERK immunostaining. For each circadian time, data were normalized to the control (i.e., no light) Syngap1 +/+ animal with the lowest pERK intensity, and the pERK intensity/value of this animal was set equal to 1. (D–F: Left panels) Representative actograms from Syngap1 +/+ and Syngap1 +/− animals that were dark-adapted at least 2 weeks before being pulsed with light (15 min; ~40 lux) at CT 6 (D), CT 15 (E) and CT 22 (F). Yellow arrows denote the time of the light pulse, and red regression lines were drawn both before and after the pulse to approximate the phase-shift. (D–F: Right panels) Group mean ± SEM phase-shifting effects of light for each time point; the phase-shifting effects for each animal are presented in scatter plot form. N = 5–6 animals per genotype/condition for the pERK light pulse experiments and N = 7–13 animals per genotype/condition for the wheel-running light pulse experiments.*: p < .05; **: p < .01; ***: p < .001; ****: p < .0001; n.s. = not significant; pERK light pulse experiment data were analyzed by two-way ANOVA followed by post hoc tests, and wheel running pulse experiments were analyzed using Student’s t-test
2.2 |. Tissue processing for immunolabeling and Western blotting
Animals were sacrificed via cervical dislocation under white light (Figure 1), or under dim red light for CT (circadian time)/light pulse experiments (Figures 3, and 6). Brains were then removed and placed in ice-cold artificial cerebral spinal fluid and cut into 600 μm-thick (SCN-containing) coronal slices using a Leica vibratome (Leica VT1200). These SCN-containing sections were placed on a rocker for 6 hr in 4% paraformaldehyde in phosphate-buffered saline (PBS) at 4°C and were then cryoprotected overnight at 4°C in 30% sucrose in PBS. For immunolabeling assays, SCN-containing sections were thin cut into 40 μm thickness using a microtome.
For Western blotting experiments (Figure 2), a similar process was used as described above. However, once the 600 μm-thick SCN-containing section was collected with the vibratome, the section was transferred to a glass slide (pre-cooled to −80°C). The SCN and cortex were then micro-dissected using a razor blade and placed in an Eppendorf tube and stored at −80°C. Tissue from three animals was pooled for each sample.
FIGURE 2.
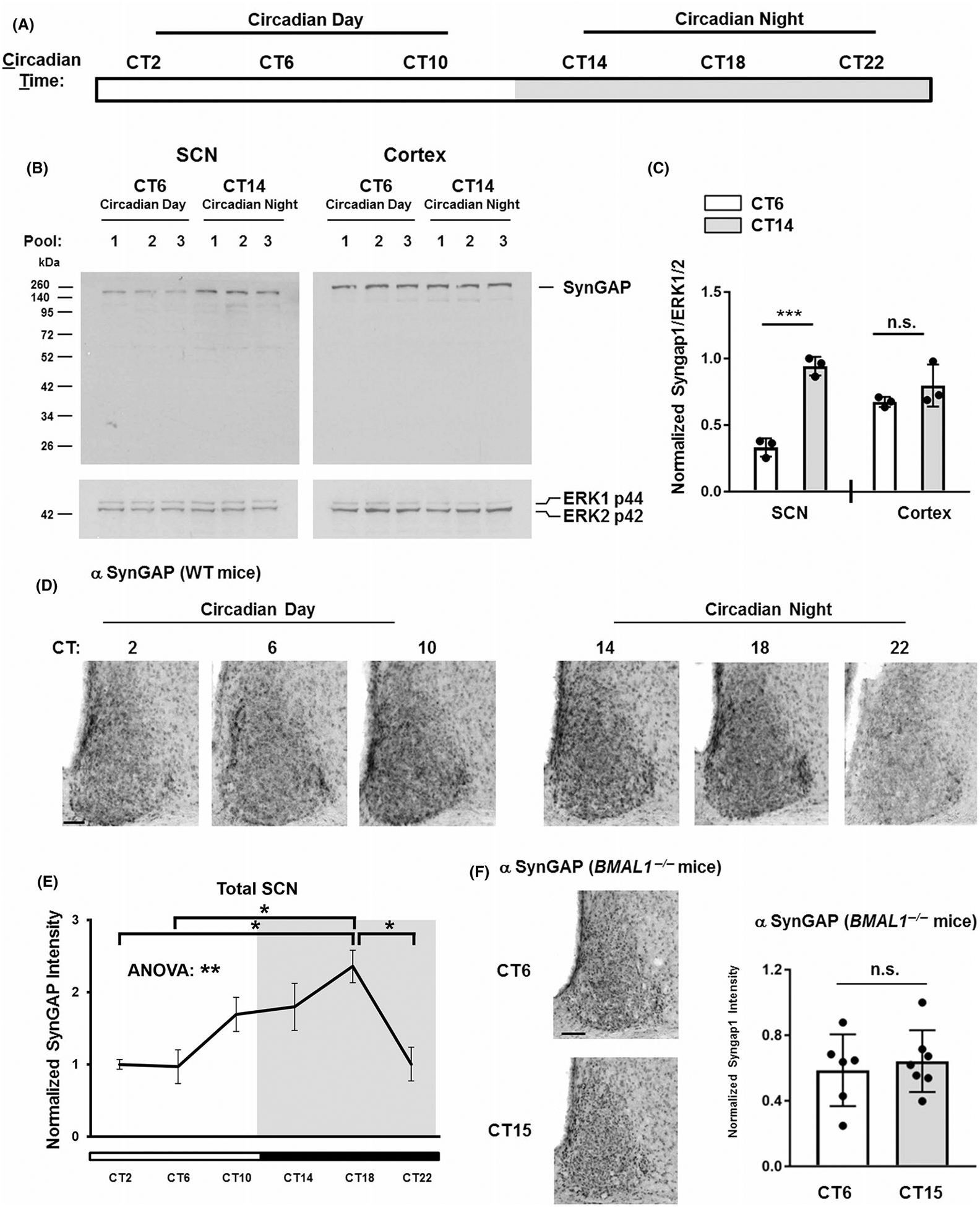
SynGAP expression across the circadian cycle. (A) Diagram illustrating the 24 hr circadian cycle under dark-adapted (no light) conditions. The white section of the horizontal bar refers to the circadian day (experimental time points are noted: CT2, CT6, and CT10), a period when lights would normally be ‘on’. The gray horizontal bar represents the circadian night (experimental time points are noted: CT14, CT18, and CT22) when lights would normally be ‘off’. (B) Representative Western blots from SCN and cortical lysates probed for SynGAP and ERK1/2 (the loading control). Samples were obtained from three separate tissue pools (biological triplicates) for the circadian day (CT 6) and the circadian night (CT 14) timepoints. (C) Quantitation of SynGAP expression. Data are presented as the normalized expression ratio of SynGAP/ERK1/2 (please see the Methods section for a description of the quantitation method). Statistical significance was assessed using the Student’s t-test. ***: p < .001; n.s.: not significant. (D) Immunohistochemical profiling of SynGAP expression in the SCN over the circadian cycle. Animals were sacrificed at 4-hr intervals beginning at CT2. Scale bar = 100 μm. (E) SynGAP expression profiled as a function of circadian time. Data were normalized to the mean value at CT2, which is set equal to a value of 1. N = 4–5 animals per timepoint; Data were analyzed using one-way ANOVA followed by post hoc tests. **: p < .01; *: p < .05. (F) Left panels: Representative images of SynGAP immunolabeling in SCN sections obtained from BMAL1 −/− animals sacrificed at CT6 and CT15 timepoints. Scale bar = 100 μm. Right panel: Densitometric analysis of SynGAP expression in BMAL1 −/− animals sacrificed at CT6 and CT15. All values were normalized to the sample with the highest expressing value of SynGAP at CT15, which was set equal to a value of 1. SynGAP expression did not differ between these timepoints. Data were analyzed using the Student’s t-test. n.s. = not significant. N = 6–7 mice analyzed per timepoint
2.3 |. Immunohistochemical labeling, imaging, and analysis
For 3,3’-diaminobenzidine (DAB)-based immunolabeling, ~40 μm-thick, free-floating tissue sections containing the SCN were washed in PBS containing 1% Triton X-100 (PBST) three times for 5 min. Tissue was then incubated in 0.3% hydrogen peroxide in PBST for 20 min at room temperature. Following this, tissue was blocked with 10% normal goat serum (NGS) diluted in 1X PBST for 1 hr at room temperature and incubated overnight at 4°C with one of the following primary antibodies: rabbit polyclonal anti-Syn-GAP (1:300 dilution; Cat. #: 19739–1-AP; Proteintech), rabbit anti-phospho-Threonine-202/Tyrosine-204 ERK (pERK; 1:3,000 dilution; Cat #: 9,101; Cell Signaling Technology) or rabbit anti-serine 1138 phosphorylated SynGAP (SynGAP pSer1138;1:350 dilution). The SynGAP pSer1138 antibody was generously provided by Dr. Richard Huganir’s laboratory and has been previously described (Araki et al., 2015). On the second day, tissue was washed in PBST and then incubated for 2 hr at room temperature in biotin-conjugated goat anti-rabbit IgG secondary antibody (1:1,000 dilution; Vector Laboratories; catalog # BA-1000). Next, tissue was processed using an ABC labeling kit (Vector Laboratories; catalog # PK-6100). The DAB signal was visualized using nickel intensified DAB processing (Vector Laboratories; Cat # SK-4100). Sections were then mounted onto gelatin-coated glass slides, washed in distilled water, and coverslipped using Permount Mounting Medium (Fisher Chemical).
Bright-field images of the SCN (15X or 20X optical magnification) were acquired using a 16-bit digital camera (Micromax YHS 1300; Princeton Instruments) on a Leica DMIR microscope with MetaMorph Software (MetaMorph Microscopy Automation and Image Analysis Software). For quantification of pERK, SynGAP, or SynGAP pSer1138 intensity, an ROI was digitally traced around the total SCN, the SCN core, or the SCN shell (a demarcation of core and shell regions of the SCN is presented in Figure 1B2) from 1 to 2 sections per animal. Intensity levels for each section were then background subtracted from the lateral hypothalamus and analyzed using ImageJ software. For statistical analysis, the intensity was averaged from each animal and displayed as the mean ± SEM for each condition/genotype.
2.4 |. Western blotting and blot quantification
For analysis of SynGAP expression during the circadian day and night timepoints (Figure 2B,C), three separate cohorts of C57/Bl6 WT animals were sacrificed at CT6 and CT14. Six hundred μm-thick coronal brain sections containing the SCN or cortex were collected from each animal (3 animals were pooled together to create a single sample). SCN tissue was lysed in 100 μl of radioimmuno-precipitation assay buffer and cortical tissue was lysed in 200 μl of buffer. Protein (10 μg/lane) was run on a 12% SDS-PAGE gel and then transferred onto polyvinylidene difluoride membranes (Immobilon-P; EMD Millipore). After transfer, the membrane was blocked for 1 hr at room temperature in 10% milk (in PBST) and incubated overnight at 4°C with the anti-rabbit SynGAP antibody in 5% NGS in PBST (1:1,000; dilution; Cat # 19739–1-AP; Proteintech). The next day, membranes were incubated in 10% milk (prepared from Bio-Rad Blotting-Grade Blocker non-fat dry milk #1706404) in PBST with goat anti-rabbit IgG horseradish peroxidase-conjugated antibody (1:1,000 dilution; PerkinElmer). A bioluminescent signal was generated using the Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate (PerkinElmer), and the signal was acquired using BioBlue-Lite Western Blot film (Alkali Scientific). PBST washes (three times, 5 min each) were carried out between each of the noted steps. Additionally, as a loading control, membranes were also probed for mouse total ERK1/2 (1:2,000 dilution in 5% NGS/PBST; catalog # 4696S; Cell Signaling Technology).
Western blot densitometric band analysis was carried out using ImageJ software. To this end, the band intensity of SynGAP was digitally traced, inverted, background subtracted, and finally divided by the ERK1/2 band signal from the same lane. Note that the intensity from both the ERK1 and ERK2 signal was averaged for each lane/sample. An average (from each of three pools) for the SCN and cortex was then obtained.
2.5 |. Visual tracking/vision tests
The visual tracking test has been previously described by our lab (Snider et al., 2016). In short, mice were placed in a rotating optokinetic response drum. The walls of this drum were covered in white and black vertical stripes. Animals were given ~3 min to habituate to the drum. Following this habituation period, each animal underwent four trials (each trial lasting ~2 s), during which period the drum was randomly turned to the left or to the right by the observer. Each turn of the head was scored as “correct” (i.e., the mouse turned following the direction of the spinning drum) or “incorrect” (i.e., the mouse turned in the opposite direction as the spinning drum). The percentage of correct head turns was then tallied for each mouse. This experiment was run during the subjective daytime (~ ZT6–10) under 500 lux white light illumination.
After the optokinetic response test was conducted, all animals were also screened for proper visual acuity by assessing whether they reach for the surface with their forelimbs before making contact with the counter.
2.6 |. Circadian clock timing and entrainment-wheel running assays and assessment
As described in our recent publication (Wheaton et al., 2018), mice were individually housed in polycarbonate cages containing running wheels (16.5 cm diameter). Wheel revolutions were recorded in 5 min binned periods using VitalView data collection software (Actimetrics). For all assays, mice were initially entrained to a 12-hr light/12-hr dark cycle (white light: 250–300 lux at cage level) for at least 10 days. For experiments that examined locomotor activity under LD conditions, data were collected from a subsequent 2 week period; activity data over a 10-day period were processed using ClockLab Analysis software.
For experiments that examined the free-running period (tau), mice were transitioned to constant darkness (DD) and were permitted to run for 17–21 days. The free-running period was then determined using ClockLab software. To this end, a regression line was drawn through the onset of daily activity for an ~2 week period during the DD experiment. Further, using ClockLab software, the average total activity per day in DD was determined from an ~2 week period. Similarly, to assess the circadian period under constant lighting (LL) conditions, mice were first entrained to standard 12-hr lights on/12-hr lights off the LD cycle, and then transferred into a constant illumination (10 lux) condition for 3–4 weeks. Analysis of period and total activity in LL was conducted using regression analysis with the assistance of ClockLab software (as described above for DD conditions). The % total activity in LL relative to LD (see Figure 5B4) was calculated on an animal-by animal basis and presented as both a group mean and as a scatter plot of individual animals: analysis was conducted over the last two weeks of LD (before mice were transferred to LL) and the last two weeks of LL.
FIGURE 5.
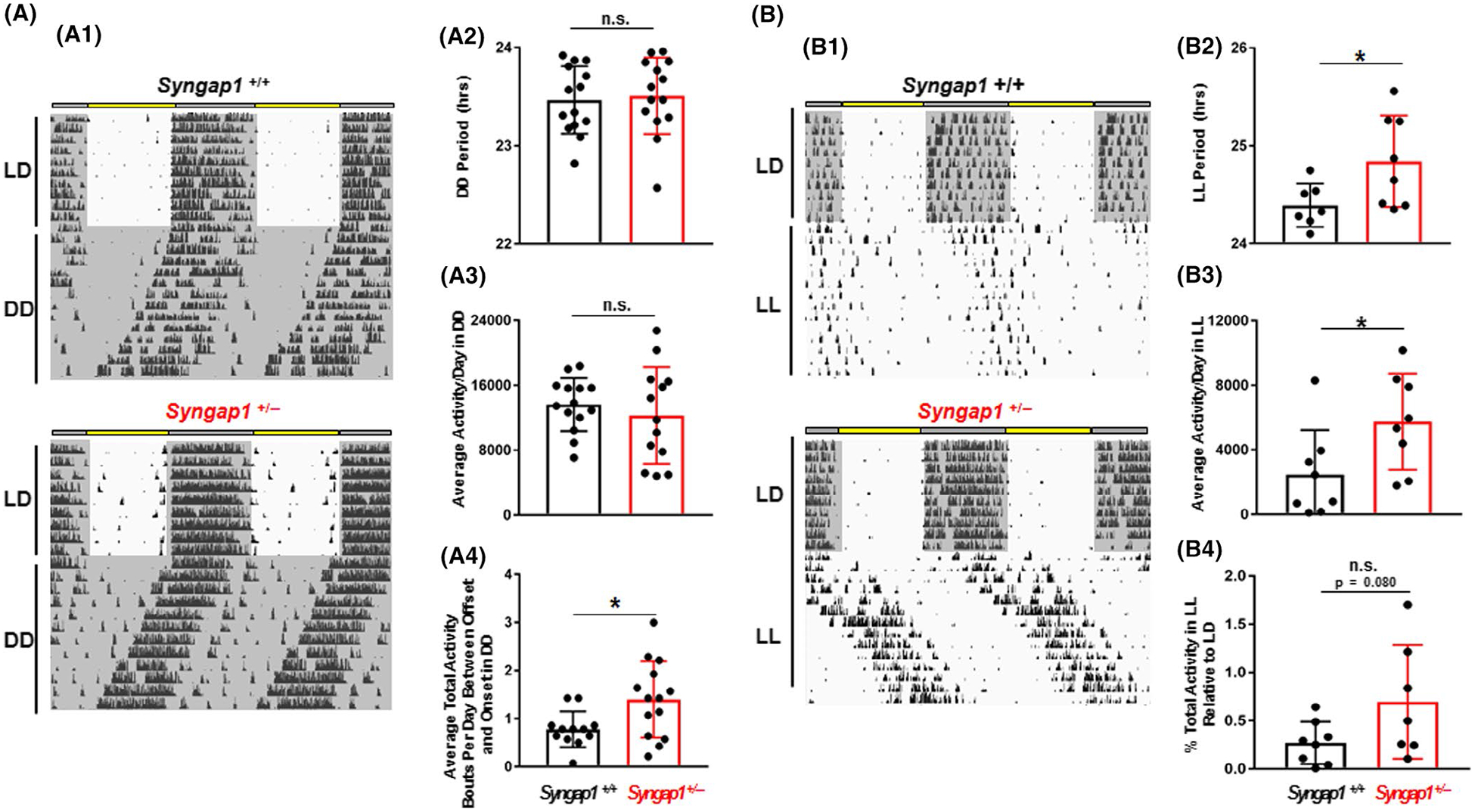
Circadian-regulated locomotor activity in Syngap1 +/− mice. (A: A1) Representative actograms of Syngap1 +/+ and Syngap1 +/− animals in free-running (DD) conditions. The mean ± SEM circadian period (tau) (A2) and activity (rotations per 24 hr period) (A3) are presented for each genotype. Mean tau and daily activity values for each animal are presented in scatter plot form in A2, and A3, respectively. (A4) Activity bouts per day during the rest period are presented as group means ± SEM; data for individual animals are presented in scatter plot form. (B: B1) Representative actograms of Syngap1+/+ and Syngap1+/− animals in constant light (LL). The group mean ± SEM circadian period (tau) (B2) and mean daily activity (rotations per day) (B3) in LL conditions for each genotype are shown. Mean tau and daily activity values for each animal are presented in scatter plot form in B2, and B3, respectively. (B4) Mean ± SEM percent total activity in LL condition relative to the LD condition is presented as both a group mean and scatter plot. Please see the Methods section for a description of the analysis approaches. All data presented in A and B were averaged from 7 to 14 animals per genotype; *: p < .05; n.s. = not significant; Student’s t-test
2.7 |. Light-evoked phase-shifting-behavioral and cellular profiling
To assess the entraining effects of light on wheel-running activity, mice were initially entrained using the noted LD paradigm, and then dark-adapted (i.e., maintained under constant darkness: DD) for at least 2 weeks before being exposed to white light (15 min; 40 lux) at circadian time (CT) 15. Mice were then returned to (DD) for at least 2 additional weeks before being pulsed again at either CT6 or CT22. The phase-shifting effects of CT6, 15, and 22 light pulses were determined utilizing the linear regression method described by Daan and Pittendrigh (1976). ClockLab Analysis software was used to generate the pre- and post-light pulse regression lines.
As a cellular-level readout of phase-shifting responses to light, mice were dark adapted for two days before being exposed to white light (15 min; 40 lux) at CT6, 15, or 22. Control (“No Light”) animals were not exposed to light. Mice were then sacrificed and SCN tissue was isolated, sectioned, and immunolabeled for pERK, using the methods described above.
2.8 |. Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Prism) software and data are presented as mean ± SEM and statistical significance ascribed to p-values <.05. Comparisons between two sets of group means were performed using Student’s t-test, while comparisons among three (or more) group means were conducted using one-way or two-way ANOVA followed by post hoc testing. Grubb’s test was conducted on data obtained from each group to determine whether outliers were statistically significant (p < .05). Grubb’s test was used to exclude the following: one WT mouse sacrificed at CT18 for SynGAP pSer1138 analysis (Figure 3C), one Syngap1 WT wheel running mouse used in LD activity experiments (Figure 4A), one Syngap1 WT wheel running mouse used in DD activity experiments (Figure 5A), one Syngap1 WT wheel running mouse used in LL experiments (Figure 5B), one Syngap1 +/− wheel running mouse used in CT15 phase-shift experiments (Figure 6E), and one Syngap1 WT mouse used in CT22 phase-shift experiments (Figure 6F).
3 |. RESULTS
3.1 |. SynGAP expression in the SCN
Initially, C57/Bl6 mice were sacrificed during the middle of the light period (~ZT 6) and coronal brain sections containing the SCN were labeled via DAB-based immunohistochemistry for SynGAP. A representative low-magnification whole-brain image in Figure 1A reveals SynGAP expression throughout the CNS, including the cortex, hippocampus, and SCN.
Higher magnification photomicrographs revealed the expression of SynGAP throughout the SCN (Figure 1B1). Consistent with this idea, SynGAP labeling was found within the two main neuroanatomical subregions of the SCN: the shell (denoted by red shading in Figure 1B2) and the core (denoted by blue shading in Figure 1B2). Of note, the omission of the primary antibody from the IHC labeling procedure led to a complete loss of the noted cellular-labeling pattern (Figure 1B3).
To further test the specificity of the SynGAP antibody used in Figure 1, Western blotting was conducted on SCN and cortical lysates. With this approach, a single major band with a mass of ~ 148 kDa was detected (Figure 2B), which is consistent with the expected molecular weight of SynGAP (Chen et al., 1998).
3.2 |. Circadian expression profile of SynGAP in the SCN
We next examined whether SynGAP expression changes as a function of circadian time (Figure 2A). To this end, C57/Bl6 WT mice were dark-adapted for 2 days, and then SCN (and cortical) lysates were collected (in three different pooled cohorts) at CT6 (circadian day) and CT14 (circadian night) (Figure 2B) and examined via Western analysis for SynGAP. Quantitative densitometric analysis revealed that SynGAP expression was higher during the circadian night compared to the circadian day within the SCN (Figure 2C; t(4) = 10.79; p = .0004; Student’s t-test). In contrast, a time-of-day difference in SynGAP expression was not detected in the cortex (Figure 2C; t(4) = 1.319; p = .2575; Student’s t-test).
To further explore the temporal profile of clock-gated SynGAP expression, C57/Bl6 WT mice were sacrificed at 4-hr intervals over a 24 hr period and SCN tissue was immunolabeled for SynGAP (Figure 2D). Densitometric analysis revealed a time-of-day difference in SynGAP expression, with peak expression occurring during the mid-subjective night (CT18) and nadir/lowest expression occurring during the mid-subjective day (CT6) (Figure 2E; F(5, 22) = 4.343; p = .0067; one-way ANOVA). Along these lines, SynGAP expression was significantly different at CT2 and CT18, CT6 and CT18, and CT18 and CT22 (Figure 2E; adjusted p-values = .0327; .0173; and .0334, respectively; Tukey post hoc tests). Further, to determine whether the SynGAP SCN rhythm was regulated by the circadian clock, SynGAP expression was examined in tissue from BMAL1−/− animals. BMAL1 deletion results in a loss of circadian clock timekeeping capacity since it is a core constituent of the molecular clock (Bunger et al., 2000). We did not detect a time-of-day difference in SynGAP expression in BMAL1 −/− mice (Figure 2F; t(11) = 0.4914; p = .6328; Student’s t-test). Together, these data support the idea that SynGAP expression in the SCN is under the control of the circadian timing system.
3.3 |. SCN clock-regulated SynGAP phosphorylation at serine 1138
Phosphorylation has been shown to function as a rapid and dynamic regulator of SynGAP-mediated signaling. Notably, SynGAP phosphorylation at serine 1138 (pSynGAP) plays a key role in coupling synaptic activity to the rapid redistribution of SynGAP from dendritic spines, which in turn facilitates Ras and Rap signaling (Araki et al., 2015). To examine whether SynGAP phosphorylation at serine 1138 is regulated by clock timing, SCN tissue from animals sacrificed at 4-hr intervals over the circadian cycle was labeled with an antibody directed against the serine 1138 phosphorylated form of SynGAP. Representative IHC labeling and quantitative profiling (Figure 3A–C) revealed that SynGAP phosphorylation varied across the circadian cycle. Interestingly, distinct temporal patterns of pSynGAP were detected within the SCN core and shell regions. For example, within the SCN core, peak pSynGAP was detected during the mid-subjective night (CT18), and the lowest expression level was detected during the late subjective night (CT22) (Figure 3C; F(5, 29) = 2.593; p = .0468; One-way ANOVA). Specifically, a significant difference was observed in SCN core pSynGAP expression at CT18 and CT22 and a trending difference was noted at CT14 and CT22 (Figure 3C; adjusted p-values = .0292 and p = .0822, respectively; Tukey post hoc tests). In contrast, within the shell, statistically significant peak levels of pSynGAP were observed during the middle of the subjective day (CT6) (Figure 3C; F(5, 28) = 2.849; p = .0335; one-way ANOVA). Specifically, a significant difference was observed in SCN shell pSynGAP expression at CT6 and CT22 and a trending difference was observed at CT10 and CT22 (Figure 3C; adjusted p-values = .0280 and .0744, respectively; Tukey post hoc tests). Together, these data indicate that SynGAP phosphorylation is regulated by the circadian clock. Further, the differential time-gated patterns of phosphorylation within the core and shell suggests distinct upstream signaling processes drive pSynGAP within each region.
3.4 |. Photic regulation of SynGAP phosphorylation at serine 1138
Next, we examined whether photic stimulation regulated SynGAP phosphorylation at serine 1138. To this end, WT C57/Bl6 mice were dark-adapted for two days and exposed to light for 15 min (~100 lux) at CT15, and then sacrificed. Immunohistochemical labeling (Figure 3D) followed by quantative densitometry (Figure 3E) revealed that photic stimulation led to an increase in pSynGAP (t(8) = 2.873; p = .0207; Student’s t-test). Of note, elevated levels of pSynGAP were concentrated within the core region (Figure 3D; high magnification panels to the right). These data indicate that light has the capacity to trigger rapid changes in the functional state of SynGAP in the SCN.
3.5 |. Syngap1 heterozygous mice
Next, we moved to an examination of a SynGAP heterozygous mouse model (Syngap1lx-st) wherein a LoxP-STOP-LoxP cassette was inserted downstream of Exon 5 on the Syngap1 gene—a design that leads to the truncation of the full-length protein, and thus inactivates SynGAP (Clement et al., 2012). Importantly, this construct parallels SYNGAP1 haploinsufficiency in human subjects, which also results from the truncation of the full-length SynGAP protein (Hamdan et al., 2011, 2009). Prior work with this mouse model has shown that Syngap1+/− mice exhibit the anticipated (~50%) reduction in SynGAP protein levels (Clement et al., 2012), in addition to profound behavioral abnormalities that have been observed with other Syngap1+/− mouse models (Clement et al., 2012; Guo et al., 2009). Of note, Syngap1−/− (null) animals were not used in our study since they die within a week after birth (Kim et al., 2003; Komiyama et al., 2002). Similar to previously published studies with this mouse line (Clement et al., 2012; Creson et al., 2019), the Syngap1+/− animals used in our study were viable and fertile, and exhibited no obvious gross anatomical abnormalities.
Given that the transmission of photic input from the retina to the SCN is critical for clock entrainment, we assessed vision in a subset of Syngap1+/− and Syngap1+/+ mice using an optokinetic drum. Importantly, we found no significant genotypic differences in the percent of correct head tracking behavior in this assay (t(19) = 0.3017; p = .7661; Student’s t-test). Further, a one-way t-test determined that both genotypes displayed visual acuity that was “above chance,” suggesting a strong response for correct head tracking (t(10) = 19.36; p < .0001 for WT and t(9) = 14; p < .0001; one-sample t-tests). Additionally, animals were screened for proper vision by reaching for the surface of the bench (with their forelimbs) before their vibrissae made contact with the bench. No visual issues were detected in any of the mice, according to this assay. Hence, these studies suggest that visual acuity is intact in Syngap1+/− mice.
3.6 |. Syngap1 heterozygous mice display alterations in locomotor activity
In the following studies, we used wheel-running activity assays to test the role of SynGAP in SCN clock timing and photic entrainment. To begin, Syngap1+/+ and Syngap1+/− mice were transferred to a 12/12 LD cycle, and activity during both the day and night time periods was examined. Representative wheel running actograms (Figure 4A1) and daily activity profiling (Figure 4B) reveal an elevated level of locomotor activity in Syngap1+/− mice. In specific, Syngap1+/− mice exhibited an overall increase in mean activity during the dark period (i.e., the “active” phase) relative to WT mice (Figure 4A2; t(26) = 2.066; p = .0490; Student’s t-test). In contrast, no difference in the ratio of light/dark period activity between Syngap1 +/− and Syngap1+/+ mice was detected (Figure 4A3), or when the daily activity data were averaged for the light period (Figure 4A4).
To assess the effects of Syngap1 heterozygosity on the period of the master clock, light-entrained mice were placed in constant darkness (DD) and permitted to free-run for a 2–3 week period (Figure 5A1). No statistically significant differences in period (Figure 5A2; t(26) = 0.3042; p = .7634; Student’s t-test), were observed between the genotypes, indicating that Syngap1 haploinsufficiency does not affect the inherent oscillatory capacity of the SCN clock under standard DD/free-running conditions. Analysis of daily locomotor activity also did not detect a phenotypic difference (Figure 5A3; t(25) = 0.7252; p = .4751), though, an increase in the number of daily bouts of transient activity during the “rest period” (i.e., the “inactive” phase) was observed in Syngap1+/−mice, relative to Syngap1+/+ animals (Figure 5A4; t(24) = 2.475; p = .0208; Student’s t-test).
We next examined the effects of Syngap1 heterozygosity on the circadian period under constant lighting (LL) conditions (Figure 5B1). In rodents, exposure to constant light has been shown to lengthen the circadian period and to suppress wheel-running behavior/activity (Aschoff, 1960; Daan & Pittendrigh, 1976). Interestingly, LL triggered an increase in tau length in Syngap1 +/− mice compared to Syngap1 +/+ animals (Figure 5B2; t(13) = 2.33; p = .0366; Student’s t-test). Further, total wheel-running activity in LL was increased in Syngap1+/− animals (Figure 5B3; t(14) = 2.284; p = .0385; Student’s t-test); however, a comparative analysis of activity under LL and LD conditions revealed that the enhanced LL activity in Syngap1 +/− animals was consistent with the increased activity observed under LD conditions (Figure 5B4).
3.7 |. SynGAP regulates light-evoked ERK/MAPK activation and light-evoked clock entrainment
The potent inhibitory effects that SynGAP has on the Ras/ERK/MAPK signaling pathway (Komiyama et al., 2002; Rumbaugh et al., 2006; Wang et al., 2013), coupled with the well-characterized role of the ERK/MAPK signaling cassette in SCN photic entrainment, led us to investigate whether light-evoked ERK/MAPK activation and clock resetting might be altered in Syngap1 heterozygous mice. To begin, Syngap1+/− and Syngap1+/+ animals were dark-adapted for two days and then exposed to a light pulse (15 min; ~40 lux) during the middle of the subjective day (CT6), the early subjective night (CT15) or the late subjective night (CT22) and immediately sacrificed. Control animals were not exposed to light. Coronal SCN sections were immunolabeled for the dual threonine and tyrosine-phosphorylated form of ERK1/2 (pERK) a marker of ERK/MAPK pathway activation (Figure 6A–C). At CT6, basal pERK levels were not significantly different between the two genotypes, and photic stimulation did not induce a significant increase in ERK activation in either genotype (Figure 6A; F(1, 20) = 0.05899; p = .8106; 2-way ANOVA). Turning to the CT15 timepoint, we found an interaction between light and genotype on pERK expression (Figure 6B; F(1, 19) = 17.07; p = .0006; 2-way ANOVA). Specifically, we found a significant difference in pERK expression between light-pulsed Syngap1 WT and heterozygous mice (Figure 6B; adjusted p-value <.0001; Tukey post hoc test). Similarly, an interaction between photic stimulation and genotype on pERK expression was also observed during the late subjective night (CT22) (Figure 6C; F(1, 16) = 13.19; p = .0022; 2-way ANOVA). Specifically, we found a significant difference in pERK expression between light-pulsed Syngap1 WT and heterozygous mice (Figure 6C; adjusted p-value = .0006; Tukey post hoc test). Together, these data suggest that Syngap1 functions as a negative regulator of ERK/MAPK signaling. In specific, light-induced ERK/MAPK activation is potentiated in Syngap1+/− mice during the subjective night time domain, but Syngap1 haploinsufficiency does not affect the temporal gating of ERK/MAPK activation (i.e., light-evoked pERK1/2 expression was not observed in Syngap1+/− mice during the subjective day).
Next, to test whether SynGAP contributes to light-evoked clock entrainment, mice were challenged using an Aschoff type 1 photic entrainment paradigm (Aschoff, 1960). To begin, Syngap1+/− mice and Syngap1+/+ littermates were dark-adapted for at least 2 weeks before being exposed to a light pulse (15 min; 40 lux) at CT6, CT15 or CT22, and the phase-shifting effects were examined using regression analysis. At CT6 (Figure 6D) phase-shifting responses to the mid-day light pulse were not observed in Syngap1 +/+ or Syngap1 +/− mice (Figure 6D; t(13) = 0.01498; p = .9883; Student’s t-test). At CT15 (Figure 6E) Syngap1 +/− mice displayed an increased light-evoked phase-delay relative to Syngap1 +/+ mice (Figure 6E; t(14) = 2.571; p = .0222; Student’s t-test). At CT22 (Figure 6F), Syngap1 +/− mice exhibited a light-evoked phase advance, while the phasing of the circadian rhythm was not altered by light in Syngap1+/+ mice (Figure 6F; t(23) = 2.526; p = .0189; Student’s t-test). Taken together, these data indicate that SynGAP functions as a negative regulator of the entraining effects of light in the SCN. As such, the deletion of a single SynGAP allele augments light-evoked MAPK activation and enhances the photic response of the SCN clock to light.
4 |. DISCUSSION
Here, we provide data showing that SynGAP functions as a regulator of light-evoked SCN clock entrainment. The key findings of our study reveal that 1) SynGAP is enriched within the SCN, 2) the circadian clock regulates total and phosphorylated SynGAP levels, and 3) SynGAP signaling decreases the responsiveness of the MAPK pathway to light.
Immunohistochemical profiling detected SynGAP expression throughout the SCN, with marked labeling in both the core and shell regions. As a downstream effector of NMDA receptor signaling, the widespread expression pattern of SynGAP suggests the NMDA receptor is expressed in these two major functional subdivisions of the SCN. Consistent with this idea, both core and shell regions of the SCN express the ion channel-forming subunit of the NMDA receptor (NR1) (Duffield et al., 2012; Stamp et al., 1997), and robust NMDA-evoked calcium transients and currents have been detected throughout the rostrocaudal extent of the SCN (Colwell, 2001). Further, SCN core neurons receive a direct monosynaptic glutamatergic input from the retina, and NMDA receptor activity has been implicated in photic entrainment (Abe et al., 1991; Colwell, 2001; Ebling, 1996; Moriya et al., 2000; Pennartz et al., 2001). SCN shell neurons receive glutamatergic inputs from the cortex, and basal forebrain (Leak et al., 1999; Moga & Moore, 1997), though the precise functional roles of these inputs are not known. Interestingly, recent work has also shown that glia-mediated glutamatergic signaling regulates the activity of the pre-synaptic NMDA receptors within the shell region and that this inter-SCN astrocytic-neuronal signaling circuit regulates SCN pacemaker activity (Brancaccio et al., 2017). Given the widespread expression of SynGAP in the SCN, these findings indicate that SynGAP could be contributing to a diversity of physiological processes.
With respect to our circadian profiling studies, we found that SynGAP exhibited peak expression during the mid-subjective night and low expression during the mid-subjective day. These results, coupled with the fact that a time-of-day difference in expression was not detected in BMAL1 null animals, indicates that SynGAP is under the control of the circadian clock. Consistent with this finding, microarray-based RNA profiling also detected a circadian rhythm in Syngap1 in the SCN (http://circadb.hogeneschlab.org/; probeset 10,443,108) (Pizarro et al., 2013). Of note, genomic database analysis did not identify a canonical E-box (CACGTG) motif within the 5′ regulatory region of Syngap1 (within 1 kbp flanking the transcription start site) (Cunningham et al., 2019). Thus, SynGAP SCN transcriptional rhythms are likely under the control of an ancillary core-clock-gated transcriptional mechanism. Consistent with this, numerous clock-regulated transcription factors (TFs) bind to the regulatory region of the Syngap1 promoter, including AP-1 and CRE binding TFs, as well as GC box element-binding TFs (Rouillard et al., 2016). Finally, recent work showing that SynGAP expression is dynamically regulated by the clock-gated RNA binding protein FUS (Jiang et al., 2018; Wang et al., 2018; Yokoi et al., 2017), raises the prospect that daily oscillations in mRNA stability could also contribute to the data reported here.
Turning to its regulation, neuronal activity-mediated changes in SynGAP phosphorylation have been shown to play a key role in its functional effects (Jeyabalan & Clement, 2016; Walkup et al., 2015). Along these lines, phosphorylation of SynGAP at serine 1138 (pSynGAP) by CaMKII leads to its rapid dispersion from dendritic spines, which in turn, results in increased Ras/MAPK signaling (Araki et al., 2015). Here we show that SynGAP phosphorylation at serine 1138 was both rhythmic and induced by light within the SCN. Interestingly, the high levels of daytime pSynGAP within the shell corresponds with the time period of elevated levels of neuronal firing (Inouye & Kawamura, 1982; for review, see Welsh et al., 2010), and increased cytoplasmic Ca2+(Brancaccio et al., 2013; Enoki et al., 2017; Noguchi et al., 2017). This, coupled with the well-characterized role of CaMKII in SCN timing (Kon et al., 2014), raises the prospect that rhythmic SynGAP phosphorylation may be a functional output of a clock-gated rhythm in CaMKII activity. It should also be noted that the increased nighttime expression of pSynGAP (within the SCN core) appears to coincide with the total expression of SynGAP (see Figure 2D); hence, the ratio of pSynGAP to total SynGAP could remain unchanged (or even decrease) during the subjective night. Further studies are required to tease apart the nature of the changes in the temporal profile of pSynGAP expression within the SCN shell and core subregions.
Similar to LTP paradigms, which show that SynGAP is rapidly phosphorylated at serine 1138 (Araki et al., 2015), we found that a brief photic stimulation paradigm triggered SynGAP phosphorylation in the SCN. Given the noted model, wherein SynGAP phosphorylation leads to enhanced Ras activity, we postulated that light-evoked ERK activation would be enhanced in Syngap1+/− mice. Consistent with this idea, we detected elevated levels of light-evoked MAPK signaling during both the early and late subjective night in Syngap1+/− mice, compared to Syngap1+/+ animals. We should mention that we did not detect a significant baseline elevation in ERK/MAPK expression in Syngap1+/− mice, though several studies have reported such increases (Komiyama et al., 2002; Rumbaugh et al., 2006). The reason for this difference is not clear, although it is reasonable to consider that ERK signaling in the SCN is modulated by a distinct set of regulatory processes from other brain regions that have shown the upregulation of basal ERK activity in Syngap1+/− mice; a salient example of one such mechanism is the tight, time-of-day, regulation of MAPK activity by the SCN timing system (Obrietan et al., 1998).
The increased sensitivity of the MAPK pathway that we observed in light-pulsed Syngap1+/− mice corresponded with an increased phase-shifting effect of light. Given prior work showing that the MAPK pathway plays a key role in light-evoked clock entrainment (Butcher et al., 2002; Coogan & Piggins, 2003; Obrietan et al., 1998), the data provided here support the idea that SynGAP functions as a modulator of light-evoked clock entrainment, via its effects on MAPK signaling. Clearly, additional experiments that examine the role of SynGAP in shaping the light-evoked PRC (phase response cure) profile, as well as inducible gene expression programs are highly merited. Nevertheless, to place our current data into a larger context, we have included a model outlining how SynGAP may be functioning to facilitate light-evoked, MAPK-dependent, circadian clock entrainment (Figure 7). Finally, it is worth noting that while a late night (~CT20–23) light pulse typically leads to a phase advance, our lab (and others) have found that phase advances are weak (and sometimes absent) in C57/Bl6 mice that receive low intensity (~30–40 lux), short duration (≤15 min), light pulses (for a few examples, see Antoun et al., 2012; Cheng et al., 2006). As such, the limited phase advancing effects of light reported here are consistent with prior studies.
FIGURE 7.
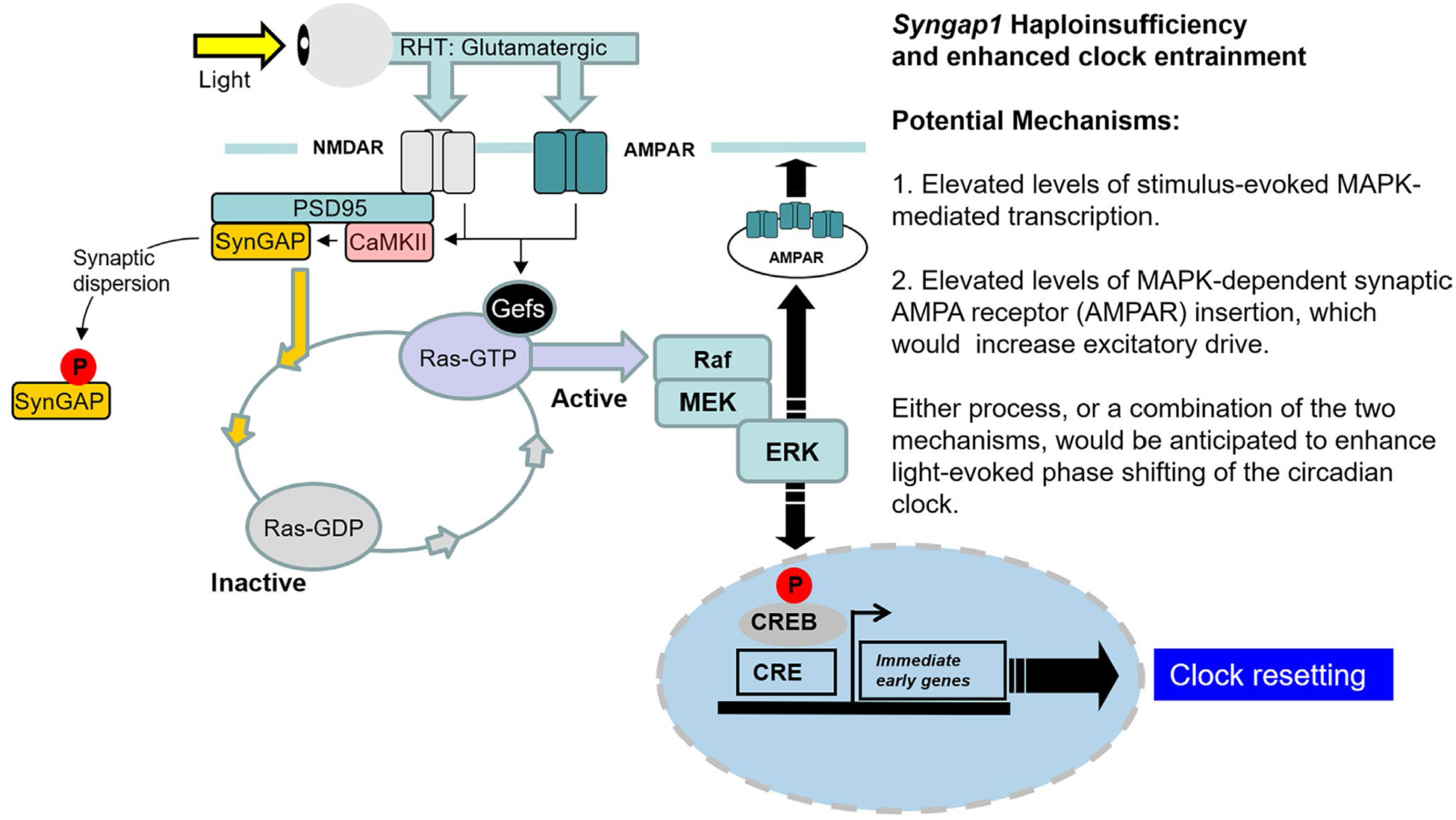
Potential mechanism by which SynGAP regulates light entrainment capacity in the SCN. In this model, photic input drives glutamate release from retinohypothalamic (RHT) nerve terminals, which triggers Gefs (guanine nucleotide exchange factors)-mediated exchange of GDP for GTP in Ras. In its activated, GTP-loaded, form, Ras stimulates MAPK pathway activation. By stimulating the conversion of GTP to GDP, SynGAP functions as a negative regulator of this glutamate/RAS/MAPK signaling cassette. This negative regulatory effect of SynGAP can be reduced via its phosphorylation by CaMKII, which leads to its dispersion from dendritic spines. In Syngap1 heterozygous mice, a reduced level of negative regulation of Ras GTP hydrolysis is predicted to trigger prolonged light-evoked Ras signaling, which in turn, would lead to enhanced MAPK activity (either peak levels or duration of activation). Potential mechanisms by which enhanced MAPK signaling leads to an increase in light-evoked clock entrainment are listed
While several groups have shown that Syngap1+/− mice display hyper-locomotion in both novel and familiar cages/spaces (Berryer et al., 2016; Clement et al., 2012; Guo et al., 2009; Muhia et al., 2012, 2010; Nakajima et al., 2019; Ozkan et al., 2014), data examining time-of-day-based activity profiling in Syngap1+/− mice is limited. Here, we observed a marked increase in wheel-running activity during the nighttime domain in Syngap1+/− mice housed in LD conditions. Of note, while our manuscript was under review, locomotor video tracking data from Sullivan et al. (2020) reported a similar increase in nigh time motor activity in Syngap1 +/− mice compared to WT animals. Interestingly, our studies show that this same hyperactive phenotype was also observed in LL conditions—wherein Syngap1+/− mice displayed increased overall activity relative to their WT littermates. Whether or not SynGAP signaling within the SCN contributes to this increased level of locomotor activity has yet to be examined.
An analysis of the circadian timing properties of the SCN did not detect an effect on periodicity (tau) in Syngap1 heterozygous mice under DD conditions. This lack of an effect on free-running periodicity in DD indicates that a reduction in SynGAP expression/signaling is not sufficient to alter SCN clock timing properties. Clearly, a more complete picture of SynGAP functionality in the SCN would require the use of mice that are homozygous for null alleles of Syngap1; unfortunately, Syngap1 null mice die early in postnatal development (Kim et al., 2003; Komiyama et al., 2002).
In contrast to the lack of an effect under DD conditions, under LL, a marked tau lengthening phenotype was detected in Syngap1+/− mice compared to WT mice. Constant light-mediated Tau lengthening is a well-characterized phenomenon (Aschoff, 1979, 1960; Daan & Pittendrigh, 1976; DeCoursey, 1959; Pittendrigh, 1960) that has been postulated to be a function of the shape of the phase response curve, with period lengthening resulting from greater phase delaying versus phase advancing effects of light. Notably, tau lengthening effects of LL increase as a function of light intensity (Aschoff, 1979, 1960; Daan & Pittendrigh, 1976; Hofstetter et al., 1995).
Placed within this mechanistic model, the most parsimonious explanation for the data reported here is that the enhanced tau lengthening in Syngap1+/− mice results from an increased photic sensitivity of the SCN to early night light (although increased photic sensitivity was also observed during the late night). Additional experiments that more fully characterize light sensitivity as a function of clock time would help clarify the mechanism. Likewise, experiments that examine MAPK signaling and clock gene rhythms within the core and shell under LL conditions could provide mechanistic insights into this tau lengthening phenotype.
In humans, de novo genetic variants in the Syngap1 gene that result in haploinsufficiency (named MRD5; OMIM#603384) are functionally characterized by a number of neurological deficits which manifest in early childhood, including reduced intellectual aptitude/cognitive capacity, delayed language development, and autistic-like tendencies (Berryer et al., 2013; Mignot et al., 2016; Parker et al., 2015). In addition to these neurodevelopmental abnormalities, 60%–70% of individuals with SYNGAP1 gene mutations also report sleep issues (Jimenez-Gomez et al., 2019; Prchalova et al., 2017; Vlaskamp et al., 2019). Interestingly, both sleep and cognition are processes that are modulated by the circadian clock (Fisk et al., 2018; Potter et al., 2016; Snider et al., 2018; Waterhouse, 2010; Wright et al., 2012). Given that the Syngap1+/− mice used in our study serve as a construct-valid/translatable model of SYNGAP1 gene haploinsufficiency in humans (Jeyabalan & Clement, 2016; Kilinc et al., 2018; Ogden et al., 2016), these mice could serve as a powerful tool to further our understanding of how disruptions in the circadian timing and/or clock entrainment capacity might contribute to the clinical characteristics observed in MRD5 patients.
ACKNOWLEDGMENTS
We thank Dr. Richard Huganir (and lab) for his donation of the phospho-SynGAP antibodies. We thank Dr. Andrew Fischer (and lab) for allowing us to use their optokinetic drum for the visual acuity assay.
Funding information
National Institutes of Health-Grant codes: GM133032, AG065830, and MH103361. National Science Foundation-Award Code: 1354612.
Abbreviations:
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- AP-1
activator protein 1
- AVP
arginine vasopressin
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CT
circadian time
- DAB
diaminobenzidine
- DD
constant darkness
- ERK
extracellular signal-regulated kinase
- GAP
GTPase-activating protein
- IHC
immunohistochemistry
- LL
constant light
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase
- NMDA
N-methyl-d-aspartate
- PCR
polymerase chain reaction
- PRC
phase response curve
- ROI
region of interest
- VIP
vasoactive intestinal peptide
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/ejn.15043.
DATA AVAILABILITY STATEMENT
Data are available from the corresponding author upon request.
REFERENCES
- Abe H, Rusak B, & Robertson HA (1991). Photic induction of Fos protein in the suprachiasmatic nucleus is inhibited by the NMDA receptor antagonist MK-801. Neuroscience Letters, 127, 9–12. 10.1016/0304-3940(91)90881-s [DOI] [PubMed] [Google Scholar]
- Antoun G, Bouchard-Cannon P, & Cheng HY (2012). Regulation of MAPK/ERK signaling and photic entrainment of the suprachiasmatic nucleus circadian clock by Raf kinase inhibitor protein. Neurosci., 32, 4867–4877. 10.1523/jneurosci.5650-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, & Huganir RL (2015). Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron, 85, 173–189. 10.1016/j.neuron.2014.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Gärtner U, Seeger G, Barmashenko G, Palm K, Mittmann T, Yan L, Hümmeke M, Behrbohm J, Brückner MK, Holzer M, Wahle P, & Heumann R (2004). Neuronal activation of Ras regulates synaptic connectivity. European Journal of Neuroscience, 19, 2953–2966. 10.1111/j.0953-816X.2004.03409.x [DOI] [PubMed] [Google Scholar]
- Aschoff J (1960). Exogenous and endogenous components in circadian rhythms. Cold Spring Harbor Symposia on Quantitative Biology, 25, 11–28. 10.1101/sqb.1960.025.01.004 [DOI] [PubMed] [Google Scholar]
- Aschoff J (1979). Circadian rhythms: Influences of internal and external factors on the period measured in constant conditions1. Zeitschrift für Tierpsychologie, 49, 225–249. 10.1111/j.1439-0310.1979.tb00290.x [DOI] [PubMed] [Google Scholar]
- Berryer MH, Chattopadhyaya B, Xing P, Riebe I, Bosoi C, Sanon N, Antoine-Bertrand J, Lévesque M, Avoli M, Hamdan FF, Carmant L, Lamarche-Vane N, Lacaille J-C, Michaud JL, & Cristo GD (2016). Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nature Communications, 7, 1–14. 10.1038/ncomms13340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, Lacaille J-C, Niu Z, Eng CM, Yang Y, Palardy S, Belhumeur C, Rouleau GA, Tommerup N, Immken L, … Di Cristo G (2013). Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Human Mutation, 34, 385–394. 10.1002/humu.22248 [DOI] [PubMed] [Google Scholar]
- Brancaccio M, Maywood ES, Chesham JE, Loudon ASI, & Hastings MH (2013). A Gq-Ca2+ axis controls circuit-level encoding of circadian time in the suprachiasmatic nucleus. Neuron, 78, 714–728. 10.1016/j.neuron.2013.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M, Patton AP, Chesham JE, Maywood ES, & Hastings MH (2017). Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron, 93, 1420–1435.e5. 10.1016/j.neuron.2017.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, & Bradfield CA (2000). Mop3 is an essential component of the master circadian pacemaker in mammals. Cell, 103, 1009–1017. 10.1016/s0092-8674(00)00205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GQ, Dziema H, Collamore M, Burgoon PW, & Obrietan K (2002). The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. Journal of Biological Chemistry, 277, 29519–29525. 10.1074/jbc.M203301200 [DOI] [PubMed] [Google Scholar]
- Cermakian N, & Sassone-Corsi P (2000). Multilevel regulation of the circadian clock. Nature Reviews Molecular Cell Biology, 1, 59–67. 10.1038/35036078 [DOI] [PubMed] [Google Scholar]
- Chen H-J, Rojas-Soto M, Oguni A, & Kennedy MB (1998). A synaptic Ras-GTPase aactivating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron, 20, 895–904. 10.1016/S0896-6273(00)80471-7 [DOI] [PubMed] [Google Scholar]
- Cheng H-YM, Dziema H, Papp J, Mathur DP, Koletar M, Ralph MR, Penninger JM, & Obrietan K (2006). The molecular gatekeeper dexras1 sculpts the photic responsiveness of the mammalian circadian clock. Journal of Neuroscience, 26, 12984–12995. 10.1523/JNEUROSCI.4253-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, Xu X, & Rumbaugh G (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting the maturation of dendritic spine synapses. Cell, 151, 709–723. 10.1016/j.cell.2012.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS (2001). NMDA-evoked calcium transients and currents in the suprachiasmatic nucleus: Gating by the circadian system. European Journal of Neuroscience, 13, 1420–1428. 10.1046/j.0953-816x.2001.01517.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan AN, & Piggins HD (2003). Circadian and photic regulation of phosphorylation of ERK1/2 and Elk-1 in the suprachiasmatic nuclei of the Syrian hamster. Journal of Neuroscience, 23, 3085–3093. 10.1523/JNEUROSCI.23-07-03085.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creson TK, Rojas C, Hwaun E, Vaissiere T, Kilinc M, Jimenez-Gomez A, Holder JL, Tang J, Colgin LL, Miller CA, & Rumbaugh G (2019). Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. eLife, 8, 10.7554/eLife.46752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, Cummins C, Davidson C, Dodiya KJ, Gall A, Girón CG, Gil L, Grego T, Haggerty L, Haskell E, … Flicek P (2019). Ensembl 2019. Nucleic Acids Research, 47, D745–D751. 10.1093/nar/gky1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daan S, & Pittendrigh CS (1976). A Functional analysis of circadian pacemakers in nocturnal rodents. Journal of Comparative Physiology, 106, 253–266. 10.1007/BF01417857 [DOI] [Google Scholar]
- DeCoursey PJ (1959). Daily Activity Rhythms in the Flying Squirrel, Glaucomys Volans, Dissertation, University of Wisconsin-Madison. [Google Scholar]
- Doi M, Cho S, Yujnovsky I, Hirayama J, Cermakian N, Cato ACB, & Sassone-Corsi P (2007). Light-inducible and clock-controlled expression of MAP kinase phosphatase 1 in mouse central pacemaker neurons. Journal of Biological Rhythms, 22, 127–139. 10.1177/0748730406298332 [DOI] [PubMed] [Google Scholar]
- Duffield GE, Mikkelsen JD, & Ebling FJP (2012). Conserved expression of the glutamate NMDA receptor 1 subunit splice variants during the development of the Siberian hamster suprachiasmatic nucleus. PLoS ONE, 7, e37496. 10.1371/journal.pone.0037496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebling FJ (1996). The role of glutamate in the photic regulation of the suprachiasmatic nucleus. Progress in Neurobiology, 50, 109–132. 10.1016/s0301-0082(96)00032-9 [DOI] [PubMed] [Google Scholar]
- Enoki R, Ono D, Kuroda S, Honma S, & Honma K (2017). Dual origins of the intracellular circadian calcium rhythm in the suprachiasmatic nucleus. Scientific Reports, 7, 1–8. 10.1038/srep41733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez DC, Chang Y-T, Hattar S, & Chen S-K (2016). Architecture of retinal projections to the central circadian pacemaker. Proceedings of the National Academy of Sciences of the United States of America, 113, 6047–6052. 10.1073/pnas.1523629113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk AS, Tam SKE, Brown LA, Vyazovskiy VV, Bannerman DM, & Peirson SN (2018). Light and cognition: Roles for circadian rhythms, sleep, and arousal. Front. Neurol, 9, 10.3389/fneur.2018.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego M, & Virshup DM (2007). Post-translational modifications regulate the ticking of the circadian clock. Nature Reviews Molecular Cell Biology, 8, 139–148. 10.1038/nrm2106 [DOI] [PubMed] [Google Scholar]
- Gamache TR, Araki Y, & Huganir RL (2020). Twenty years of SynGAP research: From synapses to cognition. Journal of Neuroscience, 40(8), 1596–1605. 10.1523/JNEUROSCI.0420-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith CS, & Bell-Pedersen D (2013). Diverse roles for MAPK signaling in circadian clocks. Advances in Genetics, 84, 1–39. 10.1016/B978-0-12-407703-4.00001-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golombek DA, & Rosenstein RE (2010). Physiology of circadian entrainment. Physiological Reviews, 90, 1063–1102. 10.1152/physrev.00009.2009 [DOI] [PubMed] [Google Scholar]
- Guo X, Hamilton PJ, Reish NJ, Sweatt JD, Miller CA, & Rumbaugh G (2009). Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of schizophrenia. Neuropsychopharmacology, 34, 1659–1672. 10.1038/npp.2008.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs M-O, Joober R, Lacaille J-C, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, & Michaud JL (2011). De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biological Psychiatry, 69, 898–901. 10.1016/j.biopsych.2010.11.015 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Perreault-Linck E, Carmant L, D’Anjou G, Fombonne E, Addington AM, Rapoport JL, Delisi LE, Krebs M-O, Mouaffak F, Joober R, … Synapse to Disease Group. (2009). Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. New England Journal of Medicine, 360, 599–605. 10.1056/NEJMoa0805392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings MH, Maywood ES, & Brancaccio M (2018). Generation of circadian rhythms in the suprachiasmatic nucleus. Nature Reviews Neuroscience, 19, 453–469. 10.1038/s41583-018-0026-z [DOI] [PubMed] [Google Scholar]
- Hattar S, Kumar M, Park A, Tong P, Tung J, Yau K-W, & Berson DM (2006). Central projections of melanopsin-expressing retinal ganglion cells in the mouse. The Journal of Comparative Neurology, 497, 326–349. 10.1002/cne.20970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstetter JR, Mayeda AR, Possidente B, & Nurnberger JI (1995). Quantitative trait loci (QTL) for circadian rhythms of locomotor activity in mice. Behavior Genetics, 25, 545–556. 10.1007/bf02327578 [DOI] [PubMed] [Google Scholar]
- Inouye ST, & Kawamura H (1982). Characteristics of a circadian pacemaker in the suprachiasmatic nucleus. Journal of Comparative Physiology, 146, 153–160. 10.1007/BF00610233 [DOI] [Google Scholar]
- Jeyabalan N, & Clement JP (2016). SYNGAP1: Mind the gap. Frontiers in Cellular Neuroscience, 10, 10.3389/fncel.2016.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Zhang T, Wang H, Wang T, Qin M, Bao P, Wang R, Liu Y, Chang HC, Yan J, & Xu J (2018). Neurodegeneration-associated FUS is a novel regulator of circadian gene expression. Transl Neurodegener, 7, 10.1186/s40035-018-0131-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Gomez A, Niu S, Andujar-Perez F, McQuade EA, Balasa A, Huss D, Coorg R, Quach M, Vinson S, Risen S, & Holder JL (2019). Phenotypic characterization of individuals with SYNGAP1 pathogenic variants reveals a potential correlation between posterior dominant rhythm and developmental progression. J. Neurodev. Disord, 11, 18. 10.1186/s11689-019-9276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilinc M, Creson T, Rojas C, Aceti M, Ellegood J, Vaissiere T, Lerch JP, & Rumbaugh G (2018). Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Molecular and Cellular Neurosciences, 91, 140–150. 10.1016/j.mcn.2018.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee H-K, Takamiya K, & Huganir RL (2003). The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. Journal of Neuroscience, 23, 1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, & Huganir RL (1998). SynGAP: A synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron, 20, 683–691. 10.1016/s0896-6273(00)81008-9 [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJC, O’Carroll CM, Martin SJ, Morris RGM, O’Dell TJ, & Grant SGN (2002). SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J. Neurosci. off. J. Soc. Neurosci, 22, 9721–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon N, Yoshikawa T, Honma S, Yamagata Y, Yoshitane H, Shimizu K, Sugiyama Y, Hara C, Kameshita I, Honma K, & Fukada Y (2014). CaMKII is essential for the cellular clock and coupling between morning and evening behavioral rhythms. Genes & Development, 28, 1101–1110. 10.1101/gad.237511.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegsfeld LJ, & Silver R (2006). The regulation of neuroendocrine function: Timing is everything. Hormones and Behavior, 49, 557–574. 10.1016/j.yhbeh.2005.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leak RK, Card JP, & Moore RY (1999). Suprachiasmatic pacemaker organization analyzed by viral transynaptic transport. Brain Research, 819, 23–32. 10.1016/s0006-8993(98)01317-1 [DOI] [PubMed] [Google Scholar]
- Lee H, Chen R, Lee Y, Yoo S, & Lee C (2009). Essential roles of CKIδ and CKIε in the mammalian circadian clock. Proc. Natl. Acad. Sci. U. S. A, 106, 21359–21364. 10.1073/pnas.0906651106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Aiba A, Yamada A, Ichise T, Sakagami H, Kondo H, & Katsuki M (2000). Regulation of long-term potentiation by H-Ras through NMDA receptor phosphorylation. Journal of Neuroscience, 20, 2504–2511. 10.1523/JNEUROSCI.20-07-02504.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q-J, Logunova L, Maywood ES, Gallego M, Lebiecki J, Brown TM, Sládek M, Semikhodskii AS, Glossop NRJ, Piggins HD, Chesham JE, Bechtold DA, Yoo S-H, Takahashi JS, Virshup DM, Boot-Handford RP, Hastings MH, & Loudon ASI (2008). Setting clock speed in mammals: The CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron, 58, 78–88. 10.1016/j.neuron.2008.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, von Stülpnagel C, Nava C, Ville D, Sanlaville D, Lesca G, Rastetter A, Gachet B, Marie Y, Korenke GC, Borggraefe I, Hoffmann-Zacharska D, Szczepanik E, Rudzka-Dybała M, Yiş U, Çağlayan H, Isapof A, Marey I, Panagiotakaki E, …Depienne C (2016). Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. Journal of Medical Genetics, 53, 511–522. 10.1136/jmedgenet-2015-103451 [DOI] [PubMed] [Google Scholar]
- Moga MM, & Moore RY (1997). Organization of neural inputs to the suprachiasmatic nucleus in the rat. The Journal of Comparative Neurology, 389, 508–534. [DOI] [PubMed] [Google Scholar]
- Moriya T, Horikawa K, Akiyama M, & Shibata S (2000). Correlative association between N-methyl-d-aspartate receptor-mediated expression of period genes in the suprachiasmatic nucleus and phase shifts in behavior with photic entrainment of clock in hamsters. Molecular Pharmacology, 58, 1554–1562. 10.1124/mol.58.6.1554 [DOI] [PubMed] [Google Scholar]
- Muhia M, Willadt S, Yee BK, Feldon J, Paterna J-C, Schwendener S, Vogt K, Kennedy MB, & Knuesel I (2012). Molecular and behavioral changes associated with adult hippocampus-specific SynGAP1 knockout. Learning & Memory, 19, 268–281. 10.1101/lm.026351.112 [DOI] [PubMed] [Google Scholar]
- Muhia M, Yee BK, Feldon J, Markopoulos F, & Knuesel I (2010). Disruption of hippocampus-regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. European Journal of Neuroscience, 31, 529–543. 10.1111/j.1460-9568.2010.07079.x [DOI] [PubMed] [Google Scholar]
- Nakajima R, Takao K, Hattori S, Shoji H, Komiyama NH, Grant SGN, & Miyakawa T (2019). Comprehensive behavioral analysis of heterozygous Syngap1 knockout mice. Neuropsychopharmacol. Rep, 39, 223–237. 10.1002/npr2.12073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi T, Leise TL, Kingsbury NJ, Diemer T, Wang LL, Henson MA, & Welsh DK (2017). Calcium circadian rhythmicity in the suprachiasmatic nucleus. Cell Autonomy and Network Modulation., eNeuro 4, 10.1523/ENEURO.0160-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, Impey S, & Storm DR (1998). Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nature Neuroscience, 1, 693–700. 10.1038/3695 [DOI] [PubMed] [Google Scholar]
- Ogden KK, Ozkan ED, & Rumbaugh G (2016). Prioritizing the development of mouse models for childhood brain disorders. Neuropharmacology, 100, 2–16. 10.1016/j.neuropharm.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JS, Manzerra P, & Kennedy MB (2004). Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. Journal of Biological Chemistry, 279, 17980–17988. 10.1074/jbc.M314109200 [DOI] [PubMed] [Google Scholar]
- Ozkan ED, Creson TK, Kramár EA, Rojas C, Seese RR, Babyan AH, Shi Y, Lucero R, Xu X, Noebels JL, Miller CA, Lynch G, & Rumbaugh G (2014). Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron, 82, 1317–1333. 10.1016/j.neuron.2014.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park S-M, Benoit V, Lederer D, Maystadt I, Study D, & FitzPatrick DR (2015). De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. American Journal of Medical Genetics. Part A, 167A, 2231–2237. 10.1002/ajmg.a.37189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz CM, Hamstra R, & Geurtsen AM (2001). Enhanced NMDA receptor activity in retinal inputs to the rat suprachiasmatic nucleus during the subjective night. Journal of Physiology, 532, 181–194. 10.1111/j.1469-7793.2001.0181g.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittendrigh CS (1960). Circadian rhythms and the circadian organization of living systems. Cold Spring Harbor Symposia on Quantitative Biology, 25, 159–184. 10.1101/sqb.1960.025.01.015 [DOI] [PubMed] [Google Scholar]
- Pizarro A, Hayer K, Lahens NF, & Hogenesch JB (2013). CircaDB: A database of mammalian circadian gene expression profiles. Nucleic Acids Research, 41, D1009–D1013. 10.1093/nar/gks1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter GDM, Skene DJ, Arendt J, Cade JE, Grant PJ, & Hardie LJ (2016). Circadian rhythm and sleep disruption: Causes, metabolic consequences, and countermeasures. Endocrine Reviews, 37, 584–608. 10.1210/er.2016-1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prchalova D, Havlovicova M, Sterbova K, Stranecky V, Hancarova M, & Sedlacek Z (2017). Analysis of 31-year-old patient with SYNGAP1 gene defect points to importance of variants in broader splice regions and reveals developmental trajectory of SYNGAP1-associated phenotype: Case report. BMC Medical Genetics, 18, 62. 10.1186/s12881-017-0425-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart JP, Stornetta RL, Seidenman K, Mack V, van Aelst L, & Zhu JJ (2005). State-dependent Ras signaling and AMPA receptor trafficking. Genes & Development, 19, 2000–2015. 10.1101/gad.342205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, & Ma’ayan A (2016). The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database, 2016, 10.1093/database/baw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Adams JP, Kim JH, & Huganir RL (2006). SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proceedings of the National Academy of Sciences of the United States of America, 103, 4344–4351. 10.1073/pnas.0600084103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangoram AM, Saez L, Antoch MP, Gekakis N, Staknis D, Whiteley A, Fruechte EM, Vitaterna MH, Shimomura K, King DP, Young MW, Weitz CJ, & Takahashi JS (1998). Mammalian circadian autoregulatory loop: A timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron, 21, 1101–1113. 10.1016/s0896-6273(00)80627-3 [DOI] [PubMed] [Google Scholar]
- Serchov T, & Heumann R (2017). Ras activity tunes the period and modulates the entrainment of the suprachiasmatic clock. Frontiers in Neurology, 8, 264. 10.3389/fneur.2017.00264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R, & Kriegsfeld LJ (2014). Circadian rhythms have broad implications for understanding brain and behavior. European Journal of Neuroscience, 39, 1866–1880. 10.1111/ejn.12593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider KH, Dziema H, Aten S, Loeser J, Norona FE, Hoyt K, & Obrietan K (2016). Modulation of learning and memory by the targeted deletion of the circadian clock gene Bmal1 in forebrain circuits. Behavioral Brain Research, 308, 222–235. 10.1016/j.bbr.2016.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider KH, Sullivan KA, & Obrietan K (2018). Circadian regulation of hippocampal-dependent memory: Circuits, synapses, and molecular mechanisms. Neural Plasticity, 2018, 7292540. 10.1155/2018/7292540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamp JA, Piggins HD, Rusak B, & Semba K (1997). Distribution of ionotropic glutamate receptor subunit immunoreactivity in the suprachiasmatic nucleus and intergeniculate leaflet of the hamster. Brain Research, 756, 215–224. 10.1016/S0006-8993(97)00199-6 [DOI] [PubMed] [Google Scholar]
- Sullivan BJ, Ammanuel S, Kipnis PA, Araki Y, Huganir RL, & Kadam SD (2020). Low-dose perampanel rescues cortical gamma dysregulation associated with parvalbumin interneuron GluA2 upregulation in epileptic syngap1+/− mice. Biol. Psychiatry, Neurodegeneration, 87, 829–842. 10.1016/j.biopsych.2019.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Hayashi S, Tamada Y, Ikeda T, Hisa Y, Takamatsu T, & Ibata Y (1997). Direct retinal projections to GRP neurons in the suprachiasmatic nucleus of the rat. NeuroReport, 8, 2187–2191. 10.1097/00001756-199707070-00020 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Ichitani Y, Okamura H, Tanaka Y, & Ibata Y (1993). The direct retinal projection to VIP neuronal elements in the rat SCN. Brain Research Bulletin, 31, 637–640. 10.1016/0361-9230(93)90134-w [DOI] [PubMed] [Google Scholar]
- Vlaskamp DRM, Shaw BJ, Burgess R, Mei D, Montomoli M, Xie H, Myers CT, Bennett MF, XiangWei W, Williams D, Maas SM, Brooks AS, Mancini GMS, van de Laar IMBH, van Hagen JM, Ware TL, Webster RI, Malone S, Berkovic SF, … Scheffer IE (2019). SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. Neurology, 92, e96–e107. 10.1212/WNL.0000000000006729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup WG, Sweredoski MJ, Graham RL, Hess S, & Kennedy MB (2018). Phosphorylation of synaptic GTPase-activating protein (synGAP) by polo-like kinase (Plk2) alters the ratio of its GAP activity toward HRas, Rap1 and Rap2 GTPases. Biochemical and Biophysical Research Communications, 503, 1599–1604. 10.1016/j.bbrc.2018.07.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup WG, Washburn L, Sweredoski MJ, Carlisle HJ, Graham RL, Hess S, & Kennedy MB (2015). Phosphorylation of synaptic GTPase-activating protein (synGAP) by Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5) alters the ratio of its GAP activity toward Ras and Rap GTPases. Journal of Biological Chemistry, 290, 4908–4927. 10.1074/jbc.M114.614420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-C, Held RG, & Hall BJ (2013). SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS ONE, 8, 10.1371/journal.pone.0083941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Symul L, Yeung J, Gobet C, Sobel J, Lück S, Westermark PO, Molina N, & Naef F (2018). Circadian clock-dependent and -independent posttranscriptional regulation underlies temporal mRNA accumulation in mouse liver. Proc Natl Acad Sci USA, 115, E1916–E1925. 10.1073/pnas.1715225115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse J (2010). Circadian rhythms and cognition. Progress in Brain Research, 185, 131–153. 10.1016/B978-0-444-53702-7.00008-7 [DOI] [PubMed] [Google Scholar]
- Welsh DK, Takahashi JS, & Kay SA (2010). Suprachiasmatic nucleus: cell autonomy and network properties. Annual Review of Physiology, 72, 551–577. 10.1146/annurev-physiol-021909-135919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton K, Aten S, Queiroz LS, Sullivan K, Oberdick J, Hoyt KR, & Obrietan K (2018). Circadian expression and functional characterization of PEA-15 within the mouse suprachiasmatic nucleus. European Journal of Neuroscience, 47, 845–857. 10.1111/ejn.13850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KP, Lowry CA, & LeBourgeois MK (2012). Circadian and wakefulness-sleep modulation of cognition in humans. Frontiers in Molecular Neuroscience, 5, 10.3389/fnmol.2012.00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi S, Udagawa T, Fujioka Y, Honda D, Okado H, Watanabe H, Katsuno M, Ishigaki S, & Sobue G (2017). 3’UTR Length-Dependent Control of SynGAP Isoform α2 mRNA by FUS and ELAV-like Proteins Promotes Dendritic Spine Maturation and Cognitive Function. Cell Rep, 20, 3071–3084. 10.1016/j.celrep.2017.08.100 [DOI] [PubMed] [Google Scholar]
- Zheng X, & Sehgal A (2012). Speed control: Cogs and gears that drive the circadian clock. Trends in Neurosciences, 35, 574–585. 10.1016/j.tins.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, & Malinow R (2002). Ras and rap control AMPA receptor trafficking during synaptic plasticity. Cell, 110, 443–455. 10.1016/S0092-8674(02)00897-8 [DOI] [PubMed] [Google Scholar]