

Shear stress induces endothelial transition to pro-inflammatory mesenchymal phenotype in a coronary artery–on–a–chip model.
Abstract
A three-dimensional microengineered human coronary artery–on–a–chip was developed for investigation of the mechanism by which low and oscillatory shear stress (OSS) induces pro-atherogenic changes. Single-cell RNA sequencing revealed that OSS induced distinct changes in endothelial cells (ECs) including pro-inflammatory endothelial-to-mesenchymal transition (EndMT). OSS promoted pro-inflammatory EndMT through the Notch1/p38 MAPK–NF-κB signaling axis. Moreover, OSS-induced EC phenotypic changes resulted in proliferation and extracellular matrix (ECM) protein up-regulation in smooth muscle cells (SMCs) through the RANTES-mediated paracrine mechanism. IL-37 suppressed OSS-induced pro-inflammatory EndMT and thereby abrogated SMC proliferation and ECM protein remodeling. Overall, this study provides insights into endothelial heterogeneity under atheroprone shear stress and identifies the mechanistic role of a novel EC subtype in promoting adverse vascular remodeling. Further, this study demonstrates that anti-inflammatory approach is capable of mitigating vascular pathobiology evoked by atheroprone shear stress.
INTRODUCTION
Atherosclerosis, a chronic vascular disorder characterized by endothelial dysfunction and formation of atheromatous plaque within the artery wall, remains the leading cause of cardiovascular morbidity and mortality worldwide (1). In adults, atherosclerotic lesions preferentially form at arterial bifurcations and branching points, where blood flow exerts low and bidirectional oscillatory shear stress (OSS), typically ranging ±4 dynes/cm2 to the vascular wall (2, 3). The endothelial monolayer in coronary arteries is constantly exposed to hemodynamic shear stress that evokes local vascular pathobiology (4). It is known that shear stress exerts a wide range of hemodynamic forces to the vascular wall and leads to regional heterogeneity of endothelial gene expression (5). However, the response of individual cells to hemodynamic shear stress and the contribution of differential cellular responses to vascular pathobiology associated with atherogenesis are not fully understood.
The initiation of atherosclerosis arises from complex interactions of circulating factors, tissue factors, and different cell types, including endothelial cells (ECs), lymphocytes, macrophages, and smooth muscle cells (SMCs). Among them, ECs have been identified as triggers for atherogenesis, and SMCs are the key players in extracellular matrix (ECM) remodeling (6). It is well known that atherogenesis involves endothelial dysfunction and dysregulated interactions between ECs and SMCs (7). Aberrant production of endothelial mediators, such as nitric oxide (8), platelet-derived growth factor (9), and extracellular vesicles (10), stimulates SMC migration and proliferation. Therefore, investigating EC-SMC interactions under the clinically relevant hemodynamic shear is needed for a better understanding of atherogenesis.
Commonly used in vitro models in atherogenesis studies are two-dimensional (2D) models that use cone-and-plate viscometers or microfluidic devices (11–13). Previous studies revealed that low shear stress can induce endothelial-to-mesenchymal transition (EndMT) (3, 14, 15). Moreover, accumulating evidence from these studies points to an important role of EndMT in vascular pathobiology associated with atherosclerosis (16). While these 2D platforms are useful and conventional tools for assessing the effect of hemodynamic shear stress, they lack a 3D microenvironment and/or EC-SMC interactions.
Anti-inflammatory cytokines have therapeutic potential for chronic inflammatory diseases, including cardiovascular diseases (17). Among anti-inflammatory cytokines, interleukin-37 (IL-37) has attracted much attention due to its potent anti-inflammatory properties (18). A number of studies demonstrate that IL-37 protects against tissue injury or disease progression in animal models of cardiovascular pathology, including ischemia/reperfusion injury (19), myocardial infarction and adverse remodeling (20), atherosclerosis (21), and vascular calcification (22). It is likely that this anti-inflammatory cytokine can exert a beneficial effect in suppressing pro-atherogenic responses in vascular cells.
Recent advances in bioengineering and microfabrication made it possible to develop 3D organ-on-a-chip models that recapitulate in vivo multicellular architecture and the physiological microenvironment (23, 24). Here, we adapted this approach to engineering a human coronary artery–on–a–chip system to investigate the mechanism by which hemodynamic shear stress induces EC phenotypic changes to promote vascular remodeling. The purposes of this study are as follows: (i) to systemically analyze EC phenotypic changes in response to OSS, (ii) to evaluate the impact of the changes in ECs on SMC atherogenic activity, (iii) to identify the molecule that is responsible for EC-SMC interaction, and (iv) to explore approaches for suppression of the pro-atherogenic activity in ECs.
RESULTS
Engineering of the human coronary artery–on–a–chip system
A three-layered coronary artery–on–a–chip device containing two parallel channels was fabricated via soft lithography and is displayed in fig. S1. Immunostaining of EC biomarker CD31 (green) and SMC biomarker α-smooth muscle actin (α-SMA; red) demonstrated the formation of desired cellular compartmentalization in this coronary artery–on–a–chip system (fig. S1, C and D). 3D microscopic analyses confirmed that ECs and SMCs were uniformly distributed on the opposite side of the membrane (fig. S1, C and D, and movie S1). When cells become confluent, OSS was introduced to the upper chamber (endothelial chamber) using a syringe pump to mimic shear stress values experienced at atheroprone regions of arteries (fig. S1, E and F). A computational fluid dynamic simulation was implemented to evaluate local shear stress distribution within a microfluidic channel. The shear stress was controlled in a range of 0 ± 4 dynes/cm2 (fig. S1G). This microsystem enabled accurate replication of the dynamic microenvironment within a human coronary artery.
scRNA-seq identifies a pro-inflammatory EndMT subpopulation induced by OSS
Emerging evidence suggests that shear stress modulates vascular tone and vascular remodeling by influencing EC function (25). To gain insight into the transcriptional changes at a single-cell level, single-cell RNA sequencing (scRNA-seq) was performed in ECs exposed to OSS for 24 hours. After stringent filtering, 4749 cells with high quality, including 2433 cells in the control group and 2316 cells in the OSS group, were obtained to conduct subsequent analyses (26). Uniform manifold approximation and projection (UMAP) analysis showed a clear separation between control and OSS groups (Fig. 1A). Four transcriptionally distinct clusters were identified on the basis of their gene expression profiles: quiescent, pro-inflammatory EndMT, EndMT, and pro-inflammatory clusters (Fig. 1B). We observed a marked shift in cell phenotype following OSS, with drastically reduced numbers of cells in the quiescent (QET) cluster (18.6% versus 86.8% in controls) and greatly increased numbers of cells in the pro-inflammatory EndMT cluster (57.9% versus 0.8% in controls; Fig. 1C).
Fig. 1. scRNA-seq analysis reveals that OSS induces pro-inflammatory EndMT in ECs.

(A) UMAP representation of merged dataset with cells color-coded by experimental condition (OSS or Ctrl). (B) UMAP representation of single-cell gene expression showing the four identified clusters. (C) Stacked bar plot showing the relative proportion of clusters in each sample, with total cell counts indicated. (D) Heatmap of unsupervised clustering analysis featuring the top 10 discriminative genes per cluster. (E) UMAP feature plots demonstrating differential expression of selected EndMT genes (PECAM1, VWF, CDH5, VIM, ACTA2, and COL1A1) and genes associated with inflammation (ICAM1, IL6, and CXCL8) in each cluster. (F) Split view of UMAP representation of the segregation of clusters under experimental conditions.
To validate each cluster, we present the expression of marker genes for each cluster in a heatmap (Fig. 1D). Four representative genes (COL4A5, COL3A1, PTX3, and NFKB1) were selected from the four clusters to confirm their characteristics (fig. S2). Further, using classical endothelial biomarkers (PECAM1, VWF, and CDH5), mesenchymal biomarkers (VIM, ACTA2, and COL1A1), and pro-inflammatory biomarkers (ICAM1, IL6, and CXCL8), we validated the identity of each cluster as assigned (Fig. 1E and fig. S3). When we projected single cells from the control and OSS groups side by side, we observed the expansion of a pro-inflammatory EndMT cluster (57.9%, green) and the reduction of a QET cluster (18.6%, red) in the OSS group (Fig. 1F).
Among the four clusters, QET and pro-inflammatory EndMT subpopulations were chosen for sequential analysis as they displayed the most notable shift after OSS (Fig. 2A). There were 2002 genes down-regulated and 1486 genes up-regulated in pro-inflammatory EndMT compared with QET (Fig. 2B). To further characterize the overarching biological process, we performed gene set enrichment analysis (GSEA) with the hallmark gene sets. We identified 20 positively enriched gene sets with false discovery rate (FDR) < 0.25, and the top sets were shown in Fig. 2C. The most significantly enriched were those for epithelial-to-mesenchymal transition (EMT) and inflammatory networks (Fig. 2D). Violin plots confirmed that mesenchymal marker genes (e.g., ACTA2 and VIM) and pro-inflammatory marker genes (e.g., CXCL8 and IL1B) are enriched in the pro-inflammatory EndMT subpopulation compared to the QET subset (Fig. 2E).
Fig. 2. GSEA uncovers enrichment of EMT and inflammatory responses in pro-inflammatory EndMT (PIM-EndMT) population.

(A) UMAP representation of two major subpopulations under experimental conditions. (B) Volcano plot showing statistical significance (P value) versus fold change for differentially expressed genes in cluster QET and cluster PIM-EndMT. (C) GSEA hallmark analysis of the top pathways significantly up-regulated in PIM-EndMT versus QET ECs. (D) GSEA enrichment plots showing that EMT and inflammatory markers are enriched in PIM-EndMT cluster. (E) Violin plots showing the distribution of specific mesenchymal and pro-inflammatory markers in QET and PIM-EndMT clusters. Each dot represents a single cell. NES, normalized enrichment score.
OSS induces pro-inflammatory EndMT through the Notch1/p38 MAPK–NF-κB signaling axis
To validate pro-inflammatory EndMT at cellular and molecular levels, we performed double immunofluorescence staining and immunoblotting. Upon OSS treatment, ECs changed gradually from a cobblestone appearance to an elongated spindle-shaped morphology (Fig. 3A), and the emergence of yellow color in 3D immunofluorescence microscopic image indicates the presence of mesenchymal biomarkers α-SMA/ACTA2 in ECs (Fig. 3, B and C, and movie S2). We further observed a significant reduction in CD31/PECAM1 and a marked increase in α-SMA and vimentin/VIM in OSS-treated ECs (Fig. 3D). These findings suggest the presence of cells in intermediate stages of EndMT (27). Immunoblotting confirmed that OSS increased the expression of α-SMA and vimentin and reduced the expression of CD31 (Figs. 3E and 4A). Concordantly, a substantial increase in intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) was observed (Figs. 3F and 4B). These results demonstrate that OSS induces pro-inflammatory EndMT in ECs and thereby elevates cellular inflammatory activity.
Fig. 3. OSS induces pro-inflammatory EndMT through Notch1/p38 MAPK–NF-κB signaling axis in ECs.

(A) Representative phase-contrast light microscopy images showing morphological changes in ECs. Scale bars, 100 μm. (B) A 3D reconstruction of cocultured human coronary artery ECs (CD31, green; α-SMA, red) and human coronary artery SMCs (α-SMA, red) after OSS exposure. Scale bar, 50 μm. (C) Confocal laser micrograph of coculture sample after OSS exposure viewed from the x-z plane. (D) Representative images of double immunofluorescence staining showing reduced levels of CD31 (green) and increased levels of α-SMA and vimentin (red) in ECs after being exposed to OSS for 24 hours. Scale bars, 20 μm. (E) Immunoblotting confirmed the changes in protein levels of CD31, α-SMA, and vimentin following OSS. (F) The expression levels of VCAM-1 and ICAM-1 were assessed by immunoblotting. (G) Gene ontology enrichment analysis showing significantly up-regulated gene sets in the pro-inflammatory EndMT cluster relative to the QET cluster. (H) Representative immunoblots showing OSS-induced p38 MAPK phosphorylation and Notch1 cleavage peaked at 2 hours of OSS, while NF-κB phosphorylation reached the highest level at 4 hours of OSS. (I) Western blots and (J) immunofluorescence staining showing attenuated NF-κB phosphorylation and nuclear translocation following OSS exposure for 4 hours in the presence of DAPT or SB203580. (K) Representative immunoblots show that inhibition of NF-κB with Bay 11-7082 (10 μM) suppresses EndMT in ECs exposed to OSS for 24 hours. Densitometric data of immunoblots are presented in fig. S4.
Fig. 4. OSS-induced EC phenotypic changes cause proliferation and ECM protein remodeling in SMCs via RANTES-mediated paracrine mechanism.
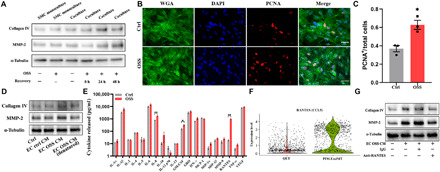
(A) ECs and SMCs were cocultured on the microchip, and the chips were incubated for another 0 to 48 hours after ECs were exposed to OSS stimulation for 24 hours. Representative immunoblotting showing increased collagen IV and MMP-2 levels in SMCs. (B) Immunofluorescence images displaying increased expression of PCNA in SMCs. (C) PCNA+ cells were counted three times in at least four high-power fields by two evaluators. Scale bars, 50 μm. Data are expressed as means ± SEM of three independent experiments. *P < 0.05. (D) SMCs were treated with different CM as indicated for 48 hours, and the protein levels of collagen IV and MMP-2 were measured by Western blot analysis. (E) Multiplex ELISA for EC pro-inflammatory cytokine and chemokine expression with or without OSS treatment. Data are expressed as means ± SEM of four independent experiments. *P < 0.05 and **P < 0.01. (F) Violin plots showing relative expression of RANTES (CCL5) in QET and pro-inflammatory EndMT clusters. (G) SMCs were treated for 48 hours with EC OSS CM preincubated with RANTES neutralizing antibody (10 μg/ml) or nonimmune mouse immunoglobulin G (IgG; 10 μg/ml) for 1 hour, and the protein levels of collagen IV and MMP-2 were measured by Western blot analysis. Densitometric data of immunoblots are presented in fig. S6.
Using pathway analysis, we found marked up-regulation of several signaling molecules in the pro-inflammatory EndMT population, including nuclear factor κB (NF-κB), p38 mitogen-activated protein kinase (MAPK), and Notch1, that could be activated by shear stress (Fig. 3G) (28–30). As shown in Fig. 3H and fig. S4C, Notch1, p38 MAPK, and NF-κB were activated by OSS. The levels of Notch1 and p38 MAPK activation peaked at 2 hours of OSS and declined with the prolongation of the exposure. In contrast, NF-κB activation peaked at 4 hours of OSS and remained at an elevated level at 12 and 24 hours of OSS (fig. S5). Because Notch1 activation and p38 MAPK phosphorylation preceded NF-κB activation, we determined the link of Notch1 and p38 MAPK with NF-κB activation. We found that inhibition of either Notch1 or p38 MAPK suppressed NF-κB phosphorylation (Fig. 3I and fig. S4D) and nuclear translocation (Fig. 3J). Inhibition of NF-κB with Bay 11-7082 suppressed EndMT in ECs exposed to OSS for 24 hours (Fig. 3K and fig. S4E). Together, these data demonstrate that OSS induces NF-κB activation through the Notch1 and p38 MAPK signaling pathways and that NF-κB activation is critical for the induction of pro-inflammatory EndMT by OSS.
To confirm that pro-inflammatory EndMT occurs in vivo, we examined aortic arch and descending thoracic aorta in mice using triple immunofluorescence staining of endothelial biomarker CD31, inflammatory biomarker ICAM-1, and mesenchymal biomarkers vimentin and α-SMA. En face image of murine aortic endothelium displays a tight monolayer of ECs recognized by CD31 immunoreactivity. The aortic arch area has higher densities of ECs with positive staining for α-SMA and vimentin in comparison to areas of descending thoracic aorta (n = 5 mice; fig. S6). In addition, cells expressing mesenchymal biomarkers have lower levels of CD31. It is noteworthy that the inflammatory biomarker ICAM-1 (gray) was colocalized in cells expressing mesenchymal biomarkers (α-SMA and vimentin, red) in the aortic arch. Quantification data confirmed the occurrence of pro-inflammatory EndMT in 12 to 15% of ECs in this area (fig. S7). Further, ECs in this area had greater levels of phosphorylated p38 MAPK (fig. S8). It has been reported that the endothelium of aortic arch sustains OSS due to constantly disturbed blood flow, and ECs in aortic arch display an atheroprone gene expression profile (28, 31). Our observations in laboratory animals confirm that pro-inflammatory EndMT occurs in vivo in this aortic segment and is associated with spontaneous p38 MAPK activation.
EC phenotypic transition induces SMC proliferation and ECM protein remodeling
Previous studies indicate a role for EndMT in plaque formation and ECM protein remodeling in atherosclerosis (16, 32, 33). To determine the impact of ECs on SMCs, we subjected ECs in coculture to OSS and examined ECM protein expression in SMCs. As shown in Fig. 4A and fig. S9A, the expression of ECM protein collagen IV and matrix metalloproteinase-2 (MMP-2) in SMCs was not changed when OSS was applied to SMC monoculture or when SMCs were cocultured with ECs in the absence of OSS. Up-regulation of collagen IV and MMP-2 was observed in SMCs when they were cocultured with ECs exposed to OSS. Further, SMC proliferation assay with staining of proliferating cell nuclear antigen (PCNA) revealed increased proliferation when SMCs were cocultured with ECs exposed to OSS (Fig. 4, B and C). Together, these data demonstrate that ECs exposed to OSS gain the ability to promote SMC proliferation and ECM protein remodeling.
Because the pore of the membrane on the coronary artery–on–a–chip was too small for cellular migration to occur, we hypothesized that ECs exposed to OSS exert an impact on SMCs through a paracrine mechanism. To test this hypothesis, we obtained culture supernatants [conditioned medium (CM)] from ECs under different conditions and used these media to treat SMCs. As shown in Fig. 4D and fig. S9B, CM from OSS-treated ECs up-regulated the expression of collagen IV and MMP-2 in SMCs. Denature of such CM by heat abrogated its effect on collagen IV and MMP-2 expression in SMCs.
As the pro-inflammatory EndMT population is the majority (57.9%) of ECs following exposure to OSS, it is likely that this previously unknown EndMT population promotes SMC atherogenic activity by a pro-inflammatory mechanism. We performed multiplex enzyme-linked immunosorbent assay (ELISA) analysis and compared 20 pro-inflammatory cytokines and chemokines in CM from control ECs and OSS-treated ECs. OSS evoked a substantial increase in the levels of IL-1β (2-fold), IL-6 (2-fold), IL-8 (2-fold), granulocyte-macrophage colony-stimulating factor (GM-CSF; 2-fold), and RANTES (55-fold). Among all pro-inflammatory cytokines and chemokines, RANTES presented the most remarkable increase in CM from OSS-treated ECs compared to CM from control ECs (Fig. 4E). Our scRNA-seq finding verified that RANTES/CCL5 was significantly up-regulated in the pro-inflammatory EndMT population (Fig. 4F). Further, we found that neutralization of RANTES by adding a specific antibody to CM from OSS-treated ECs markedly reduced the potency of this CM for up-regulation of collagen IV and MMP-2 (Fig. 4G and fig. S9C). These findings support a key role for RANTES in the paracrine mechanism by which the novel pro-inflammatory EndMT population promotes the atherogenic activity in SMCs.
IL-37 suppresses endothelial phenotypic transition to abrogate atherogenic activity in SMCs
IL-37 is an anti-inflammatory cytokine and has been used in clinical studies as an inhibitor of innate immunity (34). To explore its potential for suppression of OSS-induced pro-inflammatory EndMT in coronary ECs, we examined EC NF-κB phosphorylation in the presence of recombinant human IL-37 (rhIL-37). The results showed that rhIL-37 inhibited NF-κB activation as potently as the NF-κB inhibitor Bay 11-7082 did (fig. S10). As displayed in Fig. 5A and fig. S11A, treatment with rhIL-37 significantly reduced OSS-induced EndMT. As expected, rhIL-37 abolished the up-regulation of ICAM-1 and VCAM-1 (Fig. 5B and fig. S11B) and NF-κB intranuclear translocation (Fig. 5C) induced by OSS. Overall, rhIL-37 inhibits pro-inflammatory signaling to suppress EndMT induced by OSS.
Fig. 5. IL-37 suppresses EC phenotypic changes and thereby abolishes ECM protein remodeling caused by OSS.

(A) Representative immunoblots and (C) fluorescence imaging showing the suppression of rhIL-37 on OSS-induced expression changes in CD31, α-SMA, and vimentin. (B) Immunofluorescence staining confirming that rhIL-37 abolished up-regulation of ICAM-1 and VCAM-1. (D) Immunofluorescence images of proliferating cells in SMCs after ECs were exposed to OSS for 24 hours in the absence or presence of rhIL-37, followed by incubation for 48 hours. Scale bars, 50 μm. (E) PCNA+ cells were counted three times in at least four high-power fields by two evaluators. Data are expressed as means ± SEM of three independent experiments. *P < 0.05. (F) Same as (D) for the ECM protein (collagen IV and MMP-2) analysis by immunoblotting. (G) ECs were transfected with either IL-37 plasmid or empty control vector. Immunoblotting analysis of OSS-induced EndMT in ECs overexpressing IL-37 or an empty control vector. (H) Same as (G) for the ECM protein (collagen IV and MMP-2) analysis by immunoblotting in SMCs. Densitometric data of immunoblots are presented in fig. S8.
Then, we examined whether rhIL-37 modulates atherogenic activity in SMCs. Immunofluorescence staining revealed that rhIL-37 markedly reduced PCNA expression in SMCs cocultured with ECs exposed to OSS (Fig. 5, D and E). Immunoblotting results confirmed that up-regulation of collagen IV and MMP-2 by OSS-treated ECs was abolished by rhIL-37 (Fig. 5F and fig. S11C).
To confirm that the inhibitory mechanism of rhIL-37 was due to inhibition of phenotypic transition in ECs, we overexpressed IL-37 in ECs by transfection with IL-37–expressing plasmid vectors. As presented in Fig. 5G and fig. S11D, levels of IL-37 were markedly increased upon transfection, and overexpression of IL-37 suppressed the OSS-induced CD31 down-regulation and α-SMA/vimentin up-regulation in ECs. In addition, overexpression of IL-37 in ECs abrogated ECM remodeling in SMCs following exposure of ECs to OSS (Fig. 5H and fig. S11E). Thus, both recombinant rhIL-37 and endogenous IL-37 in ECs are capable of inhibiting EndMT to prevent OSS-elicited atherogenic activation in coronary artery SMCs.
To evaluate the effect of IL-37 in vivo, we examined the expression of endothelial, mesenchymal, and inflammatory biomarkers in the endothelium of the aortic arch and descending thoracic aorta in IL-37 transgenic mice (IL-37tg mice). Consistent with our in vitro observations, en face imaging with triple staining revealed that overexpressed IL-37 markedly reduced the density of ECs expressing α-SMA, vimentin, and ICAM-1 in the aorta, particularly in the aortic arch (fig. S6). These data confirmed that IL-37 suppresses vascular pro-inflammatory EndMT in vivo and indicated that anti-inflammatory approaches may have therapeutic potential for suppression of OSS-induced vascular atherogenic activity.
DISCUSSION
In this study, we leveraged human organ chip technology to establish an in vitro human coronary artery–on–a–chip model integrated with a flow system that mimics blood flow dynamics within atheroprone sites of the coronary arteries. Using scRNA-seq, we revealed that OSS induces heterogeneous phenotype transition in ECs and identified a novel pro-inflammatory EndMT population. We demonstrated that OSS induces pro-inflammatory EndMT through activation of the Notch1/p38 MAPK–NF-κB signaling axis. We revealed that this novel pro-inflammatory EndMT leads to SMC proliferation and ECM protein remodeling mediated mainly by secretion of RANTES (Fig. 6). Moreover, we found that anti-inflammatory cytokine IL-37 inhibits NF-κB to suppress pro-inflammatory EndMT in coronary artery ECs and thereby abrogate SMC pro-atherogenic activity.
Fig. 6. A schematic diagram depicts the mechanism underlying the interaction of stressed ECs with SMCs.

See Discussion for details.
In addition to inducing EndMT, hemodynamic shear stress evokes an inflammatory response in vascular ECs (35). Currently, it is unclear whether ECs undergoing EndMT are pro-inflammatory. In the present study, we observed that human coronary ECs display four distinct clusters after OSS. A novel subset of cells with characteristic EndMT features and pro-inflammatory property, namely, the pro-inflammatory EndMT cells, becomes the majority following OSS. Pro-inflammatory EndMT appears to occur in vivo in atheroprone segments of arteries because we observed the expression of vimentin, α-SMA, and ICAM-1 in ECs of the aortic arch in mice. In this regard, ECs in the aortic arch have been reported to display an atheroprone gene expression profile (28, 31).
Several studies found that inflammatory cytokines, particularly tumor necrosis factor–α (TNF-α) and IL-1β, induce EndMT (36). In the present study, the expression of TNF-α and IL-1β did not increase significantly in ECs exposed to OSS, while the expression of RANTES was markedly increased. Thus, the mechanism underlying EndMT, including pro-inflammatory EndMT, induced by OSS may not involve TNF-α or IL-1β, and these pro-inflammatory cytokines may not play an important role in mediating the effect of pro-inflammatory EndMT. Studies also showed that low shear stress promotes EndMT via activation of transcriptional factors GATA4, TWIST1, and Snail (3, 15). NF-κB is an essential transcriptional factor that mediates cellular inflammatory responses (37). Previous studies found that low shear stress activates the NF-κB signaling pathway (38–40). In the present study, we identified a key role of NF-κB in mediating pro-inflammatory EndMT in human coronary ECs following OSS. Inhibition of NF-κB with Bay 11-7082 markedly reduced pro-inflammatory EndMT. Further, anti-inflammatory cytokine IL-37 inhibited NF-κB and essentially abrogated pro-inflammatory EndMT. Thus, it is NF-κB activation, particularly sustained NF-κB signaling, that mediates the pro-inflammatory EndMT in coronary artery ECs exposed to OSS. As Notch1 and p38 MAPK mediate NF-κB activation in ECs exposed to OSS, our findings indicate that NF-κB is involved in converting the signals from Notch1 and p38 MAPK into the initiation of cell phenotype transition and that this master transcription factor enables the pro-inflammatory EndMT in coronary artery ECs.
SMC proliferation and excessive deposition of ECM proteins have been implicated in the pathogenesis of several cardiovascular conditions, including atherosclerosis (41, 42). Previous studies suggested that EndMT may be a source of myofibroblasts in fibrotic tissue (43, 44). In addition, several studies found that pro-inflammatory ECs can secrete cytokines, such as TNF-α and RANTES, and that these cytokines may play a role in tissue fibrosis (45, 46). Further, ECs can interact directly with SMCs to influence their function, resulting in ECM protein remodeling (47). In the present study, we observed that OSS-induced endothelial phenotypic transition enhances SMC pro-atherogenic activity with a twofold increase in MMP-2 and collagen IV levels, as well as increased density of PCNA-positive SMCs. The changes in SMCs are mainly due to the OSS-induced pro-inflammatory EndMT because they only occur when OSS is applied to cocultured ECs. The changes in SMC activity are markedly attenuated when ECs are treated with anti-inflammatory cytokine IL-37 that suppresses pro-inflammatory EndMT.
The effect of pro-inflammatory EndMT subpopulation on SMCs does not involve physical cell-cell interaction because CM from OSS-treated ECs can evoke comparable changes in SMCs. Apparently, a paracrine mechanism is involved. An important paracrine mediator from OSS-treated ECs is RANTES, and pro-inflammatory EndMT subpopulation is the main source of this cytokine. RANTES has been reported to play a role in the pathogenesis of atherosclerosis (48, 49). Our recent work and studies by others found that RANTES modulates cellular profibrotic activities, including migration, proliferation, and ECM protein production (50, 51). In this study, we observed that neutralization of RANTES suppresses the enhancement of pro-atherogenic activity in SMCs by CM from OSS-treated ECs. Thus, pro-inflammatory EndMT subpopulation enhances SMC pro-atherogenic activity mainly through the secretion of RANTES. It should be noted that neutralization of RANTES greatly reduced, but not abolished, ECM protein up-regulation, indicating that other inflammatory mediators released by ECs, such IL-6 and IL-1β, may also play a role in mediating the paracrine effect of pro-inflammatory EndMT subpopulation on SMCs. Future work should identify other mediators involved.
To explore approaches for suppression of SMC pro-atherogenic response to OSS, we applied rhIL-37 to the EC compartment and found that it potently inhibits the pro-atherogenic response. Detailed analysis revealed that rhIL-37 inhibits EC phenotypic transition, particularly pro-inflammatory EndMT, leading to attenuated SMC proliferation and ECM protein remodeling. These observations provide further support for the causality of EC phenotypic transition and SMC pro-atherogenic activation. The effect of IL-37 on OSS-induced EndMT is correlated with its inhibitory effect on NF-κB activation. In this regard, our previous study demonstrates that IL-37 is a potent NF-κB inhibitor (52). As IL-37 exerts its anti-inflammatory function through both extracellular and intracellular mechanisms, we determined the effect of expression of IL-37 in ECs on SMC pro-atherogenic activity following applying OSS to ECs. The results showed that elevation of levels of endogenous IL-37 in ECs also suppresses cell phenotype transition and prevents SMC pro-atherogenic activation. Together, the results demonstrate that either administration of rhIL-37 or up-regulation of IL-37 expression could suppress EndMT and pro-atherogenic activity in the coronary vasculature. Our observation that aortic arch in IL-37tg mice has a much lower density of ECs expressing mesenchymal and inflammatory biomarkers indicates that elevation of anti-inflammatory capacity may suppress vascular pro-atherogenic activity induced by shear stress.
Collectively, our in vitro and in vivo data demonstrate that pro-inflammatory EndMT occurs in vascular ECs under OSS, although there is a discrepancy between our scRNA-seq data in coronary artery ECs (57.9%) and en face immunofluorescence staining results in mouse aorta (12 to 15%). This apparent discrepancy is due to several differences between the in vitro and in vivo conditions. First, anti-inflammatory factors in the circulation and tissue are absent in the in vitro model. Our observation in the present study demonstrates that the anti-inflammatory cytokine IL-37 markedly suppresses pro-inflammatory EndMT in human coronary artery ECs. Second, RNA sequencing is much more sensitive than immunofluorescence staining of marker proteins for detection of cell phenotype transition. Third, coronary ECs are phenotypically distinct from ECs in the aorta. In this regard, several studies suggest that coronary ECs are susceptible to EndMT in response to physical stress (3, 33). All of the three factors, together with others, contribute to the greater occurrence of pro-inflammatory EndMT in ECs of human coronary artery–on–a–chip in response to OSS exposure. Nevertheless, the novel findings of this study demonstrate that OSS induces pro-inflammatory EndMT in coronary artery ECs through Notch 1/p38 MAPK–mediated NF-κB activation and that this phenotypic transition in ECs promotes SMC activation and subsequent ECM protein remodeling.
The human coronary artery–on–a–chip flow system is a synthetic system that enables the investigation of cellular and molecular contributors to organ-level pathophysiology and provides a novel approach for the exploration of potential therapies. It should be noted that the system used in the present study only applied OSS. The simplicity of this system enabled evaluation of the impact of OSS on coronary vascular cells in comparison to static cells, as well as the investigation of underlying molecular mechanisms. However, the simplicity is also a limitation because many factors, such as varied matrix stiffness and various tissue factors, mediate or modulate cellular stress response. In addition, different patterns of flow have distinct effects on vascular ECs (53). In this regard, laminar flow has been found to elevate extracellular signal–regulated kinase 5 (ERK5) and activate the phosphatidylinositol 3-kinase (PI3K)/AKT and Rho/Rac pathways in vascular ECs, resulting in a vascular protective effect (54, 55). While the effect of laminar flow on vascular ECs has been extensively studied, it is appealing to compare the effects of laminar flow and oscillatory flow on coronary artery ECs in this artery-on-a-chip model. Further studies are needed to address this, as well as the effect of OSS on vascular ECs in the presence of other relevant factors.
In summary, this study unraveled that OSS induces novel pro-inflammatory EndMT in coronary artery ECs via the Notch1/p38 MAPK–NF-κB signaling pathway, and this novel cell phenotype transition leads to SMC pro-atherogenic activation via a paracrine mechanism involving RANTES. Further, this study identified administration or up-regulation of anti-inflammatory cytokine IL-37 as a potential therapeutic strategy for suppression of vascular pro-atherogenic activity evoked by shear stress. Overall, the findings of this study provide new insights into the mechanism of coronary pro-atherogenic response to shear stress and offer a potential target for manipulation.
METHODS
Cell culture and cell transfection
Human coronary artery ECs were purchased from Lonza (Walkersville, MD) and Lifeline Cell Technology (Frederick, MD), maintained in endothelial basal medium-2, supplemented with the EGM-2 Bullet Kit (Lonza). Human coronary artery SMCs were obtained from Lifeline Cell Technology and grown in medium 199 (M199) growth medium (Lonza) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and penicillin/streptomycin (100 U/ml; Gibco). Donors were healthy with an age range of 25 to 50. The empty vector pIRES2–enhanced green fluorescent protein (EGFP) and IL-37 expression plasmid pIRES2–EGFP–IL-37 were provided by C. Dinarello laboratory. EC transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells from passages 3 to 6 were used for this study.
Microfluidic device fabrication
The biomimetic microfluidic device was fabricated by integrating two polydimethylsiloxane (PDMS; Sylgard 184, Dow Corning, USA) channels and a porous polyester membrane. Briefly, the upper and bottom channel molds were 3D printed (Objet350, Stratasys, USA) directly from computer-aided design files (top channel, 1.5 mm wide and 1 mm high; bottom channel, 1.5 mm wide and 1 mm high; length of overlapping channels, 40 mm). The mold replicas were obtained by pouring PDMS mixture [1:10 (w/w) ratio of curing agent to prepolymer] into the molds and cured overnight at 65°C.
For device assembly, access holes were punched in the upper channel mold with a 1-mm biopsy punch after peeling the replicas from molds. Last, the transparent polyester membrane (pores, 0.4 μm; 12 μm thick; it4ip, Belgium) was laser cut and bonded with the two-channel molds under a stereomicroscope after exposure to oxygen plasma (Harrick Scientific, NY, USA) at 18 W for 1 min. To stabilize bonding, the device was subsequently baked at 65°C for 30 min.
Microfluidic cell seeding and flow experiment
To establish a 3D coculture system, ECs and SMCs were seeded on both sides of the membrane. The assembled device was sterilized with ultraviolet irradiation and 70% ethanol. Following sterilization, microchannels were coated with collagen I solution (0.3 mg/ml) at 37°C for 3 hours, then rinsed with phosphate-buffered saline (PBS), and dried in a sterile hood at room temperature (RT) for future use.
To fabricate the coronary artery–on–a–chip, human coronary artery SMCs were first harvested and resuspended in M199 medium containing 10% FBS, penicillin/streptomycin at a density of 2 × 106 cells/ml. Cells were then slowly injected into the lower channel, and the device was immediately inverted for the adherence of cells onto the lower side of the membrane. After incubation at 37°C for 4 hours, human coronary artery ECs (4 × 106 cells/ml) were inducted into the upper channel of the chip. The device was again incubated at 37°C for cell attachment. Unattached cells were gently washed away a few hours later, and the cultures were maintained until fully confluent. The confluence of cell layers was ensured to be optimal (at least 90%) through phase-contrast microscopy before shear stress was applied. The culture medium was then exchanged with a medium that was identical to the previous medium except that it contained only 2.5% FBS. Once complete confluence for both cells was confirmed, the device was connected to a syringe pump (KD Scientific, USA) and bidirectional OSS (±4 dynes/cm2, 1 Hz) was applied to ECs for 24 hours. The shear stress was calculated using the equation τ = 6 μQ/wh2 (56), where τ is the shear stress at the bottom surface of the chamber (dyne/cm2), μ is the fluid viscosity (g/cm·s), Q is the volumetric flow rate (ml/s), h is the height of the top chamber (mm), and w is the width of the chamber (mm). Cells under static culture served as controls.
For different experiments, ECs and SMCs were monocultured or cocultured on the chip. To examine the phosphorylation of NF-κB, p38 MAPK, and Notch1 cleavage, ECs were monocultured and treated with OSS for 1 to 8 hours. To investigate NF-κB phosphorylation and nuclear translocation, ECs were preincubated with N-[N-(3, 5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester (DAPT) (Sigma-Aldrich, 10 μM) or SB203580 (Sigma-Aldrich, 10 μM) for 1 hour followed by OSS treatment for 4 hours. For NF-κB inhibition, ECs were preincubated with Bay 11-7082 (Sigma-Aldrich, 10 μM) or rhIL-37 (R&D Systems, 1 ng/ml) for 1 hour followed by OSS stimulation for 4 or 24 hours. To study the EC-SMC interaction, ECs and SMCs were cocultured on the microchip, ECs were exposed to OSS for 24 hours, and then the microchip was left static after stimulation for another 0 to 48 hours.
Droplet-based scRNA-seq library preparation and sequencing
ECs were collected 24 hours after incubation in the absence or presence of OSS and processed for scRNA library preparation using the Chromium Single Cell Platform (10x Genomics) as per the Chromium Single Cell 3′ Reagent Kits v3 user guide. Briefly, single-cell suspensions were portioned into Gel Bead-In-Emulsions in the Chromium system (10x Genomics) at the Colorado University Anschutz Genomics and Microarray Core for Genome Analysis, followed by cell lysis and barcoded reverse transcription of RNA, complementary DNA amplification and shearing, and 5′ adaptor and sample index attachment. Libraries were sequenced on NovaSeq 6000 (Illumina) for a depth of 50,000 reads per cell.
scRNA-seq data processing
scRNA-seq raw data were first processed through the 10x Genomics Cell Ranger pipeline (v3.1) and then analyzed in R (v3.6) with the Seurat package (http://satijalab.org/seurat/). Low-quality cells (<400 genes per cell and >25% mitochondrial transcript presence per cell) were excluded from analysis. After quality control, 4749 cells (2433 for control and 2316 for OSS) were included in subsequent analysis. Gene expression was log-normalized to a scale factor of 10,000 and then regressed on the number of molecules detected per cell. Highly variable genes were selected and used for principal components analysis. Cells were projected in 2D space using UMAP with default parameters. Using graph-based clustering function, 10 principal components were used in cell clusters with the resolution parameter set at 0.2, resulting in four clusters. The top 10 genes with the highest dispersion were used to construct the heatmap for each cluster.
The Broad Institute GSEA software (https://gsea-msigdb.org/gsea/) was used to run analyses on normalized gene expression data. We used the hallmark gene set database, which includes 50 MSigDB gene sets. In particular, we followed the standard procedure as described by GSEA user guide (http://broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html). The FDR for GSEA is the estimated probability that a gene set with a given normalized enrichment score represents a false-positive finding, and an FDR < 0.25 is considered to be statistically significant for GSEA.
Immunofluorescence imaging
Microfluidic chips were rinsed in PBS and fixed in 4% paraformaldehyde (Sigma-Aldrich) for 15 min at RT and then permeabilized with a methanol/acetone mixture for 15 min. Subsequently, cells were blocked with PBS (5% goat serum, 5% donkey serum, and 1% bovine serum albumin) for 1 hour at RT. The following primary antibodies (table S1) were used for EC and SMC experiments: mouse anti-CD31, rabbit anti–α-SMA, rabbit anti-vimentin, rabbit NF-κB, and mouse anti-PCNA. After being washed three times in PBS, cells were incubated with Alexa Fluor 488– or Cy3-tagged secondary antibody for 1 hour at RT. 4′,6-Diamidino-2-phenylindole (DAPI) was used for nucleus counterstaining. Alexa Fluor 488–tagged wheat germ agglutinin (Invitrogen, 1:200) was used to outline cells when needed. Devices were cut using a razor blade, and membranes were carefully separated from the PDMS and then mounted on microscope slides with mounting medium. Microscopy was performed with a Leica DM5500 B research microscope (Leica Microsystems GmbH, Wetzlar, Germany). 3D reconstitution image was carried out using a confocal laser scanning microscope (Olympus FV1000, Japan). Image processing and Z-stack reconstitution were performed using ImageJ and Imaris software.
Immunoblotting
Immunoblotting was applied to analyze CD31, α-SMA, vimentin, notch intracellular domain (NICD), phosphorylated NF-κB, total NF-κB, phosphorylated p38 MAPK, total p38 MAPK, α-tubulin, ICAM-1, VCAM-1, collagen IV, MMP-2, and IL-37. Different cells were harvested from separate channels, and protein extraction was performed using a commercial sample buffer according to the manufacturer’s instructions [100 mM tris-HCl (pH 6.8), 2% SDS, 0.02% bromophenol blue, and 10% glycerol]. The protein samples were resolved on 4 to 20% SDS–polyacrylamide gel electrophoresis gels and then transferred onto nitrocellulose membranes (Bio-Rad Laboratories, CA) using a wet transfer system. The membranes were blocked with 5% nonfat dry milk solution for 1 hour at RT. The blocked membranes were probed with primary antibodies (table S1) overnight at 4°C. After washing with PBS containing 0.02% Tween 20, the membranes were incubated with a peroxidase-linked secondary antibody specific to the primary antibody for 1 hour at RT. After further washes, the membrane was developed with enhanced chemiluminescence reagents. The signal was detected with Bio-Rad Gel Doc, and band density was analyzed using Image Lab software. Two identical chips per condition were performed at the same time.
Enzyme-linked immunosorbent assay
Quantibody human multiplex cytokine array Q1 (RayBiotech Inc., Peachtree Corners, GA) was used to quantify levels of GM-CSF, GRO (growth-regulated oncogene), TNF-α, IL-1α, IL-1β, IFN-γ (interferon-γ), IL-6, IL-8, IL-10, IL-12 p70, IL-13, IL-2, IL-4, IL-5, MCP-1 (monocyte chemoattractant protein-1), MIP (macrophage inflammatory protein)-1α, MIP-1β, MMP-9, RANTES, and VEGF-A (vascular endothelial growth factor–A) in the CM. Samples and standards were prepared following the manufacturer’s instructions. Scanning and data extraction were performed by RayBiotech Inc. By comparing signals of samples to the standard curve, cytokine concentrations in samples were determined, and results were plotted using Prism Software (GraphPad).
CM and SMC treatment
To generate varied CM, ECs were monocultured and exposed to OSS treatment or left untreated for 24 hours, and cell culture supernatants were collected afterward, described as EC OSS CM and EC control CM, respectively. Proteins in EC OSS CM were denatured by incubating with boiling water for 30 min, described as EC OSS CM (denatured).
SMCs were monocultured on-chip until approximately 90% confluence was reached, different CMs were applied and incubated for 48 hours, and medium was changed daily. Fresh SMC culture medium was used as control.
To neutralize RANTES in the EC OSS CM, the CM was treated with RANTES neutralizing antibody (R&D Systems, MN, USA) at 10 μg/ml for 1 hour at 37°C before applying the CM to SMC culture. Aliquots of the CM were treated with nonimmune immunoglobulin G (IgG) and used as controls.
Animal experiments
C57BL/6 mice of 12 to 14 months old were obtained from the National Institute on Aging (Bethesda, MD, USA). IL-37tg mice of 12 to 14 months old were from our colonies at the University of Colorado Anschutz Medical Campus. No unique abnormality in organ/tissue has been observed in IL-37tg mice. The experiments were approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver, and this investigation conforms to the Guide for the Care and Use of Laboratory Animals (National Research Council, revised 1996).
All animals in this study were acclimated for at least 14 days before the experiments in an animal facility with a 12:12-hour light-dark cycle and free access to water and regular chow diet. The heart with aorta was harvested from five mice of each genotype using the procedure reported in a previous study (57). Briefly, four steps include (i) isolation of heart and aorta, (ii) removal of fatty tissue, (iii) immobilization of the artery on a silicone layer and making longitudinal opening, and (iv) fixation and antibody staining from the lumen side of the vessel.
For immunostaining, aorta segments were fixed with 4% paraformaldehyde for 15 min. After permeabilization/blocking in 0.05% Triton X-100 in PBS and 10% goat and donkey serum in PBS for 1 hour at RT, vessels were incubated at 4°C overnight with diluted primary antibodies (table S1). After washing with PBS three times, vessels were incubated with diluted secondary antibodies (Cy3-, Cy5-, or Alexa Fluor 488–tagged; Thermo Fisher Scientific, 1:200 dilution) and DAPI for 1 hour at RT. Fluorescence signals were detected using a Zeiss LSM780 confocal laser scanning microscope, and image analysis and quantification were performed using the Zeiss Zen Lite software.
Statistical analysis
Data are presented as means ± SEM. GraphPad Prism 8.0 software was used for the statistical analyses, statistical difference between two groups was determined using Student’s t test, and differences between more than two groups was determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. Nonparametric Mann-Whitney U test was performed to confirm the difference between two-group comparison. For multiple group comparisons, nonparametric Kruskal-Wallis test was performed to confirm the differences. P values less than 0.05 were considered statistically significant.
Acknowledgments
Funding: This study was supported, in part, by NIH grants R01HL106582 and R01HL121776. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank the Genomics Core Laboratory of University of Colorado Anschutz Medical Campus for providing support and assistance in performing scRNA-seq. Imaging and image analysis were performed in the Advanced Microscopy Core of NeuroTechnology Center at the University of Colorado Anschutz Medical Campus, and this Core Laboratory is supported, in part, by Rocky Mountain Neurological Disorders Core grant number P30 NS048154 and by Diabetes Research Center grant number P30 DK116073. Author contributions: X.M., D.A.F., C.A.D., and P.Z. conceived and designed the project. P.Z., Q.Y., P.-J.Z., E.T., L.A., and Y.Z. performed the experiments. P.Z., Q.Y., and Y.Z. analyzed the data. P.Z., Q.Y., and M.J.J. drafted the manuscript. X.M., C.A.D., and D.A.F. reviewed and revised the manuscript. All authors discussed the results and approved the final version of this manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabg1694/DC1
REFERENCES AND NOTES
- 1.Benjamin E. J., Muntner P., Alonso A., Bittencourt M. S., Callaway C. W., Carson A. P., Chamberlain A. M., Chang A. R., Cheng S., Das S. R., Delling F. N., Djousse L., Elkind M. S. V., Ferguson J. F., Fornage M., Jordan L. C., Khan S. S., Kissela B. M., Knutson K. L., Kwan T. W., Lackland D. T., Lewis T. T., Lichtman J. H., Longenecker C. T., Loop M. S., Lutsey P. L., Martin S. S., Matsushita K., Moran A. E., Mussolino M. E., O’Flaherty M., Pandey A., Perak A. M., Rosamond W. D., Roth G. A., Sampson U. K. A., Satou G. M., Schroeder E. B., Shah S. H., Spartano N. L., Stokes A., Tirschwell D. L., Tsao C. W., Turakhia M. P., Van Wagner L. B., Wilkins J. T., Wong S. S., Virani S. S.; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee , Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 139, e56–e528 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Malek A. M., Alper S. L., Izumo S., Hemodynamic shear stress and its role in atherosclerosis. JAMA 282, 2035–2042 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Mahmoud M. M., Serbanovic-Canic J., Feng S., Souilhol C., Xing R., Hsiao S., Mammoto A., Chen J., Ariaans M., Francis S. E., Van der Heiden K., Ridger V., Evans P. C., Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci. Rep. 7, 3375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glagov S., Zarins C., Giddens D. P., Ku D. N., Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch. Pathol. Lab. Med. 112, 1018–1031 (1988). [PubMed] [Google Scholar]
- 5.Davies P. F., Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 6, 16–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakagami H., Kaneda Y., Ogihara T., Morishita R., Endothelial dysfunction in hyperglycemia as a trigger of atherosclerosis. Curr. Diabetes Rev. 1, 59–63 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Li M., Qian M., Kyler K., Xu J., Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Front. Cardiovasc. Med. 5, 151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu J., Zhang Y., Zhang X., Rudic R. D., Bauer P. M., Altieri D. C., Sessa W. C., Endothelium derived nitric oxide synthase negatively regulates the PDGF-survivin pathway during flow-dependent vascular remodeling. PLOS ONE 7, e31495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi Y.-X., Jiang J., Jiang X.-H., Wang X.-D., Ji S.-Y., Han Y., Long D.-K., Shen B.-R., Yan Z.-Q., Chien S., Jiang Z.-L., PDGF-BB and TGF-β1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc. Natl. Acad. Sci. U.S.A. 108, 1908–1913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hafiane A., Daskalopoulou S. S., Extracellular vesicles characteristics and emerging roles in atherosclerotic cardiovascular disease. Metabolism 85, 213–222 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Kouzbari K., Hossan M. R., Arrizabalaga J. H., Varshney R., Simmons A. D., Gostynska S., Nollert M. U., Ahamed J., Oscillatory shear potentiates latent TGF-β1 activation more than steady shear as demonstrated by a novel force generator. Sci. Rep. 9, 6065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang J., Saha A., Boo Y. C., Sorescu G. P., McNally J. S., Holland S. M., Dikalov S., Giddens D. P., Griendling K. K., Harrison D. G., Jo H., Oscillatory shear stress stimulates endothelial production of O2-from p47phox -dependent NAD(P)H oxidases, leading to monocyte adhesion. J. Biol. Chem. 278, 47291–47298 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Sei Y. J., Ahn S. I., Virtue T., Kim T., Kim Y., Detection of frequency-dependent endothelial response to oscillatory shear stress using a microfluidic transcellular monitor. Sci. Rep. 7, 10019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moonen J.-R. A. J., Lee E. S., Schmidt M., Maleszewska M., Koerts J. A., Brouwer L. A., van Kooten T. G., van Luyn M. J., Zeebregts C. J., Krenning G., Harmsen M. C., Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 108, 377–386 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Mahmoud M. M., Kim H. R., Xing R., Hsiao S., Mammoto A., Chen J., Serbanovic-Canic J., Feng S., Bowden N. P., Maguire R., Ariaans M., Francis S. E., Weinberg P. D., van der Heiden K., Jones E. A., Chico T. J., Ridger V., Evans P. C., TWIST1 integrates endothelial responses to flow in vascular dysfunction and atherosclerosis. Circ. Res. 119, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evrard S. M., Lecce L., Michelis K. C., Nomura-Kitabayashi A., Pandey G., Purushothaman K. R., d’Escamard V., Li J. R., Hadri L., Fujitani K., Moreno P. R., Benard L., Rimmele P., Cohain A., Mecham B., Randolph G. J., Nabel E. G., Hajjar R., Fuster V., Boehm M., Kovacic J. C., Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 7, 11853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinarello C. A., Anti-inflammatory agents: Present and future. Cell 140, 935–950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavalli G., Dinarello C. A., Suppression of inflammation and acquired immunity by IL-37. Immunol. Rev. 281, 179–190 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Yang Y., Zhang Z. X., Lian D., Haig A., Bhattacharjee R. N., Jevnikar A. M., IL-37 inhibits IL-18-induced tubular epithelial cell expression of pro-inflammatory cytokines and renal ischemia-reperfusion injury. Kidney Int. 87, 396–408 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Zhu R., Sun H., Yu K., Zhong Y., Shi H., Wei Y., Su X., Xu W., Luo Q., Zhang F., Zhu Z., Meng K., Zhao X., Liu Y., Mao Y., Cheng P., Mao X., Zeng Q., Interleukin-37 and dendritic cells treated with interleukin-37 plus troponin I ameliorate cardiac remodeling after myocardial infarction. J. Am. Heart Assoc. 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCurdy S., Baumer Y., Toulmin E., Lee B. H., Boisvert W. A., Macrophage-specific expression of IL-37 in hyperlipidemic mice attenuates atherosclerosis. J. Immunol. 199, 3604–3613 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Chai M., Ji Q., Zhang H., Zhou Y., Yang Q., Zhou Y., Guo G., Liu W., Han W., Yang L., Zhang L., Liang J., Liu Y., Shi D., Zhao Y., The protective effect of interleukin-37 on vascular calcification and atherosclerosis in apolipoprotein E-deficient mice with diabetes. J. Interferon Cytokine Res. 35, 530–539 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Benam K. H., Villenave R., Lucchesi C., Varone A., Hubeau C., Lee H. H., Alves S. E., Salmon M., Ferrante T. C., Weaver J. C., Bahinski A., Hamilton G. A., Ingber D. E., Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 13, 151–157 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Novak R., Ingram M., Marquez S., Das D., Delahanty A., Herland A., Maoz B. M., Jeanty S. S. F., Somayaji M. R., Burt M., Calamari E., Chalkiadaki A., Cho A., Choe Y., Chou D. B., Cronce M., Dauth S., Divic T., Fernandez-Alcon J., Ferrante T., Ferrier J., FitzGerald E. A., Fleming R., Jalili-Firoozinezhad S., Grevesse T., Goss J. A., Hamkins-Indik T., Henry O., Hinojosa C., Huffstater T., Jang K.-J., Kujala V., Leng L., Mannix R., Milton Y., Nawroth J., Nestor B. A., Ng C. F., O’Connor B., Park T.-E., Sanchez H., Sliz J., Sontheimer-Phelps A., Swenor B., Thompson G., Touloumes G. J., Tranchemontagne Z., Wen N., Yadid M., Bahinski A., Hamilton G. A., Levner D., Levy O., Przekwas A., Prantil-Baun R., Parker K. K., Ingber D. E., Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 4, 407–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peyton K. J., Liu X.-m., Durante W., Prolonged cyclic strain inhibits human endothelial cell growth. Front. Biosci. 8, 205–212 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I. W., Ng L. G., Ginhoux F., Newell E. W., Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Dong J., Hu Y., Fan X., Wu X., Mao Y., Hu B., Guo H., Wen L., Tang F., Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 19, 31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mack J. J., Mosqueiro T. S., Archer B. J., Jones W. M., Sunshine H., Faas G. C., Briot A., Aragon R. L., Su T., Romay M. C., McDonald A. I., Kuo C. H., Lizama C. O., Lane T. F., Zovein A. C., Fang Y., Tarling E. J., de Aguiar Vallim T. Q., Navab M., Fogelman A. M., Bouchard L. S., Iruela-Arispe M. L., NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 8, 1620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gee E., Milkiewicz M., Haas T. L., p38 MAPK activity is stimulated by vascular endothelial growth factor receptor 2 activation and is essential for shear stress-induced angiogenesis. J. Cell. Physiol. 222, 120–126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sumpio B. E., Yun S., Cordova A. C., Haga M., Zhang J., Koh Y., Madri J. A., MAPKs (ERK½, p38) and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J. Biol. Chem. 280, 11185–11191 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Dai G., Kaazempur-Mofrad M. R., Natarajan S., Zhang Y., Vaughn S., Blackman B. R., Kamm R. D., García-Cardeña G., Gimbrone M. A. Jr., Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. U.S.A. 101, 14871–14876 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeisberg E. M., Tarnavski O., Zeisberg M., Dorfman A. L., McMullen J. R., Gustafsson E., Chandraker A., Yuan X., Pu W. T., Roberts A. B., Neilson E. G., Sayegh M. H., Izumo S., Kalluri R., Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Chen P.-Y., Qin L., Baeyens N., Li G., Afolabi T., Budatha M., Tellides G., Schwartz M. A., Simons M., Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Invest. 125, 4514–4528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Quan Y., Yue Y., Heng X., Che F., Interleukin-37: A crucial cytokine with multiple roles in disease and potentially clinical therapy. Oncol. Lett. 15, 4711–4719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feaver R. E., Gelfand B. D., Blackman B. R., Human haemodynamic frequency harmonics regulate the inflammatory phenotype of vascular endothelial cells. Nat. Commun. 4, 1525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho J. G., Lee A., Chang W., Lee M.-S., Kim J., Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 9, 294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhatt D., Ghosh S., Regulation of the NF-κB-mediated transcription of inflammatory genes. Front. Immunol. 5, 71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganguli A., Persson L., Palmer I. R., Evans I., Yang L., Smallwood R., Black R., Qwarnstrom E. E., Distinct NF-κB regulation by shear stress through ras-dependent IκBα oscillations: Real-time analysis of flow-mediated activation in live cells. Circ. Res. 96, 626–634 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Tzima E., Del Pozo M. A., Kiosses W. B., Mohamed S. A., Li S., Chien S., Schwartz M. A., Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 21, 6791–6800 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chien S., Li S., Shyy J. Y., Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension 31, 162–169 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Chistiakov D. A., Sobenin I. A., Orekhov A. N., Vascular extracellular matrix in atherosclerosis. Cardiol. Rev. 21, 270–288 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Lan T.-H., Huang X.-Q., Tan H.-M., Vascular fibrosis in atherosclerosis. Cardiovasc. Pathol. 22, 401–407 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Piera-Velazquez S., Jimenez S. A., Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 99, 1281–1324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun X., Nkennor B., Mastikhina O., Soon K., Nunes S. S., Endothelium-mediated contributions to fibrosis. Semin. Cell Dev. Biol. 101, 78–86 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Lin C. S., Hsieh P. S., Hwang L. L., Lee Y. H., Tsai S. H., Tu Y. C., Hung Y. W., Liu C. C., Chuang Y. P., Liao M. T., Chien S., Tsai M. C., The CCL5/CCR5 axis promotes vascular smooth muscle cell proliferation and atherogenic phenotype switching. Cell. Physiol. Biochem. 47, 707–720 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Jean-Charles P. Y., Wu J. H., Zhang L., Kaur S., Nepliouev I., Stiber J. A., Brian L., Qi R., Wertman V., Shenoy S. K., Freedman N. J., USP20 (ubiquitin-specific protease 20) inhibits TNF (tumor necrosis factor)-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 38, 2295–2305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fillinger M. F., Sampson L. N., Cronenwett J. L., Powell R. J., Wagner R. J., Coculture of endothelial cells and smooth muscle cells in bilayer and conditioned media models. J. Surg. Res. 67, 169–178 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Veillard N. R., Kwak B., Pelli G., Mulhaupt F., James R. W., Proudfoot A. E. I., Mach F., Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 94, 253–261 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Braunersreuther V., Pellieux C., Pelli G., Burger F., Steffens S., Montessuit C., Weber C., Proudfoot A., Mach F., Arnaud C., Chemokine CCL5/RANTES inhibition reduces myocardial reperfusion injury in atherosclerotic mice. J. Mol. Cell. Cardiol. 48, 789–798 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Agere S. A., Akhtar N., Watson J. M., Ahmed S., RANTES/CCL5 induces collagen degradation by activating MMP-1 and MMP-13 expression in human rheumatoid arthritis synovial fibroblasts. Front. Immunol. 8, 1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li B.-H., He F.-P., Yang X., Chen Y.-W., Fan J.-G., Steatosis induced CCL5 contributes to early-stage liver fibrosis in nonalcoholic fatty liver disease progress. Transl. Res. 180, 103–117.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Zeng Q., Song R., Fullerton D. A., Ao L., Zhai Y., Li S., Ballak D. B., Cleveland J. C. Jr., Reece T. B., McKinsey T. A., Xu D., Dinarello C. A., Meng X., Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc. Natl. Acad. Sci. U.S.A. 114, 1631–1636 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Souilhol C., Serbanovic-Canic J., Fragiadaki M., Chico T. J., Ridger V., Roddie H., Evans P. C., Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 17, 52–63 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Zhou J., Li Y. S., Chien S., Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 34, 2191–2198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y., Miao H., Li S., Chen K.-D., Li Y.-S., Yuan S., Shyy J. Y.-J., Chien S., Interplay between integrins and FLK-1 in shear stress-induced signaling. Am. J. Physiol. Cell Physiol. 283, C1540–C1547 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Balaguru U. M., Sundaresan L., Manivannan J., Majunathan R., Mani K., Swaminathan A., Venkatesan S., Kasiviswanathan D., Chatterjee S., Disturbed flow mediated modulation of shear forces on endothelial plane: A proposed model for studying endothelium around atherosclerotic plaques. Sci. Rep. 6, 27304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Geemen D., Smeets M. W. J., van Stalborch A.-M. D., Woerdeman L. A. E., Daemen M. J. A. P., Hordijk P. L., Huveneers S., F-actin-anchored focal adhesions distinguish endothelial phenotypes of human arteries and veins. Arterioscler. Thromb. Vasc. Biol. 34, 2059–2067 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/34/eabg1694/DC1