

Abstract
Oxidative distress and mitochondrial dysfunction, are key factors involved in the pathophysiology of Parkinson's disease (PD). The pleiotropic hormone insulin-like growth factor II (IGF-II) has shown neuroprotective and antioxidant effects in some neurodegenerative diseases. In this work, we demonstrate the protective effect of IGF-II against the damage induced by 1-methyl-4-phenylpyridinium (MPP+) in neuronal dopaminergic cell cultures and a mouse model of progressive PD. In the neuronal model, IGF-II counteracts the oxidative distress produced by MPP + protecting dopaminergic neurons. Improved mitochondrial function, increased nuclear factor (erythroid-derived 2)-like2 (NRF2) nuclear translocation along with NRF2-dependent upregulation of antioxidative enzymes, and modulation of mammalian target of rapamycin (mTOR) signalling pathway were identified as mechanisms leading to neuroprotection and the survival of dopaminergic cells. The neuroprotective effect of IGF-II against MPP + -neurotoxicity on dopaminergic neurons depends on the specific IGF-II receptor (IGF-IIr). In the mouse model, IGF-II prevents behavioural dysfunction and dopaminergic nigrostriatal pathway degeneration and mitigates neuroinflammation induced by MPP+. Our work demonstrates that hampering oxidative stress and normalising mitochondrial function through the interaction of IGF-II with its specific IGF-IIr are neuroprotective in both neuronal and mouse models. Thus, the modulation of the IGF-II/IGF-IIr signalling pathway may be a useful therapeutic approach for the prevention and treatment of PD.
Keywords: Insulin like growth factor-II, Neuroprotection, Mitochondria, Oxidative distress, Parkinson's disease
Graphical abstract
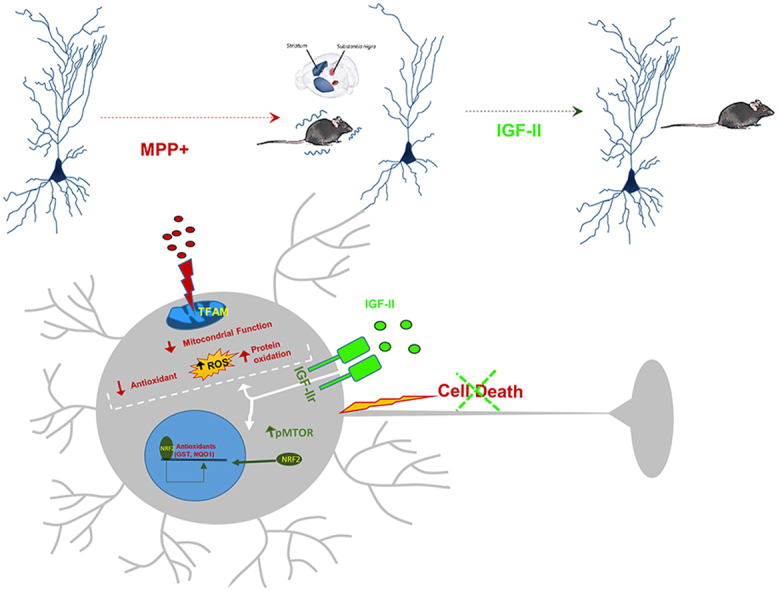
Highlights
-
•
IGF-II hampers oxidative damage and promotes survival in a cellular model of PD.
-
•
IGF-II avoids mitochondrial damage in dopaminergic cells in a model of PD.
-
•
IGF-II receptor mediates the neuroprotective effect of IGF-II in a cellular model of PD.
-
•
IGF-II prevents nigrostriatal degeneration and inflammation in a mice model of PD.
-
•
IGF-II prevents behavioural dysfunction in a mice model of PD.
1. Introduction
Parkinson's disease (PD) is a neurodegenerative disease affecting 2–3% of the population aged > 65 years. The pathogenesis of this disease is characterised by the involvement of multiple pathways and mechanisms, such as mitochondrial disfunction, oxidative stress, and neuroinflammation [1], that ultimately produce a loss of dopaminergic neurons from the substantia nigra [2,3]. Bradykinesia and other motor disorders are key for diagnosis, although the disease comes with other non-motor symptoms, such as cognitive impairment, sleep disorders, and depression, that increase disabilities [1]. To establish neuroprotective strategies, it is crucial to identify new targets leading to the creation of new therapeutic approaches in slowing progression and/or improving symptomatology [4].
Insulin-like growth factor II (IGF-II) is a pleiotropic hormone widely distributed in the Central Nervous System (CNS), where it exerts its functions through interactions with IGF-I receptors (IGF-Ir), insulin receptors (RIns) and its specific receptor, the IGFII/Mannose 6-Phosphate receptor (IGF-IIr). The genes that encode these receptors are highly conserved in vertebrates. In the nervous system of adults, we find a high expression of these receptors in the choroid plexuses, in the meninges and in the vascular network, as well as in the different nuclei of the hippocampus, cerebral cortex, cerebellum, and other areas of the midbrain [5] including the substantia nigra [6]. However, this growth factor is still poorly studied in adults. IGF-II has numerous functions throughout cellular homeostasis. Recently, it has been proposed that the effects of IGF-II, working independently or synergistically with IGF-I, may be relevant not only for energy homeostasis, growth, and development, but also for learning and memory, modulating neurotransmitter release, adult hippocampal neurogenesis, and synaptic plasticity [7,8]. In addition, new evidence is emerging that IGF-II is a key neuroprotection factor in pathological conditions. In this context, the study of IGF-II as a neuroprotection factor in neurodegenerative diseases is of increasing interest [[9], [10], [11]]. IGF-II has been shown to have neuroprotective and antioxidant actions in aging conditions [12], glucocorticoid-mediated stress situations [13,14], neurodegenerative pathologies such as Alzheimer's disease [15], and neuropsychiatric disorders such as schizophrenia [16,17] and autism [18].
The study of the mechanisms underlying this neuroprotective function of IGF-II is still in the early stages of research. A key aspect is the function of the specific IGF-IIr, the type of cell where it is found, and the nuclei that show greater expression. Associated functions include the clearance or activation of extracellular ligands, lysosome formation, the segregation and transport of lysosomal enzymes, and the activation of specific transduction signals, although the latter are not well known [[9], [10], [11]]. By activating these mediators (interaction with heteromeric G-proteins, movement of Ca2+, phosphorylation of proteins through PKC, and/or activation of glycogen synthase) and via mammalian/mechanistic target of rapamycin (mTOR), IGF-II participates in fundamental nerve functions such as regulation of proliferation/apoptosis, adult neurogenesis, release of neurotransmitters, and other synaptic factors related to memory consolidation [8,9,[18], [19], [20]].
To date, only a few studies have been published, five of which were by our research team, where the neuroprotection mechanisms of IGF-II were shown to be related to antioxidant function [10,[12], [13], [14],21,22], supporting that IGF-II could be involved in neuroprotective actions on aging conditions [12], glucocorticoid-mediated stress situations [13,14], neurodegenerative pathologies [15], and neuropsychiatric disorders [[16], [17], [18]]. In addition, PD is characterised by oxidative-mitochondrial damage, which is a major mechanism that explains the degeneration of these dopaminergic cells [2,3]. In addition, genetic association studies have shown the protective role of IGF-II in PD [23]; there is a wide distribution of IGF-IIr in different brain locations related with these pathologies [5,24].
Based on the above, we decided to study the neuroprotective effects of IGF-II against oxidative damage induced by 1-methyl-4-phenylpyridinium (MPP+) in dopaminergic neuron cultures and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mice, along with the mechanisms involved in these effects. Although pharmacological models of PD, such as 6-hydroxydopamine and/or MPTP, have been questioned by its poor ability to identify successful disease modifying therapies, they continue being a valuable tool to the study of PD [25].
2. Material and methods
2.1. Study design
In this study, we assessed the antioxidant and neuroprotective effects of treatment with IGF-II in neuronal cell cultures and the involvement of its specific IGF-IIr in this effect. We also studied the effect of treatment with IGF-II in a mouse model of progressive PD. Animals, plates, culture dishes, and slides were randomised by an investigator, and the analysis was performed by different researchers. The minimum sample size for animal experiments was 10 mice per group, based on previous experience and pilot studies. In cellular experiments, a minimum of 6 experiments in triplicate for each situation were performed. Further experimental details and technical protocols of cellular and mouse models are provided in the Supplementary Materials.
2.2. Cell culture
To develop this study, we used the SN4741 cell line of dopaminergic neurons of the substantia nigra [26] (provided by Prof. Ernest Arenas, Karolinska Institute, Stockholm). These cells express IGF-IIr as demonstrated by the immunocytochemistry of the receptor shown in Fig. S1.
These cells were incubated with the toxic MPP+ (400 μM) (Cas nº 36913-39-0, SIGMA), IGF-II (25 ng/mL) (provided by Lilly Laboratories [Madrid, Spain]), and BMS-536924 (1 μM) (Cas nº 468740-43-4, TOCRIS), which inhibit the tyrosine kinase effect of the IGF-Ir and RIns. To reverse the effects of IGF-II mediated by its own receptor, as there are no specific antagonists, the antibody AF-292 (20 ng/mL) (Cat nº: AF-292-NA, R&D System) against the IGF-IIr was used [13,27,28]. To check the involvement of IGF-IIr on the main neuroprotective effects of IGF-II, some experiments were performed using [Leu 27]-IGF-II (LEU) (GroPep, Adelaide, SA, Australia) (10 nM), a specific IGF-IIr agonist that show for the IGF-IIr more than 100-fold higher affinity than for the IGF-I or RIns, and so, at the concentration used in this study we assume that only the IGF-IIr is stimulated. If the IGF-II effects seen in this work were mediated by its interaction with the specific IGF-IIr, then would be reproducible in presence of other agonists such as LEU [29,30].
The treatments were carried out in a modified Locke's solution (NaCl 137 mM, CaCl2 5 mM, KCl 10 mM, glucose 25 mM, Hepes 10 mM, pH: 7.4) supplemented with penicillin-streptomycin and l-glutamine.
The protective effects of IGF-II at the end time (6 h) (Fig. 1A) and the mechanisms involved with redox homeostasis, as well as the neuronal function at short time (2.5 h) (Fig. 2, Fig. 3, Fig. 4, A), were studied following the scheme shown in the figures. Experiment was performed in n = 6 each group (3 independent experiment). The redox parameters studied are presented in the scheme shown in Fig. S2.
Fig. 1.

IGF-II protects SN4741 dopaminergic cells against MPP+-induced toxicity in cell cultures. (A) Experimental design for the study of the neuroprotective effects of IGF-II on neuronal survival and redox homeostasis. The measures were taken in SN4741 cells after 6 h of incubation with MPP+, in the presence or absence of IGF-II and/or BMS and AB. (B) Giemsa staining. (C) Cytotoxicity, measured by quantifying LDH release and expressed as % of control; BMS and AB are used to define the receptor involved in the IGF-II effect. (D) Lipid oxidative damage evaluated as LOOH; AB is used to define the receptor involved in IGF-II effect. (E) Protein oxidation evaluated as AOPP; AB is used to define the receptor involved in the IGF-II effect. (F) Neurodegeneration evaluated as FJ fluorescence intensity; BMS is used to define the receptor involved in the IGF-II effect. Data are expressed as mean ± SEM. n = 6 each group (3 independent experiment). #P < 0.05 versus CO and IGF-II groups; *P < 0.05, versus groups connected in bars. & P < 0.05, versus all other groups. Data were analysed by one-way ANOVA followed by Tukey's multiple comparison test.
Fig. 2.

IGF-II protects mitochondrial function and integrity in SN4741 dopaminergic cells against MPP+-induced toxicity in cell cultures. (A) Experimental design for the study of the neuroprotective effects of IGF-II on mitochondrial integrity, function, and redox homeostasis. The measures were taken in SN4741 cells after 2.5 h of incubation with MPP+, in the presence or absence of IGF-II and/or BMS and AB. (B) OCR time course after treatments expressed as % of the control. (C) Electronic microscopy. (D) Mitochondrial ROS production evaluated as MitoSOX fluorescence. (E) Mitochondrial mΔΨ evaluated as JC1 fluorescence aggregates; BMS is used to define the receptor involved in the IGF-II effect. (F) Mitochondrial COX activity; AB is used to define the receptor involved in the IGF-II effect. (G) Mitochondrial SOD activity; AB is used to define the receptor involved in the IGF-II effect. (H) Representative immunocytochemistry of DAPI, TFAM, and MTR mitochondrial stain. (I) Quantification of TFAM immunofluorescence; AB is used to define the receptor involved in the IGF-II effect. Data are expressed as mean ± SEM. n = 6 each group (3 independent experiment). #P < 0.05 versus CO and IGF-II groups; *P < 0.05, versus groups connected in bars; & P < 0.05, versus all other groups. Data were analysed by one-way ANOVA followed by Tukey's multiple comparison test.
Fig. 3.

IGF-II modulation of NRF2 and mTOR intracellular signalling pathways in SN4741 cells after MPP+-induced toxicity in cell cultures. (A) Experimental design for the study of the neuroprotective effects of IGF-II on NRF2 and mTOR intracellular signalling pathways. The measures were made in SN4741 cells after 2.5 h of incubation with MPP+, in the presence or absence of IGF-II and/or BMS and AB. (B) Representative immunocytochemistry stain for DAPI and NRF2 (C) Quantification of NRF2 immunofluorescence; BMS is used to define the receptor involved in the IGF-II effect (D) NRF2 target gene GST activity; AB is used to define the receptor involved in the IGF-II effect. (E) NRF2 target gene NQO1 activity; AB is used to define the receptor involved in the IGF-II effect. (F) Representative immunocytochemistry stain for DAPI and pmTOR (G) Quantification of pmTOR immunofluorescence; AB is used to define the receptor involved in the IGF-II effect. Data are expressed as mean ± SEM. n = 6 each group (3 independent experiment). #P < 0.05 versus CO and IGF-II groups; *P < 0.05, versus groups connected in bars; & P < 0.05, versus all other groups. Data were analysed by one-way ANOVA followed by Tukey's multiple comparison test.
Fig. 4.

IGF-II protects SN4741 dopaminergic cells against MPP+-induced toxicity. (A) Experimental design for the study of the effects of IGF-II on dopaminergic markers. The measures were taken in SN4741 cells after 2.5 h of incubation with MPP+, in the presence or absence of IGF-II and/or BMS and AB. (B) Representative Immunocytochemistry stain for DAPI and TH. (C) Quantification of TH immunofluorescence; BMS is used to define the receptor involved in the IGF-II effect. (D) Representative immunocytochemistry stain for DAPI and VMAT2. (E) Quantification of VMAT2 immunofluorescence; AB is used to define the receptor involved in the IGF-II effect. (F) DAT activity; AB is used to define the receptor involved in the IGF-II effect. Data are expressed as mean ± SEM. n = 6 each group (3 independent experiment). #P < 0.05, versus CO and IGF-II groups; *P < 0.05, versus groups connected in bars; & P < 0.05, versus all other groups. Data were analysed by one-way ANOVA followed by Tukey's multiple comparison test.
2.3. Viability, morphology, oxidative damage, and neurodegeneration
Cell death was measured as the percentage of LDH released to the medium compared to control cells. In morphology studies, cells were fixed in 100% methanol and stained with Giemsa [31]. Cells were examined for nuclear, cytoplasmic, and cell membrane changes. Oxidative damage was evaluated by quantifying LOOH and AOPP using spectrophotometric methods [14,31]. Neurodegeneration was measured using Fluoro-Jade B™ dye according to a previously published procedure [14].
2.4. Mitochondrial studies
Mitochondrial superoxide radical production was analysed by flow cytometry using a dihydroetidine derivate probe [32]. Mitochondrial function was measured as variation in OCR by Seahorse technology, mΔΨ by flow cytometry using JC1 probe, and COX activity by spectrophotometric methods. The activity of mitochondrial SOD was quantified by spectrophotometric methods. Mitochondrial morphology was studied by electron microscopy [31,33]. To identify and localise TFAM expression, a double immunocytochemical labelling experiment was performed using antibodies against TFAM following standard procedures and the mitochondrial stain MTR and was assessed by immunocytochemistry and confocal microscopy.
2.5. Intracellular signalling pathways
NRF2 and phosphorylated mTOR were analysed by immunocytochemistry and confocal microscopy. The activities of antioxidant enzymes GST and NQO1 were also quantified by spectrophotometric methods [31].
2.6. Dopamine markers
TH and VMAT-2 were analysed by immunocytochemistry and confocal microscopy. DAT activity was assessed using a fluorescence-based assay [34].
2.7. MPTP/p mouse model and pharmacological treatments
Ten-week-old male C57BL/6J mice (Janvier, Le Genest-St-Isle, France) were housed in groups of 3/4 in standard laboratory cages provided with nesting material and ad libitum access to water and food. Mice were maintained under a 12 h light/dark cycle (lights on at 8:00 a.m.) in a room with controlled temperature (22±2 °C), humidity, and ventilation. All procedures were approved by the research ethics committee of the University of Málaga (CEUMA no. 10/06/2019/104). To induce progressive damage in the dopaminergic nigrostriatal pathway, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in combination with the clearance inhibitor probenecid (MPTP/p) was administered over 5 weeks [35]. Probenecid was used because decreases MPTP renal elimination and helps to maintain high MPTP concentration in the brain [36]. Mice submitted to the MPTP/p treatment (n = 28) received intraperitoneal (ip) injections of probenecid (250 mg/kg, diluted initially in 1 M NaOH and immediately buffered in PBS at pH 7.4) followed by subcutaneous (sc) MPTP (24 mg/kg, diluted in 0.9% NaCl) twice a week at 84 h intervals. Control groups (CO; n = 16) received injections of ip vehicle (NaOH/PBS pH 7.4) followed by sc saline on an identical administration regime. MPTP/p-treated and control conditions were co-administered with either sc IGF-II (7.5 μg/kg, diluted in phosphate-buffered saline, PBS 0.1 M) or vehicle twice a day at 12 h intervals, from day 1–44 (MPTP/p + Veh, n = 8; MPTP/p+(44d) IGF-II, n = 11; CO + Veh, n = 8; CO + IGF-II, n = 8). Finally, a different group of mice was treated with IGF-II once the MPTP-induced insult has been triggered (MPTP/p+(23d) IGF-II, n = 9). The MPTP/p+(23d) IGF-II group received vehicle (PBS 0.1 M) from day 1–21, after which the IGF-II co-treatment began (days 22–44). The experimental design is depicted in Fig. 5A.
Fig. 5.

Chronic IGF-II administration protects against behavioural deficits and dopaminergic degeneration in the MPTP/p mouse model of progressive PD. (A) Experimental design. (B) Motor deficits. (C) Self-grooming deficits. (D) TH-producing dopaminergic cells in SNc. The average coefficient of error (CE) of stereological counting is shown for each group. (E) TH expression in the striatum (Str). (F) DAT expression in the striatum. (G) GFAP+ cells in SNc. (H) GFAP+ cells in Str. (I) Representative immunostaining in every experimental condition. When applicable, black arrows indicate examples of positive immunolabeling. Data are expressed as mean ± SEM. *P < 0.05, difference between groups; #P < 0.05, versus control groups; & P < 0.05, versus all other groups; Ø P < 0.05, versus basal. Data were analysed by one-way ANOVA (immunohistochemical markers) or two-way ANOVA with repeated measures (rota-rod test and self-grooming) followed by Duncan's multiple range test.
2.8. Behavioural testing
To investigate the recovery of the behavioural dysfunction induced by MPTP/p after IGF-II treatments, two different behaviours were assessed: motor coordination in the rotarod and self-grooming in the open field. Both behaviours were dependent on the integrity of the dopaminergic nigrostriatal pathway [37,38]. All mice were exposed to these two behavioural tasks under basal conditions and after the pharmacological treatments (Fig. 5A). A detailed description of the behavioural testing is available in the Supplementary Materials.
2.9. Immunohistochemistry for dopaminergic markers and astrogliosis
TH, DAT, and glial fibrillary acidic protein (GFAP) diaminobenzidine (DAB) immunostaining was performed on striatal and SN sections. Four to five sections per brain containing SN or striatum were used. Total number of TH+ cells in SNc was counted by unbiased stereology using the optical fractionator method and the newCAST software (Visiopharm, Hoersholm, Denmark) [39]. The optical density of TH+ innervation and DAT immunoreactivity in the striatum and the number of striatal and SNc GFAP-immunoreactive astrocytes were measured from the digitised images using the ImageJ software (https://imagej.nih.gov/ij/). Immunohistochemistry procedure and quantification details are provided in the Supplementary Materials.
2.10. Measurement of the striatal MPP + levels
C57BL/6J male mice were treated with sc IGF-II (15 μg/kg in PBS 0.1 M) (n = 7) or vehicle (n = 8). Twenty minutes later, all mice were injected with ip probenecid (250 mg/kg in NaOH) followed by sc MPTP (30 mg/kg, diluted in 0.9% NaCl). Two hours after the MPTP dose, the mice were euthanised, the brains were removed, and the striatum was dissected, weighted, and stored at −80 °C until analysis. Striatal MPP+ content was measured by liquid chromatography-mass spectrometry (LC-MS) as described previously [40]. Detailed procedure is provided in the Supplementary Materials.
2.11. Statistical analysis
Comparisons of multiple groups were performed using one or two-way Analysis of Variance (ANOVA) followed by post hoc test (Tukey's post hoc test or Duncan's multiple range test). Comparison between two groups was performed using Student's unpaired t-test. Statistics were conducted using Statistica 8 (StatSoft Power Solutions Inc., Tulsa, OK), IBM SPSS 20 (IBM, Armonk, NY), and Prism 8 (GraphPad Software, San Diego, CA) softwares. Differences with P < 0.05 were considered statistically significant. Data are plotted as mean ± SEM. Detailed information of the test used, including the specific post hoc test and P values, is stated in the figure legends.
3. Results
3.1. Protective role of IGF-II in a cellular model of PD
Neurodegeneration and cellular death are triggered by many factors; the major factors involved and related with oxidative distress are shown in Fig. S2. In this work, we have studied the major factors, which are grouped as follows:
3.1.1. Cellular damage: viability, morphology, oxidative damage, and neurodegeneration (Fig. 1A)
To study cell viability, we resorted to the analysis of cytosolic lactate dehydrogenase (LDH) released after incubation with MPP+ (Fig. 1C). In these experiments, we found an increase of 3.5 times in LDH levels compared to the control; coincubation in the presence of IGF-II prevents the release of LDH induced by MPP+ (P < 0.05), with values close to that of the control. Incubation in the presence of BMS, an inhibitor of RIns and IGF-Ir, does not modify the effect of IGF-II; the addition to the incubation media of the antibody against IGF-IIr abolishes the protective effect of IGF-II (P < 0.05). Control cells in the presence of IGF-II do not show any significant change compared to the control. In the study of morphology, the Giemsa image experiments showed clear changes in morphology in cells exposed to MPP+, where we found great heterogeneity in cell size and shape, with shrunken condensed pyknotic nuclei. When IGF-II was included in the treatment media, cells recovered a morphology similar to that of control cells. This effect on cell morphology was reverted in the presence of the antibody against IGF-IIr (Fig. 1B).
In order to assess the oxidative damage, lipid hydroperoxide (LOOH) and advanced oxidation protein products (AOPP) were studied in these cells. LOOH and AOPP levels were increased in cells treated with MPP+ (27% and 32%, respectively) compared to the controls (P < 0.05). The inclusion of IGF-II in the incubation media restores LOOH and AOPP levels to values close to the control cells; the presence of the antibody against IGF-IIr in the incubation media reverted these effects (Fig. 1D and E).
In the analysis of neurodegeneration assessed by Fluoro-Jade B™ (FJ) experiments, we found an increase in fluorescence in neurons treated with MPP+ (100%) compared to controls (P < 0.05). Inclusion of IGF-II in the incubation media restored values to those found in control cells. The presence of BMS in the incubation media did not modify the effect of IGF-II (Fig. 1F).
3.1.2. Mitochondrial study (Fig. 2A)
In order to evaluate mitochondrial function, oxygen consumption rate (OCR), free radical production, mitochondrial membrane potential (mΔΨ), and cytochrome c oxidase (COX) were studied in neuronal cell cultures. In the study of OCR, MPP+ produces a decrease (70%) after 2 h of incubation compared to the control (P < 0.05) (Fig. 2B). The addition of IGF-II in the incubation media reduces the decrease in OCR over time (by 50%).
Furthermore, incubation of cells in the presence of MPP+ induces an increase in free radicals (Fig. 2D), leading to a significant decrease compared to the control, in mΔΨ (48% less than the control) and COX activity (68% less than control) (P < 0.05) (Fig. 2E and F). The addition of IGF-II in the incubation media protects mitochondria against MPP+ induced free radicals and increases mΔΨ to values close to the control level (Fig. 2D and E); the blockade of RIns and IGF-Ir with BMS did not modify the IGF-II effect. Moreover, COX activity recovered values close to the control when IGF-II was included in the incubation media and this effect was blocked by the specific IGF-IIr antibody (Fig. 2F).
In the study of mitochondrial SOD, we found a great increase in SOD production (almost 150%) in cells treated with MPP+ compared to the control (P < 0.05); this increase was counteracted by the inclusion of IGF-II in the incubation media, with values close to those of control cells (Fig. 2G). The incorporation of antibody against IGF-IIr in the incubation media partially avoided the protective effect of IGF-II.
In morphological studies at the electron microscopy level (Fig. 2C), we could see some differences between control and MPP+ treated cells. Many mitochondria were characterised by swelling, partial or total loss of cristae, and electron-lucent matrix. In general, we observed that MPP+ treatment induced a mitochondrial number reduction of 16% compared to control cells. When IGF-II was present in the incubation media, cells exhibited round or elliptical-shaped mitochondria with intact cristae and outer and inner membranes and no reduction in mitochondrial number.
In the study of mitochondrial transcription factor A (TFAM) (Fig. 2H and I), we found a decrease in the cells exposed to MPP+ (38% less) compared to the control (P < 0.05); this was reverted in the presence of IGF-II to values close to the control. Incubation in the presence of antibody against IGF-IIr abolishes the effect of IGF-II. In the images, we can see a decrease in the expression of TFAM in neuronal mitochondria (identified by immunocytochemical staining with MitoTracker Deep Red FM [MTR]) compared to the control cells (P < 0.05), which was reverted in the presence of IGF-II. The presence of antibodies against the IGF-IIr abolishes the effect of IGF-II.
3.1.3. Intracellular signalling pathways (Fig. 3A)
NRF2 is a transcription factor that binds to antioxidant response elements (ARE) in the nucleus, leading to transcription of ARE genes. In this work, we assessed NRF2 by immunocytochemistry (Fig. 3B and C); in our experiments, we could see a decrease in NRF2 expression in cells treated with MPP+ (26%) compared to the control (P < 0.05); supplementation with IGF-II in the incubation media restores NRF2 expression to levels close to those of control cells (Fig. 3C). Interestingly, when studying nuclear NRF2 translocation, the decrease in MPP+ treated cells was higher (Fig. 3B); in these experiments, the inclusion of IGF-II in the incubation media produced an increase in nuclear fluorescence compared to MPP+ treated cells, recovering values close to those found in control cells. The presence of BMS did not modify this behaviour. Translocation of NRF2 to the nucleus induces the expression of some redox protective enzymes, such as NADPH quinone dehydrogenase 1 (NQO1) and Glutathione S-transferase (GST). When we analysed the activity of NQO1 (Fig. 3E), we found a decrease of this enzyme (70%) compared to the control (P < 0.05). As mentioned, the inclusion of IGF-II in the incubation media restores NQO1 values to those of the control cells. In the study of GST activity (Fig. 3D), although we do not see variations in cells treated with MPP+, the addition of IGF-II in the incubation media increases the activity compared to both MPP+ and the control cells (P < 0.05). It is important to note that IGF-II alone was able to increase the levels of GST compared to the control (P < 0.05). The inclusion of antibody against the IGF-IIr abolishes this effect of IGF-II.
We have also studied, in this work, the phosphorylated form of mTOR protein (pmTOR). Treatment of cells with MPP+ produced a decrease (25%) of pmTOR compared to control cells (P < 0.05); the addition of IGF-II to the incubation media restores pmTOR values to those found in control cells (Fig. 3F and G). In addition, the inclusion of antibody against IGF-IIr abolishes the IGF-II effect. Interestingly, incubation of cells with IGF-II in the absence of MPP+ increased the pmTOR values by 20% compared to the control (P < 0.05).
3.1.4. Dopaminergic markers (Fig. 4A)
The treatment of cells with MPP+ induced a decrease (90%) in tyrosine hydroxylase (TH) levels compared to the control (P < 0.05); the presence of IGF-II in the incubation media recovered the damage induced by MPP+ (70%) to values close to those of control cells. The inhibition of RIns and IGF-Ir by BMS in these experiments did not modify the TH expression (Fig. 4B and C).
In our cellular model, treatment with MPP+ produced a decrease (36%) in vesicular monoamine transporter 2 (VMAT2) expression compared to the control (P < 0.05), as assessed by immunocytochemistry; the addition of IGF-II in the incubation media increased the expression of VMAT2 close to control levels. The inclusion of antibody against IGF-IIr in the media abolished the IGF-II effect, as previously mentioned (Fig. 4D and E).
In the analysis of the dopamine transporter (DAT) by fluorometry, the treatment of cells with MPP+ totally abolished DAT activity; this is partially restored with the inclusion of IGF-II in the incubation media. The incorporation of antibody against IGF-IIr in the incubation media abolishes the IGF-II effect (Fig. 4F).
3.1.5. Protective role of activation of IGF-IIr by the selective agonist LEU on SN4741 dopaminergic cells against MPP + -induced toxicity (HYPERLINK \l "appsec1" \o "appsec1"Fig. 4 supplementary)
To assess the relevance of IGF-IIr in the main action of IGF-II we designed an independent study where SN4741 cells were incubated 2.5 h or 6h with MPP+, in the presence or absence of LEU an IGF-II analogue with high selective affinity for IGF-IIr. In these experiments, we found similar protective effects on the damage induced by MPP+, to those found after the stimulation with IGF-II, especially in mitochondrial ROS production, mΔΨ, the intracellular pathway NRF2, NQO1 enzyme and the dopamine marker TH after 2,5h; which results in and increase in survival and improves REDOX homeostasis after 6h. The incubation of control cell in presence of LEU did not modify ROS production (Fig. 5 supplementary).
3.2. Protective role of IGF-II in a mouse model of progressive PD
Having confirmed the neuroprotective efficacy of IGF-II in dopaminergic SN4741 cells against the toxic MPP+, we aimed to examine whether IGF-II was also neuroprotective in mammals using a chronic progressive model of PD in mice. In this study, we employed the MPTP/p model (MPTP combined with probenecid), which is characterised by behavioural impairment, loss of striatal dopamine, degeneration of TH+ dopaminergic neurons in the pars compacta of the substantia nigra (SNc), and neuroinflammation [35,41].
In this model, we first studied relevant behaviours such as motor performance in the rotarod and self-grooming in an open field, as well as the neurodegeneration of the dopaminergic nigrostriatal pathway induced by MPTP/p. Regarding behavioural testing, baseline measures were taken immediately before the initial dose of MPTP/p (or vehicle), and the behavioural effects of the treatment were tested 10 days after the last dose of the drug (Fig. 5A). At basal conditions, before starting any treatment, there are no differences in latency to fall (s) in rota-rood nor in self-grooming among the different groups of animals, whereas consistent with previous reports [35,41], mice undergoing chronic MPTP/p treatment showed impaired performance in the rotarod (time spent on the rotating rod) and a dramatic reduction in the total self-grooming time (P < 0.05; Fig. 5B and C).
As expected, MPTP/p-treated mice also exhibited a severe loss of dopaminergic neurons in the SNc (~67% less TH+ nigral neurons) and terminals in the striatum (>80% reduced TH and DAT expression) (P < 0.05; Fig. 5B and C), together with a significant increase in the number of reactive astrocytes (GFAP+ cells) in both regions (P < 0.05; Fig. 5B and C).
We then investigated whether IGF-II was able to prevent the behavioural deficits and the nigrostriatal pathway degeneration induced by MPTP/p. For this purpose, a group of mice received two doses of IGF-II daily for 44 days, covering the whole MPTP/p administration regime and extending approximately a week further (Fig. 5A). We found that IGF-II completely prevented the development of behavioural impairments (P < 0.05; Fig. 5B and C) and the loss of TH+ neurons in the SNc (P < 0.05; Fig. 5D), also promoting a significant recovery of the TH and DAT immunoreactivity in the striatum (P < 0.05; Fig. 5E and F). Moreover, IGF-II treatment was able to mitigate the reactive astrocytosis induced by MPTP/p in both SNc and the striatum (P < 0.05; Fig. 5G and H).
We then studied whether IGF–II–treatment could halt the neurodegeneration of the dopaminergic nigrostriatal pathway and its behavioural consequences once the insult has already been triggered. To do this, we treated a separate cohort of mice with IGF-II, starting 3 weeks after the first dose of MPTP/p. Remarkably, IGF-II mitigated the loss of dopaminergic neurons in the SNc (P < 0.05; Fig. 5D), as well as the reduced expression of TH and DAT in the dopaminergic terminals of the striatum (P < 0.05; Fig. 5E and F) and the overexpression of GFAP+ astrocytes in the SNc and striatum (P < 0.05; Fig. 5G and H). Together, these results suggest that IGF-II may counteract the progression of the nigrostriatal pathway degeneration induced by MPTP/p. However, behavioural studies have demonstrated that, when the progression of the degeneration is halted at this stage with the administration of IGF-II, no behavioural impairments are present in either of the two tasks (P < 0.05; Fig. 5B and C), showing that the neurodegeneration induced by MPTP/p is not sufficient to be reflected at the behavioural level.
Importantly, no side effects were found after chronic injections of IGF-II in any of the behavioural or immunohistochemistry parameters studied (time on rotating rod: P > 0.05, Fig. 5B; self-grooming time: P > 0.05, Fig. 5C; TH+ neurons in SNc: P > 0.05, Fig. 5D; TH and DAT immunoreactivity in the striatum: P > 0.05, Fig. 5E and F; SNc and striatal GFAP+ astrocytes: P > 0.05, Fig. 5G and H).
To rule out the possibility that the neuroprotection induced by IGF-II was related to the inhibition of the conversion of MPTP to its neurotoxic metabolite MPP+, we designed an independent experiment where striatal MPP+ levels were determined 2 h after the administration of MPTP/p (250 mg/kg/p; 30 mg/kg/MPTP) with or without pre-treatment with IGF-II (15 μg/kg). As expected, IGF-II pre-treatment did not alter striatal MPP+ concentrations, showing that IGF-II did not interfere with MPTP metabolism by the astrocytes (P > 0.05; Fig. S3).
4. Discussion
In PD and other neurodegenerative diseases, an increase in oxidative distress [42] and mitochondrial damage [3,43] that may contribute to increase neurodegeneration and/or cell death has been observed. The mechanism of cell death remains unclear, probably because instead of a clear apoptotic process, we faced a mixture of different processes recently denominated regulated cell death [44], including apoptosis, ferroptosis, and necrosis, as described recently in pathologies characterised by cell death and inflammation, such as in some neurodegenerative diseases including PD [45]. Interestingly, oxidative stress may also be increased by other processes linked to degeneration, making it difficult to distinguish whether oxidative distress triggers degeneration or is a consequence of it [46].
Neuronal cells are rich in lipids, which are the target of ROS, producing structural damage in membranes and leading to neuronal damage and death. In addition, LOOH and AOPP may participate in redox reactions that could increase the cellular damage [[47], [48], [49]]. The results in the cellular model indicate that this protective effect is mediated by a decrease in oxidative damage, with concomitant decrease in LOOH and AOPP, protecting dopaminergic neurons from cell death, which was assessed as LDH release (Fig. 1D and E, C). Changes in LOOH and AOPP levels agree with our previous results in a cellular model of oxidative stress induced by corticosterone [14] and with those by Dong et al. in a cellular model of PD [22], where IGF-II acting on IGF-IIr restored oxidative balance. However, it cannot be excluded that IGF-II could induce an increase in the activity of the endosomal–lysosomal system (via the IGF-IIr) to degrade abnormal intracellular proteins induced by ROS [10] and/or eliminated/neutralised by detoxifying molecules such as GST, commented below, and other phase II enzymes [50,51].
Considering that oxidative distress is a common mechanism contributing to neurodegeneration, the effect of IGF-II reported in this work would be responsible of its beneficial effect and consequent decrease in neurodegeneration (Fig. 1F).
PD and other neurodegenerative processes are strongly related with the balance in ROS production/scavenging processes [42,49], with the mitochondria being one of the major sources of energy and ROS production [3,52,53]. In our PD model, as in other mitochondrial toxic situations, such as synucleinopathies [54], we found an increase in mitochondrial superoxide production that induced an increase in mitochondrial SOD [33,55], thereby increasing H2O2 levels, which in turn can inactivate some enzymes, and after interaction with Fe2+ [56], can induce lipid oxidation. The oxidation in the membranes of the mitochondria increases ROS production, which induces more lipid damage and neurodegeneration; furthermore, COX, the main regulatory enzyme of the mitochondrial respiratory chain, also gets affected, with impairment in neuronal energy metabolism, decrease in OCR and mΔΨ, and further increase in ROS production, inducing neuronal damage and/or death [57].
The protective effect of IGF-II found in our PD model and mediated through its specific IGF-IIr could be related to a decrease in mitochondrial ROS production that prevents lipid and protein damage, which would result in the recovery of mitochondrial mΔΨ and OCR and a consequent decrease in neurodegeneration; this is consistent with our previous results and those of other authors [12,14,22,58,59]. The beneficial effect of IGF-II on mΔΨ and OCR would restore energy resources contributing to maintain neuronal integrity and function, making neurons more resistant to neurodegeneration [60].
Subsequently, we investigated the putative mechanisms by which IGF-IIr activation protects against mitochondrial oxidative damage induced by MPP+. This effect could be mediated by the increase in expression of mitochondrial transcription factors related to neurodegenerative diseases, such as TFAM, a key regulator of mtDNA abundance. TFAM protects against diseases with oxidative stress and mitochondrial disfunction [53] and has been found to be decreased in PD and other neurodegenerative diseases [61]. TFAM improves Complex I and IV activity in neurons and mitochondrial respiratory chain function, with a decrease in ROS production, increasing mtDNA and mitochondrial biogenesis [62,63]. In our PD model, we found an increase in TFAM after treatment with IGF-II; this could be a good approach to either prevent or slow the progression of PD.
One of the emerging target factors in PD and other neurodegenerative diseases, related with oxidative balance, is NRF2, which induces the expression of an array of antioxidant response element-dependent genes to protect neurons from oxidative damage [[64], [65], [66]]. We have observed that the treatment of neurons with MPP+ decreases the NRF2 translocation to the nucleus compared to the control; this effect could be attributed to a decrease in NRF2 expression as suggested by Meng and Zhao in a model of mouse PD [67,68], or to an alteration in NRF2 metabolism as suggested by Li [69] and/or to a decrease in translocation by itself induced by the toxic as suggested by Li and Kasai [70,71]; in any case, IGF-II through the IGF-IIr, recovers that decrease.
Furthermore, NRF2 induces the expression of antioxidant enzymes [64], which is in agreement with our results discussed below; moreover, activation of the NRF2 pathway improves mitochondrial bioenergetics, function [72], and cell metabolism, as discussed above. NRF2 may also be responsible for the increase in TFAM levels described in this model, consistent with previous works [73,74]. We also examined two final products of these antioxidant response dependent genes, GST and NQO1.
GST is a detoxifying phase II enzyme that plays an essential role as an antioxidant in maintaining the redox balance in neurons, catalysing the reduction of ROS by glutathione and rendering these molecules more soluble to be eliminated by the neurons [64,75]. On the contrary, in PD, over-expression of GST isoenzyme 1 in dopaminergic neurons decreased neurodegeneration, suggesting a protective effect of this enzyme [76,77]. Although no great variations in GST activity were seen after treatment with MPP+ by other authors [77], we observed an increase in GST in control neurons after treatment with IGF-II. This increase was even higher when neurons were previously exposed to MPP+; however, we do not have a clear explanation for this finding. The IGF-II effect was mediated by the interaction with its specific IGF-IIr.
NQO1 is also a detoxifying phase II enzyme that catalyses the reduction of quinone species and that can be produced during the metabolism of dopamine to the less toxic hydroquinone, suggesting that it may be a protective mechanism for oxidative damage in PD [26,78,79]. We found a decrease in NQO1 activity after treatment with MPP+, which is consistent with the findings of other authors [[79], [80], [81]]. This effect was avoided in the presence of IGF-II, thereby recovering activities close to those found in control neurons; as mentioned previously, this effect was mediated by interaction with its specific IGF-IIr.
mTOR is an important regulator of neuron metabolism, mitochondrial homeostasis, protein synthesis, and cell death [82]. Alteration in the mTOR pathway has been found in some neurodegenerative diseases such as PD, Alzheimer's disease, Huntington's disease, or amyotrophic lateral sclerosis [[83], [84], [85], [86]]. In this work, incubation of neurons with MPP+ decreased phospho-mTOR1 (pmTOR) levels, which is similar to the findings of other studies [83,87]; addition of IGF-II to the incubation media prevented the fall in pmTOR levels. This was mediated through its specific IGF-IIr. This increase in pmTOR could be due to an increase in NRF2 levels as demonstrated by others [88,89] as well as our findings. Moreover, it has been described the stimulation of pmTOR by neurotransmitters, such as dopamine [86,90], and as commented below, we have found an increase in TH, the limiting enzyme in dopamine synthesis, that could promote the increase in pmTOR. Interestingly, when we assessed the effect of IGF-II on control cells, we found an increase in pmTOR that could be related with the known metabolic and neurotrophic factor effect of IGF-II [12,91].
Decrease in dopamine synthesis is one of the hallmarks in PD [45], with TH being the limiting enzyme in dopamine synthesis; an increase in TH immunoreactivity after treatment with IGF-II has been reported [92]. The treatment of cells with MPP+ (Fig. 4B and C) induces a decrease in the level of this enzyme compared to the control; the presence of IGF-II in the incubation media not only restores but also increases TH expression above that of the control cells. Interestingly, a similar increase was seen in the control cells in the presence of IGF-II, which is in agreement with the results of the study by Pai et al., who found that increased IGF-II may induce an increase in TH and dopamine synthesis [93]. This effect must be mediated by IGF-II interaction with its specific IGF-IIr.
Once synthesised, dopamine is stored in vesicles by VMAT2 to avoid its autooxidation and deleterious effect. Treatment with MPP+ produces a decrease in VMAT2 expression and the inclusion of IGF-II in the incubation media recovers the expression of VMAT2 close to control levels, probably due to the increase in its synthesis at the trans-Golgy network, this effect is attributable to the interaction of IGF-II with its specific IGF-IIr. The increase in VMAT2 would decrease the level of cytosolic dopamine, avoiding its oxidation and ROS production [75].
Cytosolic dopamine levels are not only regulated by VMAT2 but also highly influenced by DAT, and a correct balance between VMAT2 and DAT is essential in PD for both neurotransmission and cell survival [94]. The treatment of cells with MPP+ totally abolished DAT activity (Fig. 4F), which was partially restored with the inclusion of IGF-II in the incubation media; this effect is mediated by the interaction with IGF-IIr. The mechanism of IGF-II effect on DAT could be related with the decrease in PKC activity induced by IGF-II, as previously described [14], which induces a downregulation of DAT [95]. IGF-II did not interfere with MPP+ neuronal uptake through DAT, excluding the neuroprotective role of IGF-II, which could be explained by this mechanism.
The neuroprotective properties of the IGF-II in dopaminergic neurons were also demonstrated in a mouse model of progressive PD induced by the chronic administration of MPTP/p. This model of PD involves the degeneration of the nigrostriatal dopaminergic neurons by the conversion of the MPTP into the neurotoxic metabolite MPP+ in the astrocytes, which is uptake by dopaminergic neurons and rise ROS production and induce mitochondrial dysfunction [42]; also MPTP increases synuclein (A-syn) expression in dopaminergic neurons which in turn can further enhance ROS production and impair mitochondria, leading to dopaminergic neurodegeneration and neuronal death [44,96,97].
Even when we have not studied the mechanisms of action of IGF-II in vivo, our cellular results strongly support the capacity of the IGF-II to prevent the oxidative damage and mitochondrial dysfunction induced by MPP+. In this regard, we demonstrated that IGF-II did not interfere either with the glial formation of MPP+ (Fig. S3) or with its neuronal uptake through DAT (Fig. 4F), ruling out that the neuroprotective role of the IGF-II in the nigrostriatal pathway could be explained through these mechanisms. In addition to the ability of the IGF-II to counteract the oxidative damage in the dopaminergic neurons, it may exert neuroprotective/anti-parkinsonian actions in vivo, involving other complementary mechanisms such as neuroinflammation, autophagy, and protein trafficking of damaged molecules [10,59,98] that cannot be ruled out in our study. Regarding this, our data showed an evident neuroinflammation in both the striatum and SNc in MPTP/p-treated mice (reactive astrocytes overexpression; Fig. 5G and H), and IGF-II administration was able to reduce this inflammatory response. Thus, it has also been demonstrated that IGF-II can attenuate and even normalise neuroinflammation in pathological conditions [10,99], thereby contributing to the neuroprotective effects observed in IGF–II–treated mice. Although the study of A-syn in the animal model was out of the scope of this work, we cannot rule out the involvement of A-syn in the neuroprotective effect of IGF-II on this model, as it has been reported that impairment of IGF-I and IGF-II signalling tend to increase A-syn accumulation and increase oxidative stress [100].
Due to the lack of selective pharmacological tools, the study in the animal model was limited in that we were unable to examine the involvement of the IGF-IIr in the neuroprotection of the dopaminergic pathway mediated by IGF-II; even though, based on the results obtained in the cellular experiments, including those with the IGF-IIr specific agonist LEU shown in supplementary, we would expect that most, if not all, of these effects would be linked to the activation of the IGF-IIr.
In summary, in both models of MPP+/MPTP induced PD used in this study, we have reported an increase in neurodegeneration and cell death, with impaired neuronal function as a consequence of increase in oxidative distress and mitochondrial dysfunction. Interaction of IGF-II with its specific IGF-IIr showed neuroprotective actions by promoting NRF-2 and mTOR antioxidant pathways and restoring mitochondrial and neuronal function. The involvement of IGF-IIr in these effects was demonstrated using LEU, as this specific IGF-IIr agonist was able to preproduce the IGF-II actions.
5. Conclusions
In conclusion, we demonstrated the potential neuroprotective role of IGF-II in a cellular and a mouse model of PD. In both models, we confirmed the ability of IGF-II to protect dopaminergic neurons against the oxidative damage induced by the neurotoxin MPP+. Considering the beneficial effects of IGF-II reported both in cellular and animal models; and the role of the IGF-IIr demonstrated in the cellular experiments, we propose that the modulation of the IGF-II/IGF-IIr signalling pathway be considered a promising pharmacological target for the treatment of PD.
Declaration of competing interest
The authors have declared that no conflict of interest exists.
Acknowledgments
We wish to thank Ernest Arenas for providing SN4741 cells and Silvia Claros and Vanessa de Luque for technical assistance.
This research was supported by the following projects: M.G-F.& L.J.S. Proyectos I+D+I-Programa Operativo FEDER Andalucía 2014–2020 (UMA18-FEDERJA- 004) Junta de Andalucía that also partially supported N. Valverde; CTS507 and CTS156 from Consejería de Economía Innovación Ciencia y Empresa, Junta de Andalucía; and Fondo Social Europeo (EU) supported partially N. Valverde. L.J.S.: Ministerio de Economía y Competitividad. Gobierno de España. (MINECO, Agencia Estatal de Investigación cofinanciado por FEDER -UE. (PSI2017-82604R).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102095.
Contributor Information
Jose Pavia, Email: pavia@uma.es.
Maria Garcia-Fernandez, Email: igf@uma.es.
Author contributions
M.G-F, E.M-M., J.P. and L.J.S. designed the study. M.G-F, E. M-M., N.V., E. L., C.M., performed cellular experiment. F.B. performed electronic microscopy. D.LdG-M., E, L., Y.S.R-Z., F.B., F. A-G., A.M. P–C performed animal experiment. J.L. L-G. P.G-G, performed MPP+ measure. M.G-F. and D.LdG-M performed analyses and figure preparation. L.J.S., J. P. and M.G-F. wrote the manuscript.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., Schrag A.-E., Lang A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 2.Blesa J., Trigo-Damas I., Quiroga-Varela A., Jackson-Lewis V.R. Oxidative stress and Parkinson's disease. Front. Neuroanat. 2015;9:91. doi: 10.3389/fnana.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schon E.A., Przedborski S. Mitochondria: the next (Neurode)Generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obeso J.A., Rodriguez-Oroz M.C., Goetz C.G., Marin C., Kordower J.H., Rodriguez M., Hirsch E.C., Farrer M., V Schapira A.H., Halliday G. Missing pieces in the Parkinson's disease puzzle. Nat. Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- 5.Hawkes C., Kar S. The insulin-like growth factor-II/mannose-6-phosphate receptor: structure, distribution and function in the central nervous system. Brain Res. Rev. 2004;44:117–140. doi: 10.1016/j.brainresrev.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Hawkes C., Kar S. Insulin-like growth factor-II/mannose-6-phosphate receptor: widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J. Comp. Neurol. 2003;458:113–127. doi: 10.1002/cne.10578. [DOI] [PubMed] [Google Scholar]
- 7.Stern S.A., Kohtz A.S., Pollonini G., Alberini C.M. Enhancement of memories by systemic administration of insulin-like growth factor II. Neuropsychopharmacology. 2014;39:2179–2190. doi: 10.1038/npp.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegler A.N., Levison S.W., Wood T.L. Insulin and IGF receptor signalling in neural-stem-cell homeostasis. Nat. Rev. Endocrinol. 2015;11:161–170. doi: 10.1038/nrendo.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werner H., LeRoith D. Insulin and insulin-like growth factor receptors in the brain: physiological and pathological aspects. Eur. Neuropsychopharmacol. 2014;24:1947. doi: 10.1016/j.euroneuro.2014.01.020. –1953. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y., MacDonald R.G., Thinakaran G., Kar S. Insulin-like growth factor-II/cation-independent mannose 6-phosphate receptor in neurodegenerative diseases. Mol. Neurobiol. 2017;54:2636–2658. doi: 10.1007/s12035-016-9849-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beletskiy A., Chesnokova E., Bal N. Insulin-like growth factor 2 as a possible neuroprotective agent and memory enhancer—its comparative expression, processing and signaling in mammalian CNS. Int. J. Mol. Sci. 2021;22:1849. doi: 10.3390/ijms22041849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castilla-Cortázar I., García-Fernández M., Delgado G., Puche J.E., Sierra I., Barhoum R., González-Barón S. Hepatoprotection and neuroprotection induced by low doses of IGF-II in aging rats. J. Transl. Med. 2011;9:103. doi: 10.1186/1479-5876-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martín-Montañez E., Millon C., Boraldi F., Garcia-Guirado F., Pedraza C., Lara E., Santin L.J., Pavia J., Garcia-Fernandez M. IGF-II promotes neuroprotection and neuroplasticity recovery in a long-lasting model of oxidative damage induced by glucocorticoids. Redox Biol. 2017;13:69–81. doi: 10.1016/j.redox.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin-Montañez E., Pavia J., Santin L.J., Boraldi F., Estivill-Torrus G., Aguirre J.A., Garcia-Fernandez M. Involvement of IGF-II receptors in the antioxidant and neuroprotective effects of IGF-II on adult cortical neuronal cultures. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2014;1842:1041–1051. doi: 10.1016/J.BBADIS.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Pascual‐Lucas M., Viana da Silva S., Di Scala M., Garcia‐Barroso C., González‐Aseguinolaza G., Mulle C., Alberini C.M., Cuadrado‐Tejedor M., Garcia‐Osta A. Insulin‐like growth factor 2 reverses memory and synaptic deficits in <scp>APP</scp> transgenic mice. EMBO Mol. Med. 2014;6:1246–1262. doi: 10.15252/emmm.201404228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardo M., Cheng Y., Sitbon Y.H., Lowell J.A., Grieco S.F., Worthen R.J., Desse S., Barreda-Diaz A. Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review, Neurosci. Res. 2019;149:1–13. doi: 10.1016/j.neures.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Ouchi Y., Banno Y., Shimizu Y., Ando S., Hasegawa H., Adachi K., Iwamoto T. Reduced adult hippocampal neurogenesis and working memory deficits in the dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can Be rescued by IGF2. J. Neurosci. 2013;33:9408–9419. doi: 10.1523/JNEUROSCI.2700-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinmetz A.B., Stern S.A., Kohtz A.S., Descalzi G., Alberini C.M. Insulin-like growth factor II targets the mTOR pathway to reverse autism-like phenotypes in mice. J. Neurosci. 2018;38:1015–1029. doi: 10.1523/JNEUROSCI.2010-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkes C., Amritraj A., MacDonald R.G., Jhamandas J.H., Kar S. Heterotrimeric G proteins and the single-transmembrane domain IGF-II/M6P receptor: functional interaction and relevance to cell signaling. Mol. Neurobiol. 2007;35:329–345. doi: 10.1007/s12035-007-0021-2. [DOI] [PubMed] [Google Scholar]
- 20.Benarroch E.E. Insulin-like growth factors in the brain and their potential clinical implications. Neurology. 2012;79:2148–2153. doi: 10.1212/WNL.0b013e3182752eef. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Fernandez M., Sierra I., Puche J.E., Guerra L., Castilla-Cortazar I. Liver mitochondrial dysfunction is reverted by insulin-like growth factor II (IGF-II) in aging rats. J. Transl. Med. 2011;9:123. doi: 10.1186/1479-5876-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong W., Hu L., Xu X. Neuroprotective effect of insulin-like growth factor-II on 1- methyl-4-phenyl pyridinium-induced oxidative damage in cortical neuronal cells. Trop. J. Pharmaceut. Res. 2015;14:1191. doi: 10.4314/tjpr.v14i7.10. [DOI] [Google Scholar]
- 23.Sutherland G., Mellick G., Newman J., Double K.L., Stevens J., Lee L., Rowe D., Silburn P., Halliday G.M. Haplotype analysis of the IGF2-INS-TH gene cluster in Parkinson's disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008;147B:495–499. doi: 10.1002/ajmg.b.30633. [DOI] [PubMed] [Google Scholar]
- 24.Wilczak N., De Bleser P., Luiten P., Geerts A., Teelken A., De Keyser J. Insulin-like growth factor II receptors in human brain and their absence in astrogliotic plaques in multiple sclerosis. Brain Res. 2000;863:282–288. doi: 10.1016/S0006-8993(00)02153-3. [DOI] [PubMed] [Google Scholar]
- 25.Dawson T.M., Golde T.E., Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018;21:1370–1379. doi: 10.1038/s41593-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Son J.H., Chun H.S., Joh T.H., Cho S., Conti B., Lee J.W. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. 1999;19:10–20. doi: 10.1523/JNEUROSCI.19-01-00010.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen D.Y., Stern S.A., Garcia-Osta A., Saunier-Rebori B., Pollonini G., Bambah-Mukku D., Blitzer R.D., Alberini C.M. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin-Montañez E., Pavia J., Santin L.J., Boraldi F., Estivill-Torrus G., Aguirre J.A., Garcia-Fernandez M., Martin-Montanez E., Pavia J., Santin L.J., Boraldi F., Estivill-Torrus G., Aguirre J.A., Garcia-Fernandez M. Involvement of IGF-II receptors in the antioxidant and neuroprotective effects of IGF-II on adult cortical neuronal cultures. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2014;1842:1041–1051. doi: 10.1016/j.bbadis.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Beukers M.W., Oh Y., Zhang H., Ling N., Rosenfeld R.G. [Leu 27 ] INSULIN-LIKE growth factor II IS highly selective for the TYPE-II igf receptor IN binding, CROSS-LINKING and thymidine incorporation experiments. Endocrinology. 1991;128:1201–1203. doi: 10.1210/endo-128-2-1201. [DOI] [PubMed] [Google Scholar]
- 30.Amritraj A., Posse de Chaves E.I., Hawkes C., MacDonald R.G., Kar S. Single-transmembrane domain IGF-II/M6P receptor: potential interaction with G protein and its association with cholesterol-rich membrane domains. Endocrinology. 2012;153:4784–4798. doi: 10.1210/en.2012-1139. [DOI] [PubMed] [Google Scholar]
- 31.Martín-Montañez E., Pavia J., Valverde N., Boraldi F., Lara E., Oliver B., Hurtado-Guerrero I., Fernandez O., Garcia-Fernandez M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019;137:116–130. doi: 10.1016/j.freeradbiomed.2019.04.022. [DOI] [PubMed] [Google Scholar]
- 32.Kauffman M.E., Kauffman M.K., Traore K., Zhu H., Trush M.A., Jia Z., Li Y.R. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React. Oxyg. Species (Apex, N.C.). 2016;2:361–370. doi: 10.20455/ros.2016.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasquali-Ronchetti I., Garcia-Fernandez M.I., Boraldi F., Quaglino D., Gheduzzi D., De Vincenzi Paolinelli C., Tiozzo R., Bergamini S., Ceccarelli D., Muscatello U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: possible role in the pathogenesis of clinical manifestations. J. Pathol. 2006;208:54–61. doi: 10.1002/path.1867. [DOI] [PubMed] [Google Scholar]
- 34.Jørgensen S., Nielsen E.Ø., Peters D., Dyhring T. Validation of a fluorescence-based high-throughput assay for the measurement of neurotransmitter transporter uptake activity. J. Neurosci. Methods. 2008;169:168–176. doi: 10.1016/j.jneumeth.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Carta A.R., Carboni E., Spiga S. Methods Mol. Biol. Humana Press Inc.; 2013. The MPTP/probenecid model of progressive Parkinson's disease; pp. 295–308. [DOI] [PubMed] [Google Scholar]
- 36.Petroske E., Meredith G.E., Callen S., Totterdell S., Lau Y.S. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 2001;106:589–601. doi: 10.1016/S0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 37.Shiotsuki H., Yoshimi K., Shimo Y., Funayama M., Takamatsu Y., Ikeda K., Takahashi R., Kitazawa S., Hattori N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods. 2010;189:180–185. doi: 10.1016/j.jneumeth.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 38.Kalueff A.V., Stewart A.M., Song C., Berridge K.C., Graybiel A.M., Fentress J.C. Neurobiology of rodent self-grooming and its value for translational neuroscience. Nat. Rev. Neurosci. 2016;17:45–59. doi: 10.1038/nrn.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ladrón de Guevara‐Miranda D., Moreno‐Fernández R.D., Gil‐Rodríguez S., Rosell‐Valle C., Estivill‐Torrús G., Serrano A., Pavón F.J., Rodríguez de Fonseca F., Santín L.J., Castilla‐Ortega E. Lysophosphatidic acid‐induced increase in adult hippocampal neurogenesis facilitates the forgetting of cocaine‐contextual memory, Addict. Biol. 2019;24:458–470. doi: 10.1111/adb.12612. [DOI] [PubMed] [Google Scholar]
- 40.Muñoz-Manchado A.B., Villadiego J., Suárez-Luna N., Bermejo-Navas A., Garrido-Gil P., Labandeira-García J.L., Echevarría M., López-Barneo J., Toledo-Aral J.J. Neuroprotective and reparative effects of carotid body grafts in a chronic MPTP model of Parkinson's disease. Neurobiol. Aging. 2013;34:902–915. doi: 10.1016/j.neurobiolaging.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Petroske E., Meredith G.E., Callen S., Totterdell S., Lau Y.S. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 2001;106:589–601. doi: 10.1016/s0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 42.Dauer W., Przedborski S. Parkinson's disease. Neuron. 2003;39:889–909. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 43.Pantiya P., Thonusin C., Chattipakorn N., Chattipakorn S.C. Mitochondrial abnormalities in neurodegenerative models and possible interventions: focus on Alzheimer's disease, Parkinson's disease, Huntington's disease. Mitochondrion. 2020;55:14–47. doi: 10.1016/j.mito.2020.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W., Annicchiarico-Petruzzelli M., V Antonov A., Arama E., Baehrecke E.H., Barlev N.A., Bazan N.G., Bernassola F., Bertrand M.J.M., Bianchi K., V Blagosklonny M., Blomgren K., Borner C., Boya P., Brenner C., Campanella M., Candi E., Carmona-Gutierrez D., Cecconi F., Chan F.K.-M., Chandel N.S., Cheng E.H., Chipuk J.E., Cidlowski J.A., Ciechanover A., Cohen G.M., Conrad M., Cubillos-Ruiz J.R., Czabotar P.E., D'Angiolella V., Dawson T.M., Dawson V.L., De Laurenzi V., De Maria R., Debatin K.-M., DeBerardinis R.J., Deshmukh M., Di Daniele N., Di Virgilio F., Dixit V.M., Dixon S.J., Duckett C.S., Dynlacht B.D., El-Deiry W.S., Elrod J.W., Fimia G.M., Fulda S., García-Sáez A.J., Garg A.D., Garrido C., Gavathiotis E., Golstein P., Gottlieb E., Green D.R., Greene L.A., Gronemeyer H., Gross A., Hajnoczky G., Hardwick J.M., Harris I.S., Hengartner M.O., Hetz C., Ichijo H., Jäättelä M., Joseph B., Jost P.J., Juin P.P., Kaiser W.J., Karin M., Kaufmann T., Kepp O., Kimchi A., Kitsis R.N., Klionsky D.J., Knight R.A., Kumar S., Lee S.W., Lemasters J.J., Levine B., Linkermann A., Lipton S.A., Lockshin R.A., López-Otín C., Lowe S.W., Luedde T., Lugli E., MacFarlane M., Madeo F., Malewicz M., Malorni W., Manic G., Marine J.-C., Martin S.J., Martinou J.-C., Medema J.P., Mehlen P., Meier P., Melino S., Miao E.A., Molkentin J.D., Moll U.M., Muñoz-Pinedo C., Nagata S., Nuñez G., Oberst A., Oren M., Overholtzer M., Pagano M., Panaretakis T., Pasparakis M., Penninger J.M., Pereira D.M., Pervaiz S., Peter M.E., Piacentini M., Pinton P., Prehn J.H.M., Puthalakath H., Rabinovich G.A., Rehm M., Rizzuto R., Rodrigues C.M.P., Rubinsztein D.C., Rudel T., Ryan K.M., Sayan E., Scorrano L., Shao F., Shi Y., Silke J., Simon H.-U., Sistigu A., Stockwell B.R., Strasser A., Szabadkai G., Tait S.W.G., Tang D., Tavernarakis N., Thorburn A., Tsujimoto Y., Turk B., Vanden Berghe T., Vandenabeele P., Vander Heiden M.G., Villunger A., Virgin H.W., Vousden K.H., Vucic D., Wagner E.F., Walczak H., Wallach D., Wang Y., Wells J.A., Wood W., Yuan J., Zakeri Z., Zhivotovsky B., Zitvogel L., Melino G., Kroemer G. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Przedborski S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017;18:251–259. doi: 10.1038/nrn.2017.25. [DOI] [PubMed] [Google Scholar]
- 46.Jenner P. Oxidative stress in Parkinson's disease. Ann. Neurol. 2003;53 doi: 10.1002/ana.10483. S26–S38. [DOI] [PubMed] [Google Scholar]
- 47.Guo J., Zhao X., Li Y., Li G., Liu X. Damage to dopaminergic neurons by oxidative stress in Parkinson's disease (Review) Int. J. Mol. Med. 2018;41:1817–1825. doi: 10.3892/ijmm.2018.3406. [DOI] [PubMed] [Google Scholar]
- 48.Dexter D.T., Holley A.E., Flitter W.D., Slater T.F., Wells F.R., Daniel S.E., Lees A.J., Jenner P., Marsden C.D. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov. Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- 49.Puspita L., Chung S.Y., Shim J. Oxidative stress and cellular pathologies in Parkinson's disease. Mol. Brain. 2017;10:53. doi: 10.1186/s13041-017-0340-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shang X., Chen Y., Wang N., Niu W., Guo J. Oxidation-induced generation of a mild electrophile for proximity-enhanced protein–protein crosslinking. Chem. Commun. 2018;54:4172–4175. doi: 10.1039/C8CC01639A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tew K.D., Townsend D.M. Glutathione-S-Transferases as determinants of cell survival and death. Antioxidants Redox Signal. 2012;17:1728–1737. doi: 10.1089/ars.2012.4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Area-Gomez E., Guardia-Laguarta C., Schon E.A., Przedborski S. Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J. Clin. Invest. 2019;129:34–45. doi: 10.1172/JCI120848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grünewald A., Kumar K.R., Sue C.M. New insights into the complex role of mitochondria in Parkinson's disease. Prog. Neurobiol. 2019;177:73–93. doi: 10.1016/j.pneurobio.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 54.Ganjam G.K., Bolte K., Matschke L.A., Neitemeier S., Dolga A.M., Höllerhage M., Höglinger G.U., Adamczyk A., Decher N., Oertel W.H., Culmsee C. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019;10:1–16. doi: 10.1038/s41419-019-2091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gil A., Martín-Montañez E., Valverde N., Lara E., Boraldi F., Claros S., Romero-Zerbo S.-Y., Fernández O., Pavia J., Garcia-Fernandez M. Neuronal metabolism and neuroprotection: neuroprotective effect of fingolimod on menadione-induced mitochondrial damage. Cells. 2020;10:34. doi: 10.3390/cells10010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raha S., Robinson B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000;25:502–508. doi: 10.1016/S0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 57.Wallace D.C., Fan W., Procaccio V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du L., Lin L., Li Q., Liu K., Huang Y., Wang X., Cao K., Chen X., Cao W., Li F., Shao C., Wang Y., Shi Y. IGF-2 preprograms maturing macrophages to acquire oxidative phosphorylation-dependent anti-inflammatory properties. Cell Metabol. 2019;29:1363–1375. doi: 10.1016/j.cmet.2019.01.006. e8. [DOI] [PubMed] [Google Scholar]
- 59.Wang X., Lin L., Lan B., Wang Y., Du L., Chen X., Li Q., Liu K., Hu M., Xue Y., Roberts A.I., Shao C., Melino G., Shi Y., Wang Y. IGF2R-initiated proton rechanneling dictates an anti-inflammatory property in macrophages. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abb7389. eabb7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muddapu V.R., Dharshini S.A.P., Chakravarthy V.S., Gromiha M.M. Neurodegenerative diseases – is metabolic deficiency the root cause? Front. Neurosci. 2020;14:213. doi: 10.3389/fnins.2020.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang I., Chu C.T., Kaufman B.A. The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 2018;592:793–811. doi: 10.1002/1873-3468.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morimoto N., Miyazaki K., Kurata T., Ikeda Y., Matsuura T., Kang D., Ide T., Abe K. Effect of mitochondrial transcription factor a overexpression on motor neurons in amyotrophic lateral sclerosis model mice. J. Neurosci. Res. 2012;90:1200–1208. doi: 10.1002/jnr.23000. [DOI] [PubMed] [Google Scholar]
- 63.Piao Y., Kim H.G., Oh M.S., Pak Y.K. Overexpression of TFAM, NRF-1 and myr-AKT protects the MPP+-induced mitochondrial dysfunctions in neuronal cells. Biochim. Biophys. Acta Gen. Subj. 2012;1820:577–585. doi: 10.1016/j.bbagen.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 64.Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Esteras N., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: a focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016;397:383–400. doi: 10.1515/hsz-2015-0295. [DOI] [PubMed] [Google Scholar]
- 66.Brandes M.S., Gray N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro. 2020;12 doi: 10.1177/1759091419899782. 175909141989978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao M., Wang B., Zhang C., Su Z., Guo B., Zhao Y., Zheng R. The DJ1-Nrf2-STING axis mediates the neuroprotective effects of Withaferin A in Parkinson's disease. Cell Death Differ. 2021;28:2517–2535. doi: 10.1038/s41418-021-00767-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meng F., Wang J., Ding F., Xie Y., Zhang Y., Zhu J. Neuroprotective effect of matrine on MPTP-induced Parkinson's disease and on Nrf2 expression. Oncol. Lett. 2017;13:296–300. doi: 10.3892/OL.2016.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Q L., C N., X Z., M D. Gastrodin and isorhynchophylline synergistically inhibit MPP +-Induced oxidative stress in SH-SY5Y cells by targeting ERK1/2 and GSK-3β pathways: involvement of Nrf2 nuclear translocation. ACS Chem. Neurosci. 2018;9:482–493. doi: 10.1021/ACSCHEMNEURO.7B00247. [DOI] [PubMed] [Google Scholar]
- 70.Li X., Zhang J., Zhang X., Dong M. Puerarin suppresses MPP+/MPTP-induced oxidative stress through an Nrf2-dependent mechanism. Food Chem. Toxicol. 2020;144:111644. doi: 10.1016/J.FCT.2020.111644. [DOI] [PubMed] [Google Scholar]
- 71.Kasai S., Shimizu S., Tatara Y., Mimura J., Itoh K. Regulation of nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules. 2020;10 doi: 10.3390/BIOM10020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dinkova-Kostova A.T., Abramov A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015;88:179–188. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tufekci K.U., Civi Bayin E., Genc S., Genc K. The nrf2/ARE pathway: a promising target to counteract mitochondrial dysfunction in Parkinson's disease. Parkinsons. Dis. 2011:1–14. doi: 10.4061/2011/314082. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fu M.-H., Wu C.-W., Lee Y.-C., Hung C.-Y., Chen I.-C., Wu K.L.H. Nrf2 activation attenuates the early suppression of mitochondrial respiration due to the α-synuclein overexpression. Biomed. J. 2018;41:169–183. doi: 10.1016/j.bj.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smeyne M., Smeyne R.J. Glutathione metabolism and Parkinson's disease. Free Radic. Biol. Med. 2013;62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pohl F., Teixeira-Castro A., Costa M.D., Lindsay V., Fiúza-Fernandes J., Goua M., Bermano G., Russell W., Maciel P., Kong Thoo Lin P. GST-4-Dependent suppression of neurodegeneration in C. elegans models of Parkinson's and machado-joseph disease by rapeseed pomace extract supplementation. Front. Neurosci. 2019;13:1091. doi: 10.3389/fnins.2019.01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smeyne M., Boyd J., Shepherd K.R., Jiao Y., Pond B.B., Hatler M., Wolf R., Henderson C., Smeyne R.J. GSTπ expression mediates dopaminergic neuron sensitivity in experimental parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 2007;104 doi: 10.1073/pnas.0610978104. 1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo S., Kang S.S., Wang Z.-H., Liu X., Day J.X., Wu Z., Peng J., Xiang D., Springer W., Ye K. Akt phosphorylates NQO1 and triggers its degradation, abolishing its antioxidative activities in Parkinson's disease. J. Neurosci. 2019;39:7291–7305. doi: 10.1523/JNEUROSCI.0625-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao H., Lv F., Xu W., Zhang L., Jing P., Cao X. Deprenyl prevents MPP+-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology. 2011;290:286–294. doi: 10.1016/j.tox.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 80.Armagan G., Sevgili E., Gürkan F.T., Köse F.A., Bilgiç T., Dagcı T., Saso L. Regulation of the Nrf2 pathway by glycogen synthase kinase-3β in MPP+-Induced cell damage. Molecules. 2019;24:1377. doi: 10.3390/molecules24071377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Son H.J., Choi J.H., Lee J.A., Kim D.J., Shin K.J., Hwang O. Induction of NQO1 and neuroprotection by a novel compound KMS04014 in Parkinson's disease models. J. Mol. Neurosci. 2015;56:263–272. doi: 10.1007/s12031-015-0516-7. [DOI] [PubMed] [Google Scholar]
- 82.Li M., Zhao L., Liu J., Liu A., Jia C., Ma D., Jiang Y., Bai X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell. Signal. 2010;22:1469–1476. doi: 10.1016/j.cellsig.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 83.Zhu Z., Yang C., Iyaswamy A., Krishnamoorthi S., Sreenivasmurthy S.G., Liu J., Wang Z., Tong B.C.K., Song J., Lu J., Cheung K.H., Li M. Balancing mTOR signaling and autophagy in the treatment of Parkinson's disease. Int. J. Mol. Sci. 2019;20:728. doi: 10.3390/ijms20030728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu G.Y., Sabatini D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lan A.-P., Chen J., Zhao Y., Chai Z., Hu Y. mTOR signaling in Parkinson's disease. NeuroMolecular Med. 2017;19:1–10. doi: 10.1007/s12017-016-8417-7. [DOI] [PubMed] [Google Scholar]
- 86.Bockaert J., Marin P. mTOR in brain physiology and pathologies. Physiol. Rev. 2015;95:1157–1187. doi: 10.1152/physrev.00038.2014. [DOI] [PubMed] [Google Scholar]
- 87.Xu Y., Liu C., Chen S., Ye Y., Guo M., Ren Q., Liu L., Zhang H., Xu C., Zhou Q., Huang S., Chen L. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson's disease. Cell. Signal. 2014;26:1680–1689. doi: 10.1016/j.cellsig.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bendavit G., Aboulkassim T., Hilmi K., Shah S., Batist G. Nrf2 transcription factor can directly regulate mTOR. J. Biol. Chem. 2016;291:25476–25488. doi: 10.1074/jbc.M116.760249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Torrente Y., Bella P., Tripodi L., Villa C., Farini A. Role of insulin-like growth factor receptor 2 across muscle homeostasis: implications for treating muscular dystrophy. Cells. 2020;9:441. doi: 10.3390/cells9020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Switon K., Kotulska K., Janusz-Kaminska A., Zmorzynska J., Jaworski J. Molecular neurobiology of mTOR. Neuroscience. 2017;341:112–153. doi: 10.1016/j.neuroscience.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 91.Holly, Biernacka, Perks The neglected insulin: IGF-II, a metabolic regulator with implications for diabetes, obesity, and cancer. Cells. 2019;8:1207. doi: 10.3390/cells8101207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vazin T., Becker K.G., Chen J., Spivak C.E., Lupica C.R., Zhang Y., Worden L., Freed W.J. A novel combination of factors, termed SPIE, which promotes dopaminergic neuron differentiation from human embryonic stem cells. PloS One. 2009;4 doi: 10.1371/journal.pone.0006606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pai S., Li P., Killinger B., Marshall L., Jia P., Liao J., Petronis A., Szabó P.E., Labrie V. Differential methylation of enhancer at IGF2 is associated with abnormal dopamine synthesis in major psychosis. Nat. Commun. 2019;10:2046. doi: 10.1038/s41467-019-09786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lohr K.M., Masoud S.T., Salahpour A., Miller G.W. Membrane transporters as mediators of synaptic dopamine dynamics: implications for disease. Eur. J. Neurosci. 2017;45:20–33. doi: 10.1111/ejn.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.German C.L., Baladi M.G., McFadden L.M., Hanson G.R., Fleckenstein A.E. Regulation of the dopamine and vesicular monoamine transporters: pharmacological targets and implications for disease. Pharmacol. Rev. 2015;67:1005–1024. doi: 10.1124/pr.114.010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ganguly U., Banerjee A., Chakrabarti S.S., Kaur U., Sen O., Cappai R., Chakrabarti S. Interaction of α-synuclein and Parkin in iron toxicity on SH-SY5Y cells: implications in the pathogenesis of Parkinson's disease. Biochem. J. 2020;477:1109–1122. doi: 10.1042/BCJ20190676. [DOI] [PubMed] [Google Scholar]
- 97.Vila M., Vukosavic S., Jackson-Lewis V., Neystat M., Jakowec M., Przedborski S. α-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000;74:721–729. doi: 10.1046/j.1471-4159.2000.740721.x. [DOI] [PubMed] [Google Scholar]
- 98.García-Huerta P., Troncoso-Escudero P., Wu D., Thiruvalluvan A., Cisternas-Olmedo M., Henríquez D.R., Plate L., Chana-Cuevas P., Saquel C., Thielen P., Longo K.A., Geddes B.J., Lederkremer G.Z., Sharma N., Shenkman M., Naphade S., Sardi S.P., Spichiger C., Richter H.G., Court F.A., Tshilenge K.T., Ellerby L.M., Wiseman R.L., Gonzalez-Billault C., Bergink S., Vidal R.L., Hetz C. Insulin-like growth factor 2 (IGF2) protects against Huntington's disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 2020;140:737–764. doi: 10.1007/s00401-020-02183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vafaee F., Zarifkar A., Emamghoreishi M., Namavar M.R., Shirzad S., Ghazavi H., Mahdavizadeh V. Insulin-like growth factor 2 (IGF-2) regulates neuronal density and IGF-2 distribution following hippocampal intracerebral hemorrhage. J. Stroke Cerebrovasc. Dis. 2020;29:105128. doi: 10.1016/j.jstrokecerebrovasdis.2020.105128. [DOI] [PubMed] [Google Scholar]
- 100.Tong M., Dong M., de la Monte S.M. Brain insulin-like growth factor and neurotrophin resistance in Parkinson's disease and dementia with lewy bodies: potential role of manganese neurotoxicity. J. Alzheim. Dis. 2009;16:585–599. doi: 10.3233/JAD-2009-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.