

Abstract
We previously reported that pseudoginsenoside-F11 (PF11), an ocotillol-type saponin, significantly ameliorated Alzheimer’s disease (AD)-associated cognitive defects in APP/PS1 and SAMP8 mice by inhibiting Aβ aggregation and tau hyperphosphorylation, suggesting a potential therapeutic effect of PF11 in the treatment of AD. In the present study we further evaluated the therapeutic effects of PF11 on relieving cognitive impairment in a rat model of sporadic AD (SAD). SAD was induced in rats by bilateral icv infusion of streptozotocin (STZ, 3 mg/kg). The rats were treated with PF11 (2, 4, 8 mg·kg−1·d−1, ig) or a positive control drug donepezil (5 mg·kg−1·d−1, ig) for 4 weeks. Their cognitive function was assessed in the nest building, Y-maze, and Morris water maze tests. We showed that STZ icv infusion significantly affected the cognitive function, tau phosphorylation, and insulin signaling pathway in the hippocampus. Furthermore, STZ icv infusion resulted in significant upregulation of the calpain I/cyclin-dependent protein kinase 5 (CDK5) signaling pathway in the hippocampus. Oral administration of PF11 dose-dependently ameliorated STZ-induced learning and memory defects. In addition, PF11 treatment markedly reduced the neuronal loss, protected the synapse structure, and modulated STZ-induced expression of tau phosphorylation by regulating the insulin signaling pathway and calpain I/CDK5 signaling pathway in the hippocampus. Donepezil treatment exerted similar beneficial effects in STZ-infused rats as the high dose of PF11 did. This study highlights the excellent therapeutic potential of PF11 in managing AD.
Keywords: Alzheimer’s disease, pseudoginsenoside-F11, Tau hyperphosphorylation, insulin signaling pathway, calpain I/CDK5 signaling pathway, donepezil
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and a leading cause of cognitive and behavioral dysfunctions in the aging population worldwide [1]. Epidemiological studies have shown that most patients with AD (>95%) are diagnosed with the sporadic form, which is characterized by a late onset (80–90 years of age) [2]. The currently recognized pathological and biochemical manifestations of sporadic AD (SAD) mainly include insulin desensitization/resistance state, oxidative stress, neuroinflammation, amyloid beta (Aβ) deposition, tau hyperphosphorylation, and synapse dysfunction in the brain [3]. However, as the pathogenesis of AD is extremely complicated, no curative therapy is available to date. In recent years, agents focusing on regulating Aβ levels to treat AD have successively failed in clinical trials. Therefore, multitarget drugs are preferred as effective therapeutic agents.
Streptozotocin (STZ) is a glucosamine–nitrosourea compound that can induce tau hyperphosphorylation and cognitive impairments when administered by intracerebroventricular (icv) injection [4] and has been extensively used to generate rodent models for studying SAD [5–7]. Moreover, STZ has been demonstrated to be involved in dysfunction of the cerebral insulin signaling pathway, which is also a hallmark of SAD [8]. The cerebral insulin signaling pathway is important for neuronal growth, synaptic maintenance, and neuroprotection, and it is also associated with phosphorylation of tau protein [9]. Furthermore, it has been observed that calpains are also strongly associated with the pathological processes of AD through their regulatory effects on a series of cellular kinases. Activating calpain may increase the activity of cyclin-dependent protein kinase 5 (CDK5) by cleaving p35 to generate p25 [10]. Activated CDK5 can directly stimulate tau hyperphosphorylation and aggregation as well as the loss of synaptic proteins when it associates with p25 [11]. Thus, regulation of the insulin signaling pathway and inhibition of calpain activity may ameliorate the pathological changes associated with AD.
Pseudoginsenoside-F11 (PF11), isolated from the roots and leaves of Panax pseudoginseng subsp. Himalaicus HARA, is an ocotillol-type saponin found in Panax quinquefolium L [12]. Studies have strongly suggested that PF11 exerts extensive protective effects in a series of cardiovascular and central nervous system disorders [13, 14]. Our previous studies also indicated that PF11 treatment significantly reduces the Aβ burden and therefore exerts remarkable neuroprotective effects in a number of animal models of AD [12, 15, 16]. Strikingly, we observed that PF11 treatment can also ameliorate cognitive impairment in SAMP8 mice by ameliorating PP2A-mediated tau hyperphosphorylation [17]. To comprehensively evaluate the therapeutic potential and related mechanisms of PF11 in AD, it is necessary to study the effect of PF11 on an SAD model.
In the present study, we further showed that oral administration of PF11 significantly reduced cognitive impairment, ameliorated neuronal loss, protected synapse structure and inhibited tau hyperphosphorylation by interfering with the insulin signaling pathway and calpain I/CDK5 signaling pathway in a rat model of STZ-induced SAD.
Materials and methods
Animals
Adult male Wistar rats (220–250 g) were obtained from Liaoning Changsheng Biotechnology Co., Ltd (Liaoning, China). They were housed in groups in standard individual ventilated cages (32 cm × 20 cm × 12.5 cm) containing wood shavings as litter at a controlled temperature of 20–22 °C and a controlled humidity of 45%–55% on a modified 12/12-h dark–light cycle. Food and water were available ad libitum. All efforts were made to minimize pain and suffering and to reduce the number of animals used. All experimental procedures were conducted in accordance with the Regulations of Experimental Animal Administration issued by the State Committee of Science and Technology of the People’s Republic of China. All behavioral tests were performed between 08:00 and 12:00 every day.
Stereotaxic surgery
The rats were anesthetized using intraperitoneal injection of chloral hydrate (350 mg/kg) and placed in a rat brain stereotaxic apparatus (coordinates: 0.8 mm posterior to bregma, 1.5 mm lateral to the sagittal suture, 3.6 mm below the brain surface) [18]. Afterward, the animals received a single bilateral infusion of 5 μL STZ (3 mg/kg, Sigma-Aldrich, Darmstadt, Germany) or vehicle (artificial cerebrospinal fluid, ACSF) delivered by an automatic injector at a rate of 200 nL/min. The needle was left in place for 5 min and then slowly withdrawn from the brain.
Drug administration
PF11 was isolated from the aerial parts of P. quinquefolium by the Department of Chemistry for Natural Products of Shenyang Pharmaceutical University. The purity was more than 98%, which was determined by HPLC as previously reported [12]. The chemical structure of PF11 is shown in Fig. 2a. PF11 (2, 4, 8 mg/kg) and donepezil (5 mg/kg, Meilunbio,MB1083) were dissolved in distilled water. The dose of PF11 was selected based on our previous studies [19, 20]. Donepezil is a cholinesterase inhibitor that provides cognitive and functional benefits for patients with mild-to-severe AD [21], and the dose of donepezil was chosen based on a previous study [22].
Fig. 2. PF11 ameliorated STZ-induced cognitive decline.

a Chemical structure of PF11 (C42H72O14, molecular weight = 801.02). b Self-care activity was evaluated in the nest building test. c Working memory was assessed in the Y-maze test. d–h The spatial memory of the mice was tested in the Morris water maze test. d Distance (distance traveled to find the platform), e escape latency (time to find the platform), f swimming velocity, g number of platform crossings during the probe test, and h representative swimming paths in the probe test. The data are reported as the mean ± SEM (n = 8–10). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. the sham group; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. the STZ group.
The animals were randomly divided into six groups: (1) the sham group (ACSF (icv) plus distilled water (ig), (2) the STZ group (STZ (icv) plus distilled water (ig), (3) the STZ + PF11 2 mg/kg group (STZ (icv) plus PF11 (2 mg·kg−1· d−1, ig), (4) the STZ + PF11 4 mg/kg group (STZ (icv) plus oral PF11 (4 mg.kg−1· d−1, ig), (5) the STZ + PF11 8 mg/kg group (STZ (icv) plus PF11 (8 mg·kg−1· d−1, ig), and (6) the STZ + donepezil group (STZ (icv) plus oral donepezil (5 mg·kg−1· d−1, ig).
Behavioral procedures
The behavioral tests were conducted in an isolated room with no disturbances during testing (n = 8–10 per group) (Fig. 1).
Fig. 1. Experimental design.
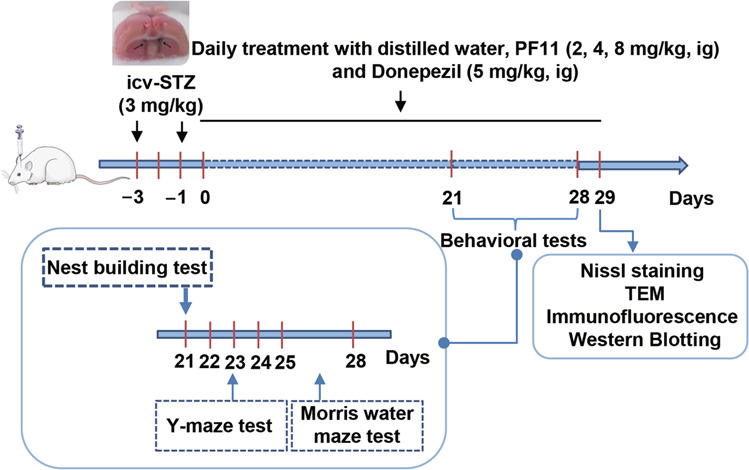
SAD was induced in rats by bilateral icv infusion of STZ (3 mg/kg). The rats were treated with PF11 (2, 4, 8 mg·kg−1· d−1, ig) or a positive control drug donepezil (5 mg·kg−1· d−1, ig) for 4 weeks. Their cognitive function was assessed in the behavioral tests. All rats were sacrificed after the behavioral tests for Nissl staining, TEM, immunochemical analysis and Western blot analysis.
Nest-building test
The nest building test is considered to evaluate behaviors analogous to “activities of daily living”, which are disrupted in AD [23]. For 3 consecutive days, rats were individually housed in cages containing sawdust in a quiet testing room, with food and water available ad libitum. Three pieces of kitchen towel (2 cm × 5 cm) were then introduced into the home cage, and nest building was assessed. After 24 h, each nest was photographed, and the quality of the nest was determined according to a point scale (pts). Images of each nest were divided equally into six grid boxes. The score was based on the number of grid boxes cleared (0.5 pts per segment) or partially cleared (1 pts per segment) of kitchen towel, as imitated by Robin Roof [24].
Y-maze test
Spatial working memory was assessed based on spontaneous alternation behavior in a Y-maze [25]. The Y-maze was made of wood and consisted of three arms (45 cm × 25 cm × 15 cm) radiating out from the center compartment. Each rat was placed in the center of the symmetrical Y-maze and allowed to freely explore the maze for 8 min. The percentage of alternations was calculated as the number of triads involving entry into all three arms (ABC, ACB, CAB, etc.) divided by the maximum possible alternations ([total number of entries minus 2] × 100). Before each trial, the maze was cleaned thoroughly using 75% ethanol.
Morris water maze test
The spatial learning and memory of the animals were tested using the Morris water maze (MWM) test [26]. The maze consisted of a white circular pool (160 cm in diameter, 55 cm in height) divided into four quadrants referred to as the southeast, southwest, northeast, and northwest quadrants. The pool also contained a moveable white circular platform (10 cm in diameter, 25 cm in height) located in the center of one quadrant 1 cm below the water surface. The water was dyed with black ink and maintained at a temperature of 22 ± 1 °C; the tank was placed in a dimly lit area. The place navigation test involved of an acquisition phase and a probe trial carried out in a sound proof test room without visual cues. The acquisition phase was performed 5 consecutive days, with four trials per day. Four points (north, south, east, and west) equally spaced along the circumference of the pool served as starting positions, and the rats were placed in the water facing the pool wall at these points. If a rat did not find the platform within 120 s, it was guided to the platform and allowed to stay on it for 10 s. If a rat found the platform within 120 s, it was allowed to remain on it for at least 10 s. The rats were dried between each trial and returned to their home cage to rest for 120 s after each trial. The escape latency and path length were recorded to evaluate the effect of PF11 on learning activity, specifically, memory retention of the platform location. After the place navigation test, the number of platform crossings and time spent searching for the hidden platform were recorded. These activities were recorded and analyzed using an Ethovision XT 8.0 computerized video tracking system (Noldus Information Technology, Wageningen, the Netherlands).
Tissue preparation
Rats were fully anesthetized using intraperitoneal injection of chloral hydrate (350 mg/kg) and transcardially perfused with saline solution. The brain tissues were then carefully detached (n = 3 per group), fixed in 4% buffered paraformaldehyde for 24 h, and transferred to 20% and 30% sucrose/phosphate-buffered saline (PBS) at 4 °C. Then, brain tissues from each rat containing the hippocampus and cortex were sectioned on a freezing microtome (AS-620; Shandon, Astmoor, UK) at a thickness of 20 μm for each region (−1.82 mm to −2.54 mm from bregma). Every 6th coronal section was used for staining, and a total of three sections were taken. Afterward, the sections were stored at −20 °C until further processing. The hippocampi of other animals (n = 3 per group) were isolated, stored at −80 °C and used for Western blotting.
Nissl staining
Nissl staining was also performed as previously described [27]. Sections (n = 3 per brain) were removed from the −20 °C freezer and allowed to adapt to room temperature for half an hour. For Nissl staining, the sections were dehydrated through graded ethanol solutions (75%, 85%, 95%, and 100%) and stained with cresyl violet. The sections were soaked in ethanol to differentiate the stain and cleared by immersion in xylene for 2 min. They were then covered with mounting medium and digitized using a 4×, 20×, or 40× objective (Olympus BX40; Tokyo, Japan) with an MCID computer imaging analysis system (Image-Pro 3D Plus Workstation; Media Cybernetics, Inc., Rockville, MD, USA). The number of Nissl-positive cells was measured in each section. The data were analyzed in a blinded manner and are presented as the number of positive cells in one field.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed as previously described [28]. Brains (n = 3 per group) were fixed using transcardial perfusion of saline followed by perfusion of and immersion in 4% paraformaldehyde and 2.5% glutaraldehyde. Small blocks from the hippocampal CA1 region were processed for electron microscopy and embedded in Epon resin. The sections were then examined using a transmission electron microscope (JEM-1200EX, Jeol Ltd, Tokyo, Japan) at an accelerating voltage of 80 kV.
Immunofluorescence
Sections (n = 3 per brain) were removed from the −20 °C freezer and allowed to adapt to room temperature for half an hour. For immunofluorescence, the sections were washed in PBS and subjected to antigen retrieval in a microwave. Subsequently, the sections were rinsed twice in 0.1 M PBS, blocked with 5% normal goat serum for 60 min at 37 °C, and incubated with a p-tau (Ser396) (1:500, Abcam, Cambridge, UK) primary antibody overnight at 4 °C. The sections were then incubated with a FITC-conjugated goat anti-mouse IgG (H + L) secondary antibody (Beyotime, Shanghai, China). The sections were incubated with DAPI (1:1000, Sigma-Aldrich, St. Louis, MO, USA), and confocal images were taken using a Nikon C2 Plus microscope with a 20× objective.
Western blotting
Hippocampal tissues were lysed using RIPA Tissue Protein Extraction Reagent (1:9, 9 μL reagent/1 mg tissue; Beyotime, Shanghai, China), 0.1% phenylmethanesulfonyl fluoride, 0.1% sodium fluoride (NaF), and 0.1% sodium orthovanadate (Na3VO4), sonicated for 3 min on ice using a probe sonicator, and centrifuged for 5 min at 12,000 × g at 4 °C. The supernatants were then extracted and boiled in a water bath. Protein extracts (20–30 μg) were separated using 10%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (n = 3 per group) and electrophoretically transferred to a nitrocellulose membrane at 4 °C. The blots were incubated for 1 h with a mixture of 5% milk in tris-buffered saline with Tween 20 (TBST) to block nonspecific-binding sites. They were then incubated with primary antibodies overnight at 4 °C followed by secondary antibodies for 1 h at room temperature. The primary antibodies were as follows: polyclonal rabbit anti-insulin receptor substrate-1 (IRS-1) (1:1000, CST, Danvers, MA, USA); polyclonal rabbit anti-p-IRS-1(serine (Ser)307) (1:1000, CST); polyclonal rabbit anti-phosphatidylinositol 3-kinase (PI3K) p85 (1:1000, CST); polyclonal rabbit anti-p-PI3K p85 (1:1000, CST); polyclonal rabbit anti-protein kinase B (AKT) (Ser473) (1:1000); polyclonal rabbit anti-p-AKT (Ser473) (1:1000, CST); polyclonal rabbit anti-glycogen synthasekinase-3β (GSK-3β) (1:500, CST); polyclonal rabbit anti-p-GSK-3β (Ser9) (1:500, CST); monoclonal mouse anti-calpain I (1:500, Santa, TX, USA); polyclonal rabbit anti-CDK5 (1:1000, CST); polyclonal rabbit anti-p35/25 (1:1000,CST); monoclonal mouse anti-p-tau (Ser199/202) (1:1000, Invitrogen, Rockford, USA); monoclonal mouse anti-p-tau (Ser396) (1:1000, CST); monoclonal mouse anti-tau 5 (1:1000, Millipore, Darmstadt, Germany); monoclonal mouse anti-β-actin (1:2000, Santa); and monoclonal mouse anti-GAPDH (1:1000, ZSGB-BIO, Beijing, China). Immunoreactive proteins were detected using a chemiluminescent substrate (Pierce ECL Western Blotting Substrate, Perkin Elmer).
Statistical analysis
The data are expressed as the mean ± standard error of the mean (SEM). MWM test data were analyzed using two-way ANOVA for repeated measures, with ‘training’, ‘day’, and their interactions as fixed factors. Other behavioral and biochemical data were analyzed using one-way ANOVA with Fisher’s least significant difference (LSD) test and Dunnett’s T3 post hoc comparison. Graphs were created using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA). All statistical analyses were performed using SPSS 17.0 software for Windows (SPSS Inc., Chicago, IL, USA). The level of significance was defined as P < 0.05.
Results
PF11 alleviated STZ-induced deficits in self-care in the nest building test
In the present study, the nest-building ability of the STZ group was significantly worse than that of the sham group, whereas the nest-building ability of the PF11 (2, 4, and 8 mg/kg) groups was significantly better than that of the STZ group. Donepezil had the same beneficial effect (Fig. 2b).
PF11 ameliorated STZ-induced working memory deficits in theY-maze test
In the Y-maze test, the number of arm entry alternations was calculated to further evaluate the effects of PF11 on STZ-induced working memory deficits. Post hoc multiple comparisons revealed that the STZ group displayed fewer alternations than the sham group and that compared with STZ, treatment with PF11 (2, 4, 8 mg/kg) significantly increased the number of alternations in a dose-dependent manner. Similar results were also observed in the donepezil-treated group. No differences in locomotor activity or total number of arm entries were observed among the groups (Fig. 2c).
PF11 ameliorated STZ-induced learning and memory deficits in the MWM test
We evaluated the effects of PF11 on STZ-induced cognitive deficits in rats using the MWM test. In the place navigation test, swimming distance was analyzed using two-way ANOVA for repeated measures. The results indicated significant effects of training (F = 171.673; P < 0.001) and group (F = 635.365; P < 0.001) but no significant effect of training × group (F = 2.150; P = 0.073). The LSD test further revealed that the STZ group consistently showed a significantly longer swimming distance than the sham group on day 4. Compared with STZ, PF11 (2, 4, 8 mg/kg) decreased the swimming distance (Fig. 2d). Moreover, two-way ANOVA for repeated measures revealed significant effects of training (F = 111.33; P < 0.001) and group (F = 666.757; P < 0.001) but no significant effect of training × group (F = 2.770; P > 0.05) on escape latency. The LSD test further revealed that the STZ group consistently exhibited a significantly longer escape latency than the sham group on days 3 and 4 and that PF11 (2, 4, and 8 mg/kg) significantly decreased the escape latency after STZ injection (Fig. 2e). No differences in swimming speed were observed among the five groups (Fig. 2f). In the spatial probe test, memory retention of the platform location was measured after the place navigation test. The number of platform crossings in the STZ group was lower than that in the sham group, but this difference was reversed by PF11 (8 mg/kg) and donepezil (Fig. 2g, h).
PF11 alleviated STZ-induced neuronal death and synaptic damage
STZ injection significantly reduced the number of surviving neurons in the DG and CA1 regions of the hippocampus. Moreover, the TEM results indicated that the synaptic ultrastructure of the CA1 region was damaged after STZ injection, with a reduction in synaptic vesicles and thinning of the postsynaptic membrane density (PSD) (Fig. 3c). Furthermore, our data showed that PF11 significantly increased the number of surviving neurons (Fig. 3a, b), the number of synapses, and PSD thickness, having similar effects as donepezil treatment. There were no differences in PSD length among the groups (Fig. 3c, d).
Fig. 3. PF11 alleviated STZ-induced neuronal death and synaptic damage.

a Nissl staining (×40, ×400) was used to evaluate neuronal death in the hippocampal DG and CA1 regions. Sections of the hippocampal DG and CA1 regions (scale bar = 50 μm). b Quantitative analysis of the number of surviving neurons in the hippocampal DG and CA1 regions with Image-Pro Plus (Media Cybernetics, Inc., Rockville, MD, USA) (n = 9). c Transmission electron microscopy (TEM) (×900) was performed to observed synapse structure (scale bar = 500 nm). The red arrow indicates a synapse. d Quantitative analysis of the number of synapses, PSD thickness and PSD length (n = 3). The data are reported as the mean ± SEM. **P < 0.01, ***P < 0.001 vs. sham rats; #P < 0.05, ##P < 0.01 vs. the STZ group.
PF11 inhibited STZ-induced tau hyperphosphorylation in the hippocampus
Hyperphosphorylation of tau protein at the specific sites Ser396 and Ser199/202 is a typical pathological change observed in the brains of AD patients [29]. To further examine the effects of PF11 on tau phosphorylation, we investigated the expression of phosphorylated tau using Western blotting and immunofluorescence. The results indicated that the expression of tau phosphorylated at the Ser396 and Ser199/202 sites in isolated hippocampal tissues was significantly upregulated in the STZ group compared with the sham group. PF11 (2, 4, 8 mg/kg) significantly inhibited the expression of phosphorylated tau without influencing the expression of total tau. Donepezil strongly inhibited tau protein phosphorylation at Ser396 (Fig. 4b, c). Visualization of tau expression showed accumulation of phosphorylated tau in the DG region of the hippocampus, which was reversed after PF11 and donepezil treatment (Fig. 4a).
Fig. 4. PF11 inhibited STZ-induced tau hyperphosphorylation in the hippocampus.

a Representative figures of coronal sections from each group after immunofluorescence staining against p-tau (scale bar = 100 μm, the red arrow indicates positive staining). b Levels of p-tau in the hippocampus were measured by Western blotting. c The integral optical density of p-tau (Ser396), p-tau (Ser199/202), and Tau5 in the hippocampus analyzed with ImageJ (NIH Image, Bethesda, MD, USA). The data are reported as the mean ± SEM (n = 3). **P < 0.01 vs. the sham group; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. the STZ group.
PF11 attenuated STZ-induced tau hyperphosphorylation by reversing the dysregulation of the IRS-1/PI3K/AKT/GSK-3β and calpain I/CDK5 signaling pathways
To further study the potential mechanism underlying the effect of PF11 on tau hyperphosphorylation, changes in the IRS-1/PI3K/AKT/GSK-3β and calpain I/CDK5 signaling pathways, which are typical pathological features of this model, were analyzed by Western blotting. STZ administration significantly downregulated the expression of p-GSK-3β (Ser9) and upregulated the expression of the autolytic form of calpain, p35, p25, and CDK5 in the hippocampus, and these effects were reversed by PF11 treatment (2, 4, 8 mg/kg). Furthermore, we observed the effects of PF11 on the expression of proteins related to the IRS-1/PI3K/AKT signaling pathway after STZ injection, as earlier findings indicated that exposure to STZ causes dysregulation of the insulin signaling pathway in the hippocampus. The expression of phosphorylated IRS-1 and p85, the regulatory subunit of PI3K, were found to be upregulated, whereas the expression of phosphorylated AKT was downregulated. PF11 and donepezil obviously reversed these changes (Fig. 5).
Fig. 5. PF11 attenuated STZ-induced tau hyperphosphorylation by reversing dysregulation of the IRS-1/PI3K/AKT/GSK-3β and calpain I/CDK5 signaling pathways.

a–d Expression of proteins related to the IRS-1/PI3K/AKT/GSK-3β and calpain I/CDK5 signaling pathways. a, c Protein was extracted from the hippocampus for each group and evaluated by Western blot analysis. b, d The integral optical densities of proteins related to the signaling pathways in the hippocampus were analyzed with ImageJ. The data are reported as the mean ± SEM (n = 3).*P < 0.05, **P < 0.01, ***P < 0.001 vs. the sham group; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. the STZ group.
Discussion
In the present study, an SAD model established by STZ icv administration was used to evaluate the effects and mechanism of PF11 on AD. We observed that STZ-induced cognitive impairment, neuronal damage, and tau hyperphosphorylation were significantly ameliorated after PF11 treatment. Moreover, we also found that the mechanism by which PF11 attenuated tau hyperphosphorylation may be related to its regulation of the insulin signaling pathway and calpain I/CDK5 signaling pathway, as indicated by its inhibition of GSK-3β (Ser9) phosphorylation and CDK5 activity. All these results showed that PF11 has the potential to treat AD by reversing tau hyperphosphorylation.
AD is characterized by progressive cognitive impairment that results from synaptic failure and neuronal death in brain regions critical for learning and memory [30]. To evaluate the therapeutic effects of PF11 on AD-associated cognitive impairment, the nest building, Y-maze, and MWM tests were applied. We observed that STZ injection resulted in typical impairments in self-care ability and spatial memory, which were reversed after PF11 treatment for 3 weeks. Furthermore, we observed that STZ-induced neuronal death and synaptic failure could also be ameliorated by PF11 administration, as evaluated by Nissl staining and TEM. Notably, PF11 treatment also exerted significant neuroprotective effects against these changes, although the underlying mechanism needs to be further explored.
Tau is a microtubule-associated protein expressed in neurons that plays an important role in the stabilization of microtubules and axonal transport [31]. However, accumulated phosphorylated tau protein in neurons produces NFTs, leading to neuronal dysfunction. Several phosphorylation sites of the tau protein have been identified in neurons in AD patients, including Ser199, Ser202/205, Ser396, threonine (Thr) 205, and Thr231, and phosphorylation at these sites is considered a biomarker of AD [29]. Previous studies have demonstrated that STZ injection causes tau hyperphosphorylation by acting on Ser199/202 [4] and Ser396 [6]. In agreement with previous studies, we observed that STZ injection led to tau hyperphosphorylation at two sites, as determined by Western blotting. We also investigated the expression of p-tau (Ser396) in the DG region by immunofluorescence, which was significantly reversed by PF11 treatment.
The insulin signaling pathway and calpain/CDK5 signaling pathway are involved in hyperphosphorylation of tau in the AD brain [9, 11]. The tau protein can be phosphorylated at many sites by several protein kinases, such as GSK-3β and CDK5 [32]. Dysregulation of insulin signaling, which is characterized by reduced brain insulin receptor (IR) sensitivity, downregulation of the IR and insulin-growth factor receptor-1 (IGF-1) and hyperphosphorylation of IRS-1, has been extensively observed in early stage and moderate AD patients [33]. Activation of the insulin signaling cascade starts with binding of the ligand insulin to the IR and affects the transduction of PI3K/AKT signaling through phosphorylation of IRS-1. The activation of AKT can further lead to a decrease in the activity of GSK-3β through phosphorylation of Ser9. Therefore, any interference in the PI3K/AKT insulin signaling pathway can promote phosphorylation of GSK-3β (Ser9), which further leads to tau phosphorylation and NFT formation [34, 35]. In contrast, when internal calcium concentrations are increased, calpain can be activated, resulting in the cleavage of p35 into p25, which can further activate CDK5 by regulating its substrate specificity [36, 37]. In addition, evidence indicates that there is crosstalk between insulin and calpain I/CDK5 signaling cascades, as activation of GSK-3β leads to activation of CDK5 through the regulation of p25 [38]. Thus, both of these signaling cascades are closely associated with abnormal phosphorylation of the tau protein and play key roles in the pathological process of AD. In the present study, we found that PF11 regulated GSK-3β (Ser9) phosphorylation by modulating the IRS-1/PI3K/AKT signaling pathway. Moreover, we observed abnormalities in the calpain I/CDK5 signaling pathway for the first time in the brains of rats that received icv injection of STZ and found that these abnormalities were reversed after PF11 treatment. Interestingly, this reversal effect can also be observed in a rat model of transient cerebral ischemia injury, as we previously reported [39], revealing that calpain I may be a potential target of PF11. These findings also suggest that dysregulated calpain I/CDK5 signaling pathway-induced tau hyperphosphorylation might be a pathological mechanism in animal models of STZ-induced AD that can be attenuated by PF11.
AD is a neurodegenerative disorder characterized by extracellular Aβ deposition and tau hyperphosphorylation, which activates microglia, triggers neuroinflammation, and leads to neuronal death and cognitive deficits [40, 41]. Numerous studies have documented that impairment of cognitive function can be ameliorated by inhibition of Aβ deposition, tau hyperphosphorylation and neuroinflammation in AD animal models [42, 43]. Based on the complexity and multicausality of AD, multitarget drugs may have better clinical therapeutic potential than single-target drugs. In our previous studies, PF11 dramatically improved the cognitive abilities of APP/PS1 and SAMP8 mice by reducing the Aβ burden and tau hyperphosphorylation [12, 17]. We have also demonstrated that PF11 treatment can significantly attenuate d-galactose-induced oxidative stress in vivo and lipopolysaccharide-induced neuroinflammation in vitro and in vivo [14, 16]. More importantly, in this study, we further found that PF11 treatment can ameliorate tau hyperphosphorylation by reversing the dysregulation of the insulin signaling pathway and calpain I/CDK5 signaling pathway. Taken together, these results combined with those of our previous works suggest that PF11 exerts excellent therapeutic effects on AD by acting on a series of signaling cascades.
Conclusion
In conclusion, we showed for the first time that PF11 treatment significantly reversed SAD-associated cognitive impairments and neuronal damage, possibly by regulating the insulin signaling pathway and calpain I/CDK5 signaling pathway-related tau hyperphosphorylation, indicating that PF11 has excellent therapeutic potential for the management of AD.
Author contributions
Designed research: LZ, XJH, JYY. Performed research: XJH, LZ, TSZ, XQL. Analyzed data: XJH, LZ. Wrote the manuscript: LZ. Revised the manuscript: XHC, JYY, CFW. All authors contributed substantially to this work and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Chun-fu Wu, Email: wucf@syphu.edu.cn.
Jing-yu Yang, Email: yangjingyu2006@gmail.com.
References
- 1.Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 2018;4:195–214. doi: 10.1016/j.trci.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Prim. 2015;1:15056. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 3.Rasheed NOA. Study of the possible protective effect of formoterol in streptozotocin-induced sporadic Alzheimer’s disease mouse model. Doctor of Philosophy degree. Cairo: Cairo University; 2019.
- 4.Chen Y, Liang Z, Blanchard J, Dai CL, Sun S, Lee MH, et al. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse) Mol Neurobiol. 2013;47:711–25. doi: 10.1007/s12035-012-8375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ponce-Lopez T, Hong E, Abascal-Díaz M, Meneses A. Role of GSK3β and PP2A on regulation of tau phosphorylation in hippocampus and memory impairment in ICV-STZ animal model of Alzheimer’s disease. Adv Alzheimer Dis. 2017;06:13–31. doi: 10.4236/aad.2017.61002. [DOI] [Google Scholar]
- 6.Du LL, Xie JZ, Cheng XS, Li XH, Kong FL, Jiang X, et al. Activation of sirtuin 1 attenuates cerebral ventricular streptozotocin-induced tau hyperphosphorylation and cognitive injuries in rat hippocampi. Age. 2014;36:613–23. doi: 10.1007/s11357-013-9592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao C, Liu YZ, Jiang YH, Ding JM, Li L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3β pathway in streptozotocin‐induced Alzheimer rat model. Brain Pathol. 2014;24:261–9. doi: 10.1111/bpa.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grieb P. Intracerebroventricular streptozotocin injections as a model of Alzheimer’s disease: in search of a relevant mechanism. Mol Neurobiol. 2016;53:1741–52. doi: 10.1007/s12035-015-9132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedse G, Di Domenico F, Serviddio G, Cassano T. Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci. 2015;9:204. doi: 10.3389/fnins.2015.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee MS, Kwon YT, Li M, Peng JM, Friedlander RM, Tsai LS. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–4. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 11.Kurbatskaya K, Phillips EC, Croft CL, Dentoni G, Hughes MM, Wade MA, et al. Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol Commun. 2016;4:34. doi: 10.1186/s40478-016-0299-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang CM, Liu MY, Wang F, Wei MJ, Wang S, Wu CF, et al. Anti-amnesic effect of pseudoginsenoside-F11 in two mouse models of Alzheimer’s disease. Pharmacol Biochem Behav. 2013;106:57–67. doi: 10.1016/j.pbb.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Liu YY, Zhang TY, Xue X, Liu DM, Zhang HT, Yuan LL, et al. Pseudoginsenoside-F11 attenuates cerebral ischemic injury by alleviating autophagic/lysosomal defects. CNS Neurosci Ther. 2017;23:567–79. doi: 10.1111/cns.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Wang C, Wang J, Zhao S, Zhang K, Wang J, et al. Pseudoginsenoside-F11 (PF11) exerts anti-neuroinflammatory effects on LPS-activated microglial cells by inhibiting TLR4-mediated TAK1/IKK/NF-κB, MAPKs and Akt signaling pathways. Neuropharmacology. 2014;79:642–56. doi: 10.1016/j.neuropharm.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Li Z, Guo Y, Wu C, Li X, Wang J. Protective effects of pseudoginsenoside‐F11 on scopolamine‐induced memory impairment in mice and rats. J Pharm Pharmacol. 1999;51:435–40. doi: 10.1211/0022357991772484. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Yang HL, Yang JY, Xie J, Xu JY, Liu C, et al. Pseudoginsenoside-F11 attenuates cognitive impairment by ameliorating oxidative stress and neuroinflammation in dgalactose-treated mice. Int Immunopharmacol. 2019;67:78–86. doi: 10.1016/j.intimp.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Yang JY, Liu C, Xie J, Qiu S, Yang X, et al. Pseudoginsenoside-F11 alleviates cognitive deficits and Alzheimer’s disease-type pathologies in SAMP8 mice. Pharmacol Res. 2019;139:512–23. doi: 10.1016/j.phrs.2018.10.024. [DOI] [PubMed] [Google Scholar]
- 18.Kosaraju J, Gali CC, Khatwal RB, Dubala A, Chinni S, Holsinger RM, et al. Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology. 2013;72:291–300. doi: 10.1016/j.neuropharm.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Wang JY, Yang JY, Wang F, Fu SY, Hou Y, Jiang B, et al. Neuroprotective effect of pseudoginsenoside-f11 on a rat model of Parkinson’s disease induced by 6-hydroxydopamine. Evid Based Complement Altern Med. 2013;2013:152798. doi: 10.1155/2013/152798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu CF, Liu YL, Song M, Liu W, Wang JH, Li X, et al. Protective effects of pseudoginsenoside-F11 on methamphetamine-induced neurotoxicity in mice. Pharmacol Biochem Behav. 2003;76:103–9. doi: 10.1016/S0091-3057(03)00215-6. [DOI] [PubMed] [Google Scholar]
- 21.Zhang N, Gordon ML. Clinical efficacy and safety of donepezil in the treatment of Alzheimer’s disease in Chinese patients. Clin Inter Aging. 2018;13:1963–70. doi: 10.2147/CIA.S159920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agrawal R, Tyagi E, Shukla R, Nath C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology. 2009;56:779–87. doi: 10.1016/j.neuropharm.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Galasko D, Schmitt F, Thomas R, Jin S, Bennett D. Detailed assessment of activities of daily living in moderate to severe Alzheimer’s disease. J Int Neuropsychol Soc. 2005;11:446–53. doi: 10.1017/S1355617705050502. [DOI] [PubMed] [Google Scholar]
- 24.Roof R, Snyder B, Yabe Y, Zaleska M. Use of ethological rodent behavior to assess efficacy of potential drugs for Alzheimer’s disease. Proc Meas Behav. 2010;2010:389. [Google Scholar]
- 25.Galeano P, Martino Adami PV, Do Carmo S, Blanco E, Rotondaro C, Capani F, et al. Longitudinal analysis of the behavioral phenotype in a novel transgenic rat model of early stages of Alzheimer’s disease. Front Behav Neurosci. 2014;8:321. doi: 10.3389/fnbeh.2014.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal R, Mishra B, Tyagi E, Nath C, Shukla R. Effect of curcumin on brain insulin receptors and memory functions in STZ (ICV) induced dementia model of rat. Pharmacol Res. 2010;61:247–52.. doi: 10.1016/j.phrs.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Wang N, Liu Y, Liu Y, Zhang T, Zhu L, et al. Yonkenafil: a novel phosphodiesterase type 5 inhibitor induces neuronal network potentiation by a cGMP-dependent Nogo-R axis in acute experimental stroke. Exp Neurol. 2014;261:267–77. doi: 10.1016/j.expneurol.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Llorens-Martín M, Fuster-Matanzo A, Teixeira CM, Jurado-Arjona J, Ulloa F, Defelipe J, et al. GSK-3 beta overexpression causes reversible alterations on postsynaptic densities and dendritic morphology of hippocampal granule neurons in vivo. Mol Psychiatry. 2013;18:451–60. doi: 10.1038/mp.2013.4. [DOI] [PubMed] [Google Scholar]
- 29.Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6:6. doi: 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:167–79. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 31.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/S0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 32.Gao Y, Tan L, Yu JT, Tan L. Tau in Alzheimer’s disease: mechanisms and therapeutic strategies. Curr Alzheimer Res. 2018;15:283–300.. doi: 10.2174/1567205014666170417111859. [DOI] [PubMed] [Google Scholar]
- 33.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–38. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akintola AA, van Heemst D. Insulin, aging, and the brain: mechanisms and implications. Front Endocrinol (Lausanne) 2015;6:13. doi: 10.3389/fendo.2015.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2006;9:309–17. doi: 10.3233/JAD-2006-9S335. [DOI] [PubMed] [Google Scholar]
- 36.Kusakawa GI, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga SI. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–72. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 37.Patrick GN, Zukerberg L, Nikolic M, de La Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–22. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 38.Chow HM, Guo D, Zhou JC, Zhang GY, Li HF, Herrup K, et al. CDK5 activator protein p25 preferentially binds and activates GSK3β. Proc Natl Acad Sci U S A. 2014;111:E4887–95. doi: 10.1073/pnas.1402627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang TY, Wu CF, Yang XW, Liu YY, Yang HL, Yuan LL, et al. Pseudoginsenoside-F11 protects against transient cerebral ischemia injury in rats involving repressing calcium overload. Neuroscience. 2019;411:86–104. doi: 10.1016/j.neuroscience.2019.05.030. [DOI] [PubMed] [Google Scholar]
- 40.Laurent C, Buée L, Blum D. Tau and neuroinflammation: what impact for Alzheimer’s disease and tauopathies? Biomed J. 2018;41:21–33. doi: 10.1016/j.bj.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–9. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin SJ, Jeong YO, Jeon SG, Kim S, Lee SK, Nam Y, et al. Jowiseungchungtang inhibits amyloid-beta aggregation and amyloid-beta-mediated pathology in 5XFAD mice. Int J Mol Sci. 2018;19:4026. doi: 10.3390/ijms19124026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grizzell JA, Patel S, Barreto GE, Echeverria V. Cotinine improves visual recognition memory and decreases cortical Tau phosphorylation in the Tg6799 mice. Prog Neuropsychopharmacol Biol Psychiatry. 2017;78:75–81. doi: 10.1016/j.pnpbp.2017.05.010. [DOI] [PubMed] [Google Scholar]